

Abstract
The site of protein folding and maturation for the majority of proteins that are secreted, localized to the plasma membrane or targeted to endomembrane compartments is the endoplasmic reticulum (ER). It is essential that proteins targeted to the ER are properly folded in order to carry out their function, as well as maintain protein homeostasis, as accumulation of misfolded proteins could lead to the formation of cytotoxic aggregates. Because protein folding is an error-prone process, the ER contains protein quality control networks that act to optimize proper folding and trafficking of client proteins. If a protein is unable to reach its native state, it is targeted for ER retention and subsequent degradation. The protein quality control networks of the ER that oversee this evaluation or interrogation process that decides the fate of maturing nascent chains is comprised of three general types of families: the classical chaperones, the carbohydrate-dependent system, and the thiol-dependent system. The cooperative action of these families promotes protein quality control and protein homeostasis in the ER. This review will describe the families of the ER protein quality control network and discuss the functions of individual members.
Keywords: endoplasmic reticulum, secretory pathway, quality control, molecular chaperones, N-linked glycosylation, oxidoreductases
Introduction
Gunter Blobel’s seminal studies on protein targeting that led to his proposal of the signal hypothesis theory in the 1970s [1–3], laid the conceptual framework for later studies on the eukaryotic secretory pathway and provided a valuable experimental system to dissect processes involving the endoplasmic reticulum (ER) [4–6]. A third of the proteome was later found to be targeted to the ER for entry into the secretory pathway for maturation and eventually secretion or delivery to various locations within the secretory/endocytic pathways [7]. With the discovery of molecular chaperones a decade later by Ellis, Hartl, Horwich, Laskey, Lorimer, Pelham and others [8–12], protein folding and assembly was found to be an assisted process within the cell. Several molecular chaperone families reside in the ER to help these early maturation events including the folding reaction for proteins that traverse the secretory pathway. As protein folding is an error-prone process, prolonged binding to molecular chaperones is also utilized in an interrogation or evaluation process to determine if the structural integrity of the protein product is sound so that native proteins can be passed along the secretory pathway and non-native products can be retained and repaired, or eventually sorted for degradation. This cellular interrogation step was termed protein quality control by Ari Helenius shortly after the discovery of molecular chaperones [13]. In this article, we will review the quality control processes that take place in the early eukaryotic secretory pathway or ER that seek to ensure that only native substrates are passed along the secretory pathway while terminally misfolded proteins are targeted for degradation.
The ER is the site of protein maturation for secretory pathway cargo. These processes are assisted by chaperone and oxidoreductase family members that help increase the efficiency of reaching the native and active state of a protein. Many of these maturation assistance factors are also central players in the protein quality control evaluation and sorting processes. There are three general categories of protein quality control factors in the ER that will be discussed below: (1) the classical molecular chaperone systems; (2) glycan-dependent molecular chaperone systems; and (3) thiol-dependent oxidoreductases. These factors play diverse but well integrated roles in maintaining protein homeostasis in the ER that involves the passage of properly folded cargo, while defective cargo is retained and subsequently degraded.
The Classical Chaperones of the ER
Chaperones from the classical heat shock (heat shock proteins, Hsp) families generally bind to hydrophobic domains on substrates in order to promote productive folding and prevent aggregation [14]. The binding cycle of the classical chaperones is regulated by adenine nucleotides. The ER contains members from the Hsp40s, Hsp70 (BiP/GRP78) and Hsp90 (GRP94) families. Of these, BiP is the most abundant and appears to play the widest role in the ER, including assisting protein folding, protein translocation, ER retention and promotion of ER-associated degradation (ERAD) of misfolded substrates, and inducing the unfolded protein response (UPR) signaling cascade [15, 16]. Here, we will focus largely on the protein quality control functions of BiP and its associated regulators.
BiP binds promiscuously to clients frequently at a number of sites on a maturing protein [17, 18]. These sites are generally hydrophobic and contain alternating aliphatic residues [19, 17]. There are algorithms that predict BiP sites on client proteins by analyzing the primary amino acid sequence [20]. However, the utilization of these predicted sites by BiP requires experimental demonstration for validation. BiP interacts with substrates via its substrate binding domain (SBD) that is regulated by its nucleotide binding domain (NBD; Figure 1A and B) [16]. When the NBD is bound by ATP, BiP exhibits a low substrate affinity due to the lid of the SBD being in an open conformation, leading to a high on-and-off rate of the substrate. Hydrolysis of ATP, leaving BiP in an ADP-bound state, increases substrate affinity by allowing the lid of the SBD to close over the substrate. In the case of some substrates, direct interaction between the lid and the substrate can occur without the lid domain closing over the substrate, therefore allowing for significant substrate tertiary structure and suggesting that BiP has a relatively flexible substrate preference [21].
Fig. 1. BiP domain architecture and binding cycle.
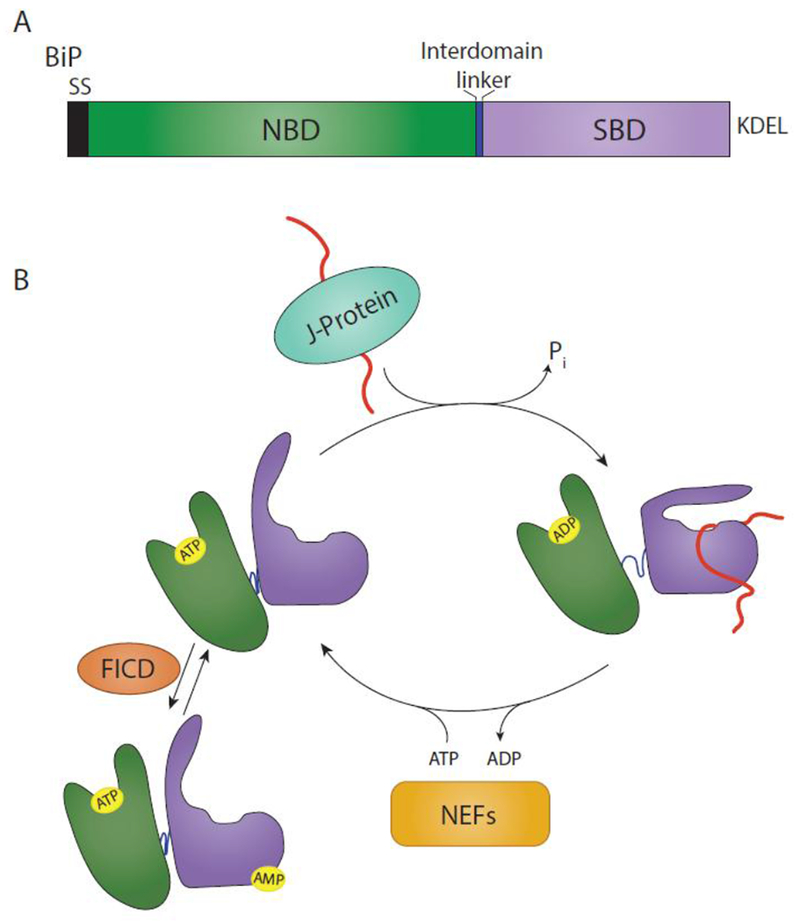
(A) BiP is targeted to the ER via a signal sequence (SS) that is cleaved in the mature form of the protein. From N- to C-terminus, BiP is comprised of a nucleotide binding domain (NBD) (green), interdomain linker (blue), and substrate binding domain (SBD) (purple). It is retained in the ER via a KDEL motif. (B) The substrate binding cycle of BiP is regulated by ATP. When the NBD is bound to ATP, BiP is in a low substrate affinity state. Interaction with a substrate bound J-protein promotes ATP hydrolysis, leading to an extended conformation of the interdomain linker, SBD lid closing and a high substrate affinity. A BiP nucleotide exchange factor (NEF) can then exchange ADP for ATP, placing BiP back in a low substrate affinity state. This process can be inhibited by the AMPylation of BiP by FICD. AMPylation places BiP in a state similar to an ATP-bound state.
BiP co-factors regulate the binding and release of substrates. Proteins that contain J-domains or HPD motifs (the tripeptide His-Pro-Asp) are known to interact with Hsp70 family members to induce their ATPase activity at locations throughout the cell [22]. The ATPase activity of BiP is stimulated by the ERdj proteins that also assist the recruitment of substrates to BiP [23]. Release of substrate occurs when ADP is exchanged for ATP, placing BiP back in a low substrate affinity conformation. Nucleotide exchange is regulated by nucleotide exchange factors (NEFs). There are two known NEFs for BiP: Sil1 and Lhs1 in Saccharomyces cerevisiae [24–26], and Bap and GRP170 are mammalian homologues [26–28, 16]. GRP170, in addition to being a NEF for BiP, also possesses a region that is homologous to Hsp70 itself [29]. However, the role for this region is uncertain.
In yeast, BiP (Kar2p), is localized to the ER translocon by its association with the J-domain protein Sec63 [30]. This positions BiP in its substrate bound state to help with the translocation and early stages of nascent chains folding to its native state [5, 31, 32]. BiP also plays a central role in targeting misfolded proteins for degradation [33–35]. These alternate fates for a BiP substrate may be directed by multiple factors. Prolonged substrate binding, as opposed to transient interactions with early folding intermediates, may target a substrate for ERAD [36, 37]. Further specificity may be conferred by the BiP cofactor J-proteins. While the role of each J-protein is currently unclear, certain J-proteins have been shown to promote distinct fates for BiP substrates. ERdj4 promotes the degradation of a natural variant of surfactant protein C (SP-C) and represses UPR by promoting BiP interaction with IRE1 and repressing IRE1 dimerization [38, 39]. ERdj5 possesses reductase activity and has been shown to promote the degradation of multiple substrates including SP-C and the 1-antitrypsin variant null Hong Kong (NHK), possibly by reduction of disulfide bonds, allowing the substrate to be threaded through the ERAD retrotranslocon [38, 40, 41]. A cellular peptide library demonstrated that ERdj4 and ERdj5 recognize peptides rich in aromatic residues, suggesting that these J-proteins recognize misfolded residues exposing aggregation prone domains and therefore support degradation of these proteins [18]. ERdj3 has a diverse set of roles, but generally promotes protein folding [16, 42]. However, ERdj3 can also promote degradation of specific substrates such as-glucocerebrosidase [43]. ERdj6 (p58IPK) appears to promote protein folding and the protection against ER stress [44]. The J-domain co-factors of BiP control the localization of BiP, its substrate selection, and activity, thereby contributing to the diversification of its roles in ER protein quality control.
The activity of BiP can also be regulated by chemical modification. In the absence of ER stress, BiP is chemically modified by AMPylation [45–47]. AMPylation of BiP by FICD (or HYPE) has been shown to be inhibitory to BiP substrate binding as it confers a substrate affinity similar to the ATP-bound state [48]. AMPylation does not inhibit ERdj protein binding but allosterically inhibits ERdj mediated ATP-hydrolysis, causing BiP to remain in a low substrate affinity state. Under stress, the level of BiP increases and it is de-AMPylated by FICD, which can act to both AMPylate and de-AMPylate BiP, depending on its functional state (Figure 1B) [47, 49]. In this manner, a large pool of BiP can be quickly converted into an active state upon ER stress, thereby decreasing the time required to respond to stress.
While the ER hsp90 family member GRP94, is absent from the yeast ER, it is found in higher eukaryotes. ERAD substrates including α-1 antitrypsin NHK and γ-aminobutyric acid type A (GABAA) receptors were stabilized by the knockdown of GRP94 in cells [50–52]. These studies also found GRP94 associated with the lectin ERAD receptor, Os-9, and the ERAD E3 ligase HRD1. However, other data demonstrates that Os-9 preferentially interacts with hyperglycosylated GRP94, and knockdown of GRP94 does not stabilize α-1 antitrypsin NHK [53]. Together these results support a possible role for GRP94 in ERAD substrate selection and targeting, but these conclusions remain unclear.
Carbohydrate-dependent protein quality control
The majority of proteins that are targeted to the ER are N-linked glycosylated [54]. N-linked glycosylation plays numerous roles during protein folding. These large, hydrophilic protein appendages promote protein solubility and alter folding energetics (Figure 2A), as well as protein function [55–59]. N-linked glycans also act as reporters of the folded state and age of the glycoprotein. As a glycoprotein folds and matures, the glycan is modified, through both trimming and addition, and these modifications affect the glycoprotein’s interaction with carbohydrate-binding proteins resident to the ER, thereby altering folding and trafficking [60–62]. At the hub of the glycan-dependent quality control system are the lectin chaperones calnexin and calreticulin. Calnexin and calreticulin substrate binding plays a central role in the folding and quality control of glycosylated cargo in the ER.
Fig. 2. Glyan dependent quality control.
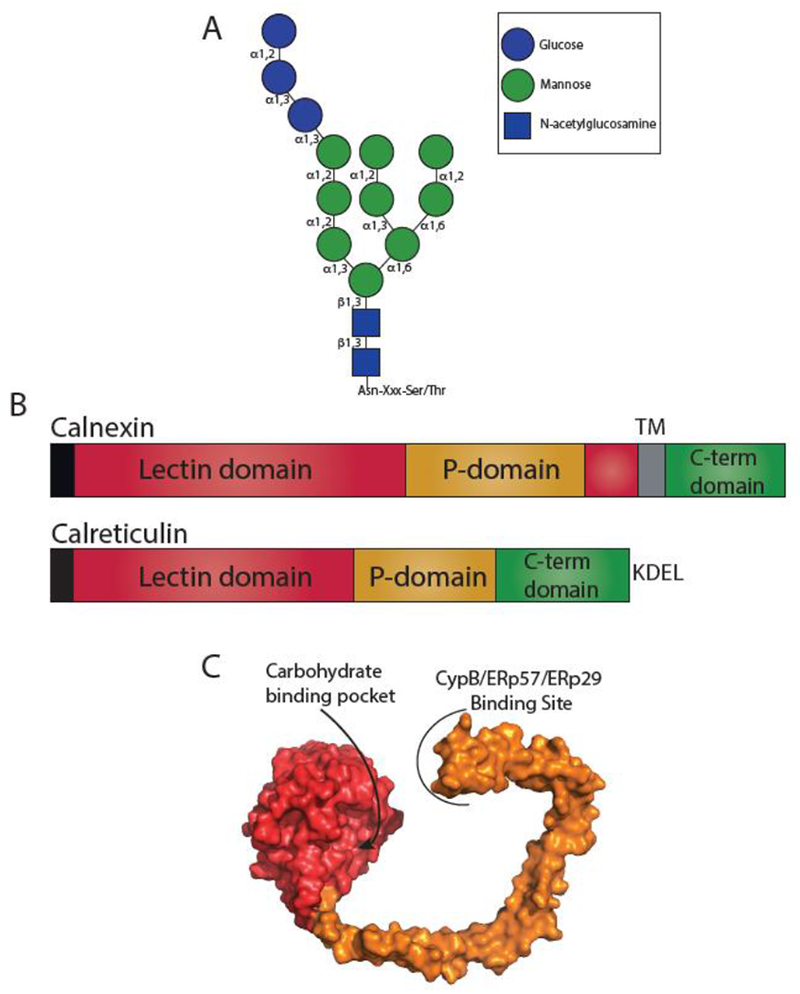
(A) Structure of an N-linked glycan. N-linked glycans are transferred en bloc to an Asn residue in acceptor sites Asn-X-Ser/Thr, where X is not a proline. Glycosidic bonds are denoted. (B) The domain architecture of calnexin and calreticulin. Both calnexin and calreticulin possess an N-terminal signal sequence (black) that is cleaved in the mature protein. Calnexin possesses a lectin domain (red) that is composed of two regions separated by the P-domain (orange), a transmembrane region (TM) (grey) and a cytosolic C-terminal domain (green). Calreticulin possesses a contiguous lectin domain, a P-domain, a C-terminal domain, and a KDEL retention motif. (C) Surface representation of the crystal structure of the luminal domain of calnexin (PDB: 1JHN). The lectin domain is shown in red and the P-domain in orange. The carbohydrate binding pocket in the lectin domain and the binding site of CypB/ERp57/ERp29 on the tip of the P-domain are designated.
The N-linked glycan (Fig 2A) is preassembled on a dolichol phosphate in the ER membrane and is appended en bloc by the oligosaccharyltransferase (OST) as GlcNAc2Man9Gluc3 to the Asn in the consensus site of Asn-X-Ser/Thr (where X is not Pro) on the substrate [62, 63]. While most glycosylation occurs co-translationally via OST complexes containing the catalytic subunit STT3A, skipped acceptor sites can be glycosylated post-translationally via STT3B-containing OST complexes [64].
After glycosylation, the glycan is then trimmed by -glucosidase I, generating a glycan with two glucoses. In this state, the glycoprotein can interact with the lectin malectin. Malectin is a membrane-associated ER-resident protein that specifically recognizes di-glucosylated glycans [65]. Malectin associates with the OST component ribophorin I and acts to retain misfolded glycoproteins in the ER [66, 67]. The ER retention role of malectin is somewhat surprising as it resides in a region of the ER where early folding steps occur. A potential answer to this conundrum may be that malectin is ER-stress induced, and therefore may only play a relevant role under stress when proteins translocating into the ER are less likely to undergo productive folding [68]. Alternatively, malectin’s association with an OST subunit may aid in glycosylation of downstream sites as most glycoproteins contain more than one site of modification.
Trimming of the second glucose by -glucosidase II creates monoglucosylated glycans. In this state, the glycan can be bound by the membrane-bound lectin chaperone calnexin or its soluble paralogue calreticulin that bind to monoglucosylated glycans with micromolar affinity (Figure 3) [69–71]. Trimming of the final glucose by -glucosidase II yields a non-glucosylated glycan that supports release of the glycoprotein from calnexin and calreticulin. In this state, natively folded glycoproteins can be further trafficked through the ER. Non-natively folded glycoproteins are recognized by the folding sensor UDP-glucose: glycoprotein glucosyltransferase 1 (UGGT1). UGGT1 reglucosylates substrates regenerating monoglucosylated glycans that then become substrates for calnexin/calreticulin rebinding, supporting retention of misfolded substrates while allowing for continued folding [6, 72]. Cycles of substrate binding to calnexin and calreticulin, trimming by -glucosidase II, and reglucosylation by UGGT1 support the retention of misfolded glycoproteins in the ER while promoting folding through interactions with chaperones and their associated co-factors. As such, UGGT1 acts as a gatekeeper of the calnexin/calreticulin cycle, determining if glycoproteins may exit the ER or must be retained.
Fig. 3. The calnexin/calreticulin substrate binding cycle.
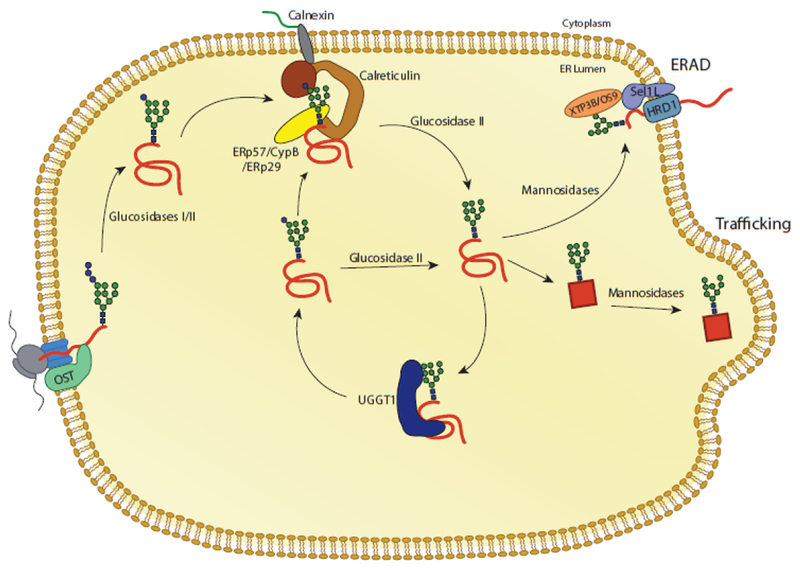
Proteins targeted to the ER receive N-linked glycans that are transferred by the OST complex to acceptor sites. The first two glucoses are trimmed by glucosidases I and II, leaving a monoglucosylated glycan. In this state, the glycan is a substrate for calnexin and calreticulin. Release from calnexin/calreticulin and trimming of the final glucose by glucosidase II leaves the glycan in a non-glucosylated state. Productive folding and adoption of a native state allows for trafficking of the glycoprotein from the ER. Glycoproteins that do not adopt a native fold can be recognized by the folding sensor UDP-glucose: glycoprotein glucosyltransferase 1 (UGGT1). UGGT1 reglucosylates substrates, allowing for rebinding to calnexin/calreticulin or trimming by glucosidase II. Glycoproteins that continue to non-productively fold can be removed from the calnexin/calreticulin cycle through trimming by mannosidases and targeting to ER associated degradation (ERAD) machinery.
While calnexin and calreticulin share 39% sequence identity, each has unique properties [73, 74]. Both calnexin and calreticulin are composed of a lectin domain, a flexible proline-rich domain (P-domain) and a C-terminal domain (Figure 2B and C) [70]. Calnexin possesses a transmembrane domain near its C-terminus, while the soluble calreticulin’s C-terminal domain has low affinity and high capacity calcium binding sites comprised of a series of acidic residues that support its role as a calcium buffer [75]. The lectin binding domain of both calnexin and calreticulin folds to a globular conformation containing a single carbohydrate binding site (Figure 2C). Isothermal calorimetry data has demonstrated that the lectin domain alone is capable of binding substrate [76]. The P-domain adopts an extended, arm-like conformation with a hairpin turn. The P-domain of calnexin contains four copies of a Pro rich motif [77], while the P-domain of calreticulin contains three such motifs, though both are structurally similar. The P-domain of both calnexin and calreticulin are interaction sites for the cofactors ERp57 and ERp29, protein disulfide isomerases; and cyclophilin B, a peptidyl proline isomerase [78–80]. By interacting with a diverse set of folding factors, the P-domains of calnexin and calreticulin function to bring substrates and folding factors in close proximity, supporting productive protein folding and quality control.
UGGT1 is a soluble ER-resident protein consisting of an N-terminal folding sensor domain and a C-terminal glucosyltransferase domain. Recent x-ray crystal structures from both Thermomyces dupontii and Chaetomium thermophilum have found that the N-terminal domain consists of four catalytically inactive thioredoxin like domains [81, 82]. Three of these domains are sequential while the fourth is non-sequential, comprised of long range interactions. The N-terminal folding sensor domain adopts a flexible, curved conformation with a prominent central cavity that contains hydrophobic patches. These findings suggest that UGGT1 interacts with high-mannose substrates primarily via hydrophobic interactions. UGGT1 has a homologue UGGT2 that is 55% identical to UGGT1. UGGT2 has been shown to possess an active glucosyltransferase domain when domain swapped with UGGT1, but biological substrates have not been identified for UGGT2 [83].
Selenoprotein 15 (Sep15/Selenof) is a UGGT binding protein with redox activity [84]. Sep15 interacts tightly with UGGT1 and 2, such that the entire pool of Sep15 is bound to UGGT1/2, though not all UGGT1/2 are bound by Sep15 [85]. In vitro, Sep15 has been shown to increase the enzymatic activity of UGGT1/2 [83]. Sep15 may modulate the selection of substrates by UGGT or act upon substrates modified by UGGT to help with their repair and folding [86]. Selenocysteines, considered the 21st amino acid, are found in the active site of Sep15. Since selenocysteines generally possess reducing or disulfide bond breaking activity, it is possible that Sep15 acts to reduce disulfides to try to repair non-native cargo. Additional work is needed to elucidate the biological role of Sep15.
The roles of the calnexin/calreticulin cycle are diverse and important for proper ER function. These roles range from promoting productive folding, limiting deleterious folding pathways and aggregation, and retention of non-native or incompletely assembled proteins [87– 90]. Indicative of these vital roles is the embryonic lethality of knocking out multiple factors in the calnexin/calreticulin pathway including calreticulin (18 days), UGGT1 (13 days) and ERp57 (13.5 days) [91–93]. Calnexin knockout mice display decreased rates of survival within 48 hr post-birth while survivors displayed multiple motor disorders [94]. Such lethality and disorders are indicative of the diverse roles of calnexin and calreticulin, and the extensive set of substrates that are dependent on this carbohydrate-dependent chaperone pathway for proper folding and trafficking.
A central role of the calnexin/calreticulin pathway is to promote proper folding of glycoprotein intermediates. Numerous substrates exhibit reduced folding efficiency and decreased rates of trafficking when interactions with calnexin and calreticulin are abrogated, including hemagglutinin, neuraminidase, vesicular stomatitis virus G protein, MHC class I, antithrombin III and corin [88, 90, 89, 95–97]. Calnexin and calreticulin act as molecular chaperones by decreasing the rate of folding of substrates in a region-specific manner [73, 98, 96, 95]. This is done by sterically blocking regions proximal to the glycan interacting with the lectin domain while leaving regions distal to the glycan free to fold or interact with other factors. In this manner, the folding pathway of a protein can be determined both by the amino acid sequence and the location of glycans.
When proper folding or multimer assembly of calnexin and calreticulin substrates does not occur, the calnexin/calreticulin pathway generally acts to retain such substrates through persistent reglucosylation by UGGT1 and rebinding to calnexin or calreticulin [99–102]. However, data from UGGTT−/− mouse embryonic fibroblasts showed that the effect of reglucosylation and interaction with the calnexin/calreticulin pathway is substrate specific [103]. There appears to exist three classes of UGGT1 substrates. The first class are proteins that require only one round of binding to calnexin or calreticulin, and therefore do not require reglucosylation by UGGT1 to fold properly. When UGGT1 is knocked out, the trafficking of these proteins is unaltered. The second class involves proteins that are secreted faster when reglucosylation does not occur, and this might be due to the decreased interaction time with calnexin/calreticulin. Alternatively, some proteins displayed decreased rates of secretion, suggesting that reglucosylation and multiple rounds of calnexin/calreticulin binding acts to increase the rate of secretion. This may be due to the calnexin/calreticulin pathway promoting folding efficiency and sequestration of structural elements that act to retain such substrates in the ER. Overall, the calnexin/calreticulin pathway alters the folding and trafficking of a diverse set of substrates, but it does not function in a common manner for all substrates. Eventually non-productively folding substrates must be extracted from the calnexin/calreticulin cycle for targeting for degradation to maintain proper ER homeostasis [102].
Mannose trimming plays a central role both in the extraction of terminally misfolded or slow folding glycoproteins from the calnexin/calreticulin cycle and the sorting for degradation to the ERAD process. Trimming of A-branch mannose residues precludes the ability of UGGT1 to modify the glycan for calnexin and calreticulin rebinding. Whereas removal of B- and C-branch mannoses creates a degradation signal that is recognized by downstream sorting lectins that target the demannosylated substrates for ERAD. The secretory pathway possesses a number of mannosidases that trim the various mannose residues including ER mannosidase I (ER Man I also called Man1B1), the EDEM family members (EDEM1-3), Golgi 1,2-mannosidases (Golgi Man IA, IB, and IC) and endomannosidase that help to support these functions (Figure 4) [104, 105, 61].
Fig. 4. The architecture of mannosidases involved in quality control.
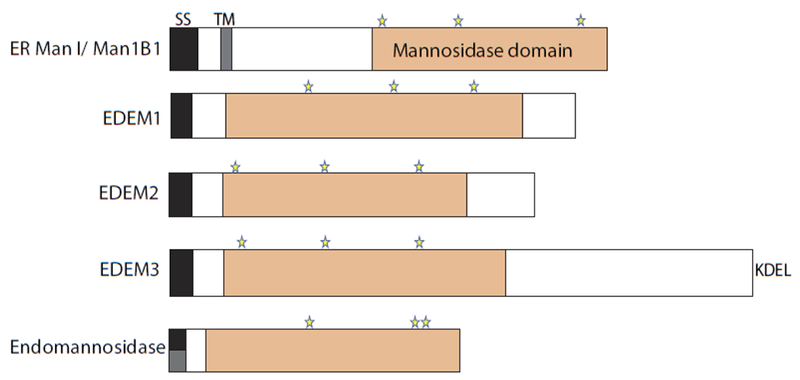
Domain architecture of mannosidases: ER Man1, EDEM1, EDEM2, EDEM3, and endomannosidase. The signal sequences (black), predicted transmembrane domains (grey), the mannosidase domains (orange), and putative catalytic residues (stars) are designated. Endomannosidase possesses a predicted non-cleavable signal sequence, as shown by a half black and half grey box.
The extraction of substrates from the calnexin and calreticulin binding cycle appears to be cell type dependent. Some cells possess an endomannosidase activity capable of cleaving the A-branch mannose residues and abolishing UGGT1 modification. However, this activity is missing from Chinese Hamster Ovary (CHO) cells and possibly other cells [106]. While EDEM1 has been shown to be able to extract substrates from the lectin chaperone binding cycle [107, 108], it is not clear if this is through direct binding to the substrate, mannose trimming of the A-branch glycans, or possibly the utilization of both properties. Further investigation is required to understand the full mechanism of extraction of misfolded substrates from the calnexin and calreticulin binding cycle.
Treatment with mannosidase inhibitors stabilizes ERAD substrates [109]. Mannose trimming on B- and C-branches is linked to creating a degradation signal on ERAD glycoprotein substrates. Initially, it was thought that the transition of the N-linked glycans to Man8GlcNAc2 sorted the protein for ERAD [110]. ER Man I/Man1B1 has been implicated in removing the B-branch terminal mannose and recognizing tertiary and quaternary structure [111, 112]. The activity of ERManI/Man1B1 is enhanced in the presence of the oxidoreductants PDI or TXNDC11 [112]. In S. cerevisiae, the ER Man I equivalent, Mnslp, removes the outermost mannose from branch B, creating an eight mannose residue glycan (termed M8B) [110, 113]. More recent studies discovered that degradation signaling required an additional ER mannosidase in yeast, Htm1p, that removes the C-branch terminal mannose, to expose an 1,6-linked mannose residue, and it is this residue that is the signal for ERAD [110, 114]. The ER lectin yos9p recognizes and binds ERAD substrates containing exposed or terminal 1,6-linked mannoses and targets them to the Hrd1p-containing dislocon complex in the ER membrane for dislocation, ubiquitination and degradation by the proteasome [115–118].
The process in metazoans appears to be more complex as the mannosidases involved have diversified. The EDEM family of three (EDEM1-3) serves as homologues to Htm1p. While all three EDEM family members have now been shown to possess mannosidase activity in cells, as well as using purified proteins more recently [119–122, 112], the precise glycans they act upon and generate remains uncertain. Following the yeast paradigm, ER ManI/Man1B1 and the EDEMs work together to create Man7-5 glycans with exposed 1,6-linked mannose residues on the C-branch [121]. These demannosylated ERAD substrates are recognized by two downstream mannose-receptor homology (MRH) domains containing lectins that reside in the ER; OS9 and XTP3B [123, 50, 124]. It is these carbohydrate-binding proteins that then target the substrate to the HRD1 dislocation/ubiquitination complex in the ER membrane. Multiple mechanisms likely protect substrates from improper trimming by mannosidases including mannosidase subcompartmentalization and regulated concentration of mannosidases in the ER [125, 126].
The mannosidases involved in ERAD do not act as traditional glycosidases that trim glycans by transiently interacting with the carbohydrate. Some of them appear to contain folding sensing properties directly or through their associated co-factors. Htm1p mannosidase activity is aided by an associated oxidoreductase, Pdi1p [127, 128]. While EDEM1 appears to stably bind ERAD substrates independently of its associated factors BiP and ERdj5, its binding appears to be bi-partite in that it has a stable interaction that survives harsh treatments that is thiol dependent and likely covalent, as well as a weaker interaction with substrates that is thiol independent [122, 129]. The associated ERdj5 appears to assist in the reduction of substrates to make them competent for dislocation but is not required for thiol dependent binding [41, 129, 130, 122]. BiP is recruited to the EDEM1 complex by the J-domain of ERdj5. BiP may also contribute to ERAD substrate selection, translocation preparation and dislocon complex delivery in some way.
The role for EDEMs in protein quality control is further complicated by the fact that they also appear to bind and select substrates in a glycan-independent manner [129, 131, 122]. In the case of EDEM1, the mannosidase-like domain can also act as a lectin and aid in the delivery of ERAD substrates to downstream dislocation complex by binding to the N-linked glycans on the HRD1 complex adapter, SEL1L (Hrd3p homologue in yeast) [129, 132]. While mannose trimming clearly marks terminally misfolded substrates for turnover through the ERAD process, the versatility and possible redundancy of lectin quality control factors has complicated the full understanding of their functions and mechanisms.
Thiol-dependent protein quality control
The ER is an oxidizing environment and the site of disulfide bond formation between proximal Cys pairs on maturing proteins. Disulfide bonds are crucial for the structure and activity of many proteins that traverse the secretory pathway. Numerous oxidoreductase proteins in the ER aid in the formation, reduction, and isomerization of disulfide bonds in order to ensure the correct native disulfides are formed, which is dependent upon the redox potential of an active site disulfide (Figure 5). As most Cys are paired in disulfides before trafficking from the ER, free thiols can act as an indicator that a protein is non-native. The ER has quality control machinery that recognizes proteins with free thiols. While extensive studies have been performed to elucidate the mechanism of disulfide bond formation and the factors involved, here we will focus on thiol-dependent protein quality control.
Fig. 5. Redox reactions catalyzed by PDI family members.
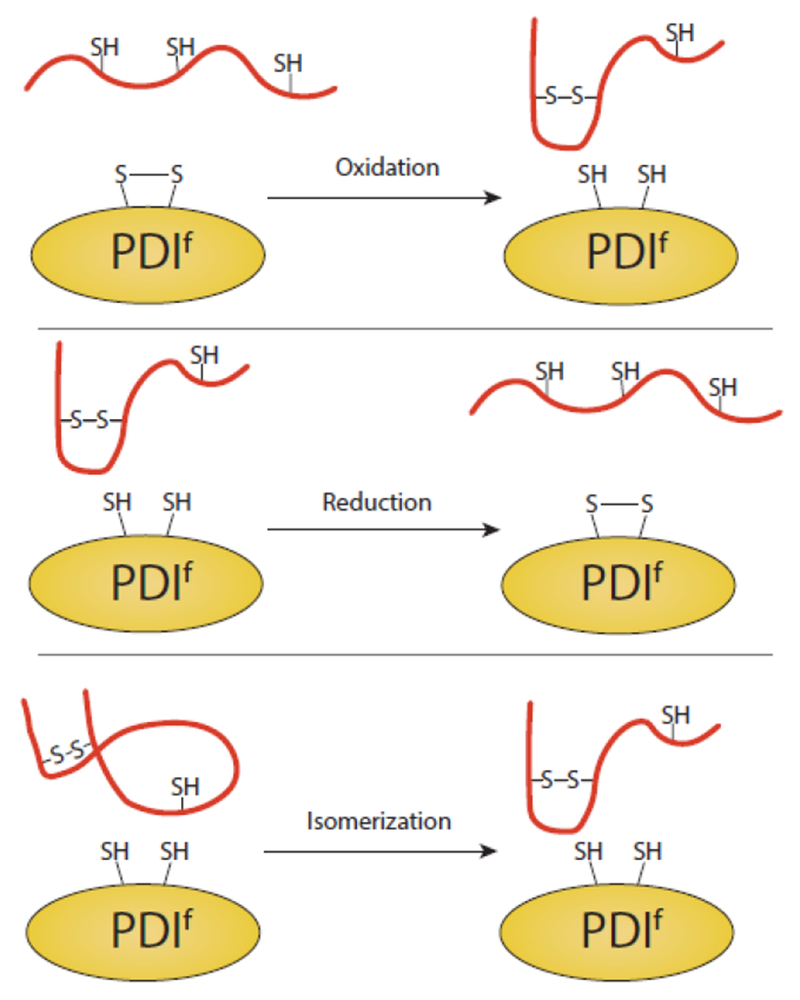
PDI, or members of the protein disulfide isomerase family (PDIf), can oxidize, reduce and isomerize disulfide bonds of substrates. In all cases, a transient intermolecular disulfide bond is formed between PDIf and the substrate (not pictured). While the various PDIf members have varying numbers of catalytic domains, a single catalytic site of PDIf is displayed for simplification.
Protein disulfide isomerase (PDIf) is a large family of ER proteins with more than twenty members that play essential roles in disulfide bond formation and maintenance [133–135, 132, 136]. We will focus on a subset of PDIf which are well characterized, though other proteins of the PDI family exist which may interact with a smaller subset of clients (i.e. TXNDC5, TXNDC15 and TMX1). A protein by the same name as the family is also used to identify the most abundant member of the family and PDI can help form, reduce, and isomerize disulfide bonds. The oxidative partner of PDI is ERO1 /, allowing PDI to remain in a redox competent state [137]. PDI is comprised of four thioredoxin like domains; a, b, b’ and a’. Of these domains, a and a’ are active thioredoxins, containing catalytically active Cys-Xxx-Xxx-Cys motifs [138]. PDI appears to be able to bind substrates in both a redox dependent and independent manner as the b’ domain of PDI binds to hydrophobic substrates [139–141]. PDI may be capable of scanning proteins for misfolded regions and potentially oxidize, reduce, or isomerize disulfide bonds of clients in order to promote proper folding [142]. PDI is a versatile protein as it has also been shown to promote the degradation of misfolded substrates [143].
Two members of the PDI family associate with the P-domain of calnexin and calreticulin, ERp57 and ERp29, for recruitment to glycosylated substrates. Similar to PDI, ERp57 is comprised of four thioredoxin-like domains of which two are catalytically active [144]. ERp57 acts as an oxidoreductase and is brought in close contact with glycoprotein folding intermediates or retained substrates through its interaction with the carbohydrate-binding chaperones, allowing for scanning of substrates to ensure proper disulfide bonding [145]. ERp29 is unusual in that it is a dimer and lacks a catalytic active site as it only possesses a single Cys residue. It is possible that this Cys is used for isomerization. Its role in polyomavirus infection and virus disassembly and penetration across the ER membrane is analogous to the preparation and translocation of aberrant proteins for ERAD but this connection will require further investigation [146].
ERp72 consists of five thioredoxin domain; a , a, a’, b, and b’, of which a , a, and a’ possess catalytically active Cys-Xxx-Xxx-Cys motifs [147]. Despite the high level of structural similarity to ERp57, ERp72 does not interact with calnexin or calreticulin, likely due to differences in exposed surface charges [148]. However, ERp72 interacts with cyclophilin B via a polyacidic stretch of amino acids on ERp72 [149]. This interaction increases the rate of in vitro folding of immunoglobulin G. ERp72 retains misfolded cholera toxin and thyroglobulin as shown by RNAi and overexpression studies [143]. ERp72 has been shown to stably interact with and retain thyroglobulin through a disulfide bond [150]. While more work is needed to fully understand the role of ERp72, these data suggest that ERp72, for a subset of clients, promotes folding and ER retention.
ERdj5 is the largest PDI family member. It has six thioredoxin domains and a J-domain that recruits BiP. Its redox state favors a role as a reductase for preparation of non-native proteins for dislocation to the cytoplasm for proteasomal degradation [41]. ERdj5 can reduce glycosylated substrates through its association with EDEM1. BiP can also capture nonglycosylated ERAD substrates independent from EDEM1 for passage to ERdj5 and delivery to the SEL1L/HRD1 dislocation complex [130]. Work from the Hendershot group using a peptide library, mapped ERdj5 binding sites to aggregation-prone sequences on a protein underscoring its role in quality control [18]. The structure of ERdj5 is dynamic as measured by high-speed atomic force microscopy and this flexibility in some way assists its ability to enhance ERAD [151]. ERdj5 plays a central reductive role in the preparation of both glycosylated and non-glycosylated substrates for degradation.
While numerous proteins facilitate proper disulfide bond formation, errors can occur that could, if progressed unchecked, lead to secretion of non-native proteins with free Cys. This is potentially problematic as reactive thiols can enhance aggregation mediated by non-native intermolecular disulfide bonds. As such, ER quality control recognizes proteins with non-native, reactive free thiols and retains them in the ER. ERp44 is a soluble chaperone of the protein disulfide isomerase family that cycles between the ER and early Golgi [152]. It contains three thioredoxin domains; a, b, and b’, of which a is catalytically active [135]. ERp44 uses Cys29 to retain non-native or unassembled substrates containing free thiols and this has been shown for substrates including unassembled IgM, adiponectin and SUMF1 [153–156]. The manner in which ERp44 binds and retains proteins in the ER is proposed to be pH dependent [157, 158]. In this model, ERp44 traffics from the ER to the Golgi, where the pH is progressively lower. The C-terminal tail of ERp44 undergoes a pH-dependent conformational change in the Golgi, exposing the Cys29 necessary for client binding while also exposing the RDEL retention/retrieval sequence at its C-terminus [159, 160]. ERp44 then traffics back to the ER by interacting with KDEL receptors with its substrate covalently attached. It is currently unclear how ERp44 releases substrates upon re-entry into the ER, though the intermolecular disulfide bond is likely reduced by another member of the PDI family. In this manner, clients with free thiols can be retained in the ER via cycling between the ER and early Golgi.
Open questions remain regarding the extent to which free thiols act as hallmarks of misfolded proteins. Numerous native proteins with free thiols exit the ER for secretion or residence in the lysosome, as in the case of lysosomal Cys proteases with active site Cys. How these proteins escape quality control recognition is unclear. One possibility is any unpaired Cys are buried and therefore not accessible to factors involved in recognizing proteins with free thiols. Also, the nearby environment or context of the Cys may contribute to its pKa and reactiveness, and thereby determine its effectiveness in supporting retention and subsequent degradation. Additionally, non-native proteins with free thiols may evade thiol dependent quality control by forming non-native, intermolecular disulfide bonds, thereby generating disulfide linked protein aggregates which are resistant to degradation.
Summary
Sequence specific chaperone and oxidoreductase binding appears to be the basis for quality control recognition. Hallmarks of misfolded proteins include exposed hydrophobic domains and free thiols. These features can also impact the N-linked glycan composition to further signal aberrancy to the quality control system. As the quality control process is responsible for monitoring thousands of proteins that traverse the secretory pathway, the sequences that signal non-nativeness must be degenerate. While some factors appear to recognize substrates that are folding intermediates and should be given additional time to fold and can be repaired, others must recognize terminally misfolded proteins to target them for degradation. Chaperones play promiscuous roles in all these functions with their precise role in some cases being determined by their associated co-factors, while others possess more specialized activities. The interplay between these diverse quality control systems that relies on exposed hydrophobic residues, free thiols or carbohydrate compositions helps to evaluate the large variety of cargo that travel through the ER. The interplay is underscored by the many complexes found between traditional chaperones, oxidoreductases and lectin chaperones or glycosidases. A deeper understanding of these quality control processes and their manipulation provides an avenue for disease intervention for the large number of diseases with etiologies rooted in quality control decisions.
Acknowledgements
This work was supported by the National Institutes of Health under award number GM086874 (to D.N.H.); and a Chemistry-Biology Interface Predoctoral training grant (T32 GM008515 to B. M. A.). We would also like to thank Jill Graham for thoughtful comments.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest There is no conflict of interest.
Ethical Approval This article does not contain any studies with human participants or animals performed by any of the authors.
References cited
- 1.Blobel G, Sabatini DD (1971) Ribosome-membrane interaction in eukaryotic cells. Biomem 2:193–195 [Google Scholar]
- 2.Blobel G, Dobberstein B (1975) Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J Cell Biol 67:835–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blobel G, Dobberstein B (1975) Transfer of protein across membranes. II. Reconstitution of functional rough microsomes from heterologous components. J Cell Biol 67:852–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulleid NJ, Freedman RB (1988) Defective co-translational formation of disulphide bonds in protein disulphide-isomerase-deficient microsomes. Nature 335:649–651 [DOI] [PubMed] [Google Scholar]
- 5.Nicchitta CV, Blobel G (1993) Lumenal proteins of the mammalian endoplasmic reticulum are required to complete protein translocation. Cell 73:989–998 [DOI] [PubMed] [Google Scholar]
- 6.Hebert DN, Foellmer B, Helenius A (1995) Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81:425–433 [DOI] [PubMed] [Google Scholar]
- 7.Huh W-K, Falvo JV, Gerke LC, et al. (2003) Global analysis of protein localization in budding yeast. Nature 425:686–691 [DOI] [PubMed] [Google Scholar]
- 8.Laskey RA, Honda B, Mills AD, Finch JT (1978) Speculations on the functions of the major heat shock and glucose-regulated proteins. Nature 275:416–420692721 [Google Scholar]
- 9.Pelham HR (1986) Speculations on the functions of the major heat shock and glucose-regulated proteins. Cell 46:959–961 [DOI] [PubMed] [Google Scholar]
- 10.Cheng MY, Hartl UF, Martin J, et al. (1989) Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature 337:620–625 [DOI] [PubMed] [Google Scholar]
- 11.Goloubinoff P, Christeller JT, Gatenby AA, Lorimer GH (1989) Reconstitution of active dimeric ribulose bisphosphate carboxylase from an unfoleded state depends on two chaperonin proteins and Mg-ATP. Nature 342:884–889 [DOI] [PubMed] [Google Scholar]
- 12.Ellis JR (1996) Discovery of molecular chaperones. Cell Stress Chaperones 1:155–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helenius A (1989) Protein oligomerization in the endoplasmic reticulum. Annu Rev Cell Biol 5:277–307 [DOI] [PubMed] [Google Scholar]
- 14.Horwich A, Neupert W, Hartl UF (1990) Protein-catalysed protein folding. Trends Biotechnol 8:126–131 [DOI] [PubMed] [Google Scholar]
- 15.Hendershot LM (2004) The ER function BiP is a master regulator of ER function. Mt Sinai J Med 71:289–297 [PubMed] [Google Scholar]
- 16.Behnke J, Feige MJ, Hendershot LM (2015) BiP and its nucleotide exchange factors Grp170 and Sill: Mechanisms of action and biological functions. J Mol Biol 427:1589–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blond-Elguindi S, Cwirla SE, Dower WJ, et al. (1993) Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell 75:717–728 [DOI] [PubMed] [Google Scholar]
- 18.Behnke J, Mann MJ, Scruggs F-L, et al. (2016) Members of the Hsp70 family recognize distinct types of sequences to execute ER quality control. Mol Cell 63:739–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flynn GC, Pohl J, Flocco MT, Rothman JE (1991) Peptide-binding specificity of the molecular chaperone BiP. Nature 353:726–730 [DOI] [PubMed] [Google Scholar]
- 20.Schneider M, Rosam M, Glaser M, et al. (2016) BiPPred: Combined sequence-and structure-based prediction of peptide binding to the Hsp70 chaperone BiP. Proteins 84:1390–1407 [DOI] [PubMed] [Google Scholar]
- 21.Schlecht R, Erbse AH, Bukau B, Mayer MP (2011) Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat Struct Mol Biol 18:345–351 [DOI] [PubMed] [Google Scholar]
- 22.Kampinga HH, Andreasson C, Barducci A, et al. (2018) Function, evolution, and structure of J-domain proteins. Cell Stress Chaperones [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otero JH, Lizák B, Hendershot LM (2010) Life and Death of a BiP substrate. Semin Cell Dev Biol 21:472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boisramé A, Kabani M, Beckerich J-M, et al. (1998) Interaction of Kar2p and Slslp is required for efficient co-translational translocation of secreted proteins in the yeast Yarrowia lipolytica. J Biol Chem 273:30903–30908 [DOI] [PubMed] [Google Scholar]
- 25.Kabani M, Beckerich J-M, Brodsky JL (2002) Nucleotide exchange factor for the yeast Hsp70 molecular chaperone Ssa1p. Mol Cell Biol 22:4677–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyson JR, Stirling CJ (2000) LHS1 and SIL1 provide a lumenal function that is essential for protein translocation into the endoplasmic reticulum. EMBO J 19:6440–6452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung KT, Shen Y, Hendershot LM (2002) BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J Biol Chem 277:47557–47563 [DOI] [PubMed] [Google Scholar]
- 28.Steel GJ, Fullerton DM, Tyson JR, Stirling CJ (2004) Coordinated activation of Hsp70 chaperones. Science 303:98–101 [DOI] [PubMed] [Google Scholar]
- 29.Chen X, Easton D, Oh H-J, et al. (1996) The 170 kDa glucose regulated stress protein is a large HSP70-HSP110-like protein of the endoplasmic reticulum. FEBS Lett 380:68–72 [DOI] [PubMed] [Google Scholar]
- 30.Matlack KE, Misselwitz B, Plath K, Rapoport TA (1999) BiP Acts as a Molecular Ratchet during Posttranslational Transport of Prepro-α Factor across the ER Membrane Author links open overlay panel. Cell 97:553–564 [DOI] [PubMed] [Google Scholar]
- 31.Helenius A, Hammond C (1994) Folding of VSV G protein: sequential interaction with BiP and calnexin. Science 266:456–458 [DOI] [PubMed] [Google Scholar]
- 32.Hebert DN, Zhang J-X, Helenius A (1998) Protein folding and maturation in a cell-free system. Biochem Cell Biol 76:867–873 [DOI] [PubMed] [Google Scholar]
- 33.Plemper RK, Bohmler S, Bordallo J, et al. (1997) Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 388:891–895 [DOI] [PubMed] [Google Scholar]
- 34.Skowronek MH, Hendershot LM, Haas IG (1998) The variable domain of nonassembled Ig light chains determines both their half-life and binding to the chaperone BiP. Proc Natl Acad Sci USA 95:1574–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brodsky JL, Werner ED, Dubas ME, et al. (1999) The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem 274:3453–3460 [DOI] [PubMed] [Google Scholar]
- 36.Farinha CM, Amaral MD (2005) Most F508del-CFTR Is Targeted to Degradation at an Early Folding Checkpoint and Independently of Calnexin. Mol Cell Biol 25:5242–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sörgjerd K, Ghafouri B, Jonsson B-H, et al. (2006) Retention of misfolded mutant transthyretin by the chaperone BiP/GRP78 mitigates amyloidogenesis. J Mol Biol 356:469–482 [DOI] [PubMed] [Google Scholar]
- 38.Dong M, Bridges JP, Apsley K, et al. (2008) ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol Biol Cell 19:2620–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amin-Wetzel N, Saunders RA, Kamphius MJ, et al. (2017) A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell 171:1625–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunnea PM, Miranda-Vizuete A, Bertoli G, et al. (2003) ERdj5, an Endoplasmic Reticulum (ER)-resident Protein Containing DnaJ and Thioredoxin Domains, Is Expressed in Secretory Cells or following ER Stress. J Biol Chem 278:1059–1066 [DOI] [PubMed] [Google Scholar]
- 41.Ushioda R, Hoseki J, Araki K, et al. (2008) ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321:569–572 [DOI] [PubMed] [Google Scholar]
- 42.Khodayari N, Marek G, Lu Y, et al. (2017) Erdj3 Has an Essential Role for Z Variant Alpha-1-Antitrypsin Degradation. J Cell Biochem 118:3090–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan YL, Genereux JC, Pankow S, et al. (2014) ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher’s disease. Chem Biol 21:967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rutkowski TD, Kang S-W, Goodman AG, et al. (2007) The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol Biol Cell 18:3681–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ham H, Woolery AR, Tracy C, et al. (2014) Unfolded protein response-regulated Drosophila Fic protein reversibly AMPylates BiP chaperone during endoplasmic reticulum homeostasis. J Biol Chem 289:36059–36069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anwesha S, Chen AJ, Nakayasu ES, et al. (2015) A novel link between Fic mediated adenylation and the unfolded protein response. J Biol Chem 290:8482–8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Preissler S, Rato C, Chen R, et al. (2015) AMPylation matches BiP activity to client protein load in the endoplasmic reticulum. eLife 4:e12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Preissler S, Rohland L, Yan Y, et al. (2017) AMPylation targets the rate-limiting step of BiP’s ATPase cycle for its functional inactivation. eLife 6:e29428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Preissler S, Rato C, Perera L, et al. (2017) FICD acts bi-functionally to AMPylate and de-AMPylate the endoplasmic reticulum chaperone BiP. Nat Struct Mol Biol 24:23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Christianson JC, Shaler TA, Tyler RE, Kopito RR (2009) OS-9 and GRP94 deliver mutant alphal-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol 10:272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong Y, Shen H, Wang Y, et al. (2015) Identification of ERAD components essential for dislocation of the null Hong Kong variant of α-1-antitrypsin (NHK). Biochem Biophys Res Commun 458:424–428 [DOI] [PubMed] [Google Scholar]
- 52.Di X-J, Wang Y-J, Han D-Y, et al. (2016) Grp94 Protein Delivers γ-Aminobutyric Acid Type A (GABAA) Receptors to Hrd1 Protein-mediated Endoplasmic Reticulum-associated Degradation. J Biol Chem 291:9526–9539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dersh D, Jones SM, Eletto D, et al. (2014) OS-9 facilitates turnover of nonnative GRP94 marked by hyperglycosylation. Mol Biol Cell 25:2220–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Apweiler R, Hermjakob H, Sharon N (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta 1473:4–8 [DOI] [PubMed] [Google Scholar]
- 55.Haraguchi M, Yamashiro S, Furukawa K, et al. (1995) The effects of the site-directed removal of N-glycosylation sites from β-1,4-N-acetylgalactosaminyltransferase on its function. Biochem J 312:273–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai G, Salonikidis PS, Fei J, et al. (2005) The role of N-glycosylation in the stability, trafficking and GABA-uptake of GABA-transporter 1. FEBS J 272:1625–1638 [DOI] [PubMed] [Google Scholar]
- 57.Skropeta D (2009) The effect of individual N-glycans on enzyme activity. Bioorg Med Chem 17:2545–2653 [DOI] [PubMed] [Google Scholar]
- 58.Culyba EK, Price JL, Hanson SR, et al. (2011) Protein native-state stabilization by placing aromatic side chains in N-glycosylated reverse turns. Science 331:571–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hebert DN, Lamriben L, Powers ET, Kelly JW (2014) The intrinsic and extrinsic effects of N-linked glycans on glycoproteostasis. Nat Chem Biol 10:902–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hebert DN, Garman SC, Molinari M (2005) The glycan code of the endoplasmic reticulum: asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol 15:364–370 [DOI] [PubMed] [Google Scholar]
- 61.Caramelo JJ, Parodi AJ (2015) A sweet code for glycoprotein folding. FEBS Letters 589:3379–3387. 10.1016/j.febslet.2015.07.021 [DOI] [PubMed] [Google Scholar]
- 62.Lamriben L, Graham JB, Adams BM, Hebert DN (2016) N-glycan based ER molecular chaperone and protein quality control system: the calnexin binding cycle. Traffic 17:308–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Breitling J, Aebi M (2013) N-Linked Protein Glycosylation in the Endoplasmic Reticulum. Cold Spring Harb Perspect Biol 5:a013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shrimal S, Cherepanova NA, Gilmore R (2015) Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin Cell Dev Biol 41:71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schallus T, Jaeckh C, Fehér K, et al. (2008) Malectin: A novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol Biol Cell 19:3404–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qin S-Y, Hu D, Matsumoto K, et al. (2012) Malectin Forms a Complex with Ribophorin I for Enhanced Association with Misfolded Glycoproteins. J Biol Chem 287:38080–38089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takeda K, Qin S-Y, Matsumoto N, Yamamoto K (2014) Association of malectin with ribophorin I is crucial for attenuation of misfolded glycoprotein secretion. Biochem Biophys Res Commun 454:436–440 [DOI] [PubMed] [Google Scholar]
- 68.Galli C, Bernasconi R, Soldà T, et al. (2011) Malectin participates in a backup glycoprotein quality control pathway in the mammalian ER. PLOS One 6:e16304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hammond C, Ineke B, Ari H (1994) Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci USA 91:913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schrag JD, Bergeron JJ, Li Y, et al. (2001) The Structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol Cell 8:633–644 [DOI] [PubMed] [Google Scholar]
- 71.Gopalakrishnapai J, Gupta G, Karthikeyan T, et al. (2006) Isothermal titration calorimetric study defines the substrate binding residues of calreticulin. Biochem Biophys Res Commun 351:14–20 [DOI] [PubMed] [Google Scholar]
- 72.Sousa M, Parodi AJ (1995) The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J 14:4196–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hebert DN, Zhang J-X, Chen W, et al. (1997) The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J Cell Biochem 139:613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vassilakos A, Michalak M, Lehrman MA, Williams DB (1998) Oligosaccharide binding characteristics of the molecular chaperones calnexin and calreticulin. Biochemistry 37:3480–3490 [DOI] [PubMed] [Google Scholar]
- 75.Li Z, Stafford WF, Bouvier M (2001) The metal ion binding properties of calreticulin modulate its conformational flexibility and thermal stability. Biochemistry 40:11193–11201 [DOI] [PubMed] [Google Scholar]
- 76.Kozlov G, Pocanschi CL, Rosenauer A, et al. (2010) Structural basis of carbohydrate recognition by calreticulin. J Biol Chem 285:38612–38620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ellgaard L, Bettendorff P, Braun D, et al. (2002) NMR structures of 36 and 73-residue fragments of the calreticulin P-domain. JMB 322:773–784 [DOI] [PubMed] [Google Scholar]
- 78.Oliver JD, van derWal FJ, Bulleid NJ, High S (1997) Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science 275:86–88 [DOI] [PubMed] [Google Scholar]
- 79.Kozlov G, Bastos-Aristizabal S, Määttänen P, et al. (2010) Structural basis of cyclophilin B binding by the calnexin/calreticulin P-domain. J Biol Chem 285:35551–35557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kozlov G, Munoz-Escobar J, Castro K, Gehring K (2017) Mapping the ER Interactome: The P Domains of Calnexin and Calreticulin as Plurivalent Adapters for Foldases and Chaperones. Structure 25:1415–1422 [DOI] [PubMed] [Google Scholar]
- 81.Roversi P, Marti L, Caputo AT, et al. (2017) Interdomain conformational flexibility underpins the activity of UGGT, the eukaryotic glycoprotein secretion checkpoint. Proc Natl Acad Sci USA 114:8544–8549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Satoh T, Song C, Zhu T, et al. (2017) Visualization of a flexible modular structure of the ER folding-sensor enzyme UGGT. Sci Rep 7:12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takeda Y, Seko A, Hachisu M, et al. (2014) Both isoforms of human UDP-glucose:glycoprotein glucosyltransferase are enzymatically active. Glycobiology 24:344–350 [DOI] [PubMed] [Google Scholar]
- 84.Ferguson AD, Labunskyy VM, Fomenko DE, et al. (2006) NMR Structures of the Selenoproteins Sep15 and SelM Reveal Redox Activity of a New Thioredoxin-like Family. J Biol Chem 281:3536–3543 [DOI] [PubMed] [Google Scholar]
- 85.Korotkov KV, Kuramaswamy E, Zhou Y, et al. (2001) Association between the 15-kDa Selenoprotein and UDP-glucose:Glycoprotein Glucosyltransferase in the Endoplasmic Reticulum of Mammalian Cells. J Biol Chem 276:15330–15336 [DOI] [PubMed] [Google Scholar]
- 86.Yim SH, Everley RA, Schildberg FA, et al. (2018) Role of Selenof as a Gatekeeper of Secreted Disulfide-Rich Glycoproteins. Cell Rep 23:1387–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rajagopalan S, Brenner MB (1994) Calnexin retains unassembled major histocompatibility complex class I free heavy chains in the endoplasmic reticulum. J Exp Med 180:407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cannon KS, Hebert DN, Helenius A (1996) Glycan dependent and independent association of Vesicular Stomatitis Virus G Protein with Calnexin. J Biol Chem 271:14280–14284 [DOI] [PubMed] [Google Scholar]
- 89.Vassilakos A, Myrna C-DF, Peterson PA, et al. (1996) The molecular chaperone calnexin facilitates folding and assembly of class I histocompatibility molecules. EMBO J 15:1495–1506 [PMC free article] [PubMed] [Google Scholar]
- 90.Hebert DN, Foellmer B, Helenius A (1996) Calnexin and calreticulin promote folding, delay oligomerization and suppress degradation of influenza hemagglutinin in microsomes. EMBO J 15:2961–2968 [PMC free article] [PubMed] [Google Scholar]
- 91.Mesaeli N, Nakamura K, Zvaritch E, et al. (1999) Calreticulin is essential for cardiac development. J Cell Biol 144:857–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Molinari M, Galli C, Vanoni O, et al. (2005) Persistent glycoprotein misfolding activates the glucosidase II/UGT1-driven calnexin cycle to delay aggregation and loss of folding competence. Mol Cell 20:503–512 [DOI] [PubMed] [Google Scholar]
- 93.Coe H, Jung J, Groenendyk J, et al. (2010) ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J Biol Chem 285:6725–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Denzel A, Molinari M, Trigueros C, et al. (2002) Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol Cell Biol 22:7398–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang N, Glidden EJ, Murphy SR, et al. (2008) The cotranslational maturation program for the type II membrane glycoprotein influenza neuraminidase. J Biol Chem 283:33826–33837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chandrasekhar K, Ke H, Wang N, et al. (2016) Cellular folding pathway of a metastable serpin. Proc Natl Acad Sci USA 113:6484–6489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang H, Li S, Wang J, et al. (2018) N-glycosylation in the protease domain of trypsin-like serine proteases mediates calnexin-assisted protein folding. eLife 7:e35672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Daniels R, Kurowski B, Johnson AE, Hebert DN (2003) N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol Cell 11:79–90 [DOI] [PubMed] [Google Scholar]
- 99.Gao B, Adhikari R, Howarth M, et al. (2002) Assembly and antigen-presenting function of MHC class I molecules in cells lacking the ER chaperone calreticulin. Immunity 16:99–109 [DOI] [PubMed] [Google Scholar]
- 100.Pearse BR, Gabriel L, Wang N, Hebert DN (2008) A cell-based reglucosylation assay demonstrates the role of GT1 in the quality control of a maturing glycoprotein. J Cell Biol 181:309–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pearse BR, Tamura T, Sunryd JC, et al. (2010) The role of UDP-Glc:glycoprotein glucosyltransferase 1 in the maturation of an obligate substrate prosaposin. J Cell Biol 189:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tannous A, Patel N, Tamura T, Hebert DN (2015) Reglucosylation by UDP-glucose:glycoprotein glucosyltransferase 1 delays glycoprotein secretion but not degradation. Mol Biol Cell 26:390–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Soldà T, Galli C, Kaufman RJ, Molinari M (2007) Substrate-specific requirements for UGT1-dependent release from calnexin. Mol Cell 27:238–249 [DOI] [PubMed] [Google Scholar]
- 104.Olivari S, Molinari M (2007) Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett 581:3658–3664 [DOI] [PubMed] [Google Scholar]
- 105.Sunryd JC, Tannous A, Lamriben L, Hebert DN (2014) Chaperones of the Endoplasmic Reticulum Associated Degradation (ERAD) Pathway In: The Molecular Chaperones Interaction Networks in Protein Folding and Degradation. Springer Science [Google Scholar]
- 106.Karaivanova VK, Luan P, Spiro RG (1998) Processing of viral envelope glycoprotein by the endomannosidase pathway: evaluation of host cell specificity. Glycobiology 8:725–730 [DOI] [PubMed] [Google Scholar]
- 107.Molinari M, Calanca V, Galli C, et al. (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299:1397–1400 [DOI] [PubMed] [Google Scholar]
- 108.Oda Y, Hosokawa N, Wada I, Nagata K (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299:1394–1397 [DOI] [PubMed] [Google Scholar]
- 109.Su K, Stoller T, Rocco J, et al. (1993) Pre-Golgi degradation of yeast prepro-alpha-factor expressed in a mammalian cell. Influence of cell type-specific oligosaccharide processing on intracellular fate. J Biol Chem 268:14301–14309 [PubMed] [Google Scholar]
- 110.Jakob CA, Burda P, Roth J, Aebi M (1998) Degradation of Misfolded Endoplasmic Reticulum Glycoproteins in Saccharomyces cerevisiae Is Determined by a Specific Oligosaccharide Structure. J Cell Biol 142:1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aikawa J, Matsuo I, Ito Y (2012) In vitro mannose trimming property of human ER α-1,2 mannosidase I. Glycoconj J 29:35–45. 10.1007/s10719-011-9362-1 [DOI] [PubMed] [Google Scholar]
- 112.Shenkman M, Ron E, Yehuda R, et al. (2018) Mannosidase activity of EDEM1 and EDEM2 depends on an unfolded state of their glycoprotein substrates. Communications Biology 1:172 10.1038/S42003-018-0174-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Słomińska-Wojewódzka M, Sandvig K (2015) The Role of Lectin-Carbohydrate Interactions in the Regulation of ER-Associated Protein Degradation. Molecules 20:9816–9846. 10.3390/molecules20069816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Quan EM, Kamiya Y, Kamiya D, et al. (2010) Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol Cell 32:870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bhamidipati A, Denic V, Quan EM, Weissman JS (2005) Exploration of the Topological Requirements of ERAD Identifies Yos9p as a Lectin Sensor of Misfolded Glycoproteins in the ER Lumen. Mol Cell 19:741–751 [DOI] [PubMed] [Google Scholar]
- 116.Kim W, Spear ED, Ng DT (2005) Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol Cell 19:753–764 [DOI] [PubMed] [Google Scholar]
- 117.Szathmary R, Bielmann R, Nita-Lazar M, et al. (2005) Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol Cell 19:765–775 [DOI] [PubMed] [Google Scholar]
- 118.Denic V, Quan EM, Weissman JS (2006) A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126:349–359 [DOI] [PubMed] [Google Scholar]
- 119.Olivari S, Galli C, Alanen H, et al. (2005) A novel stress-induced EDEM variant regulating endoplasmic reticulum associated glycoprotein degradation. J Biol Chem 280:2424–2428 [DOI] [PubMed] [Google Scholar]
- 120.Hosokawa N, Tremblay LO, Sleno B, et al. (2010) EDEM1 accelerates the trimming of α1,2-linked mannose on the C branch of N-glycans. Glycobiology 20:567–575 [DOI] [PubMed] [Google Scholar]
- 121.Ninagawa S, Okada T, Sumitomo Y, et al. (2014) EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J Cell Biol 206:347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lamriben L, Oster ME, Tamura T, et al. (2018) EDEM1’s mannosidase-like domain binds ERAD client proteins in a redox-sensitive manner and possesses catalytic activity. J Biol Chem 293:13932–13945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hosokawa N, Wada I, Nagasawa K, et al. (2008) Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J Biol Chem 283:20914–20924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.van der Goot AT, Pearce MM, Leto DE, et al. (2018) Redundant and Antagonistic Roles of XTP3B and OS9 in Decoding Glycan and Non-glycan Degrons in ER-Associated Degradation. Mol Cell 70:516–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Calì T, Galli C, Olivari S, Molinari M (2008) Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem Biophys Res Commun 371:405–410 [DOI] [PubMed] [Google Scholar]
- 126.Benyair R, Ogen-Shtern N, Mazkereth N, et al. (2015) Mammalian ER mannosidase I resides in quality control vesicles where it encounters its glycoprotein substrates. Mol Biol Cell 26:172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gauss R, Kanehara K, Carvalho P, et al. (2011) A Complex of Pdi1p and the Mannosidase Htm1p Initiates Clearance of Unfolded Glycoproteins from the Endoplasmic Reticulum. Mol Cell 42:782–793 [DOI] [PubMed] [Google Scholar]
- 128.Liu C-Y, Fujimori DG, Weissman JS (2016) Htm1p-Pdi1p is a folding-sensitive mannosidase that marks N-glycoproteins for ER-associated protein degradation. Proc Natl Acad Sci USA 113:E4015–E4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cormier JH, Tamura T, Sunryd JC, Hebert DN (2010) EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol Cell 34:627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ushioda R, Hoseki J, Nagata K (2013) Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol Biol Cell 24:3155–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tang H-Y, Huang C-H, Zhuang Y-H, et al. (2014) EDEM2 and OS-9 are required for ER-associated degradation of non-glycosylated sonic hedgehog. PLOS One 9:e92164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Saeed M, Suzuki R, Watanabe N, et al. (2011) Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J Biol Chem 286:37264–37273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hatahet F, Ruddock LW (2007) Substrate recognition by the protein disulfide isomerases. FEBS J 274:5223–5234 [DOI] [PubMed] [Google Scholar]
- 134.Appenzeller-Herzog C, Riemer J, Christensen B, et al. (2008) A novel disulphide switch mechanism in Erola balances ER oxidation in human cells. EMBO J 27:2977–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Määttänen P, Gehring K, Bergeron JJ, Thomas DY (2010) Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol 21:500–511 [DOI] [PubMed] [Google Scholar]
- 136.Sato Y, Kojima R, Okumura M, et al. (2013) Synergistic cooperation of PDI family members in peroxiredoxin 4-driven oxidative protein folding. Scientific Reports 3:2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sevier CS, Kaiser CA (2008) Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta 1783:549–556 [DOI] [PubMed] [Google Scholar]
- 138.Oka OB, Bulleid NJ (2013) Forming disulfides in the endoplasmic reticulum. Biochim Biophys Acta 1833:2425–2429 [DOI] [PubMed] [Google Scholar]
- 139.McLaughlin SH, Bulleid NJ (1998) Thiol-independent interaction of protein disulphide isomerase with type X collagen during intra-cellular folding and assembly. Biochem J 331:793–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pirneskoski A, Klappa P, Lobell M, et al. (2004) Molecular characterization of the principal substrate binding site of the ubiquitous folding catalyst protein disulfide isomerase. J Biol Chem 279:10374–10381 [DOI] [PubMed] [Google Scholar]
- 141.Denisov AY, Määttänen P, Dabrowski C, et al. (2009) Solution structure of the bb’ domains of human protein disulfide isomerase. FEBS J 276:1440–1449 [DOI] [PubMed] [Google Scholar]
- 142.Okumura M, Kadokura H, Inaba K (2015) Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radio Bio Med 83:314–322 [DOI] [PubMed] [Google Scholar]
- 143.Forster ML, Sivick K, Park Y, et al. (2006) Protein disulfide isomerase–like proteins play opposing roles during retrotranslocation. J Cell Biol 173:853–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Frickel E-M, Frei P, Bouvier M, et al. (2004) ERp57 is a multifunctional thiol-disulfide oxidoreductase. J Biol Chem 279:18277–18287 [DOI] [PubMed] [Google Scholar]
- 145.Zapun A, Darby NJ, Tessier DC, et al. (1998) Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J Biol Chem 273:6009–6012 [DOI] [PubMed] [Google Scholar]
- 146.Walczak CP, Tsai B (2011) A PDI family network acts distinctly and coordinately with ERp29 to facilitate polyomavirus infection. J Virology 85:2386–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mazzarella RA, Srinivasan M, Haugejordan SM, Green M (1990) ERp72, an abundant luminal endoplasmic reticulum protein, contains three copies of the active site sequences of protein disulfide isomerase. J Biol Chem 265:1094–1101 [PubMed] [Google Scholar]
- 148.Kozlov G, Määttänen P, Schrag JD, et al. (2009) Structure of the Noncatalytic Domains and Global Fold of the Protein Disulfide Isomerase ERp72. Structure 17:651–659 Journal home page for Structure [DOI] [PubMed] [Google Scholar]
- 149.Jansen G, Määttänen P, Denisov AY, et al. (2012) An Interaction Map of Endoplasmic Reticulum Chaperones and Foldases. Mol Cell Proteomics 11:710–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Menon S, Lee J, Abplanalp WA, et al. (2007) Oxidoreductase Interactions Include a Role for ERp72 Engagement with Mutant Thyroglobulin from the rdw/rdw Rat Dwarf. Journal of Biological Chemistry 282:6183–6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Maegawa K, Watanabe S, Noi K, et al. (2017) The Highly Dynamic Nature of ERdj5 Is Key to Efficient Elimination of Aberrant Protein Oligomers through ER-Associated Degradation. Structure 25:846–857 [DOI] [PubMed] [Google Scholar]
- 152.Anelli T, Ceppi S, Bergamelli L, et al. (2007) Sequential steps and checkpoints in the early exocytic compartment during secretory IgM biogenesis. EMBO J 26:4177–4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tiziana A, Alessio M, Bachi A, et al. (2003) Thiol-mediated protein retention in the endoplasmic reticulum: the role of ERp44. EMBO J 22:5015–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Qiang L, Wang H, Farmer SR (2007) Adiponectin Secretion Is Regulated by SIRT1 and the Endoplasmic Reticulum Oxidoreductase Ero1-Lα. Mol Cell Biol 27:4698–4707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fraldi A, Zito E, Annunziata F, et al. (2008) Multistep, sequential control of the trafficking and function of the multiple sulfatase deficiency gene product, SUMF1 by PDI, ERGIC-53 and ERp44. Hum Mol Genet 17:2610–2621 [DOI] [PubMed] [Google Scholar]
- 156.Mariappan M, Radhakrishnan K, Dierks T, et al. (2008) ERp44 Mediates a Thiol-independent Retention of Formylglycine-generating Enzyme in the Endoplasmic Reticulum. J Biol Chem 283:6375–6383 [DOI] [PubMed] [Google Scholar]
- 157.Anelli T, Sannino S, Sitia R (2015) Proteostasis and “redoxtasis” in the secretory pathway: Tales of tails from ERp44 and immunoglobulins. Free Radic Bio Med 83:323–330 [DOI] [PubMed] [Google Scholar]
- 158.Watanabe S, Harayama M, Kanemura S, et al. (2017) Structural basis of pH-dependent client binding by ERp44, a key regulator of protein secretion at the ER-Golgi interface. Proc Natl Acad Sci USA 114:E3224–E3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang L, Wang L, Vavassori S, et al. (2008) Crystal structure of human ERp44 shows a dynamic functional modulation by its carboxy-terminal tail. EMBO Rep 9:642–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vavassori S, Cortini M, Masui S, et al. (2013) A pH-Regulated Quality Control Cycle for Surveillance of Secretory Protein Assembly. Mol Cell 50:783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]