

Summary
The sympathetic nervous system drives brown and beige adipocyte thermogenesis via release of norepinephrine from local axons. However, the molecular basis underlying the higher levels of sympathetic innervation of thermogenic fat, compared to white fat, has remained elusive. Here we show that thermogenic adipocytes express a previously unknown, mammal-specific endoplasmic reticulum membrane protein, termed Calsyntenin-3β. Genetic loss or gain of Calsyntenin-3β in adipocytes reduces or enhances functional sympathetic innervation in adipose tissue respectively; Calsyntenin-3β ablation predisposes mice to obesity on a high fat diet. Mechanistically, Calsyntenin-3β promotes endoplasmic reticulum localization and secretion from brown adipocytes of S100b, a protein lacking a signal peptide. S100b stimulates neurite outgrowth from sympathetic neurons in vitro. S100b deficiency phenocopies Calsyntenin-3β deficiency, whereas forced expression of S100b in brown adipocytes rescues defective sympathetic innervation caused by Calsyntenin-3β ablation. Taken together, our data elucidate a mammal-specific mechanism of communication between thermogenic adipocytes and sympathetic neurons.
A hallmark of mammalian evolution is the emergence of brown adipose tissue (BAT), an organ specialized in performing non-shivering thermogenesis1. The presence of BAT confers an evolutionary advantage to mammals by enhancing their adaptability to cold stress and the survival of newborns. The powerful ability of thermogenic fat to oxidize substrates and increase energy expenditure has drawn growing interest as a therapeutic approach against obesity and associated metabolic disorders2.
BAT thermogenesis requires innervation by the sympathetic nervous system. Sympathetic nerves release the neurotransmitter norepinephrine (NE) from local axons3,4, which activates the β-adrenergic receptor-cAMP-PKA pathway in adipocyte to drive lipolysis and thermogenic respiration5,6. In line with its thermogenic function, BAT is much more richly innervated by sympathetic nerves than WAT7. Nevertheless, the molecular basis underlying this selective recruitment of sympathetic innervation has remained largely unexplored.
Recently, PRDM16 was identified as an important transcriptional regulator driving the thermogenic program in beige adipocytes8,9, a distinct type of inducible thermogenic adipocytes, mainly residing in subcutaneous WAT10. Interestingly, forced expression of PRDM16 leads to increased sympathetic innervation of beige adipocytes11; adipose-specific ablation of PRDM16 has the opposite effect12. These observations strongly suggest that certain adipocyte-derived factors can influence the extent of sympathetic innervation of the adipose tissue. Here we show that a previously unknown mammal-specific membrane protein, Calsyntenin-3β, promotes sympathetic innervation of both brown and beige adipocytes. Calsyntenin-3β binds to and enhances the protein expression and secretion of S100b, a protein unconventionally secreted without a signal peptide. This, in turn, acts as a neurotrophic factor to stimulate sympathetic axon growth.
Results
Clstn3β is an adipose-specific gene
We previously found that adipose-specific ablation of Lysine-specific demethylase 1 (LSD1) causes severe BAT dysfunction13. Expression of Calsyntenin-3 (Clstn3) is strongly downregulated in LSD1-deficient BAT (Extended Data Fig. 1a). Clstn3 is a plasma membrane protein promoting synaptogenesis in the central nervous system (CNS)14. In striking contrast to the known form of Clstn3, a completely distinct and unannotated form is expressed in BAT, which we have named Calsyntenin-3β (Clstn3β) (Fig. 1a). Clstn3β appears to contain three exons, with the last two shared with Clstn3, and the first large and unique exon lying within an intron of Clstn3 (Fig. 1a). Histone marker and transcription factor ChIP-seq analyses suggest the existence of distinct promoter and enhancer of Clstn3β from Clstn3 in BAT (Extended Data Fig. 1b). Taken together, these observations suggest that Clstn3β is a previously unknown gene expressed in BAT and not a splicing variant of Clstn3.
Fig. 1. Calsyntenin-3β Encodes an Adipocyte-Specific Protein.

a, RNA-seq reads distribution at the Clstn3 locus from BAT and cerebral cortex. Arrow denotes the unique exon to Clstn3β. b, Tissue specificity of Clstn3β expression as determined by qPCR (n=3 mice). c, Cloning of Clstn3β from BAT cDNA library. Red denotes nucleotides and amino acids derived from the unique exon of Clstn3β. Underline denotes peptides detected by mass spectrometry. Green denotes the peptide used to generate the Clstn3β antibody.
Clstn3β mRNA expression is highly restricted to adipose tissue, with the expression level in the interscapular BAT being 6-fold higher than both the inguinal subcutaneous and the perigonadal visceral WAT (Fig. 1b). Clstn3β expression is strongly induced in the inguinal subcutaneous WAT upon cold exposure (Extended Data Fig. 1c), suggesting it is also highly expressed in beige adipocytes.
We next attempted to clone the Clstn3β cDNA. We successfully amplified an open reading frame of 1074 bp predicted to encode a protein of 357 aa from a murine BAT cDNA library (Fig. 1c), thus confirming the existence of Clstn3β at the transcript level. Notably, the N-terminal extracellular portion of Clstn3, essential for interaction with α-neurexins and its synaptogenic activity, is completely missing in Clstn3β, strongly suggesting that these two proteins have quite distinct functions.
To detect Clstn3β at the protein level, we developed a rabbit polyclonal antibody against a C-terminal peptide of Clstn3β (aa in green, Fig. 1c). The antibody detected a doublet band of the expected size for the full-length protein in BAT (Extended Data Fig. 1d). We immunoprecipitated endogenous Clstn3β with this antibody and mass spectrometry analysis identified two peptides consistent with the predicted sequence; one derived from the region shared with Clstn3 and the other one from the unique exon, which was not included in the mouse protein database (underlined aa, Fig. 1c, Extended Data Fig. 1d), thus providing definitive evidence at the peptide level for the existence of Clstn3β.
We then examined the evolution of Clstn3β. Interestingly, unequivocal homologs can be identified only in mammals that give birth to live progeny (Extended Data Fig. 1e). Importantly, a fragment with limited homology but unproductive at protein coding exists in an intron of Clstn1 of the Chinese softshell turtle (Extended Data Fig. 1f), suggesting Clstn3β has evolved in mammals after the divergence of reptiles. In contrast, Clstn3 is conserved throughout the entire vertebrate family.
Clstn3β Localizes to the ER
We first studied the function of Clstn3β by determining its subcellular localization. Ectopically expressed Clstn3β in primary brown adipocytes was found to co-localize with KDEL, a marker for endoplasmic reticulum (ER), thus suggesting Clstn3β localizes to the ER (Fig. 2a). Endogenous Clstn3β was detected as more weakly stained puncta also positive for KDEL (Fig. 2b). To visualize Clstn3β subcellular localization at the ultrastructural level, we expressed an Apex2 reporter fused to the C-terminus of Clstn3β in brown adipocytes; Apex2 localization based on 3, 3’ diaminobenzidine tetrahydrochloride reactivity was then examined by electron microscopy. Consistent with the immunofluorescence data, Apex2 strongly labeled the ER (Fig. 2c). In contrast, we observed no Apex2 labeling of other membranous organelles (Extended Data Fig. 2a–b). The ER localization of Clstn3β was further supported by BAT fractionation and Western blot analysis (Extended Data Fig. 2c).
Fig. 2. Clstn3β Localizes to the Endoplasmic Reticulum.

a, b, Immunofluorescence analysis of localization of ectopically expressed (a) or endogenous (b) Clstn3β in primary brown adipocytes. Green, KDEL; Red; Clstn3β. Arrows denote puncta specific for Clstn3β. The region inside the white box is enlarged for clearer view. c, Electron microscopy analysis localization of Clstn3β-Apex2. Arrows denote ER positive or negative for Apex2 in infected or uninfected cells. Scale bar: 10 μm for a and b, 500 nm for c.
Clstn3β promotes adipose thermogenesis
To study the function of Clstn3β in adipose thermogenesis and whole-body energy metabolism, a global Clstn3β knockout mouse strain was generated. Deletion of Clstn3β was verified by DNA sequencing, qPCR, Western blot and immunofluorescence (Extended Data Fig. 3a–d). Expression and splicing of Clstn3 is unaffected in Clstn3β KO mice (Extended Data Fig. 3e). On chow diet, body weights of Clstn3β KO mice were not significantly different from WT mice, yet they had significantly higher body fat masses (Extended Data Fig. 3f–g). Histological analysis and triglyceride quantitation showed that Clstn3β-deficient BAT has significantly more lipid deposition than WT; this parameter often reflects BAT dysfunction (Fig. 3a). Indirect calorimetry study showed that Clstn3β KO mice had lower ORRR2RRR consumption and CORRR2RRR production rates than the WT mice (Fig. 3b, Extended Data Fig. 3h), while food intake and physical activity were unaltered (Extended Data Fig. 3i–j). Interestingly, the acute respiratory response to a β3-adrenergic agonist (CL-316,243) injection was similar between WT and KO mice (Fig. 3b, Extended Data Fig. 3k).
Fig. 3. Ablation of Clstn3β Impairs Adipose Thermogenesis.

a, Histology and triglyceride quantitation of WT and Clstn3β KO BAT (n=4 mice). b, Indirect calorimetry analysis of WT and Clstn3β KO mice (n=6 mice). c, Cold tolerance test of WT and Clstn3β KO mice (n=6 mice). d, e, f, Body weight curve (d), body composition (n=8 mice) (e) and glucose tolerance test (n=7 mice) (f) of WT and Clstn3β KO mice on high fat diet. g, h, Histology of BAT (g) and liver (h) of WT and Clstn3β KO mice on high fat diet. Scale bar: 100 μm.
To directly assess adaptive thermogenesis, we performed a cold tolerance test and found KO mice were remarkably more cold-sensitive than WT mice (Fig. 3c), providing strong evidence that Clstn3β KO mice have defective adaptive thermogenesis. Together these findings suggest that the Clstn3β KO mice are defective in energy expenditure and adaptive thermogenesis compared to WT mice, consistent with BAT dysfunction. However, Clstn3β deletion does not seem to affect the BAT response to pharmacological β3-adrenergic stimulation.
To understand the role of Clstn3β in diet-induced obesity, we challenged the mice with a high fat diet. Clstn3β KO mice gained weights more rapidly than WT mice on high fat diet (Fig. 3d). Body composition analyses revealed higher fat masses in KO than WT, yet they had comparable lean masses (Fig. 3e). The KO mice also showed significantly elevated fasting blood glucose, strongly impaired glucose tolerance, more lipid accumulation in BAT and increased liver steatosis (Fig. 3f–h). These results indicate that Clstn3β KO mice are more susceptible to diet-induced obesity and glucose intolerance than WT mice.
To complement the loss-of-function model of Clstn3β, we generated an adipose-specific Clstn3β transgenic model and performed the same set of analyses as described above. The transgenic mice displayed the opposite phenotype from the KO in all of the assays (Fig. 4a–h, Extended Data Fig. 4a–h). Taken together these data provide strong support for a pro-thermogenic function of Clstn3β.
Fig. 4. Transgenic Expression of Clstn3β Enhances Adipose Thermogenesis.

a, Histology and triglyceride quantitation of WT and Clstn3β transgenic BAT (n=4 mice). b, Indirect calorimetry analysis of WT and Clstn3β transgenic mice (n=4 mice). c, Cold tolerance test of WT and Clstn3β transgenic mice (n=6 mice). d, e, f, Body weight curve (d), body composition (e) and glucose tolerance test (n=6 mice) (f) of WT and Clstn3β transgenic mice on high fat diet. g, h, Histology of BAT (g) and liver (h) of WT and Clstn3β transgenic mice on high fat diet. Scale bar: 100 μm.
Clstn3β enhances sympathetic innervation
The observations that both the Clstn3β KO and transgenic mice showed altered cold sensitivity, coupled with their rather normal responses to the β3-adrenergic agonist, suggested that Clstn3β may affect sympathetic innervation of adipose tissue. Indeed, we observed no significant difference in norepinephrine-induced respiration between WT or KO brown adipocytes (Fig. 5a), arguing that the phenotype at the organismal level could not be explained by alterations in the intrinsic thermogenic capacity of brown adipocytes.
Fig. 5. Clstn3β Promotes Functional Sympathetic Innervation of the Adipose Tissues.

a, Respiration rates of WT or Clstn3β KO brown adipocytes (n=4 biologically independent samples). b, c, Immunostaining of cryo-section of (b) WT/Clstn3β KO and (c) WT/Clstn3β transgenic BAT (n=5 mice). d, e, f, Histology, triglyceride quantitation (d), TH staining (e) and cold tolerance test (f) of Clstn3β KO +/- adinopectin-cre mice receiving AAV-DIO-Clstn3β (n=4 mice). g, c-fos immunoreactivity and thermogenic response to CNO administration (n=8 mice for hM3Dq+CNO, n=3 mice for hM3Dq+saline and mCherry+CNO). i, j, Thermogenic response to CNO administration in WT/Clstn3β KO (n=8 mice) (i) and WT/Clstn3β transgenic mice (n=4 mice) (j). Scale bar: 100 μm.
To assess BAT sympathetic innervation, we performed Tyrosine Hydroxylase (TH) immunostaining. Strikingly, TH-immunoreactivity of the KO BAT was substantially reduced relative to the WT (Fig. 5b). Immunostaining of TUBB3, a neural-specific tubulin yielded similar results (Fig. 5b). These results indicate that KO BAT has reduced growth of sympathetic nerves relative to WT. Decreased expression of several thermogenesis-related genes in KO BAT was observed upon acute cold exposure following pre-acclimation at thermoneutrality, consistent with reduced sympathetic innervation (Extended Data Fig. 5a). BAT from Clstn3β transgenic mice showed enhanced sympathetic innervation (Fig. 5c), providing further support that Clstn3β promotes BAT thermogenesis via enhancing sympathetic innervation.
To assess whether the BAT innervation and thermogenic defects of the global Clstn3β KO mice indeed reflect deletion of Clstn3β from adipocytes, we restored Clstn3β expression specifically in brown adipocytes by crossing the Clstn3β KO mice to an adiponectin-cre line and injecting a Cre-dependent AAV-Clstn3β into the BAT15. The Cre+ BAT with restored Clstn3β expression showed significantly reduced lipid deposition and enhanced sympathetic innervation compared with the Cre- BAT (Fig. 5d–e). The Cre+ mice also showed significantly improved cold tolerance and increased energy expenditure compared with the Cre- ones (Fig. 5f, Extended Data Fig. 5b). These data show that restoring Clstn3β expression specifically in brown adipocytes is sufficient to rescue the defects of the global Clstn3β KO mice.
We next asked whether Clstn3β is important for sympathetic innervation of beige adipocytes. Similar to BAT, inguinal subcutaneous WAT from WT mice acclimated at 4°C for one week exhibited more extensive sympathetic axons than from KO (Extended Data Fig. 5c), suggesting Clstn3β plays an important role in promoting sympathetic innervation of beige adipocytes as well as in the classical BAT.
To investigate the functional significance of Clstn3β to sympathetic innervation of BAT, we chemogenetically activated sympathetic premotor neurons and assessed the downstream BAT response. Previous studies have identified medullary raphe neurons expressing vesicular glutamate transporter 3 (VGLUT3) that are proposed to activate BAT thermogenesis via direct projections to preganglionic sympathetic neurons in the spinal cord16. We crossed the VGLUT3(Slc17a8)-ires-cre mouse line17 to Clstn3β KO and transgenic lines and stereotaxically injected Cre-dependent AAV-hM3Dq-mCherry or AAV-mCherry into the medullary raphe region of brain stem to drive stable expression of the transgene specifically in the VGLUT3-expressing neurons. Injection of clozapine-N-oxide (CNO), ligand of hM3Dq, but not saline to mice receiving AAV-hM3Dq-mCherry induced c-fos expression in the medullary raphe region and an increase of 0.9°C in interscapular temperature (Fig. 5g). CNO Injection to mice receiving AAV-mCherry produced neither c-fos expression nor a temperature response (Fig. 5g), thus confirming that CNO specifically activates sympathetic premotor neurons in the medullary raphe region to trigger the thermogenic response.
We next examined the response of Clstn3β KO and transgenic mice to CNO. The response was dampened from 0.9°C in WT mice to 0.3°C in KO mice but enhanced from 0.6°C in control mice to 1.2°C in transgenic mice (Fig. 5h–i). Taken together, these findings indicate that ablation of Clstn3β impairs functional sympathetic innervation of thermogenic adipose tissue, whereas transgenic expression has the opposite effect.
Clstn3β promotes secretion of S100b
Our findings raised a critical question that how an intracellular membrane protein could regulate sympathetic innervation of thermogenic adipocytes. To gain insight into this question, we performed quantitative whole-tissue proteomic analysis of WT and Clstn3β KO BAT (Fig. 6a and Supplementary Table). The most strongly downregulated (47% down) protein in the KO BAT is S100b. Previous studies have established that S100b is protein highly expressed by astrocytes in the CNS and that it has neurotrophic activity18,19. S100b expression is much higher in BAT than in WAT and is strongly induced in the inguinal subcutaneous WAT upon cold exposure (Extended Data Fig. 6a–b). Notably, S100b transcription is positively regulated by PRDM16 (Extended Data Fig. 6c–e). We therefore examined the intriguing hypothesis that S100b might be a critical adipocyte-derived neurotrophic factor that promotes sympathetic innervation of adipose tissue.
Fig. 6. Clstn3β Promotes Secretion of S100b, an Adipocyte-derived Neurotrophic Factor.

a, Proteomics of WT and Clstn3β KO BAT. b, TUBB3 Immunostaining of sympathetic neurons +/- S100b (n=25 cells). c, d, e, Histology, triglyceride quantitation (c), TH staining (d) and cold tolerance test (e) of Clstn3β KO +/- adiponectin-cre mice receiving AAV-DIO-S100b (n=4 mice). f, TH and TUBB3 immunostaining of BAT from WT, S100b KO and Clstn3β transgenic/S100b KO mice (n=4 mice). g, h, Immunostaining (g) and Pearson’s correlation analysis (h) of S100b and KDEL in brown adipocytes (n=35 cells). i, Western blot analysis of conditioned media from brown adipocytes. j, Binding assay of S100b and Clstn3β/Clstn3. Scale bar: 100 μm.
We first asked whether S100b possesses neurotrophic activity for sympathetic neurons. S100b significantly induced neurite growth from primary sympathetic neurons (Fig. 6b), thus demonstrating that S100b can act as a neurotrophic factor for sympathetic neurons.
We next tested whether forced expression of S100b in brown adipocytes could alleviate the Clstn3β KO defects by injecting Cre-dependent AAV-S100b into BAT of Clstn3β KO mice with or without adiponectin-cre. We observed a significantly reduced triglyceride content and enhanced sympathetic innervation, only in the presence of adiponectin-cre (Fig. 6c–d). Moreover, Cre+ mice showed significantly enhanced cold tolerance and increased energy expenditure compared with Cre- mice (Fig. 6e, Extended Data Fig. 6f), indicating that forced S100b expression is sufficient to reverse the innervation and thermogenic defects caused by Clstn3β deficiency.
Whether S100b-deficient BAT phenocopies Clstn3β-deficient BAT was determined by analyzing BAT sympathetic innervation of a global S100b KO mouse line20. S100b-deficient BAT displayed a remarkable decrease in the level of sympathetic innervation (Fig. 6f), suggesting that S100b could indeed be a target of Clstn3β that mediates sympathetic-adipose communication. Importantly, we observed no difference between sympathetic innervation of the salivary gland of WT and S100b KO mice (Extended Data Fig. 6g). Moreover, ablation of S100b completely obliterates the ability of Clstn3β transgenesis to enhance BAT sympathetic innervation (Fig. 6f), suggesting that S100b works downstream of Clstn3β. Finally, how Clstn3β deficiency results in reduction in S100b protein level in BAT was examined mechanistically. We observed no difference in S100b mRNA level between WT and Clstn3β KO BAT (Extended Data Fig. 6h), suggesting S100b is post-transcriptionally regulated by Clstn3β. We then cultured primary Clstn3β KO brown adipocytes and ectopically expressed S100b with or without Clstn3β. Under this condition, we observed that Clstn3β led to a modest increase in cellular S100b protein level (Extended Data Fig. 6i). Similar results were obtained with HEK293T cells (Extended Data Fig. 6j). Importantly, Clstn3 did not have such an effect (Extended Data Fig. 6j). Of great interest, co-expression of Clstn3β significantly promotes colocalization of S100b with the ER marker KDEL in brown adipocytes (Fig. 6g–h), suggesting that Clstn3β promotes association of S100b with the ER. Importantly, the amount of S100b secreted into the media increased significantly in the presence of Clstn3β (Fig. 6i), thus indicating that Clstn3β promotes secretion of S100b. S100b is highly unusual in that it is a secreted protein with no signal peptide, and hence the pathway of its secretion has been unclear. Importantly, Clstn3β did not promote secretion of Complement Factor D/adipsin (Fig. 6i; Extended Data Fig. 6k), a secreted protein abundantly expressed in adipocytes with a signal peptide.
The observation that Clstn3β promotes ER association of S100b suggests Clstn3β may directly bind S100b. We found that Clstn3β bound S100b much more strongly than Clstn3 (Fig. 6j). Our data thus suggest that Clstn3β but not Clstn3 may enhance secretion of S100b via direct physical interaction at the ER.
Discussion
Innervation by the sympathetic nervous system is important for both the development and activation of brown and potentially beige fat as well21–24. Very little has been known about the identity of adipocyte-derived factors that promote innervation of thermogenic fat. We show here the involvement of a novel protein, Clstn3β, which is selectively and abundantly expressed in thermogenic adipocytes and directs adipose innervation. Genetic ablation of Clstn3β leads to deficiency in adipose innervation and thermogenic function. Human adipose tissue also expresses Clstn3β (Extended Data Fig. 7), suggesting Clstn3β may play a similar role of promoting adipose innervation in humans as well.
We showed that Clstn3β resides mainly in the ER, yet is also involved in innervation. It appears to do so, at least in part, through driving secretion of S100b, a protein with trophic activity on sympathetic neurons. The secretion pathway of S100b is unclear as it does not have a signal peptide25. Our findings support a chaperone-like activity of Clstn3β on S100b, explaining why ablation of Clstn3β leads to reduction in S100b protein level in BAT. Understanding the detailed mechanism of non-canonical secretion requires more analysis. However, that S100b is utilized as a physiological neurotrophic factor is supported by both gain- and loss-of-function evidence. In addition to being regulated at the protein level by Clstn3β, S100b transcription is positively regulated by PRDM16, thereby providing an explanation for the altered levels of adipose sympathetic innervation observed in PRDM16 gain- and loss-of-function mouse models11,12. Interestingly, Clstn3β expression is not as sensitive to changes in PRDM16 levels.
Previous studies have suggested Nerve Growth Factor (NGF) or Brain-derived Neurotrophic Factor (BDNF) as factors potentially promoting sympathetic innervation of thermogenic fat26,27. Nevertheless, ribosome profiling data driven by adipose-specific Cre revealed extremely low to undetectable levels of mRNA of NGF and BDNF, indicating they may not be produced by adipocytes at a physiologically relevant level28,29. In contrast, Clstn3β and S100b are abundantly and selectively expressed in thermogenic adipocytes, which strongly supports their roles as adipocyte-derived neurotrophic factors. Our data do not exclude the possibility that NGF or BDNF also contribute to adipose innervation.
Our findings should advance the understanding of the basic biology of thermogenic fat and its innervation. Moreover, the identification of a soluble protein with neurotrophic actions on the sympathetic nervous system may also provide a therapeutic opportunity for promoting thermogenic fat activity for treating obesity and associated metabolic disorders.
Methods
Mouse strains.
Clstn3β KO mice were generated with the CRISPR-Cas9 technique. Two guide RNAs (CCCCAGCAGGAAGTGTAAACTGG, ATGGCCCCGTGGCTAAGCCCAGG) flanking the unique exon of Clstn3β were synthesized by PNA-BIO. They were microinjected with Cas9 enzyme (CP02, PNA-BIO) into fertilized eggs of C57BL/6 background at the transgenic core of Beth Israel Deaconess Medical Center. F1 progenies were genotyped with the following primers: CTGCAGGGAGAGCAAGGGGTCAG, GACAGTGCTTTCAACAGTGTCCC and CCAGGCAGAAAGACAGGAAGCTTC, with the WT allele yielding a band of 368 bp and the KO allele yielding a band of approximately 480 bp. F1 progenies carrying the KO allele were crossed with C57BL/6 purchased from Jackson Laboratory to screen for germ line transmission. F2 Clstn3β +/- male mice were then crossed with Clstn3β +/− to yield Clstn3β +/+ and −/− mice for analyses. To generate Clstn3β adipose-specific mice, the murine Clstn3β cDNA was cloned into a construct for integration into the Rosa26 locus as described previously. The construct was linearized with KpnI and microinjected into C57BL/6 embryonic stem cells. Successful integration of the transgene was validated using long-range PCR (5’-forward: CCTAAAGAAGAGGCTGTGCTTTGGG; 5’-reverse: TGGGCTATGAACTAATGACCCCG; 3’-forward: TTGCAGAAGATCTCCCCAACTGGG; 3’-reverse: CAGTGGCTCAACAACACTTGGTC). Positive clones were transferred into pseudo-pregnant albino C57BL/6J mice at the transgenic core of Beth Israel Deaconess Medical Center. F1 chimeras were screened for the presence of the Clstn3β transgene (forward: CCATCAAGCTGATCCGGAACC; reverse: TACAGACCTGGACCCTGAGGG) and positive males were crossed with albino C57BL/6J females to screen for germ line transmission. F2 that have both the Clstn3β transgene and black fur color were crossed with adiponectin-cre mice to generate adipose-specific transgenic mice. The adiponectin-cre and Vglut3-IRES-cre lines were maintained at the animal facility of Beth Israel Deaconess Medical Center. The S100b KO line was recovered from cryopreservation at RIKEN Institute, Japan and genotyping was performed following the protocol in the original publication. All mice were maintained under a 12 h light/12 h dark cycle at constant temperature (23°C) unless otherwise specified with free access to food and water. All animal studies were approved by and in full compliance with the ethical regulation of the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center. Male mice of 8–12 weeks of age were used for experiments. Sample size was chosen based on literature and pilot experiment results to ensure statistical significance could be reached. Randomization was not performed because mice were grouped based on genotype.
Clstn3β Antibody.
Rabbit polyclonal antibody to Clstn3β was generated with the C-terminal peptide DSPSSDERRIIESPPHRY with an N-terminal cysteine for conjugation to the carrier protein keyhole limpet hemocyanin and affinity purified with the antigenic peptide (New England Peptide; Covance).
Cryo-section immunostaining.
Tissue was harvested immediately after euthanizing mice and fixed with 4% paraformaldehyde overnight. Tissue was then washed with PBS for 5 times, 10 min each and incubated in PBS/30% sucrose for 8 h and then frozen in Tissue-Tek O.C.T. Compound (Sakura Finetek, 4583). Frozen tissue was cut into 30 μm sections on a Leica CM3050 S cryostat. Sections were briefly rinsed with PBS and blocked with PBS/0.3% Triton X-100/5% FBS overnight. Sections were then stained with Th antibody (AB1542, EMD Millipore, 1:200) and Tubb3 antibody (ab52623, Abcam, 1:200) for 2 days. Sections were washed with PBS/0.03% Triton X-100/5% FBS for 5 times, 1 h each and then stained with anti-sheep Alexa Fluor 488 (Thermo Fisher, A-11015, 1:500) and anti-rabbit Alexa Fluor 647 (Thermo Fisher, A-21245, 1:500) for 2 days. Sections were washed with PBS/0.3% Triton X-100/5% FBS for 5 times, 1 h each and then mounted in ProLong Diamond Antifade Mountant (Thermo Fisher, P36965). Images were taken on a Nikon A1R point scanning confocal microscope.
Image analysis.
For each animal, at least six sections of adipose tissues were imaged and analyzed. Acquired images were first cleaned up in ImageJ (NIH) using the “Despeckle” function and the “Background subtraction” function (rolling ball method, radius = 20). Images were then imported into Imaris (Bitplane, Zurich, Switzerland). TH or TUBB3 immunohistochemical signals were rendered by the Surface function, which draws contour surfaces over the fluorescent signals and thus extracts 3D models of the raw 3D z-stacked images. The volume of the extracted 3D objects generated by the surface function was used as the volume of TH or TUBB3 signals. Signals on and around blood vessels were manually selected and removed from the final quantifications. Volume and intensity measurements of Th/Tubb3 signals were normalized to the volume of the imaging field. In each experiment, the function parameters were kept the same between genotypes. For Pearson’s correlation analysis, images were imported in ImageJ. Individual cells were outlined manually and colocalization between KDEL and S100b were assessed by Pearson’s correlation coefficient using the Coloc2 plugin. Images were analyzed in a blinded manner.
Superior cervical ganglia culture.
SCG was dissected out and placed in Leibovitz’s L-15 media and then transferred to DMEM/4 mg/ml collagenase Type 2/1 mg/ml DNase I/10 mg/ml BSA and incubated at 37°C for 20 min. SCG was then centrifuged down and transferred to DMEM/3 mg/ml trypsin and incubated at 37°C for 15 min. SCG was then centrifuged down and triturated in DMEM/10% FBS to yield single cell suspension. Cells were then centrifuged down, resuspended in DMEM/10% FBS with or without 100 ng/ml S100b protein (Millipore, 559290).
Primary brown adipocyte culture.
BAT was dissected, minced and digested with Krebs Ringer Bicarbonate Modified Buffer (KRBMB, 120 mM NaCl, 5 mM KCl, 1 mM MgClRRR2RRR, 1 mM CaClRRR2RRR, 0.4 mM KRRR2RRRHPORRR4RRR, 10 mM glucose, 15 mM NaHCORRR3RRR, 20 mM HEPES, pH 7.4, 4% fatty acid free BSA) and 2 mg/ml collagenase B (Worthington, CLSAFB) and 1 mg/ml soybean trypsin inhibitor (Worthington, LS003570) for 30 min at 37°C. The mixture was centrifuged at 30 g for 10 min and the infranatant and SVF were removed with a syringe. Adipocytes were washed with DMEM/10% FBS for 3 times and added to a 24-well plate. Each well was filled to top with DMEM/10% FBS and covered with a coverslip pre-coated with poly-lysine and laminin. After overnight incubation at 37°C, the coverslip was flipped and placed in DMEM/10 % FBS. The cells were then subjected to adenoviral infection and imaging studies.
Apex2 EM imaging.
Clstn3β-Apex2 localization was detected by electron microscopy as described previously30. Briefly, cells expressing Clstn3β-APEX2 were fixed and incubated in a Tris buffer containing diaminobenzidine (DAB, 0.7 mg/ml) and HRRR2RRRORRR2RRR (0.7 mg/ml) (Sigma, D4293) for 15 min or until the cytoplasm developed the brown reaction product. After extensive washing, samples were post-fixed with 1% osmium tetroxide, dehydrated and embedded in EPON resin. 48 h later, coverslips were removed and areas containing cells were randomly selected and mounted. Ultrathin sections (70 nm) were cut and examined on a JOEL electron microscope.
Stereotaxic surgery and viral injections.
For viral injections into the medullary raphe, six- to eight-week-old male mice were anesthetized with a ketamine (100 mg kg−1) and xylazine (10 mg kg−1) cocktail diluted in 0.9% saline and placed into a stereotaxic apparatus (David Kopf model 940). An incision was made to expose the skull and a small hole was drilled 5.9 mm posterior and 0 mm medial/lateral from bregma. A pulled glass micropipette (20–40 μm diameter tip) was used for stereotaxic injections of adeno-associated virus (AAV). Three injections, 6.0 mm ventral to bregma, at medial/lateral -0.15 mm, 0 mm, and +0.15 mm were used to target the medullary raphe. Virus was injected (15 nl per injection) by an air pressure system using picoliter air puffs through a solenoid valve (Clippard EV 24VDC) pulsed by a Grass S48 stimulator to control injection speed (20 nl min−1). The pipette was removed 1 minute after each injection and, upon completion of the final injection, the incision was closed using Vetbond tissue adhesive (3M). Subcutaneous injection of sustained release Meloxicam (4 mg kg−1) was provided as postoperative care. Chemogenetic experiments utilized AAV8-hSyn-DIO-hM3Dq-mCherry packaged at the Boston Children’s Hospital Viral Core (Addgene plasmid 44361; donating investigator, Dr. Bryan Roth). Animals were allowed to recover from stereotaxic surgery a minimum of 21 days prior to initiation of any experiments. Following each experimental procedure, accuracy of AAV injections was confirmed via post-hoc histological analysis of mCherry fluorescent protein reporters. All subjects determined to be surgical “misses” based on little or absent reporter expression were removed from analyses.
Histology.
Mice were terminally anesthetized with 7% choral hydrate (500 mg kg−1; Sigma Aldrich) diluted in saline and transcardially perfused first with 0.1 M phosphate-buffered saline (PBS) then 10% neutral-buffered formalin solution (NBF; Thermo Fisher Scientific). Brains were extracted and post-fixed overnight at 4° C in NBF. The next day brains were switched to PBS containing 20% sucrose for cryoprotection. Finally, brains were sectioned coronally at 30 μm on a freezing microtome (Leica Biosystems), and stored in cryoprotectant solution at -20° C until used for immunofluorescence. For immunofluorescence, Brain tissue sections were washed 3X in PBS prior to a blocking step containing 3% normal donkey serum and 0.4% Triton X-100 in PBS for one hour at room temperature. Primary antibody was prepared in the same blocking solution and incubated overnight at the following concentrations: Rat anti-mCherry (Life Technologies - M11217) 1:3,000, Rabbit anti- c-Fos (EMD Millipore - ABE457) 1:3,000. The next day sections were washed 5X in PBS, then incubated for 2 hours at room temperature in Alexa Fluor fluorescent secondary antibody (Life Technologies; 1:1,000) prepared in blocking solution. Finally, sections were washed 3X in PBS, mounted on gelatin-coated slides, and coverslipped with Vectashield mounting media containing DAPI (Vector Labs). Fluorescent images were captured using an Olympus VS120 slide-scanning microscope.
Chemogenetic activation of BAT assay.
To monitor BAT response in real time, temperature probes (Bio Medic Data Systems, IPTT-300) were implanted into the interscapular region of mice and temperature was read with a transponder (Bio Medic Data Systems, DAS-7007R). To activate sympathetic neurons, mice were i.p. injected with saline or Clozapine N-oxide (Sigma, C0832) at 1 mg/kg.
BAT fractionation and Clstn3β isolation.
BAT fraction was performed based on a procedure previously described31. Briefly, whole BAT was dissected and homogenized in a dounce homogenizer in 10 ml 20 mM HEPES, 1 mM EDTA, 250 mM sucrose, pH 7.4 (HES buffer). The homogenate was filtered with two layers of cheesecloth and centrifuged at 16,000 g for 15 min. The fat layer was removed and the supernatant (S1) was saved. The pellet (P1) was resuspended in 5 ml HES and applied on top of a 1.12 M sucrose cushion containing 20 mM HEPES, 1 mM EDTA, pH 7.4, and centrifuged at 101,000 g for 70 min to yield a pellet (P2) and fluffy material at the interface (plasma membrane fraction). P2 was resuspended in 5 ml HES, centrifuged at 700 g to yield the nuclei pellet and the supernatant was centrifuged again at 8,000 g to yield the mitochondria pellet. S1 was centrifuged at 212,000 g for 70 min to yield the microsome pellet and the supernatant was the cytosol fraction. To isolate endogenous Clstn3β, the microsome pellet was solubilized in IP buffer (25 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% IGAPEL CA-630, 5% glycerol, pH 7.4) and incubated with Dynabeads protein G pre-loaded with Clstn3β antibody at 4°C for 2 h. The beads were washed with IP buffer for 3 times, 5 min each and incubated with IP buffer containing 100 μg/ml antigenic peptide at 4°C for 30 min.
AAV injection.
Six-week old male mice were anesthetized with isoflurane and an incision was made above the interscapular area to expose the underlying adipose tissue. About 3*10^11 AAV particles were injected into each BAT lobe and the incision was closed with suture. Mice received one injection of meloxicam (2 mg/kg) 24 h before surgery and another injection immediately after surgery. Mice were allowed to recover for 3 weeks before analysis. AAV8-EF1a-DIO-Clstn3β or S100b were packaged at the Boston Children’s Hospital Viral Core (Addgene plasmid 27056; donating investigator, Dr. Karl Deisseroth).
Cold tolerance assay.
Mice were pre-acclimated at thermoneutrality (28–30°C) for two weeks and then shifted to 4°C. Body temperature was measured with a rectal probe (Physitemp, RET3) and a reader (Physitemp, BAT-12).
S100b assays.
To detect binding between S100b and Clstn3β/Clstn3, HEK293T cells transfected with pcDNA, pcDNA-Clstn3β-FLAG or pcDNA-Clstn3-FLAG were homogenized with IP buffer (25 mM HEPES 150 mM NaCl, 1 mM CaClRRR2RRR, 1% Lauryl maltose-neopentyl glycol, 5% glycerol, pH 7.4) and centrifuged at 16,000 g for 10 min. The supernatant was incubated with anti-FLAG M2 resin (Sigma, A2220) at 4°C for 1 h and the resin was washed with binding buffer (same as IP buffer except Lauryl maltose-neopentyl glycol concentration is reduced to 0.02%) for 3 times, 5 min each. The resin was then incubated with 10 μg/ml recombinant S100b (R&D Systems, 1820-SB-050) in binding buffer at 4°C for 30 min and then washed binding buffer for 3 times, 5 min each. Bound proteins were eluted with 150 μg/ml 3x FLAG peptide (Sigma, F4799) in binding buffer at 4°C for 30 min. To assess S100b secretion, media were collected from cells expressing S100b and centrifuged at 16,000 g for 20 min. The supernatant was subjected to S100b quantitation with an ELISA kit (Millipore, EZHS100B-33K). S100b antibody (Abcam, ab51642) was used for immunofluorescence and Western blot analysis. HEK293T cell line used in these experiments was purchased from ATCC and maintained in the Spiegelman laboratory. This cell line has been routinely tested negative for mycoplasma. Authentication of this cell line was not done for this particular study.
Quantitative PCR.
The following primers were used for qPCR analysis of gene expression. Clstn3β-fwd, CTCCGCAGGAACAGCAGCCC; rev, AGGATAACCATAAGCACCAG; S100b-fwd, TGGTTGCCCTCATTGATGTCT; rev, CCCATCCCCATCTTCGTCC. Primers for other genes were described previously13.
ChIPseq and metabolic assays.
All ChIP and metabolic experiments were performed as previously described13.
Whole-mount immunofluorescence.
Th whole-mount immunostaining of subcutaneous inguinal WAT was performed as described in Chi et al12. The inguinal rather than the dorsolumbar region was selected for imaging.
Mass spectrometry analysis.
Quantitative whole tissue proteomics analysis was performed as previously described13.
Extended Data
Extended Data Fig. 1. Clstn3β Encodes an Adipocyte-Specific Protein.

a, qPCR analysis of Clstn3β expression in WT and Lsd1 KO BAT (n=3 mice). b, Histone marker and transcription regulator ChIP-seq at the Clstn3 locus from BAT. c, qPCR analysis of Clstn3β expression in inguinal subcutaneous WAT from mice acclimated to RT or 4°C (n=4 mice). d, Mass spectrometry identification of Clstn3β peptides. e, Conservation of Clstn3β within the mammalian class. Red cross/green tick indicates the absence/presence of homologs of Clstn3β in subclasses. f, Sequence alignment between the unique exon of Clstn3β from human and a fragment in an intron upstream of the penultimate exon of Clstn1 in the genome of Chinese softshell turtle. Note how the position of this fragment correponds to the β-selective exon in Clstn3. All data are mean ± SEM. Statistical significance was calculated by unpaired Student’s two-sided t-test.
Extended Data Fig. 2. Clstn3β Localizes to the Endoplasmic Reticulum.

a, b, EM analysis of primary brown adipocytes expressing Clstn3β-Apex2. a, arrows denote the Golgi apparatus. b, arrows denote peroxisomes. Scale bar: 100 nm. c, Western blot analysis of the fractionation pattern Clstn3β.
* denotes a nonspecific band. For gel source data, see Supplementary Figure 1.
Extended Data Fig. 3. Ablation of Clstn3β Impairs Adipose Thermogenesis.

a, b, c, d, Sanger sequencing (a), Western blot (b), qPCR (c) (n=4 mice) and immunofluorescence (d) confirmation of CRISPR-Cas9 deletion of Clstn3β. Scale bar: 10 μm. e, qPCR analysis of Clstn3 expression in a panel of WT mouse tissues and WT/Clstn3β KO brain (n=2 mice for surveying tissue specificity in WT mouse and n=3 mice WT and KO). The primers target the junction between the third and the second last exons. f, g, Body weight curve (f) and body composition (g) of WT and Clstn3β KO mice on chow diet (n=8 mice). h, CORRR2RRR production rates from indirect calorimetry analysis of WT and Clstn3β KO mice (n=6 mice). i, j, Movement (i) and daily food intake (j) of WT and Clstn3β KO mice in metabolic chambers (n=6 mice). k, Oxygen consumption response to acute β3 agonist injection of WT and Clstn3β KO mice (n=6 mice). All data are mean ± SEM. Statistical significance was calculated by unpaired Student’s two-sided t-test.
Extended Data Fig. 4. Transgenic Expression of Clstn3β Enhances Adipose Thermogenesis.

a, b, Western blot (a) and qPCR (b) confirmation of transgenic overexpression of Clstn3β in BAT (n=5 mice). c, d, Body weight curve (c) and body composition (d) of WT and Clstn3β transgenic mice on chow diet (n=6 mice). e, CORRR2RRR production rates from indirect calorimetry analysis of WT and Clstn3β transgenic mice (n=4 mice). f, g, Movement (f) and daily food intake (g) of WT and Clstn3β transgenic mice in metabolic chambers (n=4 mice). h, Oxygen consumption response to acute β3 agonist injection of WT and Clstn3β transgenic mice (n=4 mice). All data are mean ± SEM. Statistical significance was calculated by unpaired Student’s two-sided t-test.
Extended Data Fig. 5. Clstn3β Enhances Sympathetic Innervation of Thermogenic Adipose Tissue.

a, Gene expression analysis of WT and Clstn3β KO BAT upon 5-hour acute cold exposure following pre-acclimated at thermoneutrality (n=4 mice). b, Indirect calorimetry analysis of Clstn3β KO mice with or without adiponectin-cre receiving AAV-DIO-Clstn3β AAV injection (n=4 mice). c, Whole-mount TH staining of the inguinal region of the posterior subcutaneous white adipose tissue from WT and Clstn3β KO mice acclimated at 4°C for one week. Scale bar: 50 μm. All data are mean ± SEM. Statistical significance was calculated by unpaired Student’s two-sided t-test.
Extended Data Fig. 6. Clstn3β Promotes Secretion of S100b, an Adipocyte-derived Neurotrophic Factor.
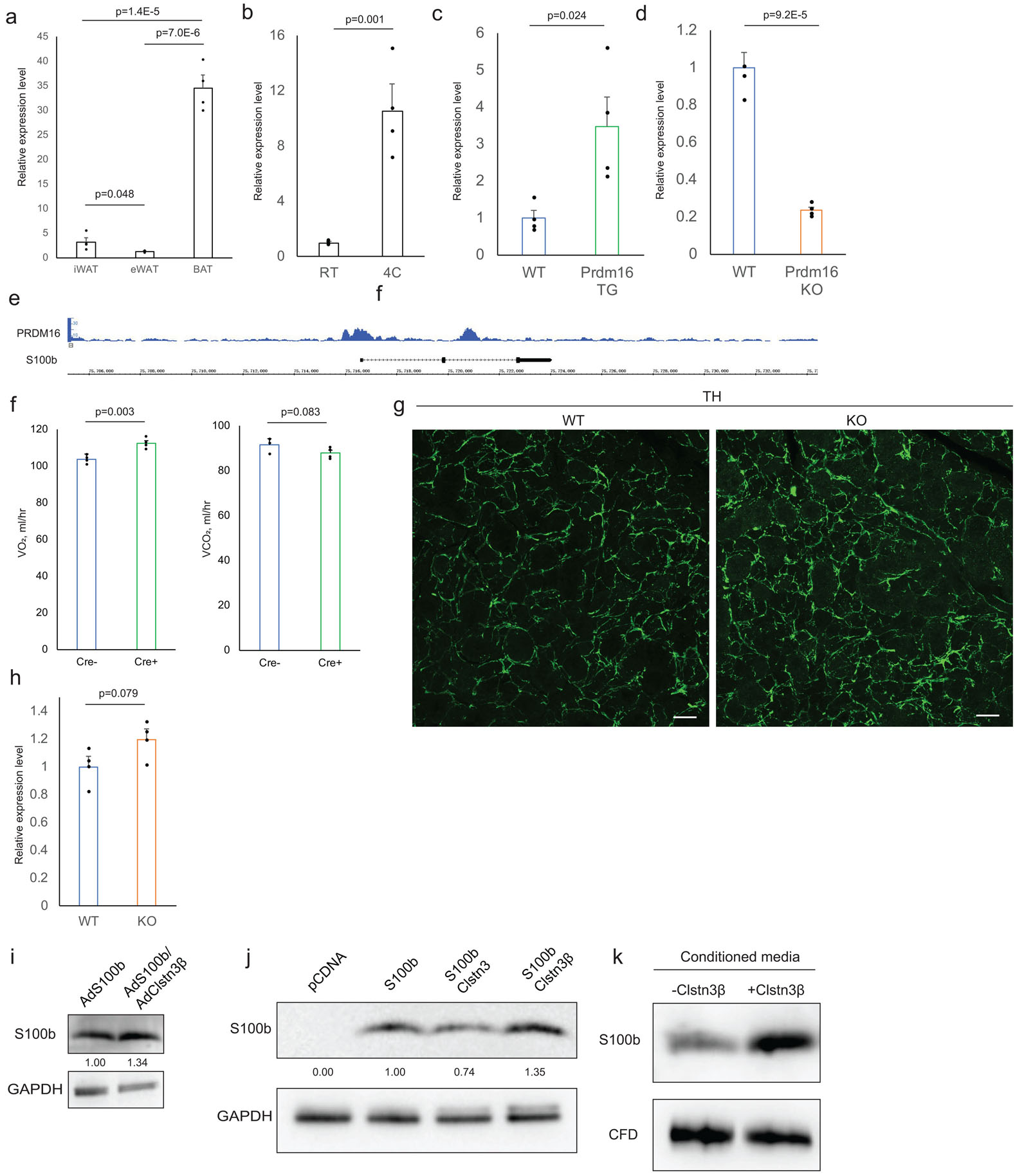
a, b, qPCR analysis of S100b expression in (a) various fat depots and in (b) inguinal subcutaneous WAT from mice acclimated to RT or 4°C (n=4 mice). c, qPCR analysis of S100b expression in control or Prdm16-transgenic inguinal subcutaneous WAT (n=4 mice). d, qPCR analysis of S100b expression in control or Prdm16-KO inguinal subcutaneous WAT (n=4 mice). e, Prdm16 ChIP-seq showing binding at the S100b locus. f, Indirect calorimetry analysis of Clstn3β KO mice with or without adiponectin-cre receiving AAV-DIO-S100b AAV injection (n=4 mice). g, TH immunostaining of salivary gland from WT and S100b KO mice. h, qPCR analysis of S100b expression in WT and Clstn3β KO BAT from mice housed at RT (n=4 mice). Note this is a different housing condition from Extended Data Fig. 5a. i, Western blot analysis of intracellular level of S100b in Clstn3β KO brown adipocytes expressing S100b lone or co-expressing with Clstn3β. j, Western blot analysis of S100b protein level in HEK293T cells transfected with various constructs as indicated. k, Western blot analysis of S100b and CFD secretion from HEK293T cells co-transfected with or without Clstn3β. All data are mean ± SEM. Statistical significance was calculated by unpaired Student’s two-sided t-test.
Extended Data Fig. 7. Clstn3β Is Specifically Expressed in Human Adipose Tissue.

Human tissue RNA-seq showing adipose-specific expression of Clstn3β. RNAseq of 13 human tissue types were analyzed for reads uniquely mapped to the Clstn3β-specific exon.
Supplementary Material
Supplementary Table Proteomics analysis of WT and Clstn3β KO BAT from mice housed at RT (n=4 mice). Statistical significance was calculated by unpaired Student’s two-sided t-test.
Acknowledgements
We thank Nikon Imaging Center at Harvard Medical School for all imaging studies; RIKEN Institute, Japan, for sharing the S100b KO strain; Zachary Herbert and the Molecular Biology Core Facilities at Dana Farber Cancer Institute for sequencing studies; the Rodent Histology Core at Harvard Medical School for histology studies; the EM Core at Harvard Medical School for Apex2 imaging studies; the viral core at Children’s Hospital Boston for AAV production; the transgenic core at Beth Israel Deaconess Medical Center for generation of mouse models; Ying Zhu for advice on sequencing data analysis. Xing Zeng was supported by the American Heart Association postdoctoral fellowship. Bo Hu is a Cancer Research Institute/Leonard Kahn Foundation Fellow. David Ginty is an investigator of the Howard Hughes Medical Institute. This study was supported by NIH grant DK31405 to B.M.S.
Footnotes
Data Availability Statement
Histone modification marker and transcription factor ChIP-seq datasets generated in this study are available at NIH SRA under the following accession code: PRJNA526243.
Statistics and Reproducibility
All data shown are mean ± SEM. Statistical significance was calculated by unpaired Student’s two-sided t-test. All experiments have been successfully repeated with similar results for at least three times.
Competing interests
The authors declare no competing interests.
References
- 1.Cannon B & Nedergaard J Brown adipose tissue: function and physiological significance. Physiological reviews 84, 277–359, doi: 10.1152/physrev.00015.2003 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Seale P & Lazar MA Brown Fat in Humans: Turning up the Heat on Obesity. Diabetes 58, 1482–1484, doi: 10.2337/db09-0622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrison SF Central neural control of thermoregulation and brown adipose tissue. Autonomic Neuroscience 196, 14–24, doi: 10.1016/j.autneu.2016.02.010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeng W et al. Sympathetic Neuro-adipose Connections Mediate Leptin-Driven Lipolysis. Cell 163, 84–94, doi: 10.1016/j.cell.2015.08.055 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Fedorenko A, Lishko PV & Kirichok Y Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413, doi: 10.1016/j.cell.2012.09.010 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kazak L et al. A Creatine-Driven Substrate Cycle Enhances Energy Expenditure and Thermogenesis in Beige Fat. Cell 163, 643–655, doi: 10.1016/j.cell.2015.09.035 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniel H & Derry DM Criteria for differentiation of brown and white fat in the rat. Canadian Journal of Physiology and Pharmacology 47, 941–945, doi: 10.1139/y69-154 (1969). [DOI] [PubMed] [Google Scholar]
- 8.Seale P et al. Transcriptional control of brown fat determination by PRDM16. Cell metabolism 6, 38–54, doi: 10.1016/j.cmet.2007.06.001 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen P et al. Ablation of PRDM16 and Beige Fat Causes Metabolic Dysfunction and Subcutaneous to Visceral Adipose Swtich. Cell (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang W & Seale P Control of brown and beige fat development. Nature Reviews Molecular Cell Biology 17, doi: 10.1038/nrm.2016.96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seale P et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. The Journal of clinical investigation 121, 96–105, doi: 10.1172/jci44271 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chi J et al. Three-Dimensional Adipose Tissue Imaging Reveals Regional Variation in Beige Fat Biogenesis and PRDM16-Dependent Sympathetic Neurite Density. Cell metabolism 27, 226–236000, doi: 10.1016/j.cmet.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Zeng X et al. Lysine-specific demethylase 1 promotes brown adipose tissue thermogenesis via repressing glucocorticoid activation. Genes & Development 30, 1822–1836, doi: 10.1101/gad.285312.116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pettem KL et al. The Specific α-Neurexin Interactor Calsyntenin-3 Promotes Excitatory and Inhibitory Synapse Development. Neuron 80, 113–128, doi: 10.1016/j.neuron.2013.07.016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saunders A, Johnson CA & Sabatini BL Novel recombinant adeno-associated viruses for Cre activated and inactivated transgene expression in neurons. Frontiers in Neural Circuits 6, 47, doi: 10.3389/fncir.2012.00047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura K et al. Identification of Sympathetic Premotor Neurons in Medullary Raphe Regions Mediating Fever and Other Thermoregulatory Functions. The Journal of Neuroscience 24, 5370–5380, doi: 10.1523/jneurosci.1219-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng L et al. Identification of spinal circuits involved in touch-evoked dynamic mechanical pain. Nature Neuroscience 20, 804–814, doi: 10.1038/nn.4549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reeves RH et al. Astrocytosis and axonal proliferation in the hippocampus of S100b transgenic mice. Proceedings of the National Academy of Sciences 91, 5359–5363, doi: 10.1073/pnas.91.12.5359 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winningham-Major F, Staecker JL, Barger SW, Coats S & Eldik VLJ Neurite extension and neuronal survival activities of recombinant S100 beta proteins that differ in the content and position of cysteine residues. The Journal of Cell Biology 109, 3063–3071, doi: 10.1083/jcb.109.6.3063 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishiyama H, Knöpfel T, Endo S & Itohara S Glial protein S100B modulates long-term neuronal synaptic plasticity. Proceedings of the National Academy of Sciences 99, 4037–4042, doi: 10.1073/pnas.052020999 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steiner G, Loveland M & Schonbaum E Effect of denervation on brown adipose tissue metabolism. The American journal of physiology 218, 566–570 (1970). [DOI] [PubMed] [Google Scholar]
- 22.Bachman ES et al. βAR Signaling Required for Diet-Induced Thermogenesis and Obesity Resistance. Science 297, 843–845, doi: 10.1126/science.1073160 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Dulloo AG & Miller DS Energy balance following sympathetic denervation of brown adipose tissue. Canadian Journal of Physiology and Pharmacology 62, 235–240, doi: 10.1139/y84-035 (1984). [DOI] [PubMed] [Google Scholar]
- 24.Cao Y, Wang H & Zeng W Whole-tissue 3D imaging reveals intra-adipose sympathetic plasticity regulated by NGF-TrkA signal in cold-induced beiging. Protein & Cell 9, 527–539, doi: 10.1007/s13238-018-0528-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donato R et al. S100B’s double life: Intracellular regulator and extracellular signal. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1793, 1008–1022, doi: 10.1016/j.bbamcr.2008.11.009 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Nisoli E, Tonello C, Benarese M, Liberini P & Carruba MO Expression of nerve growth factor in brown adipose tissue: implications for thermogenesis and obesity. Endocrinology 137, 495–503, doi: 10.1210/en.137.2.495 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Sornelli F, Fiore M, Chaldakov GN & Aloe L Adipose tissue-derived nerve growth factor and brain-derived neurotrophic factor: results from experimental stress and diabetes. General physiology and biophysics 28 Spec No, 179–183 (2009). [PubMed] [Google Scholar]
- 28.Chen Y et al. Crosstalk between KCNK3-Mediated Ion Current and Adrenergic Signaling Regulates Adipose Thermogenesis and Obesity. Cell 171, 836, doi: 10.1016/j.cell.2017.09.015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long JZ et al. A Smooth Muscle-Like Origin for Beige Adipocytes. Cell Metabolism 19, 810–820, doi: 10.1016/j.cmet.2014.03.025 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lam SS et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nature Methods 12, 51–54, doi: 10.1038/nmeth.3179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simpson IA et al. Insulin-stimulated translocation of glucose transporters in the isolated rat adipose cells: characterization of subcellular fractions. Biochimica et biophysica acta 763, 393–407 (1983). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table Proteomics analysis of WT and Clstn3β KO BAT from mice housed at RT (n=4 mice). Statistical significance was calculated by unpaired Student’s two-sided t-test.