

Abstract
Adoptive cell therapy with chimeric antigen receptor (CAR)‐engineered T cells produced lasting remissions in the treatment of advanced, so far refractory B‐cell malignancies; however, the elimination of solid tumors remains so far elusive. The low efficacy of CAR T cells is thought to be due to the immune‐repressive milieu within the tumor lesion, predominantly mediated by transforming growth factor‐β (TGF‐β) that represses effector T‐cell activities and drives differentiation towards regulatory T cells (Tregs). Seeking to boost antitumor immunity, TGF‐β is currently targeted by different means in pre‐clinical studies. While a recent clinical trial showed the utility of shielding CAR T cells from TGF‐β repression, further strategies in counteracting TGF‐β in the adoptive cell therapy warrant exploration. We here discuss the most recent advances in the field and draw future developments to make CAR T‐cell therapy more potent in the treatment of solid cancer.
Keywords: adoptive T‐cell therapy, chimeric antigen receptor, immunosuppression, immunotherapy, TGF‐β
Introduction
The outstanding success of chimeric antigen receptor (CAR)‐redirected T‐cell therapy has been noticeably achieved since its introduction in the treatment of hematologic malignancies, such as lymphoma or leukaemia; complete and lasting remissions were induced after transfer of CD19‐specific CAR T cells. However, unfortunately the same cannot be noted for the treatment of solid tumors.1 In the case of a solid tumor and in comparison with hematological cancer, CAR T cells have to infiltrate the stromal elements and to reach and eliminate the recognised cancer cell. While a number of targetable tumor antigens on solid cancer cells are known, the current pre‐clinical research and first clinical observations revealed low antitumor efficacy of the specifically redirected CAR T‐cell therapies.
In the event of CAR T cells successfully locating cognate cancer cells within the solid tumor lesion, they turn into dysfunctional cells. There are three known significant reasons for this. Firstly, the liberated intratumoral environment is presented around the tumor cells with hypoxia, oxidative stress, acidic pH and low levels of nutrients providing unsuitable conditions for a sustained T‐cell antitumor response.2, 3 Secondly, it is due to the presence of suppressive immune cells such as regulatory T cells (Tregs), myeloid‐derived suppressor cells (MDSCs), M2‐polarised tumor‐associated macrophages (TAMs) and N2‐polarised tumor‐associated neutrophils (TANs). These suppressive cells hold responsibility for the production of T‐cell inhibitory soluble factors and inhibitory cytokines. Among these inhibitory cytokines is transforming growth factor‐β (TGF‐β) produced by tumor cells, Tregs, MDSCs, M2‐polarised TAMs and N2‐polarised TANs accumulating to substantial concentrations within the tumor lesion4, 5 TGF‐β is a key regulator of immune homeostasis; however, the presence of this cytokine is considered a major reason for the lack of success in the immunotherapy for solid tumors. This is based on the following observations: (1) human T cells are susceptible to TGF‐β‐mediated immune suppression; (2) elevated levels of TGF‐β occur in a variety of solid tumors; and (3) TGF‐β production correlates with poor prognosis. The last and the third significant reason believed to be upholding the success of tumor treatment is the induced regulatory mechanisms on the infiltrating, activated CAR T cells, including upregulation of cytoplasmic and surface inhibitory receptors such as PD‐1 or CTLA‐41, 6, making these cells susceptible to repression by tumor‐expressed inhibitory ligands. TGF‐β directly inhibits T‐cell activity through binding to the TGF‐β receptors TGFBR1 and TGFBR2. TGF‐β binding induces hetero‐dimerization of the respective receptors and phosphorylation of the major TGF‐β signal mediators SMAD2 and SMAD3. Phosphorylated SMADs induce a suppressive transcriptional programme that results in reduced cytokine production, cytotoxicity and T‐cell amplification upon antigen engagement. TGF‐β also7 drives T‐cell differentiation into regulatory T cells8 that, in turn, produce TGF‐β and further promote immune repression and tumor tolerance.
Immunosuppression by TGF‐β is thought to be the major cause of unsuccessful antitumor activity
Extensive research has gone into understanding the crucial role of TGF‐β in tumor progression, metastasis and treatment. Within these effects, TGF‐β is responsible for the regulation of cancer cell amplification, the contribution to epithelial‐to‐mesenchymal transition (EMT), the ability to compromise immune response by the suppression of pro‐inflammatory immune cells, the conversion of fibroblasts to myofibroblasts and the increased production of extracellular matrix (ECM) in the tumor. Although, under physiological conditions, a continuous basal release of TGF‐β enables sustainable normal tissue homeostasis. However, under a change of conditions, for example tissue injury, the local TGF‐β secretion sourced from stromal cells and blood platelets increases rapidly in order to facilitate wound repair and to prevent uncontrolled regenerative cell proliferation and inflammation. This situation is commonly repeated in pre‐malignant tumors, and TGF‐β is secreted in the microenvironment with the intention to control cell proliferation and cancer progression. However, accumulated TGF‐β is utilised by the cancer cells for the promotion of their malignant properties. The local release of TGF‐β impacts the tumor microenvironment, which is advantageous to amplification, invasion and metastasis of cancer cells.9 Therefore, the interest of research about this cytokine is high in order to prevent immunosuppression.
Transforming growth factor‐β is a multifunctional polypeptide that holds importance within proliferation, differentiation, angiogenesis, embryonic development and wound healing along with many other functions within different cell types.10 TGF‐β comes along in three known isoforms, TGF‐βI, TGF‐βII and TGF‐βIII, and is home to a vast superfamily with 33 known human family participants, including bone morphogenetic proteins (BMPs), activins and inhibins, growth and differentiation factors (GDFs) and11 the members of the family are evolutionary conserved proteins, and there is 70‐80% homology among the TGF‐β isoforms. Among the isoforms, TGF‐βI is responsible for the largest impact on the regulation of the immune response by binding to the specific receptors on the respective immune cells.
There are three known types of TGF‐β receptors responsible for initiating downstream signalling, and these are TGF‐βRI, TGF‐βRII and TGF‐βRIII. There are seven known TGF‐βRI, five known TGF‐βRII and two known TGF‐βRlll that are established thus far. TGF‐βIs include activin receptor‐like kinases 1‐7 (ALK1‐7), TGF‐βRlls contain bone morphogenetic protein receptor‐2 (BMPRll) and activin receptor 2 (ACTR2). Finally, beta‐gycan and endoglin belong to TGF‐βRIII, and this is mainly responsible for functioning as co‐receptors to enhance activin signalling. In the majority of tissues, TGF‐β functions through the heteromeric complex formation between two TGF‐βRI and two TGF‐βRII molecules. Although both receptors inhibit Ser/Thr kinase activity, TGF‐βRIIs’ responsibility is to function as the activator, whereas TGF‐βRI holds the signalling propagating component.12 The TGF‐β and TGF‐β‐like cytokines mediate downstream intracellular signalling via the SMADs family of proteins, consisting of eight structurally related members in humans.13, 14 There are three known functionally classified groups of SMADs: the receptor‐activated SMADs (R‐SMADs), which include SMAD1, 2, 3, 5 and 8; the common mediator SMAD (Co‐SMAD), SMAD4; and the inhibitory SMADs (I‐SMADs), which include SMAD6 and 7. The TGF‐β signalling pathway can be categorised, one being the canonical or SMAD‐dependent and the second, the non‐canonical or SMAD‐independent pathway.
Because of the antagonistic function within immune cells, TGF‐β upholds immunosuppressive effects on all arms of the immune system.5 Therefore, as a result the initial pro‐inflammatory antitumor immune response of activated CD4+ and CD8+ T cells is compromised, resulting in failure to identify and kill cancer cell. More so, TGF‐β affects the function of natural killer (NK) cells, macrophages, neutrophils, dendritic cells (DCs), mast cells and B cells.15 More specifically, a tumor microenvironment rich in TGF‐β is a potent suppressor of T‐cell proliferation, which both reduces their effector function and inhibits the maturation of T helper cells.16, 17 TGF‐β within the tumor microenvironment also has the ability to induce macrophage polarisation from type M‐1 to an M‐2 phenotype, which hinders suppression of monocyte‐mediated cell death, reduces effector functions and increases chemotaxis.18 Also, TGF‐β induces an N2 neutrophil phenotype; this along with macrophages has the ability to reduce effector functions along with an increased secretion of pro‐inflammatory cytokines.19 Moreover, high levels of TGF‐β increase entry into apoptosis of B cells and inhibit the maturation of DCs and NK cells, and there is also the possibility of inducing chemotaxis of mast cells. Finally, these combined immunosuppressive effects of TGF‐β affect the ability of the host to counteract tumor progression and thereby create a barrier to immunotherapy.
Strategies to overcome TGF‐β repression
Research has indicated the suppressive tumor microenvironment inhibits CAR T‐cell functions, preventing the execution of antitumor reactions and the induction of a pro‐inflammatory immune response. The need to overcome immune suppression by counteracting TGF‐β signalling in order to provide a more supportive microenvironment and to increase the success of effector T cells in an antitumor attack has increased because of a better understanding of the effect of TGF‐β directly on CAR T cells and indirectly on other immune cells within the microenvironment. We here outline, describe and compare the current suppressive techniques using CAR T cells in the treatment of solid cancer lesions (Figure 1). The details of each technique along with a comparison will lead to an understanding of the differences, similarities, limitations, advantages and disadvantages of each technique; the review discusses what potentially holds the future for CAR T‐cell therapy in association with TGF‐β expressing solid tumors.
Figure 1.
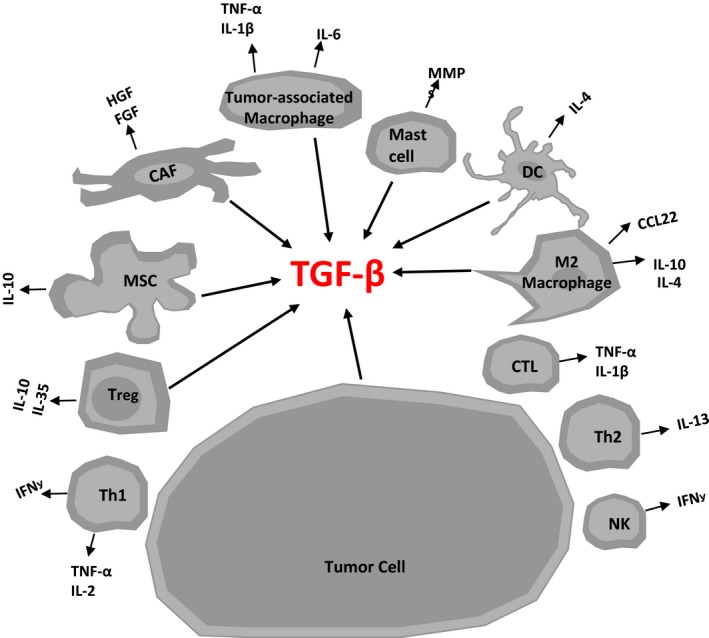
Schematic illustration identifying the major sources of transforming growth factor‐β (TGF‐β) within the tumor microenvironment. Major cell compartments producing TGF‐β in a solid tumor lesion. The figure also shows a visual representation of other cytokines secreted in tumor microenvironment from various cells. CAF, cancer‐associated fibroblast; CTL, cytotoxic lymphocytes; DC, dendritic cell; MSC, mesenchymal stem cells; NK, natural killer cell; Th1, T helper 1 cell; Th2, T helper 2 cell; Treg, T regulatory cell.
CD28 signalling CAR
It has been previously shown that CAR T‐cell repression through TGF‐β can be overcome by CD28 co‐stimulation provided by a CD28‐ζ CAR upon engagement of cognate antigen while 4‐1BB‐ζ CAR and first‐generation CD3ζ CAR T cells suffered suppression.20 In order to identify how CD28‐ζ CAR T cells were able to overcome TGF‐β, Golumba‐Nagy et al. (2018)21 identified the importance of LCK activation and the subsequent IL‐2 release along with the autocrine IL‐2 receptor signalling, all crucial components in overcoming TGF‐β repression. Since IL‐2 released by activated CD28‐ζ CAR T cells would sustain tumor‐infiltrating Treg cells, which are a major source of TGF‐β, and thereby furthermore augment immune repression, research explored the possibility of replacing IL‐2 with other γ‐cytokines and introducing a hybrid receptor that binds the γ‐cytokine and transmits the IL‐2 receptor signal, thereby mimicking IL‐2 signalling. CAR‐induced IL‐2 release can be prevented by deleting the LCK binding domain within the intracellular CD28 signalling chain.22 Using such IL‐2‐deficient CAR, an additionally engineered autocrine loop with the release of transgenic IL‐7, replacing IL‐2 and IL‐2 receptor β chain signalling upon IL‐7 binding to the extracellular IL‐7 receptor α chain, has the ability to improve antitumor efficiency of CAR T cells in a TGF‐β environment independently of IL‐2.21 With the artificial autocrine loop, replacing IL‐2 with IL‐7 and a hybrid receptor binding IL‐7 and signalling through IL‐2 receptor, T cells become activated upon binding to antigen via the CAR, and amplify and execute cytolysis along with releasing of pro‐inflammatory cytokines such as IFN‐γ (Figure 2).
Figure 2.
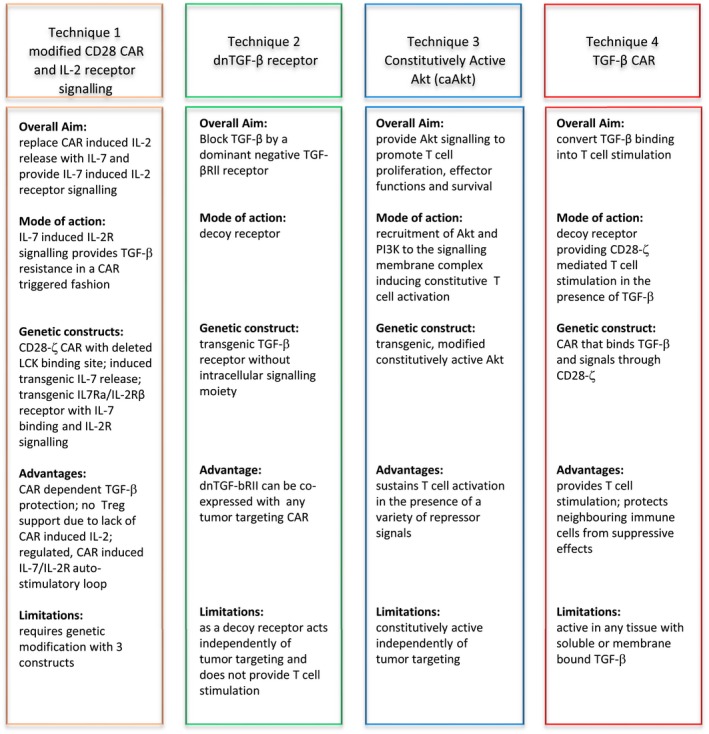
Techniques to overcome transforming growth factor‐β‐mediated repression of chimeric antigen receptor T cells in solid tumors.
Conclusive findings from this study include the suggestion of a co‐release of IL‐7 by CAR T cells is the most likely safer clinical method. The reason being is a systematic administration of this cytokine in patients with cancer for the treatment of lymphopenia did not show any severe side effects, but indeed provided a beneficial increase in CD4+ and CD8+ T‐cell numbers that resulted in a decrease in Treg cells.23 The hybrid IL‐7Rα/IL‐2Rβ receptor is advantageous in the way it enables constitutively the expression of the receptor and translates extracellular IL‐7 binding into intracellular IL‐2 signalling. Therefore, CAR T cells with an IL‐7 release and hybrid IL‐7Rα/IL‐2Rβ receptor portrayed improved survival over a continuous period.21 A technical disadvantage of the strategy is engineering an IL‐7 expression cassette together with the hybrid IL‐7Rα/IL‐2Rβ receptor and engineering the respective CAR. T‐cell modification with a high capacity vector will be required in the near future to conform to good manufacturing practices.
Along this line, Boyerinas et al. (2017) reported the co‐expression of TGFBR2 and TGFBR1 receptor variants where the TGF‐β‐binding domain of each receptor is fused to the intracellular signalling domains of a T‐cell‐stimulating interleukin receptor that induce STAT transcription factor signalling pathways. Using IL‐12 receptor signalling domains in such chimeric receptors, STAT4 signalling, and finally increase in IFN‐γ release, is induced, resulting in superior T‐cell persistence and expansion in the presence of TGF‐β.7
Co‐expression of the dnTGF‐β receptor
In contrast to CD28‐ζ CAR, T cells with the 4‐1BB‐ζ CAR are sensitive to repression by TGF‐β, which makes an alternative strategy necessary. The strategy established by Kloss et al. (2018)24 was that co‐expressing a dominant‐negative (dn) TGF‐βRII along with a PSMA‐specific CAR in T cells results in enhanced infiltration, proliferation, persistence and efficiency in the presence of TGF‐β.24 The dnTGF‐βRII thereby acts as a decoy receptor binding TGF‐β without downstream signalling. The study also identified a distinct transcriptional programme in the T cells expressing PSMA CAR and the dnTGF‐βRll in the presence of TGF‐β, indicating a successful protection from TGF‐β‐mediated repression of T‐cell effector functions. This is moreover sustained by enhanced secretion of a panel of pro‐inflammatory cytokines by dnTGF‐βRll CAR T cells that are expected to enhance innate and acquired immunity and finally antitumor response. The cytokine profile demonstrates a balanced TH2 phenotype with IL‐4, IL‐5 and IL‐13 expression, as well as TH1 phenotype with IL‐2, IFN‐γ and chemokines MIP1‐α, MIP1‐β and RANTES. Consequently, such engineered CAR T cells show a prolonged amplification in the presence of TGF‐β. However, such cytokine‐driven proliferation of CAR T cells can result in lymphoproliferative syndromes as observed in pre‐clinical mouse models25; the actual risk in a clinical situation still needs to be explored. A clinical trial has begun to infuse dnTGF‐βRII and PSMA‐specific 4‐1BB‐ζ CAR T cells in a first‐in‐human study in patients with refractory castration‐resistant metastatic prostate cancer (ClinicalTrials.gov: NCT03089203) with the aim to sustain and augment CAR T‐cell proliferation and effector function within the solid tumor lesions.
One significant difference between this technique and the CD28 CAR is that CARs with any signalling chain can be used within this technique like 4‐1BB‐ζ and CD3ζ signalling CARs. The study reports success in the way that the PSMA‐specific, TGF‐β‐insensitive CAR T cells exhibit a proliferative advantage over wild‐type PSMA CAR T cells that fully transmit TGF‐β signalling, suggesting a possibility of clinical success because of the known difficulty of CAR T‐cell expansion in patients with solid tumors.26
Constitutively active Akt (caAkt)
The serine/threonine kinase Akt/protein kinase‐B regulates T‐cell amplification, effector functions and survival; the constitutively active Akt was hypothesised to counteract a variety of tumor‐associated repressive mechanisms. The hypothesis is based on the crucial role of Akt as a major component of the phosphatidylinositol 3‐kinase (PI3K) family and thereby acts downstream of both the TCR and the cytokine receptor signalling pathways.27 On the one hand, CD28 signalling recruits PI3K and Akt to the membrane and activates tyrosine kinases required for T‐cell activation. On the other hand, Akt repression is a crucial step in the signalling pathway of multiple inhibitory receptors including PD‐1 and CTLA‐4 by counteracting tyrosine phosphatases. The activation balance of the PI3K/Akt pathway is a crucial determinant to a number of inhibitors including TGF‐β. Consequently, engineered T cells with constitutively active Akt, also named MF‐ΔAkt and being dual‐acetylated and targeted to the lipid rafts, showed sustained NF‐kB expression and increased amplification, cytokine release and resistance to apoptosis. Along with the highly activated state, caAkt T cells showed superior persistence and cytolytic activities in the presence of TGF‐β+ tumor cells and resistance to repression by cytokine deprivation, TGF‐β and the presence of Treg cells.27 With respect to safety, caAkt‐engineered T cells did not amplify autonomously, but were still dependent on cytokine and antigen stimulation; however, they showed prolonged survival and elongated telomers.27 While caAkt can compensate for subthreshold activation signals, caAkt did not furthermore increase T‐cell functions upon adequate stimulation.
Transforming growth factor‐β in the presence of stimulatory signals promotes the generation of inducible Treg cells; Akt is a strong repressor into the Treg phenotype.28 Consequently, caAkt T cells showed a low frequency of conversion to Treg cells in the presence of TGF‐β, which, in the context of prolonged survival, provides improved elimination of cognate cancer cells in the long‐term.
TGF‐β CAR T cells
Recently, Chang et al. (2018)29 reported a CAR that binds TGF‐β by a scFv‐binding domain and stimulates the engineered T cell through CD28‐ζ signalling, thereby converting the binding of an inhibitory ligand into a T‐cell‐activating signal. The so‐called ‘switch’ CARs were previously reported for PD‐130 and other. TGF‐β CAR T cells amplify and secrete pro‐inflammatory TH1 cytokines in the presence of TGF‐β. More importantly with respect to shaping the immune environment, such TGF‐β switch CAR T cells can also protect neighbouring immune cells from repression; tumor‐targeted CD8+ T cells execute their cytolytic activities against tumor cells in the presence of TGF‐β.31 Moreover, CD4+ T cells do not convert into Treg cell phenotype. An advantage of the TGF‐β CAR l lies in the ability to induce T‐cell effector functions that interface with other immune cells in the near vicinity because of at least two mechanisms. Firstly, the expression of a TGF‐β‐binding receptor can sequester TGF‐β and thereby can reduce TGF‐β‐mediated suppression of cytolytic CD8+ T cells. The effect is also obtained with dnTGF‐βRII‐engineered T cells32 and with soluble TGF‐β binding proteins. Secondly, the activated TGF‐β CAR T cells release stimulatory cytokines that help bystander immune cells to become activated and resist TGF‐β repression.
Unresolved issues and future aspects
The TGF‐β pathway is amenable to pharmacological blockade by small molecule inhibitors; however, a systemic TGF‐β blockade is likely associated with a variety of side effects given the central role of TGF‐β in regulating immune homeostasis. A blockade of TGF‐β specifically in the redirected T cell is preferable. Basically, engineering T cells with the dnTGF‐βRII or with the TGF‐β CAR was explored; the common mechanism is the sequestration of soluble TGF‐β by binding. The lower levels of TGF‐β in the tumor tissue likely reduce the TGF‐β‐mediated promotion of tumor metastasis by the induction of epithelial–mesenchymal transition.33 With the release of pro‐inflammatory cytokines and activating bystander immune cells, including tumor‐infiltrating T cells (TILs), NK cells or macrophages, the TGF‐β CAR seems to be superior to the dnTGF‐βRII, which sequesters TGF‐β but does not transmit an activating signal; the latter can be provided by co‐expressing a CAR recognising the respective cancer cell.
The techniques available to make CAR T cells resistant to TGF‐β showed feasibility, safety and efficacy in pre‐clinical models and moreover implied that they can be used to promote adoptive CAR T‐cell therapy of advanced solid tumors. Given the fact that elevated serum levels of TGF‐β are found in a broad variety of human solid tumors,15, 34 the TGF‐β blockade potentially represents a more general approach to enhance the efficacy of redirected immune cells against tumors. As long as the safety of the approach has not been fully documented in clinical trials, caution should be taken since T‐cell lymphomas developed in mouse models in which the dnTGF‐βRII was expressed under control of the CD2 or CD4 promoter.35, 36, 37 A clinical application, in contrast to the mouse model, should therefore more tightly control the dnTGF‐βRII expression in time and only when the CAR T cell enters the tumor tissue. In line with this concept, the CAR T‐cell modification with a TGF‐β‐antagonizing construct may be more tailored, for instance with a CAR‐inducible dnTGF‐βRII expression regulated by an NFAT‐IL‐2 promoter element. In a recent study,21 the release of transgenic IL‐7 was increased upon CAR engagement of antigen allowing TGF‐β resistance preferentially upon tumor entry. Alternatively, interference with the TGF‐β signalling pathway can be performed by adding a small interfering RNA38 and the RNA may be produced from a CAR‐inducible transgenic construct by the CAR T cell itself. Gene editing, in contrast, would result in permanent silencing of the specific genes.
The application of a dominant‐negative TGF‐β type 2 receptor with a purpose to inhibit TGF‐β signalling and allow effector T cells to resist otherwise inhibitory concentrations of TGF‐β, which leaves their proliferation, cytokine secretion and cytolytic functions unchanged was introduced in a clinical aspect in a study conducted by Bollard et al. (2018).39 DNRll‐modified T cells were infused into patients with Epstein–Barr virus (EBV)‐associated lymphomas. The reason being is that up to 40% of Hodgkin lymphomas (HLs) inhibit EBV genomes and express the type 2 latency proteins, latent membrane protein (LMP)‐1, LMP‐2, BARF1 and EBNA1. The study reports the effects of the administration of such engineered T cells to express a DNRll (DNRll‐LSTs) to eight patients with relapsed EBV‐positive HL.39 Significantly, the study reports that after infusion, DNRll‐LSTs expanded and persisted for up to ≥ 4 years after infusion. It was recorded that no patient had previously undergone lymphodepleting chemotherapy before infusion; however, DNRll‐LSTs alone produced therapeutic effects, such as tumor clinical responses in two of the seven patients with active disease. Additionally, there was one recorded clinical response after CD35‐targeted radio immunotherapy, one PR for a total of 19 months and stable disease in three patients for 4, 10 and 13 months, and no toxicity was recorded in association with DNRll‐LSTs.39 Overall, the findings from this report demonstrate the safety of the technique but also emphasise the importance of strategies to overcome tumor immune evasion by TGF‐β. From this, it is clear to underline the feasibility and therapeutic potential of counteracting a single but common tumor immune editing mechanism, although future applications may include exploiting this strategy to enhance the immunotherapy of many other tumors that exploit TGF‐β for survival.
The approach in using modified CAR T cells with an inducible resistance mechanism potentially avoids deleterious systemic side effects, which is likely occurring during adjuvant therapies using cytokine supplementation, small drug inhibitors or blockade of T‐cell checkpoints. A tightly regulated and fully efficient induction of TGF‐β resistance needs to be implemented within the CAR‐modified T‐cell, making them more efficacious when entering the hostile tumor tissue. The same applies for TCR‐modified T cells or any immune effector cell in general used for the treatment of solid tumors.
Transforming growth factor‐β is certainly one of the most dominant suppressive factors in the tumor tissue; however, other repressive mechanisms towards infiltrating T cells are active independently of TGF‐β and single genetic modifications of T cells may have limited effect. A number of suppressive mechanisms need to be additionally addressed, for example by blocking PD‐1 and CTLA‐4 or by co‐expressing dnPD‐1 or using a PD‐1 switch receptor that provides a CD28 signal when engaging PD‐1L. Co‐expression of IDO or PKA inhibitors or of CD40L may also be required to shield CAR T cells from repression. Taken all known mechanisms together, TGF‐β is certainly a major suppressive cytokine, but not the only one. Multiple inhibitory factors impact the PI3K/Akt pathway; increase in Akt levels, constitutively or inducible upon CAR signalling, may result in a more favorite PI3K/Akt pathway response, resulting in a balance counteracting for a broad range of tumor‐associated repressive mechanisms by regulating diverse signalling pathways associated with T cell activation,27 thereby replacing the need for multiple other manipulations to confer effector CAR T‐cell resistance. An inducible active Akt driven by CAR signalling either by increase in transgenic Akt levels or the possibility to create higher levels of active Akt within the pathway would improve the safety of such modified T cells in order to reduce the risk of signalling independently of T‐cell amplification.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
The work in the authors’ laboratory is supported by the Deutsche Forschungsgemeinschaft, Bonn; Deutsche Krebshilfe, Bonn; Deutsche José Carreras‐Leukämie Stiftung, Munich; Wilhelm Sander‐Stiftung, Munich; Else Kröner‐Fresenius Stiftung, Bad Homburg v.d.H.; the Bundesministerium für Bildung und Forschung, Berlin; and the European Commission through the Enacti2 ng consortium.
References
- 1. Newick K, O'Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med 2017; 68: 139–152. [DOI] [PubMed] [Google Scholar]
- 2. Hatfield SM, Kjaergaard J, Lukashev D et al Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med 2015; 7: 277ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fischer K, Hoffmann P, Voelkl S et al Inhibitory effect of tumor cell‐derived lactic acid on human T cells. Blood 2007; 109: 3812–3819. [DOI] [PubMed] [Google Scholar]
- 4. Massagué J. TGFβ in cancer. Cell 2008; 134: 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer 2013; 13: 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. John LB, Devaud C, Duong CPM et al Anti‐PD‐1 antibody therapy potently enhances the eradication of established tumors by gene‐modified T cells. Clin Cancer Res 2013; 19: 5636–5646. [DOI] [PubMed] [Google Scholar]
- 7. Boyerinas B, Miller SM, Murray RC et al A novel TGF‐β2/interleukin receptor signal conversion platform that protects CAR/TCR T cells from TGF‐β2‐mediated immune suppression and induces T cell supportive signaling networks. Blood 2017; 130: 1911.28835438 [Google Scholar]
- 8. Liu VC, Wong LY, Jang T et al Tumor evasion of the immune system by converting CD4+ CD25− T Cells into CD4+ CD25+ T regulatory cells: role of tumor‐derived TGF‐beta. J Immunol 2007; 178: 2883–2892. [DOI] [PubMed] [Google Scholar]
- 9. Papageorgis P, Stylianopoulos T. Role of TGFβ in regulation of the tumor microenvironment and drug delivery (Review). Int J Oncol 2015; 46: 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nagaraj NS, Datta PK. Targeting the transforming growth factor‐β signaling pathway in human cancer. Expert Opin Investig Drugs 2010; 19: 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morikawa M, Derynck R, Miyazono K. TGF‐β and the TGF‐β family: context‐dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol 2016; 8: a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF‐β receptor. Nature 1994; 370: 341–347. [DOI] [PubMed] [Google Scholar]
- 13. Derynck R, Zhang Y, Feng X‐H. Transcriptional activators of TGF‐β responses: smads. Cell 1998; 95: 737–740. [DOI] [PubMed] [Google Scholar]
- 14. Nakao A. TGF‐beta receptor‐mediated signalling through Smad2, Smad3 and Smad4. EMBO J 1997; 16: 5353–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor‐ and the immune response: implications for anticancer therapy. Clin Cancer Res 2007; 13: 5262–5270. [DOI] [PubMed] [Google Scholar]
- 16. Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov 2012; 11: 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor‐β controls T helper type 1 cell development through regulation of natural killer cell interferon‐γ. Nat Immunol 2005; 6: 600–607. [DOI] [PubMed] [Google Scholar]
- 18. Gong D, Shi W, Yi S, Chen H, Groffen J, Heisterkamp N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol 2012; 13: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fridlender ZG, Sun J, Kim S et al Polarization of tumor‐associated neutrophil phenotype by TGF‐β: “N1” versus “N2” TAN. Cancer Cell 2009; 16: 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koehler H, Kofler D, Hombach A, Abken H. CD28 costimulation overcomes transforming growth factor‐β–mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res 2007; 67: 2265–2273. [DOI] [PubMed] [Google Scholar]
- 21. Golumba‐Nagy V, Kuehle J, Hombach AA, Abken H. CD28‐ζ CAR T cells resist TGF‐β repression through IL‐2 signaling, which can be mimicked by an engineered IL‐7 autocrine loop. Mol Ther 2018; 26: 2218–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kofler DM, Chmielewski M, Rappl G et al CD28 costimulation impairs the efficacy of a redirected T‐cell antitumor attack in the presence of regulatory T cells which can be overcome by preventing Lck activation. Mol Ther 2011; 19: 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosenberg SA, Sportès C, Ahmadzadeh M et al IL‐7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T‐regulatory cells. J Immunother 2006; 29: 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kloss CC, Lee J, Zhang A et al Dominant‐negative TGF‐β receptor enhances PSMA‐targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther 2018; 26: 1855–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wartewig T, Kurgyis Z, Keppler S et al PD‐1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 2017. 10.1038/nature24649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lewis KA, Gray PC, Blount AL et al Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 2000; 404: 411–414. [DOI] [PubMed] [Google Scholar]
- 27. Sun J, Dotti G, Huye LE et al T cells expressing constitutively active Akt resist multiple tumor‐associated inhibitory mechanisms. Mol Ther 2010; 18: 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haxhinasto S, Mathis D, Benoist C. The AKT–mTOR axis regulates de novo differentiation of CD4+ Foxp3+ cells. J Exp Med 2008; 205: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T‐cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol 2018; 14: 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD‐L1 co‐stimulates primary human CD8+ cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immunol 2012; 51: 263–272. [DOI] [PubMed] [Google Scholar]
- 31. Hou AJ, Chang ZL, Lorenzini MH, Zah E, Chen YY. TGF‐β‐responsive CAR‐T cells promote anti‐tumor immune function. Bioeng Transl Med 2018; 3: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang L, Yu Z, Muranski P et al Inhibition of TGF‐β signaling in genetically engineered tumor antigen‐reactive T cells significantly enhances tumor treatment efficacy. Gene Ther 2013; 20: 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF‐β in homeostasis and cancer. Nat Rev Cancer 2003; 3: 807–820. [DOI] [PubMed] [Google Scholar]
- 34. Dong M. Role of transforming growth factor‐beta in hematologic malignancies. Blood 2006; 107: 4589–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ishigame H, Mosaheb MM, Sanjabi S, Flavell RA. Truncated form of TGF‐ RII, but not its absence, induces memory CD8+ T cell expansion and lymphoproliferative disorder in mice. J Immunol 2013; 190: 6340–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lucas PJ, Kim S‐J, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell‐specific dominant negative transforming growth factor β II receptor. J Exp Med 2000; 191: 1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L, Kerkar SP, Yu Z et al Improving adoptive T cell therapy by targeting and controlling IL‐12 expression to the tumor environment. Mol Ther 2011; 19: 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Borkner L, Kaiser A, van de Kasteele W et al RNA interference targeting programmed death receptor‐1 improves immune functions of tumor‐specific T cells. Cancer Immunol Immunother 2010; 59: 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bollard CM, Tripic T, Cruz CR et al Tumor‐specific T‐cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J Clin Oncol 2018; 36: 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]