

Abstract
The goal of Binding MOAD is to provide users with a dataset focused on high-quality X-ray crystal structures that have been solved with biologically relevant ligands bound. Where available, experimental binding affinities (Ka, Kd, Ki, IC50) are provided from the primary literature of the crystal structure. The database has been updated regularly since 2005, and this most recent update has added nearly 7000 new structures (growth of 21%). MOAD currently contains 32,747 structures, composed of 9,117 protein families and 16,044 unique ligands. The data is freely available on www.BindingMOAD.org. This paper outlines updates to the data in Binding MOAD as well as improvements made to both the website and its contents. The NGL viewer has been added to improve visualization of the ligands and protein structures. MarvinJS has been implemented, over the outdated MarvinView, to work with JChem for small molecule searching in the database. To add tools for predicting polypharmacology, we have added information about sequence, binding-site, and ligand similarity between entries in the database. A main premise behind polypharmacology is that similar binding sites will bind similar ligands. The large amount of protein-ligand information available in Binding MOAD allows us to compute pairwise ligand and binding-site similarities. Lists of similar ligands and similar binding sites have been added to allow users to identify potential polypharmacology pairs. To show the utility of the polypharmacology data, we detail a few examples from Binding MOAD of drug repurposing targets with their respective similarities.
Introduction
Structure-based drug design has benefited from the creation of several databases which combine structural information from the Protein Data Bank (PDB)[1, 2] with biochemical affinity.[3–14] These databases all have varying requirements for inclusion and provide users with a wide range of information regarding the proteins, ligands and/or the protein-ligand complexes. Early protein datasets were small enough to exist only as a list of relevant PDBids inside of their corresponding publication. As the amount of data utilized in these types of studies has increased from mere tens of structures to the hundreds or even thousands of structures employed in more modern publications, the list sizes are too large to be included in their main body-text. This has resulted in datasets presented as separate downloadable entities or even hosted on the web as publicly accessible tools. Publicly available resources are of unquestionable utility to the scientific community, so long as they are maintained regularly and transparently described in their original publication as to be reproducible and appropriately utilized.
Binding MOAD[6] was originally published in 2005 as a database of carefully curated, high quality, protein-ligand crystal structures of biologically interesting small molecules. This database includes binding data for many of the ligand-protein pairs, curated from their primary citation. The database is accessible via the web at www.BindingMOAD.org. Data is presented to users on a per-structure basis, but the proteins are also grouped by various sequence-based cutoffs to facilitate finding similar structures. Different versions of the dataset are available for download. These include a version with only the structures for which there exist curated binding data, as well as a fully compressed and zipped copy of the collective biological unit files for all entries. The database has been updated on a near-annual basis.
PDBbind[14] is the only true competitor to Binding MOAD, providing a similar collection of protein data. The entrance criteria are similar and the provided subsets of data showcase where the databases differ. The Binding MOAD dataset falls somewhere between PDBbind’s general set and refined set, as PDBbind allows for non-X-ray structures and structures with poorer than 2.5 A resolution in their general set.[15] The HiQ dataset[16] available from Binding MOAD is not restricted to proteins with multiple complexes as in PDBbind’s core set, and thus represents a larger number of protein targets. Both approaches of refining a stringent dataset of high-quality structures are equally valid, users are encouraged to choose a dataset based on the agreement between the curation criteria and the needs of their own experimental procedures. An update for Binding MOAD’s HiQ set is anticipated for the latter half of 2019. The sc-PDB[7] is the most similar after PDBbind, but the pre-processed nature of its dataset puts it into a docking/in silico pre-prep niche that sets itself apart. ChEMBL[17] and BindingDB[10] provide a tremendous amount of binding data for a significant number of protein targets. The majority of the ligand-target pairs in these two databases do not have corresponding experimentally determined structural data, resulting in a different category of database than Binding MOAD or PDBbind.
The rise in popularity, understanding, and availability of machine-learning techniques has resulted in an all-time high for production of new prediction-based algorithms, leading to even greater demand for data collections such as Binding MOAD.[18, 19] Both Binding MOAD and the HiQ dataset have been utilized by the community in training and benchmarking of various predictive algorithms and scoring functions. As an example, MOAD was recently utilized in training a method for assessing scoring function performance in binding affinity prediction.[20]
Binding MOAD’s large collection of small-molecule ligands and binding sites, combined with new features and presented data, allows for researchers to investigate more complex relationships, such as polypharmacology. Polypharmacology is when a small molecular ligand binds to multiple protein targets. Some of the practical applications of polypharmacology are drug repurposing and identifying the off-target binding behind drug side effects. Drug repurposing, or “repositioning,” is the identification of new therapeutic uses for existing drugs.[21] Drug repurposing has emerged as an efficient and inexpensive approach, through which the early stages of drug development can be bypassed by discovering a new therapeutic area for an approved drug.[21–23] A computational technique commonly used in this repurposing is to identify similar ligands and binding sites with the hypotheses that: 1) the chemical similarity between ligands of different targets can identify potential new targets for those molecules[24], and 2) the binding-site similarity of the protein targets can also be used to broaden the identification of new targets for those drugs. Web-based tools are beginning to emerge which allow users to browse similar ligands and targets.[21, 22, 25, 26]
The most recent success in the area of drug repurposing has been in the development of eRepo-ORP to identify new drugs to combat rare orphan diseases.[27] The authors of eRepo-ORP generated models of the binding sites of drug-target pairs from DrugBank[28] using eThread[29] and eFindSite[30] and compared them to models generated for the Orphanet[27] database of targets associated with orphan diseases using binding site similarity determined by eMatchSite[31]. Their method identified 18,145 potential drug candidates for repurposing.[27] As an example of their success, a new inhibitor of KRAS was identified due to the binding site similarity to PTK6, which bound the known drug vandetanib.[27] Naderi et al. have used the same method as eRepo-ORP to generate eModel-BDB, which are binding site models generated from ligand-target pairs derived from BindingDB.[32]
With polypharmacology and drug repurposing in mind, we have introduced ligand similarity data and binding-site similarity data to the Binding MOAD website. This work aims to update the community on details of the current structures in Binding MOAD along with additions and improvements made to the BindingMOAD.org website since the previous publication in 2015[33]. We have migrated to Javascript applications of JChem, MarvinJS, and the NGL Viewer for performance and security reasons. We have also added data regarding sequence similarity, ligand similarity, and binding-site similarity. Lastly, we have expanded our collection to a total of 32,747 protein-ligand crystal structures, composed of 9117 protein families and 16,044 unique ligands.
Methods
Other protein-ligand databases such as ChEMBL and BindingDB cultivate their data in a “bottom-up” course, starting with the literature and available binding information for important ligands, and gathering structural data along the way if it is available. Since we are only interested in interactions where corresponding structural data exist, we operate along a “top-down” approach which starts with the PDB. We first import the entire PDB, remove inappropriate structures, and use the remaining structures to guide our literature searches in a systematic fashion. Since almost all protein structures are annotated with the authors’ names and the appropriate reference, obtaining the reference for the literature portion of the search is straightforward.
Condensing the PDB and Hand Curation
Starting from the PDB (133,344 structures on 9/27/2017), our data pipeline assesses whether each protein structure is an appropriate entry for Binding MOAD, see Figure 1. The specific contents and functions of this data pipeline have been detailed, previously.[3, 6, 33] The condensed description is as follows: Structures must be X-ray crystal structures of 2.5 A resolution or better, contain at least one protein chain with a corresponding, non-covalently bound, biochemically relevant (valid) ligand. Structures emerging from the pipeline meeting these criteria are then hand curated for final entry into the database.
Figure 1:

Binding MOAD Update Process [6]
We emphasize that no protein-ligand structure is automatically processed from the PDB into our database without undergoing hand curation at least once. Literature citations for all final structures to be included in Binding MOAD are used to confirm the validity (biological relevance) of the ligands, as well as extract binding data. Our order of preference for affinity data is: Kd > Ki > IC50. Great care is taken to ensure that ligands entered into Binding MOAD are biochemically significant and are of relevant function in the crystal structure being considered (e.g., structures with only “invalid” crystallographic additives are not included in MOAD).
Addressing Redundancy by Sequence
Grouping proteins by similar sequence allows users to find multiple related structures, which makes various types of comparison and dataset construction much easier. Enzyme classification (EC) numbers are used to group enzymes that perform similar catalytic reactions. Binding MOAD clustering was based on EC groupings in the past, but this method was abandoned for numerous reasons. The EC number listed in PDB files is not always correct, or present at all. In the latter case, filling in the missing data gaps is convoluted. But, most importantly, there still exist massive variation within enzyme classifications, so grouping into homologous protein families by sequence has proven to be more beneficial, straightforward, and reproducible. Structure sequences are compared using BLAST,[34] and proteins are grouped into families by 90% sequence identity. Each family contains a “leader” complex, typically the complex with the tightest binding ligand, i.e. the lowest Ki, Kd, or IC50 value with Kd preferred. In cases where a family has no entry with binding data, complexes of ligand-protein or ligand-cofactor-protein are chosen over protein-cofactor complexes. When multiple complexes are available without affinity data, leaders are chosen by the following criteria in order:
Best resolution (complexes with ligands preferred over cofactor-only complexes)
Wild-type over structures with site mutations
Most recent deposition date
Factors such as R or Rfree values
If all the above criteria are identical, the entries are likely from the same paper, which will be used to help in the tie-breaker
Addressing Redundancy Using Unified Binding Sites
To compare binding sites across all the proteins in MOAD, we needed to create a robust definition of each binding site in each protein. Studies utilizing structural data will often define binding sites specific to each ligand-bound structure using a distance threshold to establish which residues are in contact with the bound ligand. As some protein targets contain massive binding sites, a significant amount of data is therefore missed by only considering residues in immediate contact with a bound small-molecule ligand of a single structure. More elegant approaches are available for proteins which there exist multiple protein-ligand structures, to incorporate more data. Here, we introduce unified binding sites.
Unified binding sites represent the union of all protein residues in contact with any ligand of a given protein family (defined by protein sequence). The contacts are derived from the biounit files using a heavy-atom-to-heavy-atom threshold (4.5 A in this case). In a family where there are N protein-ligand structures, the contacts from each of those N structures are assembled in one unified binding site which describes the entire family. All ligands in the “leader” structure are identified and independent union binding sites are constructed based on each ligand. This is a redundant set and ensures that each binding site is separate unless an overlap in residues exists. Sites are combined if there is an overlap of residues between the sites when creating the unified set of residues.
The most difficult aspect of assembling these unified binding sites resides in the protein numbering. Protein numbering is rarely always accomplished the same way in a group of more than a few structures. It is exceedingly difficult to identify and fix examples where numbering issues arise when using automated scripts for data processing. There are many examples of well resolved, high-quality crystal structures that unfortunately suffer from multiple numbering disagreements. Therefore, we addressed this issue by renumbering protein structures prior to the assembly of unified binding sites. We stress that the renumbering was only used on the backend of the database to generate the unified binding-site data and compare the binding-site similarity. The original PDB numbering is used for any datafile a user might download.
To start, a similarity matrix of all protein chains in Binding MOAD was constructed using BLAST.[34] Using this similarity matrix, chains were then annotated for similarity (for example in dimer of heterodimers, chains A and C are often identical and chains B and D are often identical). These PDBid/chain similarity indices are critical for the renumbering process, as knowing which chains within each PDB file are identical, and which chains are identical between files (different members of a same family with a 100% sequence identical chain). PDB SWS was used for renumbering templates.[35] PDB structures were then renumbered using the following framework:
If the PDBid/chain combo is found in PDB SWS, renumber it accordingly.
If the PDBid is found in PDB SWS, but not for the current chain, use any sequence-identical chain within the same PDBid that is found in PDB SWS.
If the PDBid is not found in PDB SWS, use another structure with a 100% sequence-identical chain as the renumbering template.
If no structures in a homologous family are found in PDB SWS, check to see if their numbering already matches up.
In cases where multiple renumbering frameworks were provided by PDB SWS for a single homologous family, the mapping for the family leader was chosen and the whole family was renumbered in the same manner.
Ligand and Binding-Site Similarities
The most popular and well-established measure of chemical similarity is the Tanimoto coefficient (Tc), which uses fingerprints for comparing two small-molecules. We have used PipelinePilot[36] and two different fingerprints, ECFP6[37] and MDL[2] keys, to calculate the pairwise similarity between all ligands in Binding MOAD. Our experience with exploring chemical diversity has shown a cutoff of Tc > 0.4 for ECFP6 provides a reasonable definition of chemical similarity. However, our analyses determined that ECFP6 fingerprints yield many false negatives in the forms of Tc < 0.4 for similar molecules, or Tc L 1 for identical molecules. These results may be due to ECFP6 fingerprints’ inherent sensitivity to SMILES strings (e.g., tautomers). Therefore, we have also used MDL keys in addition to ECFP6 and included all similarities with MDL score > 0.8 (a customary cutoff also verified by manual inspection).
To further extend the ligand-target associations beyond ligand similarity, we applied binding-site similarity calculations to find all target pairs that share similar binding sites. These target-target associations are based on the idea that similar binding sites accommodate similar ligands. To compare binding sites, we first assembled unified binding sites to represent entire protein families as a single, condensed entity. The unified binding site is determined by all protein residues that contain a heavy atom within 4.5 A of any ligand heavy atom within the family. Then, pairwise comparisons of unified binding sites between all combinations of family leaders in Binding MOAD were conducted. The presence of family leaders here is necessary as a physical manifestation of the unified binding site to be used in calculations, as a simple list of residues present in the binding sites is not sufficient.
Binding-site similarities between the unified binding sites of the family leaders were calculated using APoc. APoc[38] (Alignment of Pockets) is an efficient program for large-scale structural comparison of protein pockets. We used the default parameters in APoc of 100 grid points for the pocket volume and at least 10 residues in a pocket. Only the unified binding sites were given to the program, therefore global structure alignment was used. There were 14,916 unique unified binding sites in 9,117 leaders using all available biounit files for each leader. Roughly, 111 million comparisons of pairs of binding sites were performed on 132 processors, simultaneously, which took 8 weeks of computational time. To extract the statistically significant binding-site associations, only p-values below 0.05 in Apoc were considered, which resulted in 3,510,682 target-target matches (32%). The p-value and PS_Score from the Apoc output are reported on bindingmoad.org. It is already known that proteins with similar sequences have similar binding sites, so we only report binding-site matches for protein pairs that differ by more than 50% sequence similarity. As the unified binding sites are used in these calculations, the similarities between leaders inherently represent all structures contained within each of their families. Collapsible tables for both ligand similarity and binding site similarity are located in the polypharmacology section for each complex (indexed by PDBid) deposited in Binding MOAD.
Results and Discussion
The most recent update of Binding MOAD was derived from the version of the PDB extracted on September 27, 2017 (133,344 entries); a total of 32,747 valid protein-ligand complexes were obtained. Binding MOAD contains 16,044 unique, valid ligands within the 32,747 complexes. Comparatively, this updated dataset contains 11,507 structures overlapping with PDBbind’s[14] collection, representing 71.2% of their 16,151 total protein-ligand structures. These 11,507 overlapping structures contain 8538 unique ligands. Additionally, 3385 of our 16,044 ligands are found as entries in Drugbank[28]. Figure 2 provides the distribution of the valid ligands in our collection by molecular weight. The ligands range from 4–278 heavy atoms, with an average molecular weight of 433 g/mol; an example of the average ligand is adenosine-5’-diphosphate (ADP), which has a molecular weight of 427 g/mol. Figure 2 shows that the number of large ligands (>500 g/mol) drops off quickly. The largest ligands are sugar chains, peptide chains (≤10 amino acids), and nucleic acid chains (≤4 nucleic acids).
Figure 2:

Distribution of the current 16,044 unique ligands by molecular weight. The average ligand size in Binding MOAD is 433 g/mol. The largest are polysaccharides, peptides, and nucleic acid chains.
Binding MOAD also contains 12,098 binding data across the 32,747 complexes (37% coverage of affinity data). These binding data are composed of 4128 Kd or Ka, 3788 Ki, and 4182 IC50 values. These binding affinities range over 16 orders of magnitude; Table 1 presents the range of binding values for each type of binding data, and the distribution of the three types of binding is further detailed in Figure 3.
Table 1:
The distribution of binding data within Binding MOAD
| Binding Data | Tightest | Lower Quartile | Median | Upper Quartile | Weakest |
|---|---|---|---|---|---|
| Kd, Ka (as 1/Ka) | 10 fM | 110 nM | 2.51 μM | 50.0 μM | 1.4 M |
| Ki | 11 pM | 10.2 nM | 250 nM | 12.0 μM | 0.837 M |
| IC50 | 3.9 pM | 15.0 nM | 150 nM | 2.56 μM | 0.355 M |
Figure 3:

The distribution of binding-affinity data within Binding MOAD. Data is available as Kd (orange), Ki (blue), or IC50 (grey). For this histogram, any Ka values were converted to Kd.
As mentioned previously, we are committed to the growth of Binding MOAD as a quality data resource in the community. Since being introduced in 2004 Binding MOAD has regularly expanded its collection with new data. Early updates brought in ~1500 new structures each year, but the rapid growth of the PDB has afforded us with many more structures in recent years. The growth of Binding MOAD is presented in Table 2.[33] The delay in this most recent release stems from a two-year lapse in funding.
Table 2:
Growth Data for Binding MOAD (2004–2017)
| Release (version, PDB download date) | PDB release size | Protein-ligand complexes | Protein Families | Unique ligands | Binding affinity coverage |
|---|---|---|---|---|---|
| Initial release in 2004 [6] | 5331 | 1780 | 2630 | 1375 (25.8%) | |
| Prior to website in 2005 | 8250 | 2732 | 3932 | 2374 (28.8%) | |
| 1st (v2006, 12/31/2006)[3] | 32963 | 9836 | 3151 | 4665 | 2950 (30.0%) |
| 2nd (v2007, 12/31/2007) | 41093 | 11366 | 3583 | 5348 | 3452 (30.4%) |
| 3rd (v2008, 12/31/2008) | 48168 | 13138 | 4078 | 6210 | 4146 (31.6%) |
| 4th (v2009, 12/31/2009) | 56466 | 14720 | 4624 | 7064 | 4782 (32.5%) |
| 5th (v2010, 12/31/2010) | 65344 | 16948 | 5198 | 8140 | 5630 (33.2%) |
| 6th (v2011, 12/31/2011) | 74594 | 18764 | 5772 | 9048 | 6311 (33.6%) |
| 7th (v2012, 12/31/2012) | 84566 | 21109 | 6443 | 10156 | 7284 (34.5%) |
| 8th (v2013, 12/31/2013) | 95132 | 23269 | 6960 | 11173 | 8156 (35.0%) |
| 9th (v2014, 1/02/2015) | 105496 | 25759 | 7599 | 12432 | 9138 (35.5%) |
| 10th (v2017, 9/27/2017) | 133344 | 32747 | 9117 | 16044 | 12098 (37.0%) |
Clustering Binding MOAD into Homologous Protein Families
As noted above, the protein sequences of the entries in Binding MOAD are grouped into homologous protein families. Clustering at 90% sequence identity results in 9117 protein families, which is the default clustering, and individual data files for each of these families are available for download. In addition to families binned at 90% sequence identity, frequently it is necessary to think of a protein family at less strict cutoffs, like 70% or 50% sequence similarity. These families have been added to the data page for each of the entries in Binding MOAD. There are 7542 families when binned by 70% sequence similarity and 5768 families when binned by 50% sequence similarity. The clustering algorithm is a greedy algorithm and not all entries within a family are necessarily within the similarity threshold to every other entry in the family.
New Features and Functionalities
Improved Molecule and Protein Viewing
In order to improve the functionality of Binding MOAD and its web accessibility, programs which utilize JavaScript have been implemented for the viewing of binding sites and ligands. The MarvinJS[39] webservice from ChemAxon has replaced MarvinView to provide users with the ability to sketch small molecules and submit a query to JChem[40] (also from ChemAxon) for searching the small molecules in Binding MOAD. The current version utilizes ChemAxon’s MarvinJS webservice to stay up-to-date with newest releases of MarvinJS and avoid the need to update a license, since MarvinJS is free to academic institutions.
The NGL Viewer[41] is written in JavaScript and replaces the JMol viewer, which was HTML5-based. The NGL Viewer provides additional functionality to draw the molecular and solvent-accessible surfaces for individual protein and ligand residues. It also gives the user the ability to visualize the surface of interest. The solvent-accessible surface area of the ligand and unified binding sites (described below) are displayed in gray and blue respectively, by default (Figure 4).
Figure 4.

Display of NGL Viewer in Binding MOAD with Aliskiren bound to Renin (PDBid 3D91). The blue surface is the solvent accessible surface area of the unified binding site and the gray transparent surface in the center is the solvent accessible surface area of the ligand shown in black sticks.
Unified Binding Sites
Many proteins bind a variety of ligands that differ in both size and chemical functionality, often resulting in different contacts with the protein. Using only one bound conformation to represent the binding site has the potential to miss important functional information, which leads to difficulty in exploring new chemical space. The unified binding site combines all binding-site residues from all structures in a protein family to obtain a more complete picture of the binding site. Visual representations of these binding sites have been added to the Binding MOAD website via the NGL viewer[42] (Figure 4).
Ligand Similarity
Ligand similarity is noted on each datapage in Binding MOAD. This information can be used to identify new targets for existing drugs in DrugBank.[28] Ligand similarities are presented in a table on the webpage associated with the structure it is bound. Figure 5 displays the ligand table for Nilotinib (HET code NIL) associated with PDBid 3CS9’s datapage in Binding MOAD. These tables can be collapsed or expanded as desired by the user.
Figure 5.

Ligand similarity table for ligand NIL associated with PDBid 3CS9 in Binding MOAD. The Tanimoto coefficient (Tc) for each match is given.
Table 3 highlights three examples of polypharmacology that can be identified using ligand similarity. In each case, a known drug is similar to a ligand (HET group) in MOAD which is crystallized to a target other than the traditional target. In each case, the secondary target has been confirmed in the literature, and in some instances, they have also been reported in DrugBank[28]. In the first example, Simvastatin is a cholesterol-lowering drug which traditionally binds to HMG-CoA reductase. Simvastatin is crystallized bound to HMG-CoA reductase in PDBid 1HW9, and it is chemically similar to the HET group AB6 (Tc=0.51 by ECFP6) which is a Lovastatin derivative bound to Integrin-a-L. Literature indicates that Simvastatin is indeed an inhibitor of Integrin-a-L and is shown to induce apoptosis in lymphomas caused by the Epstein-Barr Virus.[28, 43]
Table 3:
Examples of similar ligands binding to two very different targets, validated by DrugBank[28]
| Drug (HET name) and Therapeutic Use |
Traditional Target and Complex in MOAD | HET Name of Similar Ligands in MOAD | Target of the Similar Ligand | Evidence that Drug Binds to Alternate Target |
|---|---|---|---|---|
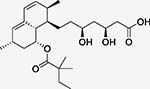 Simvastatin (SIM) A cholesterol lowering agent |
HMG-CoA reductase PDBid: 1HW9 |
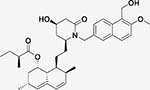 AAY (Tc = 0.48) 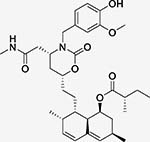 AB8 (Tc = 0.51) Both Lovastatin derivatives |
Integrin alpha- L (CD11 antigen-like family member A) PDBids: 1XDD and 1XDG |
Integrin alpha-L- (Simvastatin) in DrugBank[28]. |
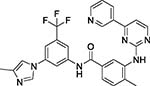 Nilotinib (NIL) Treatment of imatinib-resistant chronic myelogenous leukemia |
Tyrosine-protein kinase ABL1 PDBid: 3CS9 |
 STI (Tc = 0.46) Imatinib |
c-Kit Tyrosine Kinase PDBid: 1T46 |
Both targets in DrugBank[28] |
 Progesterone (STR) Steroid hormone |
Progesterone receptor PDBid: 1A28 |
 TES (Tc = 0.62) Testosterone |
Androgen receptor PDBid: 2AM9 |
Both targets in DrugBank[28] Binding between Androgen Receptor and Progesterone is 5.71 nM[44] |
Binding-Site Similarity
We have examined over 40 published applications to compare two binding sites. The source codes for many of these applications were either unavailable or too computationally expensive for application on the scale of MOAD. A more complete list of binding-site comparison methods is found in a review by Jalencas and Mestres.[45] There were five methods which were able to successfully accommodate calculations of large datasets, Apoc[38], G-LoSA[46], ProBis[47], FuzCav[48], and PocketMatch[49]. Apoc was chosen due to its efficiency, as it is prohibitive to utilize all four programs on the entire dataset simply to benchmark a best fit. However, we intend to add multiple binding-site comparison methods, the same way we use multiple ligand similarity measures. Pairwise calculations on 14,918 binding sites from 9,117 families result in 111,183,872 total similarity calculations. Only protein families which have <50% sequence similarity and Apoc p-value < 0.05 are listed in the table to ensure unique protein targets are being identified. The data is displayed on the website as exemplified by the table for PDBid 1OPK (Figure 6). Complex 1OPK is ABL kinase binding an inhibitor P16, and the binding site matches are for other ATP-binding sites. All of these proteins bind ATP, ADP, and the inhibitor ANP. The matched binding sites are listed in a table sorted by sequence similarity with the least similar sequences listed first so that the straightforward matches to closer homologues are listed last. This allows the user to identify the most unique matches first and the related homologues second.
Figure 6.

Polypharmacology section of the datapage for complex PDBid 1OPK on the Binding MOAD website.
To validate our approach, we examined the binding-site matches for known drug repurposing examples. Below we describe three examples of utilizing binding-site similarity in Binding MOAD to identify drugs which bind multiple targets.
Aliskiren is a Renin inhibitor which is commonly used in the treatment of hypertension[50]. Renin has been crystallized with Aliskiren (PDBid 2VOZ) and shares less than 50% sequence similarity with the AIDS target HIV-1 protease (PDBid 3T3C) despite both proteins being aspartic proteases. Using Apoc we have identified that Renin and HIV-1 protease have similar binding sites (Apoc PS_Score: 0.52). It was recently confirmed that Aliskiren is a dual inhibitor of both Renin and HIV-1 protease.[51] Additional aspartic protease structures which show binding-site similarity larger than 0.5 include Lysosomal Aspartic Protease (PDBid 1LYB, PS-Score: 0.75), Secreted aspartic protease (PDBids 2H6T, 1J71, and 2QZX, PS-Score: 0.70, 0.68, and 0.67 respectively), BACE-1 (PDBid 4GID, 0.66), BACE-2 (PDBids 3ZKN, 3ZKI, and 3ZLQ 0.60, 0.59, and 0.59 respectively), HIV-2 protease (PDBids 5UPJ and 6UPJ, 0.53 and 0.52 respectively), EIAV protease (PDBid 1FMB, 0.53) and FIV protease (PDBids 5FIV and 6FIV, 0.52), HTLV-1 protease (PDBid 3WSJ, 0.51). Note that these are all aspartic proteases. Tzoupis et al. also performed docking studies with BACE-1 and HTLV-1 which suggest Aliskiren could inhibit these proteases as well.[51]
Radicicol is an anti-tumor agent categorized as “experimental” in DrugBank. It is a known inhibitor of the chaperone Grp94 and was crystallized in complex with the protein (PDBid 1U0Z).[52] Topoisomerase VI, which shares only 16.5% sequence similarity, has a similar binding site to Grp94 (PS_Score = 0.53) and has been solved bound to radicicol (PDBid 2HKJ).[53]
Imatinib was first marketed as a potent and specific Bcr-Abl kinase inhibitor in 2002.[54] Though the initial structural characterization of its interactions was published, the actual crystal structure was not publicly deposited.[55] Since that time, numerous X-ray crystal structures of Imatinib bound to Bcr-Abl have been deposited and four are present in Binding MOAD (PDBids 3MSS, 2HYY, 3K5V, 1IEP). Imatinib was later characterized by its interactions with p38a (PDBids 3HEC).[56] Our calculations successfully identified similarity (PS_Score: 0.43) in the unified binding sites of Bcr-Abl and p38a, which may be expected as both proteins are kinases. It should be noted that imatinib has a case of extreme polypharmacology in that it also binds to human quinone reductase 2.[57] Visual examination of the binding sites show that they are very dissimilar (Apoc PS_Score = 0.30), but it is still identified through MOAD’s ligand-similarity polypharmacology feature.
Here, we have presented just a few examples of previously observed polypharmacology for various targets in the literature. These examples were able to be identified via similarity of ligands, binding sites, or both. There surely exist many more examples which are still buried in the data, and every new update of extra structures will exponentially increase the number of possible target combinations to be investigated in the future.
Conclusions
We have detailed the further development and expansion of Binding MOAD. In the future, we aim to continue our annual updates to keep pace with the growth of the PDB. Binding MOAD has >32,000 hand-curated, protein-ligand X-ray crystal structures that contain ligands of biological relevance. Binding data are available for 37% of the entries, and this coverage has only increased with every update of the database. The value of Binding MOAD is not necessarily present in the quantity of its data, but more-so in the quality. Maintaining this data quality is only achievable due to the considerable amount of effort placed in the update process and hand-curation. We have added similarity-based metrics to search the dataset, both in terms of ligand similarity as well as protein similarity.
Our datasets are available online at http://www.BindingMOAD.org. This web-accessible resource is available to the research community, and our web interface also allows for users to contact us if they find any aspects of our curated data to be incorrect. Each structure’s webpage includes details about ligands (both valid and invalid), available binding data, PDBid for structural coordinates, EC class, homologous protein families with links to related structures at multiple sequence cutoffs (90%, 70%, 50%), a 3D-visualization of the ligand bound in the unified binding site (using the NGL viewer[42]), as well as polypharmacology data presented as tables of ligand similarities and binding-site similarities.
Highlights.
Binding MOAD (BindingMOAD.org) is one of the largest databases of protein-ligand crystal structures from the Protein Data Bank, many augmented with binding affinity data from the literature
The available data has been expanded to include nearly 7000 new structures (growth of 21%)
A new viewer and improved the tools for searching ligands by chemical substructures have been added
We have added new relationships across the database that link similar ligands and binding sites, which allow users to identify potential polypharmacology pairs
We give several examples of similar compounds and binding sites that facilitate drug repurposing
Acknowledgements
This work has been supported in part by the National Institutes of Health (R01 GM124283). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rose PW, Prlic A, Altunkaya A, Bi C, Bradley AR, Christie CH, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017;45:D271–D81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Benson ML, Smith RD, Khazanov NA, Dimcheff B, Beaver J, Dresslar P, et al. Binding MOAD, a high-quality protein-ligand database. Nucleic Acids Res. 2008;36:D674–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bergner A, Günther J, Hendlich M, Klebe G, Verdonk M. Use of Relibase for retrieving complex three-dimensional interaction patterns including crystallographic packing effects. Biopolymers. 2001;61:99–110. [DOI] [PubMed] [Google Scholar]
- [5].Golovin A, Dimitropoulos D, Oldfield T, Rachedi A, Henrick K. MSDsite: a database search and retrieval system for the analysis and viewing of bound ligands and active sites. Proteins. 2005;58:190–9. [DOI] [PubMed] [Google Scholar]
- [6].Hu L, Benson ML, Smith RD, Lerner MG, Carlson HA. Binding MOAD (mother of all databases). Proteins: Structure, Function, and Bioinformatics. 2005;60:333–40. [DOI] [PubMed] [Google Scholar]
- [7].Kellenberger E, Muller P, Schalon C, Bret G, Foata N, Rognan D. sc-PDB: an annotated database of druggable binding sites from the Protein Data Bank. Journal of chemical information and modeling. 2006;46:717–27. [DOI] [PubMed] [Google Scholar]
- [8].Kufareva I, Ilatovskiy AV, Abagyan R. Pocketome: an encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012;40:D535–D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016;44:D1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Michalsky E, Dunkel M, Goede A, Preissner R. SuperLigands-a database of ligand structures derived from the Protein Data Bank. BMC bioinformatics. 2005;6:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Puvanendrampillai D, Mitchell JB. Protein Ligand Database (PLD): additional understanding of the nature and specificity of protein-ligand complexes. Bioinformatics. 2003;19:1856–7. [DOI] [PubMed] [Google Scholar]
- [13].Wang R, Fang X, Lu Y, Wang S. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J Med Chem. 2004;47:2977–80. [DOI] [PubMed] [Google Scholar]
- [14].Liu Z, Li Y, Han L, Li J, Liu J, Zhao Z, et al. PDB-wide collection of binding data: current status of the PDBbind database. Bioinformatics. 2015;31:405–12. [DOI] [PubMed] [Google Scholar]
- [15].Liu Z, Su M, Han L, Liu J, Yang Q, Li Y, et al. Forging the Basis for Developing Protein-Ligand Interaction Scoring Functions. Acc Chem Res. 2017;50:302–9. [DOI] [PubMed] [Google Scholar]
- [16].Dunbar JB Jr., Smith RD, Yang CY, Ung PM, Lexa KW, Khazanov NA, et al. CSAR benchmark exercise of 2010: selection of the protein-ligand complexes. J Chem Inf Model. 2011;51:2036–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ain QU, Aleksandrova A, Roessler FD, Ballester PJ. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip Rev Comput Mol Sci. 2015;5:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wojcikowski M, Ballester PJ, Siedlecki P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci Rep. 2017;7:46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xavier MM, Heck GS, Avila MB, Levin NMB, Pintro VO, Carvalho NL, et al. SAnDReS a Computational Tool for Statistical Analysis of Docking Results and Development of Scoring Functions. Comb Chem High Throughput Screen. 2016;19:801–12. [DOI] [PubMed] [Google Scholar]
- [21].Karaman B, Sippl W. Computational Drug Repurposing: Current Trends. Curr Med Chem. 2018. [DOI] [PubMed] [Google Scholar]
- [22].Hurle MR, Yang L, Xie Q, Rajpal DK, Sanseau P, Agarwal P. Computational drug repositioning: from data to therapeutics. Clin Pharmacol Ther. 2013;93:335–41. [DOI] [PubMed] [Google Scholar]
- [23].Li YY, Jones SJ. Drug repositioning for personalized medicine. Genome Med. 2012;4:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Awale M, Reymond JL. The polypharmacology browser: a web-based multi-fingerprint target prediction tool using ChEMBL bioactivity data. J Cheminform. 2017;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Awale M, Reymond JL. Web-Based Tools for Polypharmacology Prediction. Methods Mol Biol. 2019;1888:255–72. [DOI] [PubMed] [Google Scholar]
- [27].Brylinski M, Naderi M, Govindaraj RG, Lemoine J. eRepo-ORP: Exploring the Opportunity Space to Combat Orphan Diseases with Existing Drugs. J Mol Biol. 2018;430:2266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Brylinski M, Lingam D. eThread: a highly optimized machine learning-based approach to meta-threading and the modeling of protein tertiary structures. PLoS One. 2012;7:e50200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Feinstein WP, Brylinski M. eFindSite: Enhanced Fingerprint-Based Virtual Screening Against Predicted Ligand Binding Sites in Protein Models. Mol Inform. 2014;33:135–50. [DOI] [PubMed] [Google Scholar]
- [31].Brylinski M eMatchSite: sequence order-independent structure alignments of ligand binding pockets in protein models. PLoS Comput Biol. 2014;10:e1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Naderi M, Govindaraj RG, Brylinski M. eModel-BDB: a database of comparative structure models of drug-target interactions from the Binding Database. Gigascience. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ahmed A, Smith RD, Clark JJ, Dunbar JB Jr., Carlson HA. Recent improvements to Binding MOAD: a resource for protein-ligand binding affinities and structures. Nucleic Acids Res. 2015;43:D465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Martin AC. Mapping PDB chains to UniProtKB entries. Bioinformatics. 2005;21:4297–301. [DOI] [PubMed] [Google Scholar]
- [36].Pipeline Pilot. 9.1.0.13 ed. Accelrys Software Inc., San Diego, CA: Accelrys Software Inc., San Diego, CA; 2013. [Google Scholar]
- [37].Rogers D, Hahn M. Extended-Connectivity Fingerprints. Journal of Chemical Information and Modeling. 2010;50:742–54. [DOI] [PubMed] [Google Scholar]
- [38].Gao M, Skolnick J. APoc: Large scale identification of similar protein pockets. Bioinformatics. 2013:btt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Marvin JS. 18.26.0 ed. Budapest, Hungary: ChemAxon Ltd.; 2018. [Google Scholar]
- [40].Dakshanamurthy S, Issa NT, Assefnia S, Seshasayee A, Peters OJ, Madhavan S, et al. Predicting new indications for approved drugs using a proteochemometric method. J Med Chem. 2012;55:6832–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rose AS, Hildebrand PW. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015:gkv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rose AS, Hildebrand PW. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015;43:W576–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Katano H, Pesnicak L, Cohen JI. Simvastatin induces apoptosis of Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines and delays development of EBV lymphomas. Proc Natl Acad Sci U S A. 2004;101:4960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].van der Burg B, Winter R, Man HY, Vangenechten C, Berckmans P, Weimer M, et al. Optimization and prevalidation of the in vitro AR CALUX method to test androgenic and antiandrogenic activity of compounds. Reprod Toxicol. 2010;30:18–24. [DOI] [PubMed] [Google Scholar]
- [45].Jalencas X, Mestres J. Identification of Similar Binding Sites to Detect Distant Polypharmacology. Mol Inform. 2013;32:976–90. [DOI] [PubMed] [Google Scholar]
- [46].Lee HS, Im W. G-LoSA: An efficient computational tool for local structure-centric biological studies and drug design. Protein Sci. 2016;25:865–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Konc J, Janezic D. ProBiS-2012: web server and web services for detection of structurally similar binding sites in proteins. Nucleic Acids Res. 2012;40:W214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Weill N, Rognan D. Alignment-free ultra-high-throughput comparison of druggable protein-ligand binding sites. J Chem Inf Model. 2010;50:123–35. [DOI] [PubMed] [Google Scholar]
- [49].Yeturu K, Chandra N. PocketMatch: a new algorithm to compare binding sites in protein structures. BMC Bioinformatics. 2008;9:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sanoski CA. Aliskiren: an oral direct renin inhibitor for the treatment of hypertension. Pharmacotherapy. 2009;29:193–212. [DOI] [PubMed] [Google Scholar]
- [51].Tzoupis H, Leonis G, Megariotis G, Supuran CT, Mavromoustakos T, Papadopoulos MG. Dual inhibitors for aspartic proteases HIV-1 PR and renin: advancements in AIDS-hypertension-diabetes linkage via molecular dynamics, inhibition assays, and binding free energy calculations. J Med Chem. 2012;55:5784–96. [DOI] [PubMed] [Google Scholar]
- [52].Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J Biol Chem. 2003;278:48330–8. [DOI] [PubMed] [Google Scholar]
- [53].Corbett KD, Berger JM. Structural basis for topoisomerase VI inhibition by the anti-Hsp90 drug radicicol. Nucl Acids Res. 2006;34:4269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493–502. [DOI] [PubMed] [Google Scholar]
- [55].Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–42. [DOI] [PubMed] [Google Scholar]
- [56].Namboodiri HV, Bukhtiyarova M, Ramcharan J, Karpusas M, Lee Y, Springman EB. Analysis of imatinib and sorafenib binding to p38alpha compared with c-Abl and b-Raf provides structural insights for understanding the selectivity of inhibitors targeting the DFG-out form of protein kinases. Biochemistry. 2010;49:3611–8. [DOI] [PubMed] [Google Scholar]
- [57].Winger JA, Hantschel O, Superti-Furga G, Kuriyan J. The structure of the leukemia drug imatinib bound to human quinone reductase 2 (NQO2). BMC Struct Biol. 2009;9:7. [DOI] [PMC free article] [PubMed] [Google Scholar]