

Abstract
EvoDesign (https://zhanglab.ccmb.med.umich.edu/EvoDesign) is an online server system for protein design. The method uses evolutionary profiles to guide the sequence search simulation and demonstrated significant advantages over physics-based approaches in terms of more accurately designing proteins that adopt desired target folds. Despite the success, the previous EvoDesign program focused only on monomer protein design, which limited its ability and usefulness in terms of designing functional proteins. In this work, we propose a new EvoDesign server, which extends the principles of evolution-based design to design protein-protein interactions. Starting from a two-chain complex structure, structurally similar interfaces are identified from known protein-protein interaction databases. An interface evolutionary profile is then constructed from a multiple sequence alignment of the interface analogies, which is combined with a newly developed, atomic-level physical energy function to guide the replicaexchange Monte Carlo simulation search. The purpose of the server is to redesign the specified complex chain to increase its stability and binding affinity for the other chain in the complex. With the improved scope and accuracy of the methodology, the new EvoDesign pipeline should become a useful online tool for functional protein design and drug discovery studies.
Introduction
Proteins are complex molecular machines that ubiquitously perform the cellular tasks necessary to sustain life. Nevertheless, despite the impressive role of natural proteins, only a tiny portion of the total possible amino acid sequences appear in nature. Computational protein design can be used to more thoroughly explore the sequence space in order to design artificial proteins with increased stability and/or enhanced functionality compared to their natural counterparts. Since many protein functions are mediated by protein-protein interactions (PPIs) [1, 2], an effective strategy to enhance the function of proteins is to redesign their interfaces to increase or alter the binding affinity and binding mode of PPIs [3]. This approach has been successfully applied to the redesign of various protein systems [4–8], and holds tremendous potential for the development of novel therapeutics, enzymes, and other useful proteins.
Most current protein design methods utilize physical energy functions to search for low free energy states in the sequence space. This approach is, however, often limited by the inability of physical energy functions to accurately recapitulate inter-atomic interactions or recognize correct folds, which has also been manifested in various protein folding and structure prediction studies [9, 10]. To partially address the inaccuracies of computational protein design using physics-based energy functions, we previously developed an evolution-based method, EvoDesign [11]. EvoDesign utilizes evolutionary profiles collected from analogous protein folds to help guide the sequence search simulation. Large-scale design and folding experiments demonstrated that the combination of evolutionary profiles with physical energy terms, where the latter is included mainly to accommodate the local atomic-level packing interactions, is more effective than purely physics-based methods in terms of designing proteins that adopt a desired target fold [12]. Despite the success, the previous version of EvoDesign focused solely on the design of monomeric proteins, and could not be used to design PPIs, which considerably limited its usefulness in terms of functional protein design.
In this work, we extend the use of evolutionary-profile guided design to the design of PPIs. For this purpose, a new strategy has been developed to extract PPI profiles from structurally analogous protein interfaces, which are then used to guide the interface design search [13]. Furthermore, the former EvoDesign pipeline utilized an external program, FoldX [14], to calculate the physical energy of a protein. Although it worked reasonably well, the procedure of calling an external program was prohibitively time-consuming. We developed a new physical energy function, EvoEF (EvoDesign Energy Function), which shows an improved ability to recognize inter-molecular binding interactions, while significantly speeding up the design process. Overall, the new EvoDesign server contains two design protocols: monomer fold design and dimer interface design, each with its own online interface.
It should be noted that the focus of the new dimer interface design protocol is on the redesign of one specific chain in the complex structure, termed the scaffold, so as to increase its stability and binding affinity towards the other chain in the complex, termed the binding partner. The sequence of the binding partner is unchanged during the simulation, although its side-chain conformations are allowed to move in order to accommodate the designed interface residues. This interface design protocol can be used for various applications that allow for a variable scaffold protein but call for a fixed binding partner. One such application is the design of protein therapeutics, where the therapeutic can be redesigned to increase its affinity for a fixed target in the body. The EvoDesign pipeline is fully automated and freely available at https://zhanglab.ccmb.med.umich.edu/EvoDesign. In addition to the online server, the source code for our newly developed physical energy function, EvoEF, can be downloaded at https://zhanglab.ccmb.med.umich.edu/EvoDesign/EvoEF.tar.gz.
Methods and Results
Overview of the EvoDesign Protocol
In order to incorporate functional protein design into EvoDesign, the evolution-based design method has been extended to the design of PPIs, where an overview of the new EvoDesign pipeline is depicted in Fig. 1. Starting from a two-chain complex structure of interest, its interface is structurally aligned to interfaces in the non-redundant interface library (NIL) [13] using iAlign [15]. A profile is then constructed from the interface multiple sequence alignment (iMSA), based on the structures that have a high similarity score (IS-score [15]) to the query complex interface. Finally, the evolution-based binding affinity change for each mutation at the interface is determined by the logarithm of the relative probability of each mutant amino acid compared to the wild type amino acid in the interface profile [13, 16]. This evolutionary energy term is combined with the physical energy score calculated by EvoEF to determine the total binding energy. Complementing the interface profile, a monomer structural profile is constructed from the multiple sequence alignment of monomer proteins that have a similar fold to the scaffold chain as identified by TM-align [17] from the PDB library. Overall, the information from both the monomer and interface profiles, as well as the physical energy function, are used as the composite energy function to guide the replica-exchange Monte Carlo (REMC) simulation in order to search for low free energy sequences.
Figure 1.
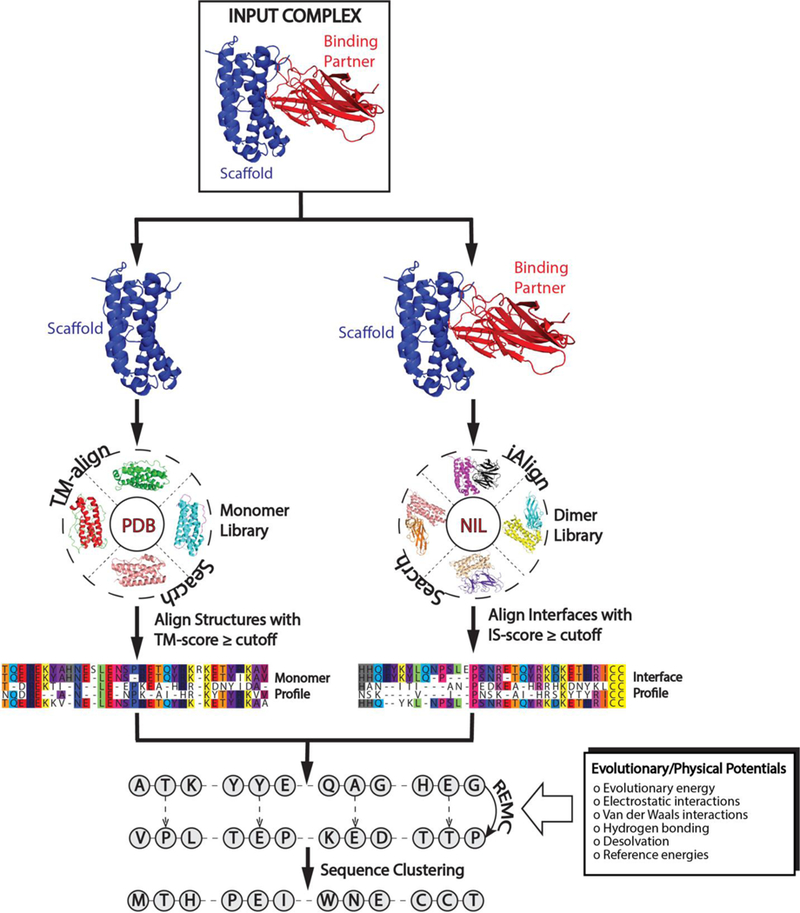
Flowchart of the EvoDesign pipeline for PPI design. Starting from a given protein complex, similar monomer and interface structures are identified from monomer and dimer structure libraries for the scaffold and protein complex, respectively. Alignments of the structural analogies are used to create evolutionary profiles. These profiles are used as energy terms in conjunction with a physical energy function, EvoEF, to guide the replica-exchange Monte Carlo simulation. After clustering the sequence decoys generated during the simulation, the final designs are selected from the lowest free energy sequences in the largest clusters.
Following the REMC simulation, the generated sequence decoys are clustered by SPICKER [18] based on the distance matrix defined by their BLOSUM62 sequence similarity. The final designs are selected from the lowest free energy sequences in the largest clusters. Here, it is important to note that EvoDesign provides an option for users to specify which chain in the complex is the scaffold and which chain is its binding partner. As stated previously, EvoDesign only focuses on the redesign of the scaffold, leaving the sequence of its binding partner unchanged, although the side-chain rotamer conformations of both chains are repacked during the design simulation.
Evolutionary Profile-Based Potentials
The evolutionary energy is composed of two terms: EevoMonomer and EevoInterface. The first term, EevoMonomer, is used to capture the information from the multiple sequence alignment (MSA) generated by TM-align based on the scaffold structure. The derivation of EevoMonomer was discussed previously [12]. For the web server description, we will focus on the new evolutionary interface potential. However, a full explanation of EevoMonomer is provided in Text S1 in the Supporting Information (SI).
The second term, EevoInterface, captures the information from the iMSA collected by the iAlign search:
| (1) |
where P(aaDes,i,i) and P(aaScaff,i, i) are the probabilities that the designed and scaffold amino acids, respectively, appear at position i in the interface. The probabilities are determined by the number of times that either the designed, Nobs(aaDes,i,i), or the wild-type scaffold, Nobs(aaScaff,i,i), amino acids appear at the ith position in the iMSA, where the corresponding position-specific pseudocounts, Npseudo(aaDes,Scaff,i), are used to help compensate for the small size of the interface library and takes into consideration gaps in the iMSA as well as amino acids that are related to the wild-type/mutant residues in the interface alignment. A full description of the pseudocount is contained in Text S2.
EvoEF Energy Terms
The energy function of EvoEF is designed to describe the atomic interactions in proteins and contains five terms:
| (2) |
The first term, Evdw(i,j), is the van der Waals energy, which is modified from the Lennard-Jones (LJ) 12–6 potential [19, 20]:
| (3) |
where
| (4) |
Here, dij is the distance between the two atoms i and j; σij = σi + σj is the sum of their van der Waals atomic radii and εij is the combined well-depth parameter for atoms i and j, which are all taken from the CHARMM19 force field [21]. The attractive and repulsive components of the van der Waals potential are split at dij = 0.89090σij. To increase the computational efficiency of EvoEF, we set a maximum distance cutoff of 6.0 Å and use a cubic function to make continuous transition of the LJ energy from its value at 5.0 Å to zero at the cutoff distance. For the repulsive component, the maximum energy cutoff is set to 5.0εij; this helps alleviate possible clashes, while not overly penalizing them due to the discrete rotameric conformations used in protein design. An example of the overall shape of the van der Waals energy between an amide N and a carbonyl C is shown in Fig. S1.
The second term in Eq. (2), Eelec(i,j), is used to determine the electrostatic interactions between partially charged atoms:
| (5) |
where qi and qj are the partial atomic charges, which are calculated using the PARSE method [22]. Furthermore, C0 = 332 Å kcal mol−1e−2, where e is the elementary charge, and ε(dij) is the distance-dependent dielectric constant, i.e., ε(dij) = 40dij. When computing the electrostatics term and dielectric constant, if the distance between two atoms, dij, is less than 0.8 σij, dij is set to 0.8 σij, to restrict the electrostatics energy to a reasonable, finite value. Again, for the sake of computational efficiency, a maximum distance cutoff is set to 6.0 Å, beyond which the value of the electrostatics term is zero.
The third term in Eq. (2), EHB(i,j), is used to calculate the hydrogen-bonding interactions. EHB(i,j) is a linear combination of three energy terms that depend on the hydrogen-acceptor distance , the angle between the donor atom, hydrogen and acceptor , and the angle between the hydrogen, acceptor and base atom :
| (6) |
where
| (7) |
The optimal distance between the hydrogen and its acceptor is set to 1.9 Å, which is taken from Kortemme et al. [23]. Additionally, dmin = 1.4 Å and dmax = 3.0 Å are the lower and upper bounds on the distance between the hydrogen-acceptor pair. The optimal value is set to either 150° or 135°, depending on the acceptor hybridization (sp2 or sp3) and the locations of the donor and acceptor atoms (from backbone-backbone, HBbb; sidechain-backbone, HBsb; or sidechain-sidechain, HBss).
The fourth term in Eq. (2), Esolv(i,j), describes the desolvation energy following the model introduced by Lazaridis and Karplus [24]:
| (8) |
where , and are the atom volumes, reference solvation energies, and correlation lengths, respectively, which are all taken from the Lazardis and Karplus paper [24]. The desolvation energy for both polar and nonpolar atoms is calculated using this method; however, the contribution from polar atoms is weighted differently from non-polar atoms. Specifically,
The last term in Eq. (2), Eref, is the reference energy of a protein sequence and is used to approximate the energy of the unfolded state ensemble:
| (9) |
where L is the length of the protein sequence, Er(aai) is an amino acid specific parameter to be optimized. The reference energy is used to choose sequences that have a large energy gap between the folded and unfolded states.
EvoEF Parameter Optimization and Benchmark Tests
EvoEF contains a total of 36 weights and 20 reference energies (see Tables S1 and S2 for detailed list). These parameters are decided by optimizing the energy function’s ability to predict protein stability and binding affinity change upon mutation. Since EvoEF’s energy calculation is split into three parts: the non-bonded atomic interactions within a residue (EintraResidue), those between different residues within the same chain (EinterResidueSameChain), and those between different residues from different chains (EinterResidueDiffChain) (see Eq. S6 in SI), the parameterization of EvoFF was performed in two steps. First, the reference energies and weighting factors for EintraResidue and EinterResidueSameChain were optimized by minimizing the difference between experimental and predicted values for mutation-induced protein monomer stability change The experimental data consisted of 3,989 non-redundant mutation samples from 210 monomeric proteins taken from the FoldX and STRUM datasets [25, 26]. Second, the 14 weights for EinterResidueDiffChain were determined using the mutation- induced protein-protein binding affinity change data which contained 2,204 non-redundant mutant samples from 177 dimeric complexes collected from the latest version of the SKEMPI database [27]. Each dataset was randomly split in half into training and test sets. A detailed description of the data construction and EvoEF optimization procedure is provided in Text S3, and the optimized parameters are listed in Tables S2 and S2. We note that the contributions from some terms (such as the electrostatics) are negligible following the parameter optimization, a phenomenon that was also observed by other studies in the ΔΔG based energy parameterizations [28].
The performance of EvoEF was evaluated using the above test datasets by calculating the Pearson correlation coefficients (PCCs) and root mean square errors (RMSEs) between the experimental and predicted and in control with FoldX version 4. The results showed that the PCC between and for EvoEF was 0.472 with an RMSE of 1.751 kcal/mol (Fig. 2A). As a comparison, FoldX obtained a PCC of 0.465 with an RMSE of 2.010 kcal/mol for the same dataset (Fig. 2B). Furthermore, the PCC between and for EvoEF was 0.514 with an RMSE of 2.109 kcal/mol (Fig. 2C), while the PCC for FoldX was 0.490 with an RMSE of 2.248 kcal/mol (Fig. 2D). The data shows that EvoEF slightly outperforms FoldX for both and prediction.
Figure 2.
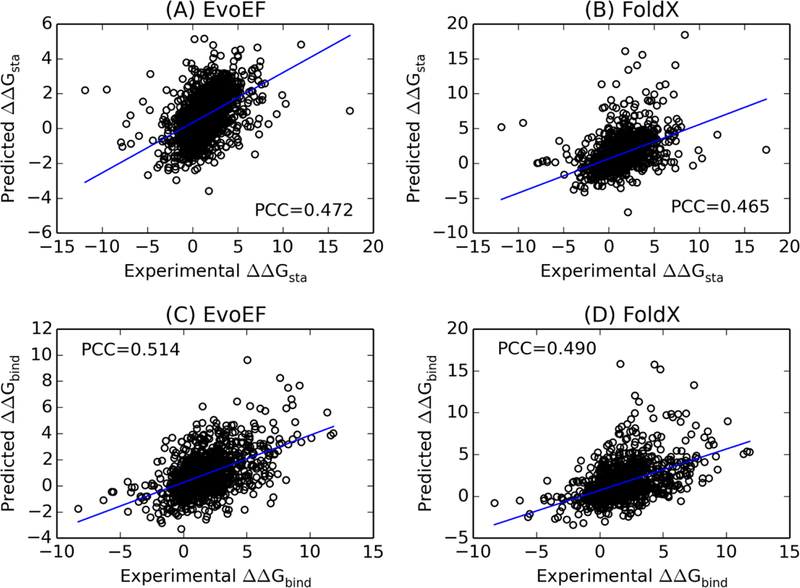
Correlation between predicted and experimental values for mutation-induced folding stability and binding affinity changes. (A, B) Folding stability changes upon mutation, for monomer proteins predicted by EvoEF (A) and FoldX (B) versus the experimental data for 1,994 test proteins. (C, D) Binding affinity changes upon mutation in the interface of protein-protein complexes, , predicted by EvoEF (C) and FoldX (D) versus the experimental data for 1,102 test proteins.
We also tested EvoEF’s ability to recognize the native structure from non-native decoys using the 3DRobot Decoy Set [29], which contains decoys from 200 non-homologous proteins. Among the 200 decoy sets, EvoEF is able to properly rank the native as the lowest energy in all the sets, while FoldX does so in 198 cases. In the second more stringent test, we calculated the energy gap between the near-native decoys (top 10% of decoys of the lowest RMSD) and the remainder of the decoys. The average Z-score (i.e., the energy gap normalized by the standard deviation) for EvoEF and FoldX is 1.959 and 1.844, respectively. If we define a successful case as that with a Z-score >1, EvoEF can successfully recognize the near-native structural decoys in 198 out of the 200 cases, while FoldX does so in 193 of the cases. These data suggest that EvoEF has a relatively better ability to distinguish nativelike monomer structures from other structural decoys (see Text S4 for a detailed description).
Furthermore, based on our tests on identical computational cores, EvoEF is about three times faster than FoldX at computing stability energy and approximately five times faster at computing protein-protein binding energy, indicating that using EvoEF can significantly increase the speed of our design simulations.
Replica-Exchange Monte Carlo Simulation for Sequence Space Search
Starting from a random sequence, REMC is used to search the sequence space, where random mutations are made on a set of randomly selected residues at each step, which are accepted or rejected based on the Metropolis criterion [30]. The composite energy function used to guide the REMC simulation is as follows:
| (10) |
where EevoMonomer and EevoInterface are the evolutionary energies from the monomer and interface profiles and EEvoEF is the physical energy calculated by EvoEF. For interface design, the weight parameters wevoInterface and wEvoEF are set to 3.0 and 2.0, respectively. These weights were selected in order to balance the average contribution from each energy term based on design simulations for a training set composed of 625 monomers and 177 protein-protein complexes.
Within REMC, four parameters need to be carefully considered. First, the highest temperature (Tmax) should be high enough to enable the simulation to overcome energy barriers, while the lowest temperature (Tmin) should be low enough to ensure the simulation sufficiently scans the low-energy states. Second, the number of replicas (Nrep) should be large enough to ensure sufficient chance for the adjacent replicas to communicate with each other. Third, the number of local movements (Nsweep) before the global swaps should be selected to make the local Metropolis search achieve satisfactory equilibrium. After successive rounds of optimization, the final parameters were selected as: Tmax = 15 Tmin = 0.5, Nrep = 40, and Nsweep = 100.
Server Input
The only input to the EvoDesign server is the monomer (for monomer design) or protein-protein complex (for interface design) structures of interest in PDB format. For monomer design, the input structure may be full-atomic or a Cα trace, while for interface design, it must be full-atomic given the sensitivity of the design to the shape of the binding pocket. In addition, for interface design, the user is able to upload the scaffold structure and its binding partner as a preformed complex structure or as two separate chains. If the two chains are uploaded separately, the user is given an option to dock the two chains together using ZDOCK [31], a state-of-the-art fast Fourier transform-based protein-protein docking software.
Several advanced options are provided to further tailor the EvoDesign simulation to suit users’ needs. This is achieved by allowing users (i) to select the structural similarity cutoff (TM-score) used during profile construction, (ii) to select the type of energy function used during the design simulation (either evolution-based only design or combined physics- and evolution-based design), (iii) to exclude residue types at specific locations, (iv) to prevent the mutation of residues at specific locations (such as interfaces), and (v) to model the structures of the final designed sequences using I-TASSER [32].
It should be noted that the default EvoDesign setting for PPI design is to redesign the entire sequence of the scaffold chain. The rationale behind designing non-interface residues is that introducing mutations in the interface may have destabilizing effects on the whole protein or lead to suboptimal packing [8, 33, 34]. However, for some large complexes with specific folding architectures such as antibody-antigen complexes, it might be beneficial to focus the design only on the interface regions. Thus, for interface design, users are given an additional option to either redesign the entire scaffold protein or to redesign only its interface residues, which are defined as residues within 5 Å of the opposite chain.
Server Output
Immediately following submission of a design job, an output page with a private URL for the job is created, which users are able to bookmark for future visit. When the EvoDesign simulation finishes, users will be notified by e-mail with a link to the results page. The results in the output page contain: (i) a summary of the input to the server (see Fig. S2 for an illustrative example), (ii) the top structural homologs used for monomer and interface profile construction as well as links to download the full multiple sequence alignment and evolutionary profile (Fig. S3), (iii) the clustering results of sequence decoys generated during the REMC simulation (Fig. S4), (iv) a summary of the top ten designed sequences and the local feature assessment parameters (Fig. S5), (v) a detailed overview of the top ten designed sequences including the sequence alignments between the scaffold and designs, and (vi) the I-TASSER folding results for the top ten designs (Fig. S6).
Discussion
The EvoDesign server is a fully automated, online tool for protein design and has the ability to design new proteins either as a free monomer (monomer design) or as a receptor in a protein-protein complex (interface design). Starting from the structural coordinates of a monomeric protein or complex, EvoDesign first collects homologous folds and protein interfaces from the PDB, from which monomeric and complex profiles are constructed separately. Next, the evolutionary profiles are combined with a newly developed physical energy function, EvoEF, to guide the replica-exchange Monte Carlo simulation in order to design new sequences. Finally, the designed sequences are clustered, and the final designs are chosen from the lowest free energy sequences in the largest clusters.
It is important to note that the core algorithm of EvoDesign has been preserved from previous iterations of the program. This algorithm was validated in a large-scale, in silico redesign experiment of >300 soluble protein folds [12]. Moreover, from this experiment, five designed domains with variable fold types and sequence lengths were experimentally tested through circular dichroism and NMR spectroscopy. All five proteins (including heterogeneous nuclear ribonucleoprotein K domain, thioredoxin domain, light oxygen voltage domain, translation initiation factor 1 domain, and the CISK-PX domain) were soluble and possessed secondary structure as determined by circular dichroism, and three of the designed domains had stable folds as shown by 1D NMR data. The follow-up X-ray crystallography study [35] showed that the crystal structure of the EvoDesign designed CISK-PX domain is very similar (1.32 Å) to the target model generated by I-TASSER structure prediction.
In this work, we have extended the EvoDesign pipeline to enable the design of PPIs by incorporating an evolutionary interface potential and a new physical energy function into the program. Previous benchmark studies of our evolutionary interface potential demonstrated that its predicted values, binding affinity change of protein complexes upon amino acid mutation, showed superior correlation with experimental values [13]; the correlation was significantly higher than that produced by leading physics- and statistical-based methods. Most recently, we applied the new EvoDesign program to the redesign of the BIR3 domain of the X- linked inhibitor of apoptosis protein (XIAP) [8], whose primary function is to suppress cell death by inhibiting caspase-9 activity. However, the suppression of cell death by wild type XIAP can be eliminated by the binding of Smac peptides. Multiple biophysical experiments such as NMR chemical shift perturbation and isothermal calorimetry binding assays demonstrated that the redesigned XIAP domains can bind the Smac peptide with dissociation constants in the low nanomolar range, but do not inhibit the caspase-9 proteolytic activity in vitro. Detailed mutagenesis analyses demonstrated that the major driving force behind the successful redesign of the XIAP-Smac interaction was the interplay of the evolutionary profiles and physical potential [8].
The physical energy function utilized by the previous version of EvoDesign was FoldX. FoldX was originally developed and optimized to predict protein stability change upon mutation and has been widely used in the protein science community. Our benchmark tests show that the newly developed EvoEF generates more accurate predictions than FoldX for both stability and binding affinity change upon mutation, where the latter is critical to PPI design/engineering. In addition to the improved model accuracy, EvoEF is significantly faster than FoldX when it comes to energy calculation. This is particularly important in extensive protein design simulations like EvoDesign, where the physics-based energy computation is one of the most time-consuming parts of the pipeline. FoldX’s inefficient energy computation is partly due to the fact that, currently, only executables are provided for the software and the computational speed cannot be fully optimized by users. Therefore, an effective and efficient physical energy function should be very helpful to the protein science community. The EvoEF source code is made freely available at https://zhanglab.ccmb.med.umich.edu/EvoDesign/EvoEF.tar.gz, where users can optimize the code and parameters according to their own needs. Text S5 in the SI provides a detailed description of the commands and functions implemented in EvoEF.
Despite the effectiveness and efficiency, the evolutionary components of the EvoDesign potential can be partly limited by the availability of structural homologs in the PDB; in particular, the number of protein interface homologs identified by iAlign may be low. In a previous study, we found that the average number of interface homologs identified for a set of test complexes was approximately five [13]. To address this issue, we recently tested a new method, SSIP, to construct interface profiles by combining the structural iMSA with sequence homologs from sequence-based PPI databases. Based on the preliminary data, the method shows promise to significantly increase binding affinity prediction accuracy and we plan to integrate it into EvoDesign after further optimization.
Finally, as one of the essential difficulties in computer-based protein design is the expensive cost of experimental validations, the EvoDesign server aims to provide various transparent intermediate data to allow for detailed annotation and analysis of the confidence of the designed sequences. With the continuous effort on the development and improvement of the scope and accuracy of the methodology, we believe the new EvoDesign pipeline should become a useful tool to the community, especially for scientists who have known protein structures but want to design new sequences with enhanced foldability and biological functionality.
Supplementary Material
Highlights:
New method to design protein-protein interactions using evolutionary profiles
New physical force field with improved speed and binding recognition power
Composite web interface for designing both monomer and complex proteins
Detailed output with transparent intermediate data and annotations
Pipeline and source code of force field are freely available to the community
Acknowledgement
The work was supported in part by the National Institute of General Medical Sciences (GM083107 and GM116960), the National Institute of Allergy and Infectious Diseases (AI134678), and the National Science Foundation (DBI1564756).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature.2001;411:41–2. [DOI] [PubMed] [Google Scholar]
- [2].Szilagyi A, Grimm V, Arakaki AK, Skolnick J. Prediction of physical protein-protein interactions. Phys Biol. 2005;2:S1–S16. [DOI] [PubMed] [Google Scholar]
- [3].Karanicolas J, Kuhlman B. Computational design of affinity and specificity at protein-protein interfaces. Curr Opin Struct Biol. 2009;19:458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chevalier A, Silva DA, Rocklin GJ, Hicks DR, Vergara R, Murapa P, et al. Massively parallel de novo protein design for targeted therapeutics. Nature. 2017;550:74-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP- binding peptides. Nature. 2009;458:859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, et al. Computational Design of Proteins Targeting the Conserved Stem Region of Influenza Hemagglutinin. Science. 2011;332:816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, Andre I, et al. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shultis D, Mitra P, Huang X, Johnson J, Y Z. Changing the Apoptosis Pathway through Evolutionary Protein Design. Journal of molecular biology. 2019:in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–6. [DOI] [PubMed] [Google Scholar]
- [10].Progress Zhang Y. and challenges in protein structure prediction. Curr Opin Struct Biol. 2008;18:342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mitra P, Shultis D, Zhang Y. EvoDesign: De novo protein design based on structural and evolutionary profiles. Nucleic acids research. 2013;41:W273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mitra P, Shultis D, Brender JR, Czajka J, Marsh D, Gray F, et al. An Evolution-Based Approach to De Novo Protein Design and Case Study on Mycobacterium tuberculosis. PLoS computational biology. 2013;9:e1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xiong P, Zhang C, Zheng W, Zhang Y. BindProfX: Assessing Mutation-Induced Binding Affinity Change by Protein Interface Profiles with Pseudo-Counts. Journal of molecular biology. 2017;429:426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic acids research. 2005;33:W382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gao M, Skolnick J. iAlign: a method for the structural comparison of protein-protein interfaces. Bioinformatics. 2010;26:2259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brender JR, Zhang Y. Predicting the Effect of Mutations on Protein-Protein Binding Interactions through Structure-Based Interface Profiles. PLoS computational biology. 2015;11:e1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang Y, Skolnick J. SPICKER: A clustering approach to identify near-native protein folds. J Comput Chem. 2004;25:865–71. [DOI] [PubMed] [Google Scholar]
- [19].Jones JE. On the determination of molecular fields.—I. From the variation of the viscosity of a gas with temperature. Proc R Soc Lond A. 1924;106:441–62. [Google Scholar]
- [20].Jones JE. On the determination of molecular fields.—II. From the equation of state of a gas. Proc R Soc Lond A. 1924;106:463–77. [Google Scholar]
- [21].Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. Journal of computational chemistry. 1983;4:187–217. [Google Scholar]
- [22].Sitkoff D, Sharp KA, Honig B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. The Journal of Physical Chemistry. 1994;98:1978–88. [Google Scholar]
- [23].Kortemme T, Morozov AV, Baker D. An Orientation-dependent Hydrogen Bonding Potential Improves Prediction of Specificity and Structure for Proteins and Protein-Protein Complexes. Journal of Molecular Biology. 2003;326:1239–59. [DOI] [PubMed] [Google Scholar]
- [24].Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins: Structure, Function, and Bioinformatics. 1999;35:133–52. [DOI] [PubMed] [Google Scholar]
- [25].Guerois R, Nielsen JE, Serrano L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. Journal of Molecular Biology. 2002;320:369–87. [DOI] [PubMed] [Google Scholar]
- [26].Quan L, Lv Q, Zhang Y. STRUM: structure-based prediction of protein stability changes upon singlepoint mutation. Bioinformatics. 2016;32:2936–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jankauskaite J, Jimenez-Garcia B, Dapkunas J, Fernandez-Recio J, Moal IH. SKEMPI 2.0: An updated benchmark of changes in protein-protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Deng HY, Jia Y, Zhang Y. 3DRobot: automated generation of diverse and well-packed protein structure decoys. Bioinformatics. 2016;32:378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E. Equation of State Calculations by Fast Computing Machines. J Chem Phys. 1953;21:1087–92. [Google Scholar]
- [31].Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nature Methods. 2015;12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Procko E, Berguig GY, Shen BW, Song Y, Frayo S, Convertine AJ, et al. A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell. 2014;157:1644–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kastritis PL, Rodrigues JP, Folkers GE, Boelens R, Bonvin AM. Proteins feel more than they see: fine- tuning of binding affinity by properties of the non-interacting surface. Journal of molecular biology. 2014;426:2632–52. [DOI] [PubMed] [Google Scholar]
- [35].Shultis D, Dodge G, Zhang Y. Crystal structure of designed PX domain from cytokine-independent survival kinase and implications on evolution-based protein engineering. J Struct Biol. 2015;191:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.