

The growth rate G of the crystalline bodies appearing in each of a set of 35 characterized polyethylene fractions ranging from 3600 to 807,000 in molecular weight has been measured as a function of the undercooling ΔT. In isothermal crystallization, only axialites were found from Mw = 3600 to 18,000. (For these runs, ΔT < 17.5 °C.) From Mw = 18,000 to Mw ≅ 115,000 coarse-grained non-banded spherulites were found for ΔT > 17.5 °C, and axialites for ΔT < 17.5 °C; a rather sharp break occurred in the logio G versus T data at ΔT ≅ 17.5 °C. The morphologic al changes were more gradual. Above Mw = 115,-000, only nearly structureless “irregular” spherulites were found at all undercoolings corresponding to isothermal growth. Typical ringed spherulites were obtained only on quenching. Wide-angle x-ray data showed that the usual orthorhombic subcell predominated in all the morphologies encountered. Low-angle x-ray data showed that the specimens exhibited lamellar crystallization irrespective of the particular gross morphology involved. The growth rate data on each fraction were analyzed using
where f ≅ 1 to obtain values of Kg, and G0. The value of Y in Kg=Ybσσe/(Δhf)k was obtained for each morphology by applying the “Z” test of Lauritzen. Y=4 for regime I crystallization (single surface nucleus leads to completion of substrate) and Y=2 for regime II crystallization (numerous surface nuclei involved in substrate completion). It was found that the axialites obeyed regime I kinetics (Y =4), the coarse-grained spherulites regime II kinetics (Y=2), and the irregular spherulites “mixed” kinetics (Y ~ 3). The assumption that the substrate length L in Lauritzen’s regime theory was ~ 5 μm led to the prediction of a rather sharp regimeI → regime II transition (corresponding to a break in the log10 G versus T data) at ΔT ≅ 17.5 °C, in accord with experiment. The σσe value calculated from Kg and Y for Mw ≥ 20,000 was approximately constant with molecular weight and independent of morphology; the limiting value of σσe from kinetic measurements was about 1285 erg2/cm4, corresponding to σe(∞) = 90.5 erg/cm2 and σ=14.2 erg/cm2. (This value of σe(∞) compares favorably with σe(eq) = 93±8 erg/cm2 from melting point experiments.) The increase of σσe and σe that took place at low molecular weights on up to ~ 20,000 was treated using an expression given by Hoffman, viz, σe = σe(∞)[(v+ßi) /(v+ 1)] where v = number of folds per molecule, ßi=σe(cilium)/σe(∞): Intermittent high and low values of σe were found experimentally in this region, showing that βi varied with increasing molecular weight between 0.15 and ~ 0.7. Theoretical estimates of these upper and lower bounds for βi are given. The variation of σe between its upper and lower bounds was tentatively explained in terms of the alternate appearance of short and long terminal cilia. Estimates of the initial lamellar thickness were made from σe, and compared with the appropriate low-angle x-ray spacings. A theoretical estimate of the ratio of the pre-exponential factors G0(I) and G0(II)) for regimes I and II was compared with experiment with satisfactory results. The value of G0 is not strongly dependent upon the viscosity of the melt. The work of chain folding deduced from the growth rate data is close to 4.1 kcal/mol, which is in good agreement with other estimates.
1. Introduction
The primary objective of this study was to determine the nucleation constants that describe the rate of growth of polyethylene crystals from the subcooled melt as a function of undercooling and molecular weight, and to interpret these in terms of predictions arising from the kinetic theory of nucleation-controlled growth with chain folding. Characterized fractions of linear polyethylene ranging in molecular weight from Mw= 3600 to Mw = 807,000 were used in the investigation. An analysis of the experimentally determined nucleation constant Kg allows the surface free energy product σσe to be obtained for each fraction. This product can be separated into its components σ, the lateral surface free energy, and σe, the fold surface free energy. The value of σe obtained in this way may be called a “kinetic” value, since it is determined from growth rate data. One of our primary interests was to determine the dependence of σσe, and therefore the effective value of σe, on molecular weight.
According to certain current theories [1–3]1 the effective value of σe should be nearly independent of molecular weight if the folded surface nuclei leading to growth contain enough folds to be essentially free of the effects of chain ends. At low molecular weights, some chain end effects must be expected to be included in the effective value of σe. The present study was designed in part to determine where the value of σe leveled off and became approximately constant with increasing molecular weight. The initial upswing of σe and the leveling off in this quantity that occurs as the molecular weight is increased is treated in terms of a simple model that allows extension of the theory of nucleation with chain folds from quite high molecular weights to cases where the surface nuclei have only a few folds per molecule.
As the work progressed, a somewhat surprising richness of morphology appeared, the details of which did not seem to have been foretold in the literature in a systematic manner. Several types of spherulites and one type of axialite were found. Except at large under-coolings accessible only by quenching, none of the spherulites seemed to closely correspond in any detail to the usual “ringed” type with numerous concentric bands so commonly reported in other investigations (see for example [4]). Meanwhile, it was discovered that axialites and spherulites sometimes occurred in different temperature regions in the same specimen, and that in these two regions the growth rate data gave decidedly different nucleation constants. Therefore, a secondary objective became the searching out of the regions of undercooling and molecular weight where these particular morphological entities occurred.
Part of the method of preparation of the specimens involved removal by filtration in solution of a very substantial fraction of the heterogeneities that initiated crystallization, with the result that each spherulite or axialite grew to relatively large size as it crystallized from the melt, thus allowing its morphology to be clearly revealed in an optical microscope. By this technique we were able to obtain large spherulites or axialites without heating the specimen far above the melting point prior to crystallization. Excessive heating can cause oxidation or degradation (see later).
It will prove useful to be more specific concerning the significance of the nucleation constants that were determined. By so doing, an important issue regarding the interpretation of not only the present data but also other growth rate data in the literature may be brought out. According to the kinetic theory of crystallization with chain folding, the growth rate G at a temperature T near the equilibrium melting temperature T°m relevant to the molecular weight under consideration depends on the undercooling ΔT=T°m-T in a manner that is proportional to [1, 2]2 exp [—Kg/T(ΔT)f] where Kg is the nucleation constant. (The quantity f is a factor that is close to unity near T°m that corrects for the change in heat of fusion with temperature.) Accordingly, a measurement of G as a function of ΔT allows Kg to be determined experimentally with considerable precision, and it is this parameter that was obtained from the growth data on the polyethylene fractions as a function of molecular weight and morphology.
An important point concerning Kg is that its theoretical interpretation, which affects the value of σσe and the resultant value of σe, depends critically on the assumptions used in relating the flux describing the surface nucleation rate i to the actual lineal growth rate G. The theory of the flux for chain-folded surface nuclei gives i ∝ exp [—4bσσeT°m/(Δhf) (ΔT)kTf], where b is the layer thickness and (Δhf) the heat of fusion. In one limit (regime 1, single surface nucleus causes substrate completion), G is directly proportional to i, and Kg is to be interpreted correctly as 4bσσeT°m/(Δhf)k. In the other limit (regime II, numerous surface nuclei involved in substrate completion), G varies as i1/2 as was pointed out by Sanchez and DiMarzio [7], and Kg is 2bσσeT°m/(Δhf)k. Thus, a question arises in analyzing experimental Kg data as to whether they should be used with regime I or regime II kinetics to estimate σσe-
Criteria delineating where regime I or regime II kinetics should be applied have been developed by Lauritzen [8], and discussed in detail elsewhere [1]. In the present work, application of these criteria suggested that the axialites followed regime I behavior and that most of the coarse-grained non-banded spherulites appearing in the same specimens at different undercoolings followed regime II behavior. The result was that, despite a difference of a factor of approximately two in the Kg values observed for spherulites and axialites, respectively, the resultant σσe values were very similar in the intermediate molecular weight range where both types of objects appeared in the same specimen. High molecular weight specimens exhibited “irregular” spherulites where mixed regime I and II behavior was suggested by the criteria. When analyzed in this light, they indicated a σσe rather similar to the axialites or coarse-grained non-banded spherulites. The regime effect is discussed in some detail for the polyethylene fractions, and an indication is given of the importance of correctly identifying the regime of crystallization in other polymers.
Attention is given to an experimental determination of the pre-exponential factors G0 that govern the absolute growth rates of the axialites and spherulites. Theoretical predictions suggest that the G0 value for regime I should be very much larger than that for regime II [1, 2].
A number of other topics are discussed briefly. These include: (1) some details of the different morphological entities that were encountered, i.e., axialites, coarse-grained non-banded spherulites, and “irregular” spherulites; (2) a discussion of the birefringence and lamellar nature of the aforementioned objects, including some information on the so-called “L1” and, “L2” low angle x-ray spacings for certain of the fractions; (3) the work of chain folding q obtained from the kinetic estimate of σe; (4) the crystallization behavior of “whole” polymer polyethylene; and (5) the relationship between this work and earlier work on the growth rate of crystallites in polyethylene and other polymers as a function of molecular weight.
2. Experimental Detail
Because the results obtained here are more extensive and somewhat different than those reported in the literature for polyethylene, both as regards Kg and morphology, we consider it of importance to describe the basic elements of our procedures and the reasons for employing them.
Most of the samples used in this investigation were prepared by column elution (CE) or preparative gel permeation chromatography (PGPC) of NBS Standard Reference Material (SRM) 1475 linear polyethylene. This SRM is a linear polyethylene that has the following molecular weight distribution characteristics: Mn= 18,310±360, Mw=53,070±620, Mz= 138,000± 3,700, and Mw/Mn= 2.90. Full details of the characterization of this source material have been published elsewhere [9].
The sample designation that will be used throughout this paper is based on the weight average molecular weight, Mw. Thus the sample with the weight average molecular weight of Mw= 3600 is designated “3.6 K”, that with Mw= 30,600 as “30.6 K”, and so on.3
The source, method of preparation, and characterization parameters of the fractions are shown in table 1. Some of the fractions were prepared from SRM 1475 by the Waters Corporation4 in PGPC columns 12.2 m in length, achieving an Mw/Mn ratio in a few cases in the vicinity of 1.05. The samples fractionated by the Waters Corporation are marked (W) in table 1. All specimens denoted CE were fractionated at NBS. Three samples were prepared by the PGPC method and supplied by the Monsanto Chemical Company, and these are denoted (M) in table 1. The latter were not prepared from SRM 1475.
Table 1.
Method of preparation and characterization parameters for the polyethylene fractions
| Sample designation | Mw | Mn | Mw/Mna | (°C) (calculated)b | Method ofc preparation |
|---|---|---|---|---|---|
| Low molecular weight region | |||||
| 3.6 K | 3,600 | 3,190 | 1.13 | 133.3 | PGPC (W) |
| 4.2 K | 4,200 | 3,330 | 1.26 | 135.0 | CE |
| 5.10 K | 5,100 | 4,640 | 1.10 | 137.0 | PGPC (W) |
| 5.62 K | 5,620 | 5,350 | 1.05 | 137.7 | PGPC (W) |
| 6.15 K | 6,150 | 5,640 | 1.09 | 138.3 | PGPC (W) |
| 6.29 K | 6,290 | 5,380 | 1.17 | 138.5 | CE |
| 6.35 K | 6,350 | 5,990 | 1.06 | 138.7 | PGPC (W) |
| 7.84 K | 7,840 | 7,610 | 1.03 | 139.9 | PGPC (W) |
| 8.06 K | 8,060 | 7,200 | 1.12 | 140.1 | CE |
| 8.56 K | 8,560 | 7,850 | 1.09 | 140.4 | PGPC (W) |
| 8.59 K | 8,590 | 7,740 | 1.11 | 140.5 | CE |
| 9.70 K | 9,700 | 9,150 | 1.06 | 141.1 | PGPC (W) |
| 11.4 K | 11,370 | 10,430 | 1.09 | 141.8 | PGPC (W) |
| 11.67 K | 11,670 | 10,710 | 1.09 | 141.9 | CE |
| 11.74 K | 11,740 | 10,970 | 1.07 | 142.0 | PGPC (W) |
| 17.0 K | 16,950 | 15,690 | 1.08 | 143.2 | CE |
| Intermediate molecular weight region | |||||
| 18.1 K | 18,120 | 13,040 | 1.39 | 143.4 | PGPC (M) |
| 19.8 K | 19,830 | 19,530 | 1.07 | 143.7 | CE |
| 23.7 K | 23,680 | 22,130 | 1.07 | 144.1 | CE |
| 24.6 K | 24,640 | 23,030 | 1.07 | 144.2 | CE |
| 30.0 K | 30,020 | 26,800 | 1.12 | 144.6 | PGPC (W) |
| 30.6 K | 30,600 | 25,710 | 1.19 | 144.6 | PGPC (W) |
| 37.6 K | 37,630 | 34,210 | 1.10 | 144.9 | PGPC (W) |
| 42.6 K | 42,580 | 34,900 | 1.22 | 145.1 | PGPC (W) |
| 62.8 K | 62,770 | 45,820 | 1.37 | 145.5 | CE |
| 68.6 K | 68,570 | 58,610 | 1.17 | 145.6 | PGPC (W) |
| 74.4 K | 74,440 | 66,460 | 1.12 | 145.7 | CE |
| 115.0 K | 114,500 | 71,120 | 1.61 | 145.9 | PGPC (M) |
| High molecular weight region | |||||
| 119 K | 119,200 | 94,600 | 1.26 | 145.9 | PGPC (W) |
| 134 K | 134,300 | 79,000 | 1.70 | 146.0 | CE |
| 210 K | 210,000 | 146,900 | 1.43 | 146.2 | CE |
| 266 K | 265,500 | 146,700 | 1.81 | 146.3 | PGPC (M) |
| 323 K | 323,200 | 200,700 | 1.61 | 146.3 | CE |
| 500 K | 500,400 | 313,000 | 1.60 | 146.4 | CE |
| 807 K | 807,400 | 507,800 | 1.59 | 146.4 | CE |
Corrected for instrumental broadening effects (see text).
Calculated using equation given later in the text.
PGPC =preparative gel permeation chromatography; CE = column elution.
All of the specimens listed in table 1 were characterized in a Waters Model 200 (analytical scale) gel permeation chromatographic (GPC) apparatus. Each column was 1.22 m long and 0.953 cm in diameter, and consisted of a stainless steel tube packed with beads of rigid cross-linked polystyrene gel. The gel was prepared and packed in the columns by the manufacturer. Five such columns having nominal exclusion limits of 107, 106, 105, 104, and 103Å were connected in series to form the set used for analysis.5
The apparatus was calibrated using fractions whose Mw and Mn values were known to about ± 5 percent from light scattering and osmotic pressure measurements. GPC instrumental broadening effects were estimated by measuring nearly pure samples of n- C32H66 and n-C94H190 for which Mw/Mn was known to be about 1.00. These samples gave Mw/Mn ≅ 1.05 in the GPC analytical apparatus. Therefore, a correction of — 0.05 was applied to the original Mw/Mn data for the fractions characterized with the analytical GPC columns. Detailed studies carried out on some specimens suggested that the distribution of molecular weights P(M) was closely approximated by the customary “log normal” function.
We turn now to the topics of the removal of heterogeneities, and other items that relate to obtaining consistent morphological and growth rate data.
Polyethylene “as received” usually contains an extremely large number of heterogeneities (≥ 109/cm3) that initiate growth centers and cause any crystalline bodies that are formed in a subsequent crystallization to impinge on one another before they attain sufficient size to be critically examined in an optical microscope. Frequently, all that is seen with an optical microscope with crossed polarizers in such material is something that may be described as a grainy fog. Samples prepared directly from SRM 1475 are no exception to this. The usual procedure used to obtain large spherulites is to inactivate most of the heterogeneities by raising the specimen to a temperature T1 that is far above the melting point prior to cooling down to the isothermal crystallization temperature T. The procedure of heating to a high T1 to inactivate most of the heterogeneities is well suited to polymers of high thermal stability such as poly(chlorotrifluoroethylene), where this technique was successfully applied [10]. In the case of polyethylene, the use of a high T1 is successful in inactivating many of the heterogeneities, allowing large spherulites to form in a subsequent crystallization, but the sample is degraded or otherwise deteriorated at the same time, often even showing a distinct brown coloration. (GPC studies on specimen 30.6 K showed no deterioration when stored at T1 = 160 °C for long periods of time between cover slips, but definite changes were noted when the specimen was held at a high T1 (190 °C) for two hours, again between glass cover slips.) The result is that the spherulites seen at the usual isothermal crystallization temperatures (circa 122 to 127 °C) in specimens where high T1 values were used are almost certainly characteristic of polymer that is degraded. A T1 of 155°C was generally used in the present study, which avoided difficulties resulting from degradation.
In order to avoid the problem of heating to an excessively high T1 to obtain large spherulites or axialites, each of the fractions was dissolved in xylene at ~ 135°C (0.1 % solution by weight) and filtered hot three times through a 0.2 μm micro pore filter to remove a considerable portion of the heterogeneities. The filtrate was then cooled to ~ 85°C and the polyethylene crystals precipitated and filtered off. The precipitate was then treated by heating in vacuum at 100 °C for 24 hours or more to remove the xylene. Tests using a gas chromatograph showed that the resultant crystal mats contained less than 10 ppm of xylene. Specimens after filtration generally showed less than 104 heterogeneous nuclei per cm3, which allowed the formation of large spherulites or axialites.
The aforementioned step of crystallization from solution certainly removed a considerable portion of any extremely low molecular weight material that might have been present in the fractions. We also mention that the characterization of the specimens by analytical GPC was carried out subsequent to the filtration and crystallization from solution steps noted above.
A dried and characterized sample was prepared for examination in an optical microscope by lightly pressing 100 to 300 mg of the fraction between glass cover slips at a temperature close to 150 °C in a vacuum oven to make a sandwich where the polymer layer was roughly 40 μm thick. The central region of the specimens was thus protected against oxidation. The cleaning of the cover slips was important, since if they were used as supplied, excessive crystallization began at the polymer-glass interface. The cover slips were first cleaned in chromic acid solution, rinsed thoroughly in distilled water, and then cleaned further in distilled water using conventional ultrasonic scrubbing techniques. After drying, the cover slips were ready for use. When treated as noted above, the cover slips caused little or no nucleation at the polymer-glass interface.
The actual observation of the growth rates were made by heating the specimen in a specially designed microscope hot stage [11] to T1 = 155 °C, which is above the equilibrium melting temperature, and then quickly cooling to a predetermined isothermal crystallization temperature T, usually somewhere in the range of about 118° to 131 °C, and controlling this temperature to ±0.001 °C. Photomicrographs (with the sample between crossed nicol prisms) were made at suitable intervals as the crystallization proceeded at T, and these were analyzed to obtain the growth rates as G = dx/dt where t is the time, and x the radius of a spherulite, or alternatively, one-half the longest dimension of an axialite (see photomicrographs shown later). In practice, the value of G at a given temperature was obtained by analysis of typically 10 points on an x versus t plot constructed from the photomicrographs taken during the isothermal growth process.
The following comments are pertinent to the reliability of the observations of the rate of growth. First, repeated heating to T1 ≅ 155 °C (or any T1 within 5 °C of this value) never caused any noticeable degradation or oxidation of the central part of glass-enclosed specimens. Some discoloration was occasionally noticed at the extreme edge of the cover slips where air could come in contact with the polyethylene, but optical measurements were always confined to regions where this did not occur. Second, the growth rates obtained at various T values on a given fraction were highly reproducible no matter what the T of the previous run. Third, the growth rate G did not depend on the residence time at T1, or the number of times it had been heated to T1. Fourth, the growth rates at any selected temperature were independent of the thickness of the specimen. Finally, we mention that care was taken to assure that the heat of crystallization did not influence the actual temperature of crystallization by restricting the measurements to growth rates that did not exceed about 5× 10−5 cm/s.
Measurements of the sign of the birefringence were carried out using well-known techniques for various specimens to determine the orientation of the polymer chains with respect to the growth direction. Wide angle x-ray (WAXR) measurements on specimens of suitable dimensions were made using fractions in the low, intermediate, and high molecular weight ranges to determine if the usual orthorhombic subcell appeared. Low-angle x-ray (LAXR) measurements were made with a Kratky camera to determine if the crystalization in selected fractions of widely different molecular weights was basically lamellar in character, and to find the values of the various lamellar spacings that were present.
3. Results: Growth Rate and Morphology
3.1. Growth Rate Curves and Determination of Tb and ΔTb
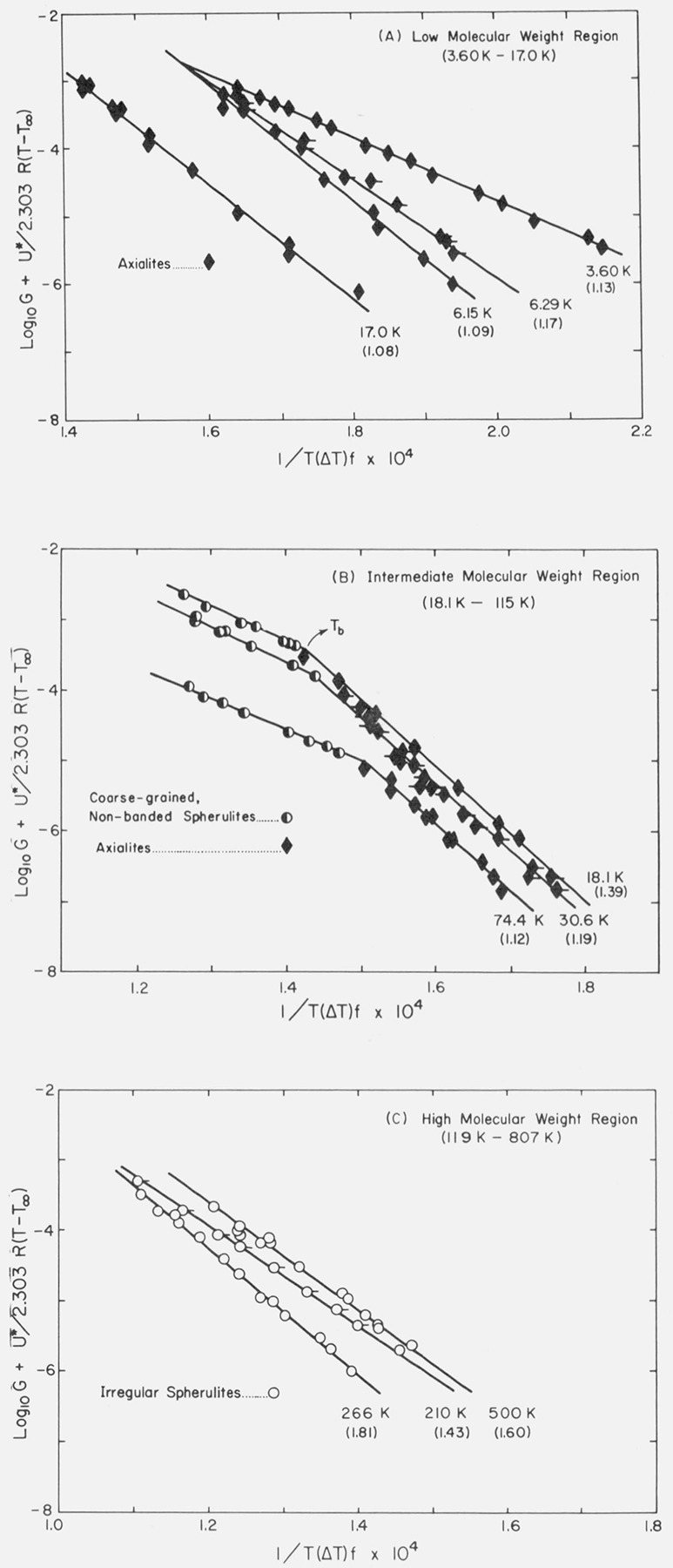
Typical growth rate data for certain fractions between 3.60 and 17.0 K are shown in figure 1A. For convenience, we refer to samples in this general category as being in the “low” molecular weight range. In this region only axialites are seen in the temperature range where isothermal growth can be reliably measured, and the slopes of the growth rate curves change rather markedly with molecular weight. The apparently irregular variations in slope seen in figure 1A as a function of molecular weight are real and reproducible, and will be dealt with in detail after the Kg and σe values have been reported for each specimen. Data for a number of other specimens in the low molecular weight range have been omitted to avoid cluttering the diagram.
Figure 1. Logarithm of spherulite or axialίte growth rate as a function of isothermal growth temperature for low, intermediate and high molecular weight specimens.
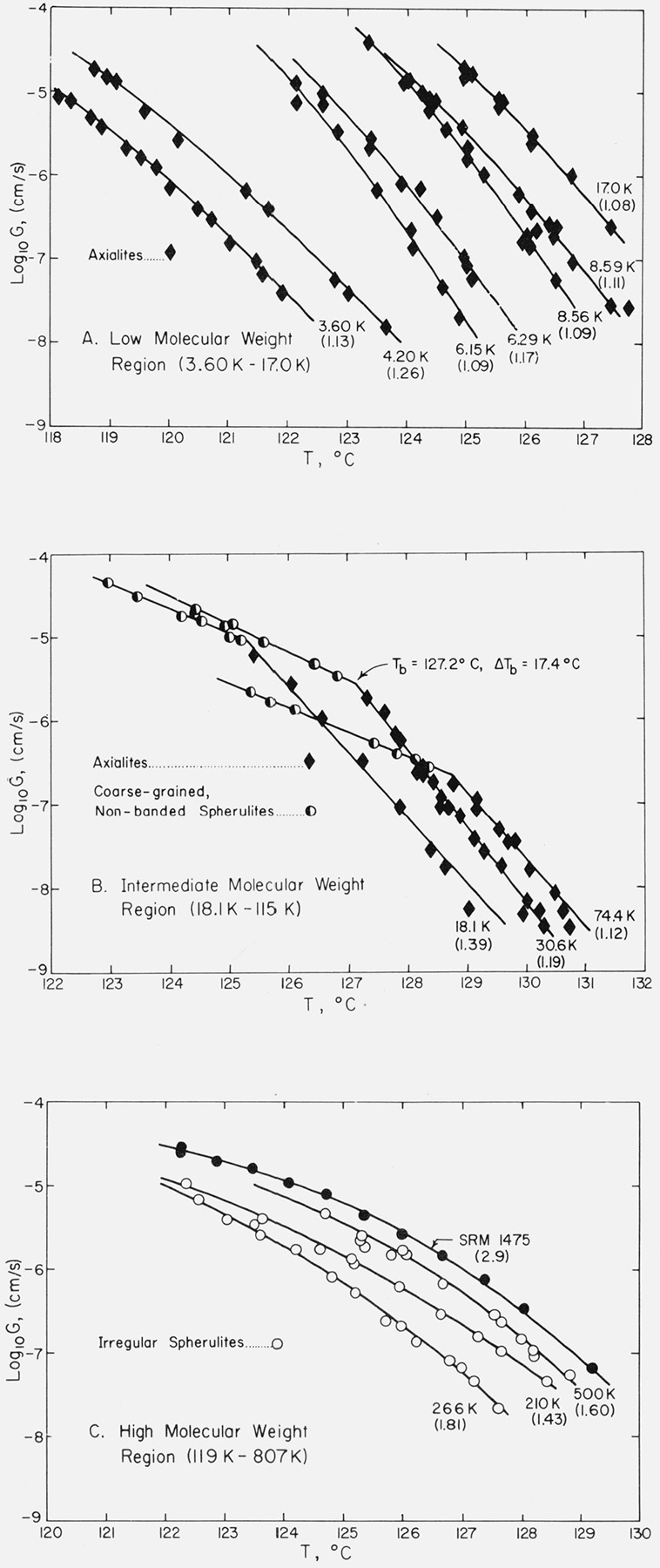
Numbers in parentheses ( ) indicate Mw/Mn for the specimen.
Data are shown for fractions 18.1 to 74.4 K in figure 1B. These specimens are typical of what we term the “intermediate” molecular weight range, which extends from 18.1 to 115 K. It is seen that a definite break occurs in the log10 G versus T data at a temperature that we denote Tb (fig. 1B). Somewhat above Tb, axialites are formed, and somewhat below it only spherulites are seen. (Details of the morphology will be given subsequently.) In the region of intermediate molecular weight, the slopes of the log10 G versus T plots for the axialites are more nearly constant with molecular weight than they were in the low molecular weight range. As in the case of the low molecular weight region, data for a number of other samples were obtained, but are not shown.
In this paper we adhere to the convention of showing axialite data as solid diamonds (♦), coarse-grained non-banded spherulites as half-filled circles (◐), and “irregular” spherulites (see below) as open circles (○).
Before discussing the growth rate data for the “irregular” spherulites that appear in the high molecular weight region, it is advantageous to show that the undercooling at which crystallization is carried out determines whether axialites or spherulites appear in the low and intermediate molecular weight regions.
The variation of Tb with molecular weight as determined from plots of log10 G versus T is shown in figure 2A. It is seen that the break in the growth curves can be clearly detected for samples in the intermediate molecular weight range of Mw= 18,100 to Mw=114,500. This feature is clarified if the under-cooling ΔTb at which the transition takes place is plotted as a function of molecular weight. The result is that the transition occurs at essentially a fixed undercooling of 17.5 ± 1 °C as is shown in figure 2B. Although the rate transitions at Tb are sharper than the corresponding morphological changes (see below), it is nevertheless found that if the undercooling is about 1 °C or more lower than ΔTb axialites are formed, and if it is about 1 °C or more greater than this, spherulites are formed. On the basis of this finding, we surmised that spherulites might be found in the specimens witha molecular weight below Mw ≅ 18,000 if crystallization with an undercooling larger than 17.5 °C could be achieved. Runs could not be made on samples lower in molecular weight than 18,000 at ΔT > 17.5 °C because the growth rates became too rapid to measure with any certainty and there is some question as to whether the growth is isothermal under such conditions, but quenching of specimens of such low molecular weight from the melt to room temperature did in fact lead to (nonisothermal) growth of spherulites. The finding that ΔΓ/, = 17.5 °C approximately separates the axialitic and spherulitic growth modes does not applytosamples exceeding about 115 Kin molecular weight. At these high molecular weights, a different type of morphology appears over the entire range where isothermal growth can be effected (“irregular” spherulites), and there is no break in the logjoG versus T curve.
Figure 2. Growth temperature and undercooling; corresponding to break in log G versus T data.
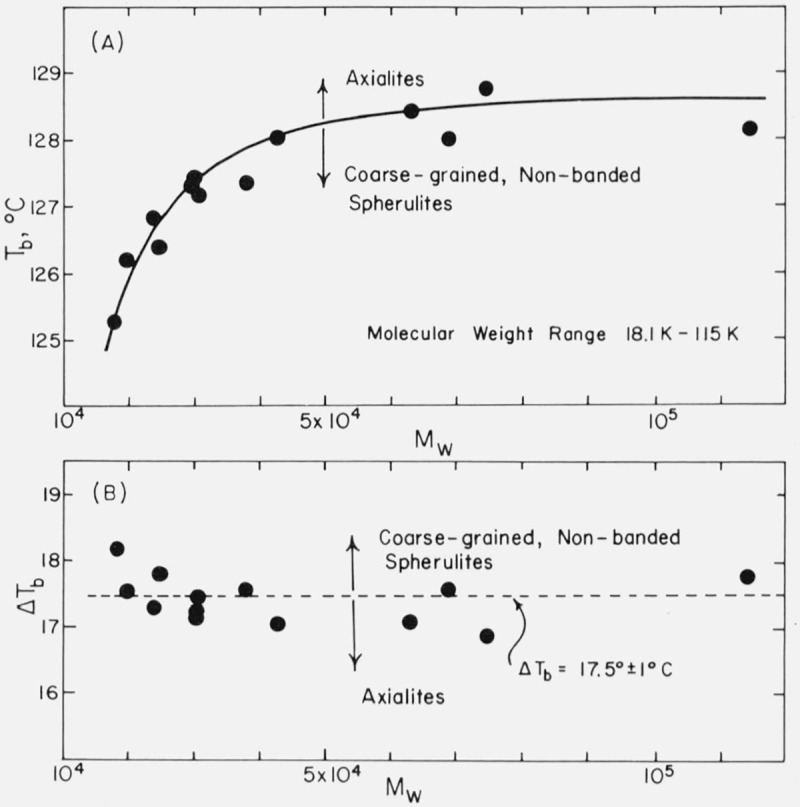
Tb and ΔTb correspond within about 1 °C to the temperatures and undercoolings that separate the axialitic and spherulitic growth modes.
The values of T°m as a function of molecular weight used to estimate ¿>b for the various specimens shown in figure 2B can be obtained from a modified Broadhurst [12] or Flory-Vrij [13] equation (discussed later).
The growth rate of the axialites and coarse-grained non-banded spherulites studied in the low and intermediate range of molecular weight was in every case lineal, i.e., dx|dt at a given temperature is a constant regardless of the size of the spherulite or axialite up to the point of impingement. (The measurements leading to values of G were generally confined to values of x far short of actual impingement.) There was no significant induction period — the x, t data passed through the origin on an x versus t plot within acceptable statistical limits.
Typical growth rate data for the “irregular” spherulites that appear in the high molecular weight region (119 to 807 K) are shown in figure 1C.
A notable feature of the irregular spherulites is that no break is found in the log10G versus T data near ΔTb=17.5 °C. Instead, each log10G versus T plot exhibits considerable curvature and has an average slope that is between that for the axialities and the coarse-grained nonbanded spherulites (compare figs. 1B and 1C).
It is possible that the absence of a relatively sharp break in the log10G versus T data for the specimens of high molecular weight exhibiting “irregular” spherulites may be associated in part with a broad distribution of molecular weights. This can be seen from a comparison of the data in figure 1 and table 1. Most of the specimens in the “intermediate” range in table 1 possess an Mw/Mn ratio of ~ 1.1 to ~ 1.4 and virtually all exhibit a clear-cut break in the log10G versus T data of the type shown in the examples depicted in figure 1B. On the contrary, most of the specimens in the high molecular weight range where irregular spherulites appear have an Mw/Mn ratio of ~ 1.4 to ~ 1.8, and specimens of this class show an overall large curvature rather than a distinct break in a plot of log10G versus T. One possible implication is that a broad distribution of molecular weights may cause the break at Tb to become quite diffuse in the high molecular weight specimens. Note in figure 1C that the SRM from which the fractions were made, for which Mw ≅ 53,000 and Mw/Mn ≅ 2.90, exhibits a distinct curvature rather than a break. It may be recalled that a narrow molecular weight fraction with Mw=53,000 would be expected to have a distinct break in the log10G versus T curve (fig. 1B). However, it must be pointed out that specimen 119 K, which has a moderately narrow distribution (Mw/Mn = 1.26), shows no break in log10G versus T, while specimen 115 K with a broad distribution of molecular weight (Mw/Mn= 1.61) exhibits the break. This suggests as an alternative that the onset of the appearance of irregular spherulites and the absence of a rather sharp break in the log10G versus T data may be an inevitable result of increasing molecular weight rather than mainly a function of the distribution. The question concerning how high in molecular weight the sharp break at Tb actually occurs and where irregular spherulites appear will probably only be answered when fractions with Mw/Mn ~ 1.1 to 1.2 are available in the high molecular weight range, and measurements made over a considerable temperature range.
The overall radial growth of the irregular spherulites is lineal with time, but possesses an apparent small scale (circa 10 μm) sporadic character in that small sectors at the outer boundary of the irregular spherulite appear to grow for a time, stop, and then begin again. This is possibly a result of the details of the extinction pattern that accompany the outward growth of the lamellae rather than an actual starting and stopping of the growth process.
3.2. Morphology
We first examine the question of morphology for samples in the low and intermediate ranges of molecular weight. Figure 3 shows representative axialites in specimens 7.84, 18.1, 30.6, and 42.6 K.6 Note that in each case the undercooling is less than 17.5 °C. Although differences are detectable, it is clear that these structures are rather similar. Figure 4A, 4B, and 4C depicts typical coarse-grained non-banded spherulites in specimens 18.1, 30.6, and 62.8 K. Observe that the undercooling exceeds 17.5 °C in each case. The “maltese cross” effect is present in the extinction pattern, but the spherulites exhibit a coarse texture and are either non-banded or alternatively, have a few very coarse bands. Typical banded spherulites with many concentric rings can be formed by strong quenching from the melt of the samples in the intermediate molecular weight range. An example is shown in figure 4D for specimen 30.6 K, which crystallizes by formation of such spherulites when quenched rapidly from T1 = 155° to 25 °C. The distinctive feature of the present work resides in the fact that both axialites and spherulites were found in different temperature ranges in the same specimen in the intermediate molecular weight range, together with the fact that these objects exhibited significantly different growth rate constants.
Figure 3. Axialites in specimens of low and intermediate molecular weight at ΔT < 17.5 °C (optical micrographs, crossed nicol prisms).

The axialites were grown under isothermal conditions.
Figure 4. Spherulites in specimens of intermediate molecular weight at ΔT > 17.5 °C ,optical micrographs, crossed nicol prisms).

A, B, and C show coarse-grained non-banded spherulites resulting from isothermal growth, ΔT > 17.5 °C. Micrograph D shows typical banded spherulites obtained in specimen 30.6 K by rapid quenching, from T1= 155 to 25 °C.
From a morphological standpoint, the transitions from axialitic to spherulitic structures observed in the intermediate molecular weight range are not completely abrupt as the undercooling at which the crystallization occurs passes through ΔTb. However, there are sufficient differences in the appearance of the crystallizing objects 1 °C above and below Tb to warrant classification as a spherulite or axialite. There are smaller variations in the details of structure within each morphology, with the result that the appearance of the spherulites or axialites in an optical microscope can actually be used by an experienced viewer to estimate the crystallization temperature within a degree or so. A set of photomicrographs illustrating the changes of structure with temperatures and degree of undercooling are shown in figure 5 for specimen 30.6 K. The steady change from the coarse-grained non-banded spherulitic morphology at ΔT > 17.5 °C to the axialitic morphology at ΔT < 17.5 °C is clearly apparent. (At the lowest isothermal growth temperature (T= 123.2 °C, ΔT= 21.4 °C) the spherulites show a few coarse bands.) Samples ranging from 18.1 to 74.4 K show rather similar variations of morphology with decreasing undercooling.
Figure 5. Transition from spherulitic to axialitic morphology in specimen 30.6 K with decreasing undercooling (optical micro-graphs, crossed nicol prisms).
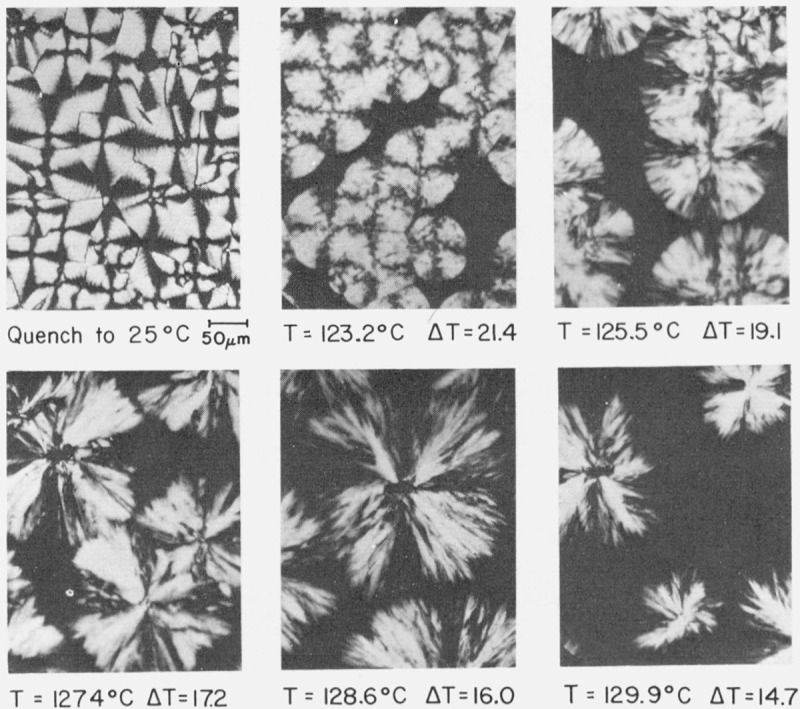
Normal banded spherulites are formed at the large undercoolings effected by quenching. In the ΔT=21.4 °C run there is some evidence of coarse bands. Typical coarse-grained non-banded spheruüties are formed at ΔT= 19.1 °C. The object formed at ΔT=17.2 °C is close to the transition at Tb(see fig. 1B). Axialites are formed at ΔT=l6.0 °C.and ΔT= 14.7°C. Isothermal growth applies in all cases except that denoted quench.
It is worth noting the somewhat unusual structures and phenomena that occur in the specimens in the “high” molecular weight range, i.e., those from 119,000 to near 807,000 in molecular weight. Figure 6 shows the crystalline objects that appear in specimens 119, 210, 323, and 807 K. The clear-cut axialitic morphology characteristic of somewhat lower molecular weights does not appear at undercoolings less than 17.5 °C, and the type of coarse-grained non-banded spherulite seen at intermediate molecular weights at undercoolings greater than 17.5 °C is also absent. Instead, somewhat irregular objects with either a weak or nonexistent “maltese cross” extinction effect appear that in many respects resembles a cauliflower (fig. 6). Some of them show evidence of being banded. We refer to these objects as “irregular” spherulites. The unusual spherulites found for specimen 119 K seem to be a transitional type; the optical micrographs seen in figure 6 for specimens 210, 323, and 807 K are more typical of the “high” molecular weigh, range.
Figure 6. Irregular spherulites in specimens of high molecular weight (optical micrographs, crossed nicol prisms).
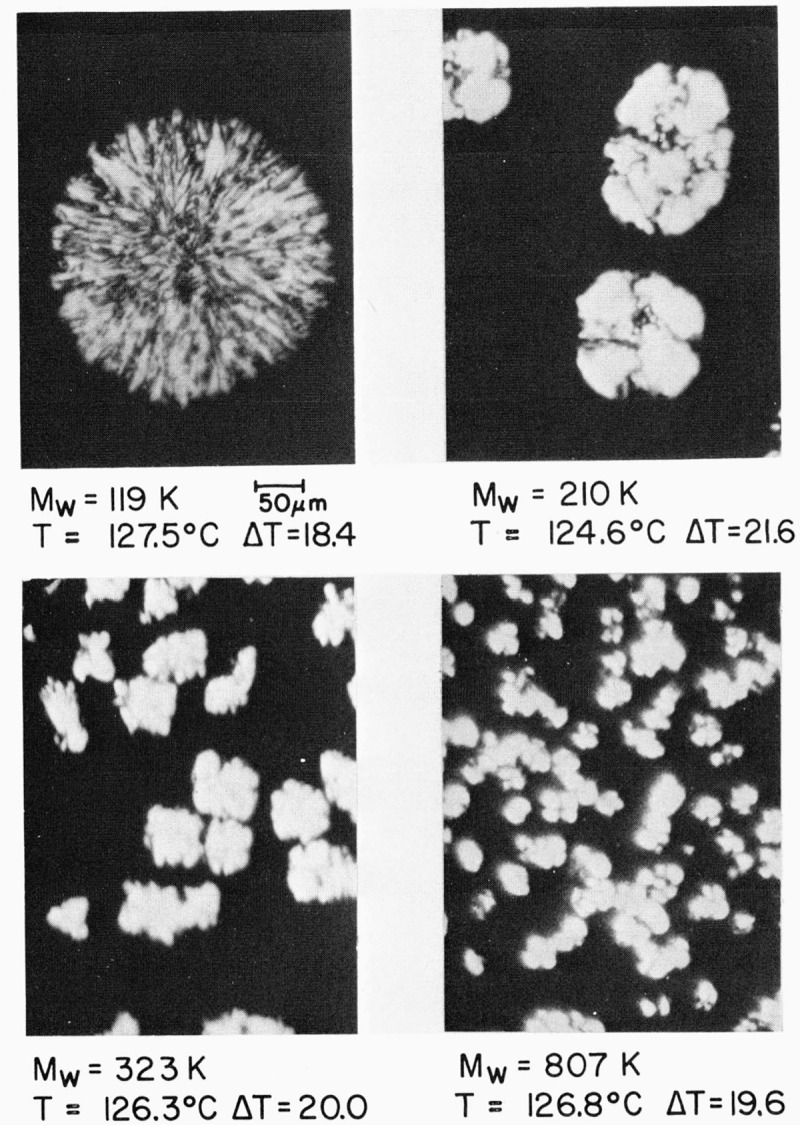
The spherulite in 119 K appears to represent a transition between the intermediate and high molecular weight types. The irregular spherulites in 210 and 323, and 807 K are typical of those found in the high molecular weight region. Growth is under isothermal conditions in all cases.
Some very high molecular weight specimens where Mw > 106 were prepared and examined (not listed). No objects clearly identifiable in an optical microscope with crossed nicol prisms appeared at all despite the fact that differential thermal analysis (DTA) measurements clearly showed that significant crystallization had taken place. Heating of such specimens even for long periods of time to T1 = 155 °C or even T1 = 190 °C failed to remove the birefringence so that any crystalline object that appeared on subsequent cooling was impossible to observe properly with an optical microscope. The highest available molecular weight specimen showing any object whose growth rate could be measured was 807 K.
3.3. Birefringence and X-Ray Studies
Birefringence measurements were made on specimens 11.67 K (axialites), 30.6 K (both axialitic and spherulitic regions), and 500 K (irregular spherulites). In each case the sign of the birefringence showed that the polymer chain axes were approximately perpendicular to the direction of growth. This result has been adduced previously for spherulites in polyethylene.
An investigation of sample 30.6 K was made in both the axialitic and spherulitic regions using wide-angle x-ray (WAXR) techniques. The WAXR studies of specimen 30.6 K showed that both the axialitic and spherulitic forms consisted principally of crystals with the usual orthorhombic subcell. The WAXR lines of the axialitic and spherulitic specimens were almost indistinguishable even when examined in detail. The transition from axiahtic to spherulitic growth centering around Tb therefore does not involve crystaUization into different unit subcells above and below Tb
Each specimen showed weak extra WAXR lines that have been assigned to either the monoclinic or the triclinic subcell by various authors [14–16]. These weak “extra” lines are commonly found in melt-crystallized polyethylene [14–16]. Similar WAXR studies were made on the axialites in a low molecular weight specimen (11.74 K) and on the irregular spherulites in a high molecular weight specimen (500 K) with the finding that the orthorhombic subcell was by a wide margin the predominant one present.
Low-angle x-ray (LAXR) studies were also made on sample 30.6 K crystallized at undercoolings corresponding to the axialitic and spherulitic regions. For LAXR studies on specimens 11.74 K and 500 K, the samples were crystallized in 1 to 1.5 mm capillary tubes at a known temperature to a high degree of crystallinity, cooled to room temperature, and the LAXR lines measured there. In one case (30.6 K) the LAXR lines were also measured in a specially designed and thermostated beryllium cell at the actual temperature of crystallization.
The results of the LAXR studies are shown in table 2. In general, the specimens showed two categories of LAXR spacings. The first and most prominent was L1 (which was usually circa 400 to 600 Å), and its higher orders. The L1 line is relatively narrow and intense, and its higher orders, when reported in table 2, are also distinct and easily detected. (We note, however, that the L1 line for 500 K is considerably broader than that found in the specimens of lower molecular weight.) In sample 30.6 K, crystallized at 128.3 °C and cooled to 30.2 °C, four orders of L1 could be clearly identified (table 2). The second category was the very broad and rather weak so-called “L2” line, which does not appear to be a higher order of L1 [17]. Later we will tentatively identify L2 with the initial lamellar thickness. Melting point data on the specimens discussed here suggest that lamellar thickening (approximately a doubling and sometimes more) occurs in the fractions during crystallization, so it is not surprising that evidence of two different thickness ranges for the lamellae is found in a given specimen (see L1 and L2 values in table 2). (In brief, we postulate that L1 represents thickened (i.e., aged) stacks of lamellae, while L2 represents the younger unthickened lamellae characteristic of the initial thickness.) Observe that the values of L2 are mostly in the range of ~ 180 to ~ 220 Å. We note that this is quite close to the theoretical value of the initial lamellar thickness of lg* = {2σeT°m/(Δhf)(ΔT)} + C2 calculated using the effective value of σe obtained from the growth rate data (see later).
Table 2.
Low angle x-ray spacings for selected fractions
| Specimen | Crystallization temp. °C | Measurement temp. °C | Morphology | X-ray spacingsa | |
|---|---|---|---|---|---|
| L1 (Å) | L2 (Å)b | ||||
| 11.74 K | 122.0 | 30.2 | Axialites | c368(1) | 186(1) |
| 30.6 K | 128.3 | 128.3 | Axialites | 670(1) | 258(1) |
| 30.6 K | 128.3 | 30.2 | Axialites | 454(4) | 195(1) |
| 30.6 K | 124.8 | 124.8 | Coarse-grained nonbanded spherulites | 396(2) | 199(1) |
| 30.6 K | 124.8 | 30.2 | Coarse-grained nonbanded spherulites | 367(1) | 194(1) |
| 500 K | 124.0 | 30.2 | Irregular spherulites | 470(1) | 210(1) |
The numbers in parentheses ( ) indicate the number of orders that were detected.
The error in L2 is rather large, since the line is broad and its value somewhat sensitive to the method used to subtract out the base line.
A weak shoulder at ~ 1200±200 Å was observed for this specimen. This appears to correspond to an extended chain structure, and probably results from thickening.
From the LAXR studies noted in table 2 it is seen that both the axialites and coarse-grained non-banded spherulites in the molecular weight range noted (and probably somewhat outside it) are basically lamellar in character. It is also apparent that the irregular spherulites have a lamellar texture.
It has been demonstrated that the spherulites in rather broad molecular weight distribution samples of melt-crystallized polyethylene possess a birefringence consistent with the chain axes in the crystal being perpendicular to the direction of growth [18]. (This same orientation is shown by microbeam x-ray data [19, 20].) Further, it is known that the spherulites in broad distribution material are basically lamellar in character [21], and the same appears to hold for certain fractions [22]. The axialites and spherulites found in the fractions used in the present study show these same general characteristics, which are thus consistent with lamellar crystallization with chain-folding. In order for the argument for chain-folding to be logically complete, it would be necessary to show that the lamellae in each of the morphological variations were parallel to the direction of growth, or to in some other manner demonstrate that the chain axes in the crystal were perpendicular to the large surfaces of the thin lamellae. This has been shown for spherulitic structures in bulk polyethylene with a rather broad molecular weight distribution [23, 24]. Also, Bank and Krimm indicate that the usual type of spherulites in polyethylene exhibit mostly adjacent reentry type chain folding [25]. (For a full discussion of these topics, see the recent review by Khoury and Passaglia [26].)
Because of the parallelism of the optical behavior and lamellar character of the axialites and spherulites found in the present investigation of fractions with the optical and lamellar nature of spherulites where chain folding has been substantiated for broader distribution material, we proceed with some confidence under the assumption that the fractions exhibit chain-folding with mostly adjacent reentry.
4. Analysis of Data to Obtain Kg and Go
The kinetic theory of nucleation-controlled polymer crystal growth with chain folds leads to the expression [1–3]
| (1) |
for the growth rate G. Here U* is the activation energy for transport of segments to the site of crystallization, R the gas constant, T the crystallization temperature, T∞ a temperature somewhat below the glass transition temperature Tg, ΔT the undercooling T°m-T, and f factor near unity that accounts for the slight diminution of the heat of fusion Δhf as the temperature falls below T°m.7 By plotting log10G + U*/2.303R(T-T∞) against 1/T(ΔT)f, the numerical value of the nucleation constant Kg can be obtained from the slope, and the pre-exponential factor G0 can be determined from the intercept on the log10G + U*/2.303R(T-T∞) coordinate.
It has been demonstrated by Suzuki and Kovacs [27] that eq (1) quantitatively fits the growth rate data for isotactic polystyrene for a range of 100 °C. In a recent review [1], this result has been confirmed, and it was shown further that eq (1) can be used to fit data on a number of polymers crystallized from the melt over a wide range of temperature with considerable accuracy, including nylon 6, poly(tetramethyl-p-silphenylene) siloxane (hereafter denoted TMPS) fractions, and poly(chlorotrifluoroethylene), correlation coefficients ranging from 0.985 to 0.999 being found. In the aforementioned review, and in the work of Suzuki and Kovacs, it was found that U* ≅ 1500 cal/mol and T∞ ≅ Tg - 30 °C, with one exception, allowed the best fit of the rate of crystallization data at low temperatures. (The values of U* and T∞ that describe bulk viscous flow in polymers between Tg and Tg +100 °C are U* ≅ 4100 cal/mol and T∞ ≅ Tg — 51.6 °C. The fact that U*= 1500 cal and T∞ ≅ Tg — 30 °C for crystallization has been interpreted in terms of segmental motions in an adsorbed polymer layer on the lateral face of the crystal [1].) In the case of the present analysis to obtain Kg and G0 for various polyethylene fractions, the crystallization rate is controlled much more strongly by the variation with temperature of the term involving Kg/T(ΔT)f than that involving U*/R(T—T∞), since the region of observable crystallization rates in polyethylene is near the melting point. The variation of U*/R(T—T∞) is small and that of Kg/T(ΔT)f quite large in this region. Therefore in the case of polyethylene, little error in Kg results from rather large variations in the assumed value of U* or T∞, and we proceed on the basis of U* = 1500 cal/mol and T=Tg-30 °C, where Tg = −40 °C = 233.2 K [28]. (The assumption that Tg was —80 °C or 193.2 K would lower Kg for a given set of data by only about 2 percent, and the use of U* = 4120 cal instead of 1500 cal would raise Kg by only 5 percent.) The value of f is approximated [1] by f=2T/(T°m + T). This correction is also quite small for polyethylene, again because the observable isothermal crystallization occurs so near the melting point.
One important parameter that requires discussion is the equilibrium melting temperature T°m, which is a function of molecular weight. This must be known with the best possible accuracy, since Kg, through its relationship to the undercooling T°m—T, is rather sensitively dependent upon it. In figure 7 is shown a plot of T¤m against molecular weight M calculated using a modified form of Broadhurst’s equation [12]. The expression used to calculate the curve is
| (2) |
where T¤m is in kelvins (K), and n=M/14.026 is the number of carbon atoms in the polymer chain. This equation was derived from Broadhurst’s work by setting T¤m at n → ∞ at T¤m(∞) = 419.7K = 146.5 °C, and adjusting the constants accordingly. The curve calculated with eq (2) passes through the melting point data for the short chain hydrocarbons in the orthorhombic form with acceptable accuracy. Therefore, if the value of Tm(∞) can be justified within certain limits, considerable confidence can be placed on the interpolated values of interest here. Broadhurst’s equationis based on a treatment due to Flory and Vrij [13], and the two treatments differ only in minor details.
Figure 7. Variation of T°m with molecular weight according to equation (2).

The experimental justification for selecting T¤m(∞) = 146.5 °C is as follows: (1) Huseby and Bair [29] found T¤m = 145.8 °C± 1 from a plot of Tm versus 1/l where l is the lamellar thickness for polyethylene single crystals with Mw ~ 20,000 to 100,000, implying a T¤m (∞) that is about 0.5 to 1 °C higher, (2) a plot of Tm versus Tx obtained by Weeks [30] for melt crystallized polymer suggests T¤m =145.5±1 °C for a specimen of finite molecular weight, again suggesting that T¤m(∞) for polyethylene of very high molecular weight is slightly higher, and (3) Rijke and Mandelkern [31] have found a melting point for fibrillar polyethylene of T¤m = 146.0 ±0.5 °C. We note also that our suggested value for T°m(∞) lies within the limits given by Flory and Vrij, who give T°m(∞)ι=145.5±l °C. In any event, it seems reasonable on the basis of the above to assume that Tm(∞) is within about 1 °C or so of 146.5 °C. No basic conclusion of this paper would be changed if a different value of T°m(∞) in this range were used or even a value somewhat outside of it. The melting point T°m for each fraction was estimated using the assumption that the M value implicit in eq (2) corresponds to Mw. Numerical values of T°m calculated using eq (2) are given for each of the fractions in table 1.
Typical plots of log10G+U*/2.303R(T-T∞) against l/T(ΔT)f constructed from low molecular weight data of the type shown in figure 1A are shown in figure 8A. As noted earlier, only axialites are found in low molecular weight samples in the range of undercooling where isothermal runs can be made. Similar plots for a number of the samples of intermediate molecular weight based on data of the type shown in figure 1B are depicted in figure 8B. Both axialites and coarse-grained non-banded spherulites appear in this molecular weight region at the undercoolings noted. It is clear from the slopes that the axialites and spherulites have different nucleation constants. Finally, a plot is shown for some of the irregular spherulites found at high molecular weight in figure 8C.
Figure 8. Typical plots of log10 G + U*/R(T—T∞) versus 1/T (ΔT)f in low, intermediate, and high molecular weight regions. Numbers in parentheses () indicate Mw/Mn.
The growth rate G is in units of cm/s.
Values of the nucleation constants Kg obtained from plots such as those shown in figure 8 are shown for all specimens studied in figure 9A. Values of Kg and log10 G0 are given in table 3. The error in the Kg for a given fraction was generally about 3 to 5 percent (calculated as one standard deviation), depending on the number of points and the temperature range involved. To test the reproducibility of our procedures, a portion of the original supply of sample 30.0 K was rerun, including all filtration, optical sample preparation, and growth rate measurement steps. The Kg for the axialites was within 1.7 percent of the previous run, while that for the coarse-grained non-banded spherulites was within 2.6 percent (table 3). Based on this and other reruns (table 3) it is believed that the Kg values are in general correct to within about 3 to 5 percent. In a few cases the error is larger, and these values of Kg are enclosed in parentheses in table 3.
Figure 9. Experimental values of Kg and log10G0 as a function of molecular weight. G0 is in cm/s and Kg is in units of K2.
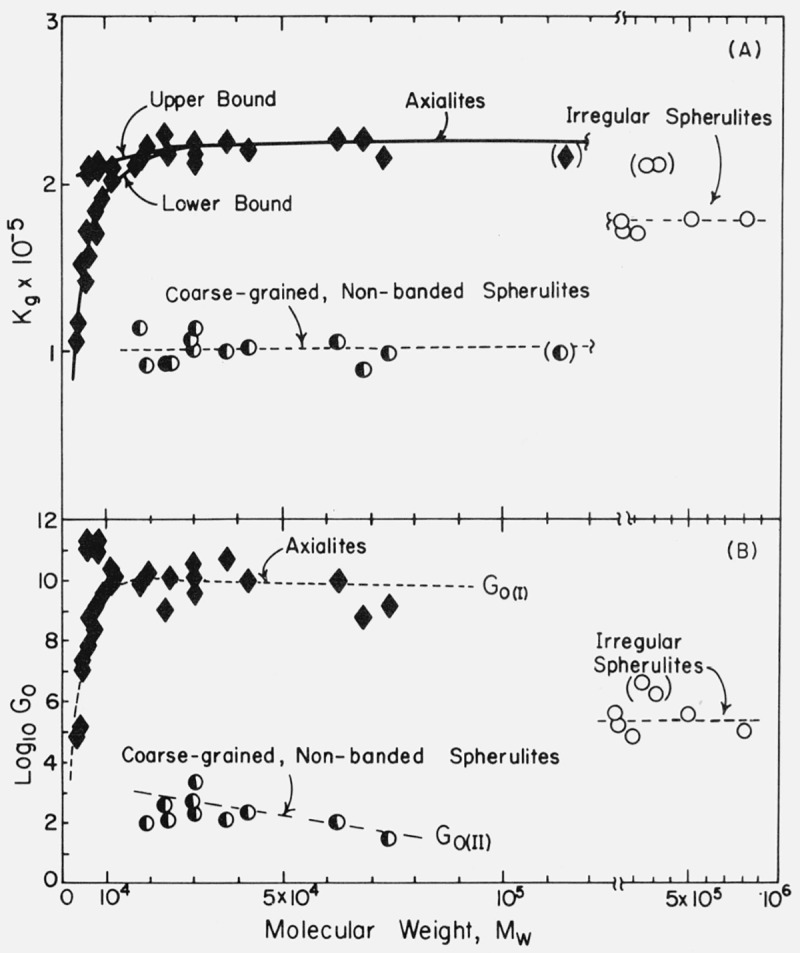
Table 3.
Values of Kg, log10G0 and σσe for various polyethylene fractions
| Sample designation | Mw/Mn | Temperature range | Kg × 10−5 | Log10 G0 | σσe calc. with b=4.15 Å using |
|---|---|---|---|---|---|
| A. Low molecular weight region (axialites) | |||||
| °C | K2 | (G0 in cm · s−1) | |||
| 3.6 K | 1.13 | 117.6–121.9 | 1.069 | 4.97 | 611 |
| 4.2 K | 1.26 | 118.7–123.7 | 1.172 | 5.14 | 668 |
| 5.10 K | 1.10 | 119.9–124.3 | 1.516 | 7.37 | 861 |
| 5.62 K | 1.05 | 119.8–124.7 | 1.434 | 7.15 | 812 |
| 6.15 K | 1.09 | 122.1–124.9 | 2.101 | 11.28 | 1188 |
| a6.15K(R) | 1.09 | 122.1–124.9 | 2.078 | 11.10 | 1176 |
| 6.29 K | 1.17 | 122.6–125.1 | 1.725 | 8.81 | 976 |
| 6.35 K | 1.06 | 120.0–125.2 | 1.578 | 7.86 | 892 |
| 7.84 K | 1.03 | 121.7–126.7 | 1.714 | 8.41 | 966 |
| 8.06 K | 1.12 | 122.7–126.3 | 1.830 | 9.07 | 1031 |
| 8.56 K | 1.09 | 123.4–126.5 | 2.127 | 11.23 | 1197 |
| a8.56 K(R) | 1.09 | 123.4–126.5 | 2.098 | 11.01 | 1181 |
| 8.59 K | 1.11 | 124.3–127.8 | 1.822 | 9.30 | 1025 |
| 9.70 K | 1.06 | 124.3–127.5 | 1.935 | 9.56 | 1087 |
| 11.4 K | 1.09 | 124.2–127.7 | 2.078 | 10.39 | 1166 |
| 11.67 K | 1.09 | 125.0–127.6 | 2.0% | 10.01 | 1175 |
| 11.74 K | 1.07 | 125.3–128.5 | 2.036 | 10.17 | 1142 |
| 17.0 K | 1.08 | 125.0–128.9 | 2.098 | 9.75 | 1173 |
| B. Intermediate molecular weight region (axialites, ΔT < 17.5 °C) | σσe calc. with b=4.15 Å using | ||||
| 18.1 K | 1.39 | 125.2–129.1 | 2.152 | 9.88 | 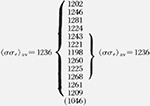 |
| 19.8 K | 1.07 | 126.2–129.1 | 2.231 | 10.23 | |
| 23.7 K | 1.07 | 126.8–130.0 | 2.296 | 9.04 | |
| 24.6 K | 1.07 | 126.4–129.6 | 2.195 | 10.07 | |
| 30.0 K | 1.12 | 127.4–128.9 | 2.230 | 10.50 | |
| a30.0 K(R) | 1.12 | 127.4–129.4 | 2.192 | 10.12 | |
| 30.6 K | 1.19 | 127.2–130.3 | 2.150 | 9.60 | |
| 37.6 K | 1.10 | 127.3–130.0 | 2.263 | 10.70 | |
| 42.6 K | 1.22 | 128.0–129.8 | 2.202 | 10.00 | |
| 62.8 K | 1.37 | 128.4–130.5 | 2.280 | 9.98 | |
| 68.6 K | 1.17 | 128.0–129.7 | 2.268 | 8.77 | |
| 74.4 K | 1.12 | 128.8–130.8 | 2.175 | 9.21 | |
| 115 K | 1.16 | 128.1–130.7 | (1.883) | (6.76) | |
| C. Intermediate molecular weight region (coarse-grained non-banded spherulites, ΔT < 17.5 °C | σσe calc. with b=4.15 Å using | ||||
| 18.1 K | 1.39 | 122.9–125.2 | 1.142 | 3.63 | 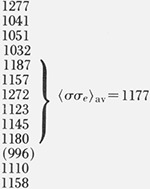 |
| 19.8 K | 1.07 | 125.8–126.2 | 0.932 | 2.00 | |
| 23.7 K | 1.07 | 124.6–126.8 | .942 | 2.60 | |
| 24.6 K | 1.07 | 124.8–126.4 | .925 | 2.12 | |
| 30.0 K | 1.12 | 124.8–127.4 | 1.065 | 2.71 | |
| a30.0 K (R) | 1.12 | 125.0–127.4 | 1.038 | 2.34 | |
| 30.6 K | 1.19 | 124.4–127.2 | 1.141 | 3.33 | |
| 37.6 K | 1.10 | 124.6–127.3 | 1.008 | 2.13 | |
| 42.6 K | 1.22 | 124.8–128.0 | 1.028 | 2.36 | |
| 62.8 K | 1.37 | 125.9–128.4 | 1.061 | 2.06 | |
| 68.6 K | 1.17 | 125.6–128.0 | (0.896) | (0.51) | |
| 74.4 K | 1.12 | 125.4–128.8 | 0.998 | 1.49 | |
| 115 K | 1.61 | 125.6–128.1 | 1.042 | 1.66 | |
| D. High molecular weight region (irregular spherulites) | σσe calc. with b=4.15 Å using | ||||
 |
|||||
| 119 K | 1.26 | 125.3–129.4 | 1.777 | 5.64 | |
| 134 K | 1.70 | 123.1–128.5 | 1.725 | 5.21 | |
| 210 K | 1.43 | 122.4–128.4 | 1.710 | 4.86 | |
| 266 K | 1.81 | 122.6–127.6 | 2.113 | (6.63) | |
| 323 K | 1.61 | 120.3–126.3 | 2.113 | (6.31) | |
| 500 K | 1.60 | 125.0–129.1 | 1.795 | 5.61 | |
| 807 K | 1.59 | 124.5–129.3 | 1.800 | 5.08 | |
(R) denotes complete rerun.
Calculated with regime I kinetics. (See text.)
Attention is specifically drawn to the apparent scatter of the Kg data in figure 9A and table 3 at closely spaced molecular weight values at low molecular weights, especially near Mw= 6200 and Mw = 8600. Note particularly specimen pairs (6.15, 6.29 K) and (8.56, 8.59 K), together with the confirmatory reruns of the high points, 6.15(R) and 8.56(R). This “scatter” is definitely outside the estimated experimental error of the Kg data. The large variation in Kg at these low molecular weights is believed to be associated with cilium length effects, and will be discussed in section 6.
For the axialites, the value of Kg generally rises at first and then tends toward a limiting value at about Mw = 20,000, and remains approximately constant within experimental error up to the highest molecular weight where an accurate axialite run can be made (Mw ~ 72,400). In the case of the coarse-grained non-banded spherulites, the Kg values are approximately constant in the molecular weight range where their growth can be observed (Mw ≅ 18,000 to Mw ≅ 115,000). From an experimental standpoint, the most accurate values of Kg for the coarse-grained non-banded spherulites lie between Mw =30,000 and Mw=62,800. The Kg value for the axialites in sample 115 K shows some evidence of being affected by the transition to high molecular weight behavior; sample 119 K exhibits no axialites and has no Tb at all.
The values of log10G0 obtained from the intercepts on the ordinate of plots of the type shown in figure 8C are shown in figure 9B. Because of the long extrapolation, the scatter in the log10G0 data is rather large. It is estimated that each G0 value is correct to within about one order of magnitude. However, there are differences between the results for axialites, non-banded spherulites, and irregular spherulites that are far outside this error limit, and which are amenable to theoretical interpretation (see sec. 6).
5. Regime I and Regime II Growth: Calculation of σσe from Kg
5.1. Relationship Between Surface Nucleation Rate i and Regime I and Regime II Crystal Growth Rate Laws
The rate of deposition i of chain-folded nuclei on a unit length of a substrate is given by nucleation theory as [1, 2]
| (3) |
where β is a retardation factor measured in units of “events” or “nuclei” per second that has a temperature dependence of the form exp [-U*/R(T-T∞)], C is a constant, in units of cm−1, that is essentially independent of temperature, and (Δf) the free energy difference between the subcooled liquid and the crystal. In general, (Δf) is given by (Δhf)(ΔT)f/T°m, where for a number of polymers including polyethylene, f ≅ 2T/(T°m + T). (In the case of crystallization of polyethylene from the melt, the factor f never differs more than 3 percent from unity. However, in the case of homogeneous nucleation, where ΔT is typically 60 °C, this factor is of considerable importance in obtaining correct estimates of σ2σe.) Thus the surface nucleation rate may be written [1, 2]8.
| (4) |
where for polyethylene z is the number of —CH2 — units corresponding to the initial lamellar thickness l*g. In rough calculations we may take l*g as 200 Å, giving z ~ 158; the quantity a is the molecular width, which is 4.55 Å for polyethylene for growth where folds form along the (110) planes (i.e., the long diagonal) of the unit cell. This corresponds to a layer thickness b of 4.15 Å.9
With such a surface nucleation rate, one can imagine two limiting cases for the rate of growth normal to the substrate which depend on the nucleation rate itself and the rate g that the folded nucleus spreads on the substrate. It is important to examine these possibilities, since it will be found that both occur in polyethylene.
5.2. Regime I (Single Surface Nucleus Causes Completion of Substrate)
The customary assumption is that each nucleation act causes the substrate or “persistence” length L to be completed before a new surface nucleus appears, leading to the accession of a new layer of thickness b (fig. 10). The growth rate G normal to the substrate is in this case given by G=biL. The quantity L is given by nsa, where ns is the number of available stems where crystallization may begin on the substrate (fig. 10).10 In any event, one readily finds in cases where G ∝ i that [1, 2]
| (5) |
Figure 10. Regime I and regime II growth (schematic).
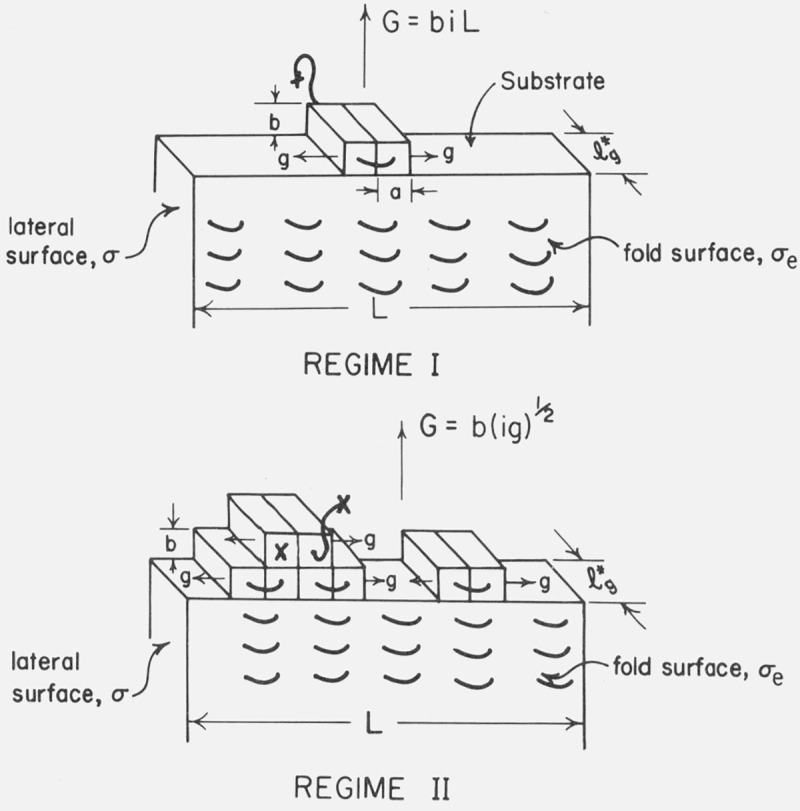
The quantity L is equal to nsa, where ns is the number of stems of length l*g comprising the substrate, and a the width of each molecule.
To a sufficient approximation the pre-exponential factor is given by
| (6) |
where J1 is a factor that was anticipated in previous work [l, 2] to be within perhaps two orders of magnitude of 10−3. Observe that G0(I) contains the factor ns which depends on the mean substrate or “persistence” length L. In previous work ns was estimated to be 103 to 106 [2]. Note that nsz is the number of —CH2 — units on the substrate, i.e., the number of “sites” where crystallization of a new layer can be initiated.
By comparison with eq (1) and eq (5), it is seen that
| (7) |
5.3. Regime II (Numerous Surface Nuclei Involved in Formation of Substrate)
Under certain circumstances to be discussed shortly, it is not reasonable to suppose that the substrate will be completed before many other nuclei impinge on it (fig. 10). Thus we consider the other limit where the surface nucleation rate i is very large compared to the spreading rate g for a specified L. Sanchez and DiMarzio [7] and more recently Lauritzen [8] have shown that the growth rate normal to the substrate in this case is (omitting a numerical factor of the order of unity) given by G=b(ig) 1/2. Noting that the spreading rate is to a sufficient approximation given by [2]
| (8) |
it can be shown that
| (9) |
where to the same approximation that was used for G0(i),
| (10) |
Note that G0(II) does not contain the factor L or ns. The ratio
| (11) |
shows that G0(I) must under comparable conditions always be larger than G0(II).
The value of Kg for regime II has the value
| (12) |
which differs from that for regime I by a factor of two. This arises fundamentally from the fact that GI∝ i, while GII∝i1/2.
We turn now to the question of which formula, eq (7) or eq (12), should be used to estimate σσe from an experimentally known value of Kg.
5.4. Criteria for Regimes I and II
Lauritzen [8] has shown that when the dimensionless quantity
| (13) |
is 0.01 or less that regime I holds with high precision, in which case eqs (5–7) apply. If Z = 0.1, the Kg for regime I is correct to within about 10 percent. Conversely, we estimate that when Z ≥ 1 that regime II is entered, and is closely followed when Z >> 1. Thus for a given substrate length L, the ratio i/g determines which regime of crystallization is applicable. When i/g is large, regime II kinetics hold, and when i/g is small, regime I kinetics obtain. The principal variation of Z with temperature is a result of the large variation of i with undercooling.
The test for regime I and regime II behavior is as follows. We consider the experimental value of Kg, as obtained by analysis of data with eq (1) to be known. We may now write [1, 2]
| (14a) |
where
| (14b) |
and
| (14c) |
In the above expression for Z, it is found in one approximation that [1, 2]
| (14d) |
With these expressions, it is possible to estimate the range of L values that are consistent with regimeI or regime II behavior. What frequently happens is that such a procedure suggests a reasonable value of L for one regime, and an impossible one for the other, allowing the correct choice of regime to be made by reductio ad absurdum. Examples that illustrate this are cited below. The value of L cannot exceed the dimensions of a small spherulite, which places an upper limit of perhaps 25 to 50 μm on this quantity. The lower limit is at present a matter of speculation, but values of L below about 0.1 μm or 1000 Å are suspect: a predicted value of L that is on the order of the molecular width a is clearly inadmissable.
5.5. Application of Regime Test to Spherulites and Axialites in Polyethylene
We select Kg values for the test that correspond to the molecular weight in the intermediate range of ~ 30,000 to ~ 70,000 where Kg is reasonably near its limiting value (fig. 9 and table 3). Values of the parameters useful in the calculations to follow have been collected in table 4 in the appropriate units.11
Table 4.
Input data for calculations on polyethylene
| Quantity | Value | Remarks |
|---|---|---|
| Heat of fusion, Δhf | 2.80 × 109 erg/cm3 | See [1] for original reference |
| Molecular width, a
Layer thickness, b |
4.55 × 10−8 cm 4.15 × 10−8 cm |
Valid at 125 °C for (110) type growth facea |
| Cross-sectional area of chain. ab = A0 | 18.9 × 10−16 cm2 | Valid at 125 °C |
| Fold surface free energy. σe(eq) from Tm vs 1/l plot | 93 erg/cm2 | Ref. [29] |
| Number of —CH2— units in stem. z | ||
| (calc. for l = 200 Å) | 158 | |
| (calc. for l=190 Å) | 150 | |
| Product z exp(2abσe/kT)=zW in eq (14a) | 8.96 × 104 |
If the growth corresponds to (200) type layers, b = 3.82×10−8 cm and a = 4.95×10−8 cm at 125 °C.
For the coarse-grained non-banded spherulites in the range 30,000 to 70,000 we use ΔT=20 °C, T=400 K, and X= Kg = 1.05 × 105 K2. Then for these objects to obey regime I, it is found with eqs (14a) and (14b) that L≤2Å, which must be regarded as completely unrealistic — no growth could occur on such a substrate. The test for regime II using eqs (14a) and (14c) for these spherulites indicates with X = 2Kg = 2.1 × 105 K2 that regime II applies when L ≥ 1.4 μm, which is an entirely reasonable substrate length. It is therefore possible to draw the definite conclusion that the coarse-grained non-banded spherulites in the molecular weight region mentioned grow according to regime II kinetics, in which case Kg is given by 2bσσeT¤m/(Δhf)k.
Consider now the corresponding calculations for the axialites in the intermediate molecular weight range, where we may use Kg= 2.2 × 105 K2, ΔT= 16 °C and T=400 K to make estimates. It is found that for these objects to obey regime I kinetics (Z≤0.01) then L≤~ 8.1 μm. If the condition Z = 0.1 is used then L≤~ 26 μm. Either way, these are reasonable substrate lengths. A test for regime II behavior with X = 2Kg=4.4 × 105 and Z ≥ 1 gives L≥~ 105 cm, which is absurd. We conclude that the axialites grow according to regime I kinetics, so that Kg=4bσσeT°m/(Δhf)k must apply.
It is of special interest to comment on the regime that applies to the axialites in the low molecular weight range where Kg falls significantly below the value ~2.2×105 K2 typical of the intermediate molecular weight range. Recall that in the intermediate range that the axialites follow regime I kinetics. Theoretically, the low Kg at low molecular weights could be a result of two quite different causes. The first hypothesis is that regime I applies as it does at higher molecular weights and that it is σe (viewed as the average surface free energy of cilia and folds) that actually falls, leading to the low Kg value. The second hypothesis is that Kg falls at low molecular weight because regime II is entered, this process taking place in such a way that σe is approximately constant. These hypotheses are easily checked using the most extreme axialite case, sample 3.6 K, for which Kg= 1.069 × 105 K2. A simple calculation using Z ≥ 1 shows that L would have to be larger than 0.14 cm for regime II kinetics to apply. This is clearly impossible, so pure regime II behavior can be discounted. A test for regime I kinetics using Z ≤ 0.1 indicates that regime I is applicable if L is 1250 Å or less. (In making this estimate, it was assumed in calculating W in eq (14d) that σe had fallen to a value of 45 erg/cm2.) This may be regarded as perhaps a borderline case of regime I behavior, but specimens of somewhat higher molecular weight give results that clearly imply the applicability of regime I kinetics for the axialites. In all these instances, the occurrence of regime I can be traced to the low undercooling at which axialites grow in specimens of low molecular weight. The average undercooling for specimen 3.6 K is only 13.6 °C. Thus, in the expression for Z, a low Kg is compensated for by a low value of . It is our conclusion that the falling off of the value of Kg for the axialites in the low molecular weight region is not a result of any serious variation of regime with molecular weight, but is instead caused mostly by a genuine lowering of σe as the molecular weight falls. The subject of the variation of σe with molecular weight will be dealt with in more detail in subsequent sections.
The case of the “irregular” spherulites that appear from 119 to 807 K is of interest. Using the test value Kg≅ 1.75 × 105 K2 from table 3 as X in eqs (14), it is found that the substrate length L is such as to suggest that the samples are crystallizing between regimes I and II. At ΔT = 20, for regime I to apply exactJy (Z ≤ 0.01), then L would have to be less than 150 Å. This is too small a substrate to be fully credible. Under the same conditions of undercooling, L would have to be 0.83 cm or larger to have regime II kinetics. This is much too large to be acceptable. The true value of L almost certainly lies between these extremes, and the samples therefore behave in a manner intermediate between regimes I and II. In this situation we apply the approximate formula
| (15) |
where the subscript (I, II) means mixed regime I and II behavior, and where it is understood that the factor of 3 contains an inherent uncertainty of perhaps ±1/2.
Two of the irregular spherulite specimens (266 and 323 K) are close to regime I. The rest all fall between regimes I and II as noted above.
5.6. Sharpness of the Regime l → Regime II Transition: An Estimate of L
The relative sharpness of the transition between regimes I and II is evident in figures 1B and 8B, and requires comment. This effect can be understood by plotting Z versus ΔT using
| (16) |
with the constants appropriate to polyethylene given in table 4. As noted earlier, the temperature dependence of Z is controlled principally by the nucleation rate i, which according to eqs (3), (4) and (16) depends on σσe. Taking σσe to have the nominal value of 1250 erg2/cm4, which corresponds to Kg(I)≅2.2×105K2, it is easy to calculate Z versus ΔT for various assumed values of L. The results are shown in figure 11 for L values between 1 and 30 μm.
Figure 11. Plot of Z versus ΔT for polyethylene for various substrate or persistence lengths L (theoretical).

Calculated with σσe = 1250 erg2/cm4, z=158 and other data from table 4 using eq (16). Regime I kinetics are substantially in effect for Z ≤ 0.l; and regime II kinetics are approximately obeyed for Z = 1 and are fully in effect for Z ≥ 10. The shaded area shows region of Z and ΔT where the bulk of regime I → regime II transition takes place.
Consider the curve for L = 5μm. It crosses the line Z = 0.1 at ΔT=17.6 °C, and intersects Z=1 at ΔT=19.2 °C. Regime I is valid within 10 percent for Z=0.1, and regime II is substantially obeyed for Z=1, and obeyed with considerable precision for Z ≥ 10. Thus, we estimate that the bulk of the transition takes place in a temperature range no larger than ~ 1.6 °C. It is also interesting to note that the treatment outlined above suggests that for a fixed value of L the transition should take place at a fixed undercooling, which is what is observed for specimens of different molecular weight (ΔTb=17.5±l °C, fig. 2B). Assuming that ΔTb corresponds to Z ~ 0.3, it is found that ΔTb is about 17.5 °C for L= 10 μm, which is in good accord with the experimental value of ΔTb=17.5±l °C.
It may be surmised from the above that the value of L near the regime I→ regime II transition is within about a factor of about three or so of 5 μm for polyethylene in the intermediate molecular weight range. The width of the fibrils in polyethylene crystallized from the melt has been reported to be of about this magnitude [22, 32].12 The value of L may be connected with the existence of a fibrillar habit in polyethylene spherulites — in such a case the width of the fibrils might be governed by L interpreted as a persistence length. Possibly this conception deserves further consideration, but we shall not pursue it further here.
5.7. Calculation of σσe as a Function of Molecular Weight from the Kg Values for Various Fractions
The K g data for the axialites in the low and intermediate range have been analyzed using regime I kinetics according to eq (7) to obtain σσe, and the results given in table 3 and plotted in figure 12A. For the axialites, the value of b in the expression for Kg(I) was taken to be equal to b(110), i.e.,4.15 Å (table 4). (The symbol b(110) refers to the thickness of the layer growing along the (110) planes; this is d(110) in the customary crystallographic notation.) This tentative assignment is based mostly on the physical appearance of the axialites, inasmuch as they at least superficially resemble aggregates of single crystals of the type grown from solution. By the use of a linear molecular weight scale, figure 12A emphasizes those results that correspond to the molecular weight range 18,100 to 72,400 where Kg and hence σσe is nearly constant with molecular weight. Because of the logarithmic molecular weight scale, figure 12B stresses detailed values of σσe calculated for the axialites in the region of low molecular weight where Kg increases noticeably with increasing molecular weight, and where a large variation in σσe occurs because of the variation in Kg.
Figure 12. σσe and σe as a function of molecular weight.
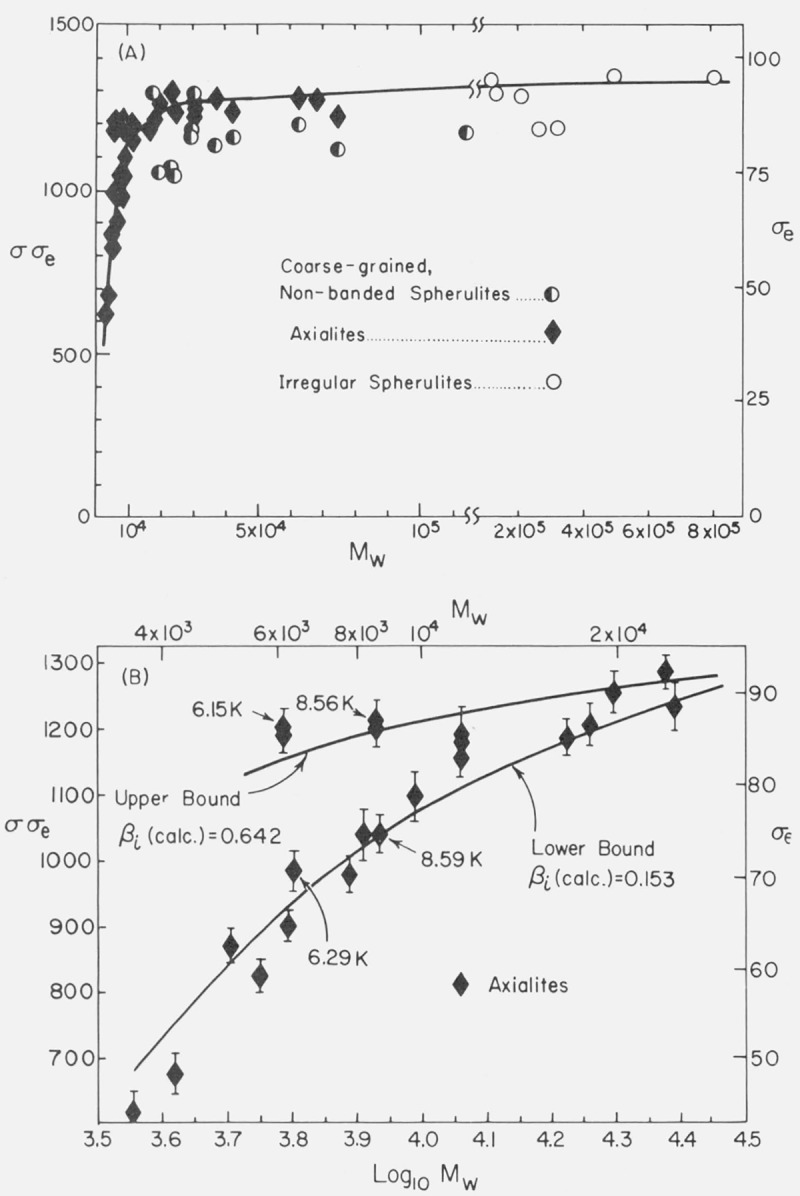
The Kg data for the coarse-grained non-banded spherulites in the region Mw=18,100 to 115,000 were analyzed to obtain σσe using regime II kinetics with eq (12). These results are shown in table 3 and in figure 12A. For these spherulites, the value of b in the expression for Kg(II) was taken to be equal to b(110), i.e., 4.15 Å (table 4). This assignment of b might seem to contradict the work of Bank and Krimm [25] who found predominantly (200) type folds for spherulites grown at ΔT> 17.5 °C in polyethylene with a broad molecular weight distribution. Actually these folds are parallel to the direction of overall growth, the latter being colinear with the b axis of the unit cell. This leaves open the definite possibility that the leading growth front is really of the (110) type. The work of Bank and Krimm does not preclude the existence of a certain fraction of (110) type folds.13
It is evident from figure 12A and table 3 that the σσe values for the axialites and coarse-grained non-banded spherulites that occur in the same specimens between 18.1 and 72.4 K are approximately the same despite the fact that the experimental Kg values from which these results were calculated differ by a factor of about two. This is mostly a result of correctly identifying the regime of crystallization in the two cases. In addition, it is clear from figure 9 and table 3 that, in qualitative accord with expectation, the G0 values for the axialites (regime I) are very much larger than those for the spherulites (regime II) in the same specimens.
With the two exceptions, the Kg, data for the irregular spherulites found between 119 and 807 K were analyzed for the approximate value of σσe using eq (15) with b = 4.15 Å and the results given in table 3 and plotted in figure 12A. The exceptions to the use of “mixed” regime kinetics are samples 266 and 323 K, which approximate regime I. Note that the σσe values for all these specimens are quite close to what was found for the axialites above Mw ≅ 20,000. The G0 values for the irregular spherulites are in all cases between those of the axialites and the coarse-grained non-banded spherulites, again suggesting a mixed regime I and II growth mode.
It should be recalled that the numerical value 3 in eq (15) possesses an inherent uncertainty of up to 15 to 20 percent, so the σσe values for the “mixed” regime irregular spherulites are not as accurate as they are for the axialites and non-banded spherulites. Nevertheless, it is perhaps somewhat surprising that the advent of interlamellar links on a fairly large scale, which is thought to occur between Mw= 105 and 106, does not affect σσe and the corresponding average value of σe in a more obvious manner. (An interlamellar link of substantial length must be expected to exhibit a high local surface free energy.) Polyethylene exhibits a marked reduction in degree of crystallinity in this range [33, 34] which has been interpreted partly in terms of the occurrence of a considerable amount of “amorphous” material consisting at least partly of interlamellar links [1]. One possible implication of the above is that the number of such links is small and the amount of material in each one rather large at high molecular weights.
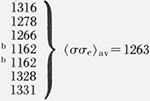
To a fair approximation, the various ranges of Kg values shown in figure 9 for the different morphologies lead to a single master curve for σσe for Mw ≥ ~ 20,000 when Kg, is analyzed by taking proper account of the regime of crystallization (fig. 12A). This is especially true of the data for the axialites and the irregular spherulites. The results for the coarse-grained non-banded spherulites appear to be slightly lower than those for the axialites. However, it may be noted that a statistical analysis of the “best” data, which lie between 30.0 and 62.8 K, shows that the coarse-grained spherulites and axialites have slightly overlapping values calculated as one standard deviation: coarse-grained non-banded spherulites, <σσe>av=1177±53 erg2/cm4; axialites in the same range, <σσe>av= 1236±26 erg2/cm4. The value of <σσe>av for the irregular spherulites is 1263± 58 erg2/cm4.
5.8. The Pre-exponenti al Factors for Regimes I and II
Before proceeding to analyze σσe to get the molecular weight dependence of σe, it is worthwhile to briefly examine the pre-exponential factors for regime I and II. The ratio G0(I)/G0(II) as given by eq (11) is independent of J1, and will be dealt with first. The best data on this ratio encompasses specimens 30.0 to 62.8 K. Here regime I and regime II crystallization occur in the same specimen over a reasonable temperature range for each regime, and the data exhibit good internal consistency within each regime. The ratio log10[G0(I)/G0(II)] obtained from the data in table 3 is shown in table 5A, together with the theoretical value calculated using eq (11) for the case L = 10−3 cm (10 μm). The result may be regarded as satisfactory in view of the fact that the value of the ratio G0(I)/G0(II) is from an experimental standpoint probably not reliable to better than an order of magnitude. In any event, the theory certainly predicts the bulk of the difference in the pre-exponential factors in regimes 1 and II. Most of this difference is a result of the factor ns, which occurs only in the expression for G0(I).
Table 5.
Estimates of pre-exponential factors
| A. The ratio G0(I)/G0(II) | Log [G0(I)/G1(II)] | |
|---|---|---|
| Experimental value (30.0 to 62.8 K) | 7.66 ± 1.0 | |
| Theoretical value for L = 10 μm (eq 11) | 6.86 | |
| B. Absolute valuesa of G0(I) and G0(II) | G0(I), cm/s | G0(II), cm/s |
| Theoretical value (with ΔF† = 4.5 kcal/mol;.J1 = 3.6 × 10−3) | 0.52 × 1010 | 0.72 × 103 |
| Experimental value (30.0 to 62.8 K) | 1.4 × 1010 | 0.3 × 103 |
G0(I) calculated with L = 10 μm.
The absolute values of G0(I) and G0(II) depend on J1 = exp(−ΔF✝/RT) (see eqs (6) and (10)). For the choice L ≅ 10 μm, which is within the range estimated previously, the most consistent results are obtained when J1 is set equal to 3.6 × 10−3, which corresponds to ΔF✝ = 4500 cal/mol. Then the absolute values of G0(I) and G0(II) are predicted within a factor of no worse than three (table 5B). The factor J1 was inserted in β = (kT/h) J1 exp[- U*/R (T- T∞)] - from which position it enters the absolute growth rate - to account for any orientational or “parking” problem effects that might occur as polymer segments migrate across the substrate to the site of crystallization [1, 2]. The value of ΔF✝~ 4500 cal/mol is undoubtedly subject to considerable uncertainty. From a theoretical standpoint little can be said of J1 without a much more detailed model of the surface segmental motions involved in crystallization except to note that it should be substantially less than unity. In the case of the crystallization of copolymers or partially atactic polymers, a “parking” problem arises which should cause J1 to become considerably smaller than was estimated for polyethylene.
Finally, we comment on the dependence of G0 on molecular weight. If the absolute growth rate depended on the melt viscosity, one would expect G0 to vary as M−3.4. Between Mw ≅ 8500 and Mw= 74,400, the value of G0(I) for the axialites shows some scatter, but no persistent trend with molecular weight (see fig. 9B and table 3). In this region the melt viscosity increases as M3.4, i.e., it rises by a factor of (74,400/8500)3.4 or about 1600 times. It is therefore possible to state with considerable certainty that the absolute growth rate of the axialites, as reflected in the value of G0(I), is not markedly affected by changes in the macroscopic viscosity. (Recall also that Kg and σσe are approximately constant over much of this range.) The theory for β was constructed in such a way as to refer to the “segmental” or “local jump” rate, which depends but little on molecular weight, and this accounts for the fact that G0 is not predicted to vary strongly with molecular weight [1, 2]. Evidently this assumption was a good one for axialites in the molecular weight range cited. Specimen 115 K shows a low value of log10G0 in the axialitic region (see table 3), but this sample is very close to the transition region where irregular spherulites appear, and has rather broad distribution.
The situation with the molecular weight dependence of G0 for the coarse-grained non-banded spherulites that occur between 18.1 and 115 K is not so clear-cut because of the large scatter (fig. 9B). On examination of the data, one nevertheless gains the impression that G0 for these structures falls as the molecular weight increases. A statistical analysis of all the points suggests that G0 ∝ Mw−1.5±0.5. It is extremely unlikely that the variation can be as large as Mw−3.4, which again means that the melt viscosity does not play the dominant role in the molecular weight dependence of G0.
The pre-exponential factor for the irregular spherulites is insensitive to changes in molecular weight from 119 to 807 K. It is evident that the macroscopic viscosity does not lead to a change of G0 in the high molecular weight region.
Our overall conclusion is that the theory gives a reasonable explanation of the order of magnitude of the ratio of the pre-exponential factors for regime I and regime II crystallization in the intermediate molecular weight range, and provides some insight into the factors that control the absolute magnitude of the pre-exponential. Another conclusion is that the macroscopic viscosity does not directly govern the pre-exponential factor or the absolute crystallization rate.
6. The Variation of σe With Molecular Weight
6.1. Experimental Determination of σe
Our first objective in this section is to extract the average value of σe from the product σσe for each fraction, and thereby establish the “kinetic” value of σe as a function of molecular weight. Then a simple theoretical treatment will be given which elucidates the variation of σe with molecular weight, and thereby allows the theory of chain folding to be applied over practically the entire range of molecular weight studied.
Basically what we shall do is establish a reasonable value of σ, and find the σe relevant to each molecular weight by calculating it as σσe/σ.
In the first method of obtaining σ, we estimate the value of σσe(∞) by noting that <σσe>av for the axialites in the intermediate molecular weight range (30.0 to 62.8 K), which corresponds to an average of v ≅ 22 per molecule, is 1236±26 erg2/cm4. We then correct this value slightly upward by the factor (v+l)/(v + βi) = 1.04 with ßi = 0.15 (see later) to get the estimated limiting value at high molecular weights, σσe(∞)= 1285 erg2/cm4 (table 6, item A.1). The value of σ=13.8 erg/cm2 is then estimated from σσe(∞) using σσe(eq) as shown in table 6, item B.1. In making this estimate of σ, axialite data have been emphasized because of their rather low standard deviation, and because the explanation of the large variation of σσe and σe in the axialites at low molecular weights is our main objective. However, we note that no basic conclusion of this paper would be altered if one of the other estimates of σσe(∞) in table 6A were employed in the estimation of cr.
Table 6.
Values of <σσe>aν and estimates of σσe(∞), σ and other parameters
| A. Input Data | Valuea | Estimated value of σσe(∞) (v → ∞) | |
|---|---|---|---|
| erg2/cm4 | erg2/cm4 | ||
| 1. 〈σσe〉av, axialites (30.0 to 62.8 K; | 1236 ± 26 | 1285 | |
| 2. 〈σσe〉av, coarse-grained non-banded spheru-lites (30.0 to 62.8 K; | 1177 ± 53 | 1224 | |
| 3. 〈σσe〉av, irregular spherulites (119 to 807 K) | 1263 ± 58 | 1263 | |
| B. Estimates of σ and σe | σ | σe | σe(∞) |
| erg/cm2 | erg/cm2 | erg/cm2 | |
| 1. Calculate σ as σe(∞) axialites/σe(eq) = 1285/93 | 13.8 | - | - |
| 2. Calculate σ for polyethylene using α = 0.12 obtained from homogeneous nucleation in n-octadecane using σ = α(Δhf)(ab)1/2 | 14.6 | - | - |
| 3. Combine 〈σσe〉av = 1236 erg/cm2 (axialites 30.0 to 62.8 K) with σ2σe = 18,000 erg3/cm6 from homogeneous nucleation data on polyethylene in same M.W. range [35]. | 14.6 | 84.9 | 88.3b |
| C. Best values —summary | |||
| Value | Remarks | ||
| 1. σ = 14.2 ± 0.4 erg/cm2 | Mid-range value for B.1, B.2, and B.3 above. | ||
| 2. α = 0.117 ± 0.006 | Calc. from α = σ/(Δhf)(ab)1/2 | ||
| 3. σe(∞) ≅ 90.5 erg/cm2 | Calc. as 1285/14.2 | ||
Errors quoted as one standard deviation.
Calculated as σe × 1.04.
The second estimate of σ is made by using the empirical relation [1, 2]
| (17) |
where α was determined by homogeneous nucleation studies on n-C18H38 (table 6, item B.2). (For details see reference [1].)
Finally we obtain another estimate of σ by combining σ2σe from homogeneous nucleation data on polyethylene fractions [35] and σσe from growth data (table 6, item B.3). Notice that this calculation also gives a value of σe(∞) in reasonable accord with the equilibrium value. The homogeneous nucleation data were confined to the same molecular weight range as the axialite data in A.1 in table 6, e.g., 30.0 to 62.8 K.
From these results it is found that σ=14.2±0.4 erg/cm2 (mid-range value for the three estimates). A very similar estimate of cr was given in a recent review [1]. Values of σe calculated by dividing the σσe value for each fraction by 14.2 erg/cm2 are shown in figure 12.
The foregoing procedure involves the implicit assumption that σ does not depend on molecular weight, and that the variation of σσe is due to changes in the average value of σe. Combination of σ2σe from homogeneous nucleation data of Ross and Frolen [35] and σσe from growth data taken on the fractions studied here at low molecular weights suggests that σe is the quantity that varies with molecular weight.
The existence of a master curve at moderate to high molecular weights for the variation of σσe with molecular weight that is approximately independent of morphology implies of course that there is a similarly shaped master curve for σe versus Mw (see fig. 12A, right-hand scale).
6.2. Theory for Variation of σe With Molecular Weight
Insofar as the variation of σσe or σe is concerned, it is seen in figure 12 that it is only at fairly low molecular weights — say from about 20,000 or so downward for the axialites — that we need seriously concern ourselves with a theoretical treatment for the variation of σe. Above this molecular weight, the theory of growth with chain folds appears to be applicable with a nearly constant value of σe or σσe. Three morphologically distinct species appear from a molecular weight of 20,000 and upward, but the temperature dependence of their growth is in each case governed by practically the same value of σσe in the expression for the surface nucleation rate i. As the “regime” analysis showed, the temperature dependence of the axialite growth rate varies as i, the growth of non-banded spherulites varies as i1/2, and the growth of the irregular spherulites changes with temperature as i3/4. but the σσe or σe value that controls i is about the same for each modification at moderate and high molecular weights.
We turn now to the low molecular weight region, and ask two questions: (1) why does the value of σσe and σe generally fall at low molecular weights and (2) why do the data show such strong variations for samples of practically the same molecular weight (fig. 12B)? That these variations are real and far outside experimental error can be traced through the analysis beginning with the log10G versus T data in figure 1A. (See for example specimen pairs (6.15, 6.29 K) and (8.56, 8.59 K) in fig. 1A and note how these data lead through the Kg values in table 3, to high and low points at nearly the same molecular weight in fig. 12B.) Note also that the reruns of the high points 6.15 and 8.56 K, denoted 6.15 K(R) and 8.56 K(R) in table 3, gave practically identical results for σσe.
The simple model shown in figure 13A will be helpful in discussing the change of σe with molecular weight and the intermittent high and low values of σe shown in fig. 12B. (Note curves denoted “upper bound” and “lower bound” in fig. 12B.)
Figure 13. Model for variation of σe with molecular weight.
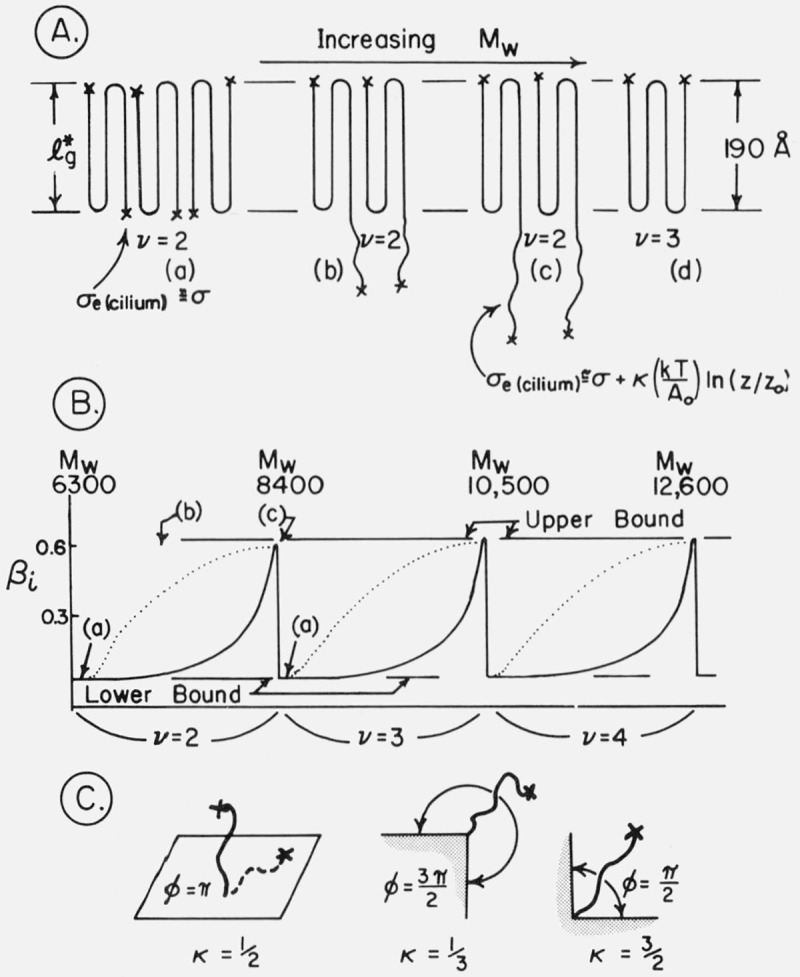
Item B calculated for case l*g = 190 Å.
Treating the observed surface free energy of the surface nucleus (and subsequent steps) as the sum of the effects of cilia and folds we may write [3]
| (18) |
where σe(∞) is the limiting value of σe at high molecular weights corresponding to a strictly folded surface, and v the number of folds per molecule on a substrate of width l*g. The quantity βi is
| (19) |
where σe(cilium) is the local contribution associated with a cilium (or chain end in the surface)to the average surface free energy. We will take σe(∞) to be 93 erg/cm2 for preliminary calculations. This will prove to be within a few percent of the “best” value.
Specific attention is drawn to the fact that eq (18) was derived [3] on the assumption that the first chain end is situated essentially flush with the fold surface as shown schematically in figure 13A. One basic reason for making this assumption (entirely apart from its simplicity) is that it minimizes the local surface free energy associated with a molecule that is forming a nucleus on the substrate. This assumption involves the implication that the molecule to be attached has time to sample various configurations before it becomes a surface nucleus, or participates in the subsequent strip completion process, Evidence exists suggesting that molecules are physically adsorbed to some extent prior to crystallization, and crystallize after transport across the surface [1, 2]. Such a process would allow ample opportunity for the assumed configuration to occur. In practice, it is sufficient that the first chain end be associated with a quite short cilium rather than one that is always exactly flush with the fold surface.
As a useful simplification, we will assume that the width of the substrate, l*g, is approximately constant in the region of interest. (It is apparent in table 2 that L2 generally has a value in the range of 200±20 Å; later we shall calculate that l*g is within the same limits.) In the case of a nearly constant substrate width, it is seen in figure 13A that as the molecular weight is increased (a) through (d) that the pendant cilium at first gets longer as the molecular weight increases [(a) through (c)]. Then as the cilium becomes as long as l*g, a new fold appears at v+l with a short cilium, and the process repeats itself, (d). This process will cause σe(cilium), and hence βi to vary in some manner (solid and dotted lines in fig. 13B) between the lower and upper bounds of βi.
The experimental σe data in figure 12B exhibit rather definite upper and lower bounds. This shows that βi in eqs (18) and (19) has a corresponding upper and lower bound. Let us now attempt on a theoretical basis to estimate the upper and lower bounds of βi. We obtain the lower bound by setting σe(cilium) ≅ σ = 14.2 erg/cm2, which with σe(∞) = 93 erg/cm2, gives βi, = 0.153. This leads to
| (20a) |
Assuming that l*g = 190 Å, each stem has a molecular weight close to 2100. The molecular weight M at each integral v value is 2100 (v+ 1), which leads to
| (20b) |
This function is plotted in figures 12B and 14. It is seen to give a reasonable description of the lower bound of the data from Mw ≅ 5,000 upwards.
Figure 14. σe as a function of molecular weight for axialites showing effects of long and short cilia as v → v + 1.
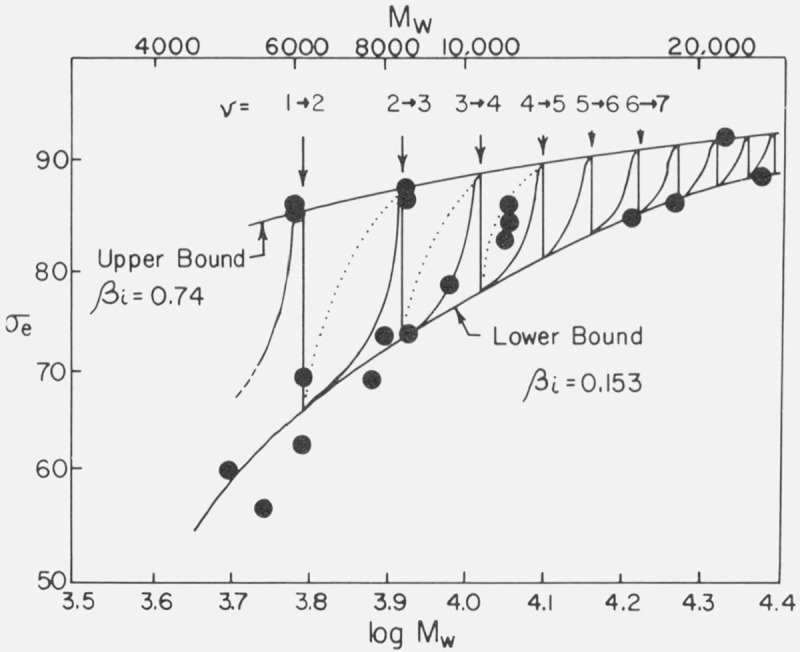
The solid and dotted lines are due to variation of βi within a given v as shown in figure 13B.
Consider now the upper bound. We have heretofore introduced the concept that the initial surface nucleus (and subsequent molecules in the strip completion process) may exhibit a pendant cilium (fig. 13A). There is considerable independent justification for the assumption that cilia can exist in unthickened crystals. (For example, Keller and Priest have demonstrated that a substantial fraction of the end groups in polyethylene single crystals are external to the body of the crystal [36].) With this concept, we proceed by assuming that the upper bound of σe corresponds to a surface containing the largest cilium that can occur, which has a length closely approaching l*g; a cilium that is any longer will fold over, giving a chain end in the surface at v+l. Lauritzen and DiMarzio [37] (LD) have devised an expression for the surface free energy contribution of a long cilium where, in its entropic wandering, configurations outside of a wedge angle ϕ are excluded (fig. 13C). Taking account of the fact that a “zero” length cilium has a surface free energy of approximately σ, the LD expression may be written
| (21a) |
where
| (21b) |
In this expression z0 is the “effective” or “statistical” segment length, which is about 6.7 for polyethylene [38], z the number of —CH2— units in the cilium, and ab the cross-sectional area A0 of the chain. The quantity kT/ab is 29.3 erg/cm2 at T = 400 K for polyethylene. The expression is only valid for values of z/z0 that are in excess of about 5. (It also gives the correct result σe(cilium)=σ for z/zo=l.) Equation (21a) is a more general case which extends the earlier calculations of DiMarzio [39] demonstrated only for the situations where ϕ = π/2 and ϕ = π.
Assuming that the cilium emanates from a plane, i.e., ϕ = π, which gives κ= 1/2, we calculate using σ = 14.2 erg/cm2 and l*g = 190 Å (z ≅ 150) in eqs (19) and (21a) that
| (22) |
Because of the uncertainty in ϕ, and other factors, this must be regarded as only a rough estimate. Also, the effect of the volume excluded by nearby cilia is not considered. Nevertheless, the point remains that it is reasonable that a long cilium has a high local surface free energy associated with it, which leads to a βi that is much higher than that of a short cilium or chain end in a surface.
Using the above estimate, we may calculate
| (23a) |
or, using M=2100 (v+l):
| (23b) |
A plot of this function is shown in figure 12B. The agreement is such as to suggest that the surface nuclei that are associated with the high values of σe (e.g., 6.15, 6.15 K(R) and 8.56, 8.56 K(R)) possess long terminal cilia approximating l*g in length. Meanwhile, it appears highly probable to us that those data points that lie near the lower bound are associated with quite short initial and terminal cilia, or initial and terminal chain ends actually in or nearly in the same plane as the folds (see fig. 13A). An analysis of the high points in ßi = 0.74 (fig. 14).
The “best” value of σe(∞) is close to 90.5 erg/cm2 (table 6, item C.3). The foregoing analysis could be repeated with this value instead of our original estimate of 93 erg/cm2, but the change in both the theoretical and experimental estimates of βi (upper bound) and βi (lower bound) would be insignificant.
Sanchez and DiMarzio have developed a theory of crystallization with chain folding that deals with the variation of growth rate with molecular weight and polymer concentration [40, 41]. They employ the assumption that the initial chain end is rather long— about one-third to one-sixth of the whole molecule forms the initial cilium — in contrast to our simplified assumption that the initial chain end is practically flush with the fold surface (fig. 13A). If their assumption were to be used in the present work, a large effective value of βi would be calculated with eq (21) for the initial cilium that would give σe values that greatly exceeded our calculated and experimentally determined lower bound (βi ≅ 0.15) and which would possibly even exceed the upper bound. While we thus have good reason for holding to our particular assumption regarding the nature of the placement of the initial chain end in bulk crystallization, it must be remembered that other aspects of the treatment of Sanchez and DiMarzio confine its validity to rather dilute solution. Nevertheless, an extension or revision of their theory using the assumption that the first chain end is quite short is probably worth some consideration.
The experimental values of σe and βi vary intermittently between rather definite upper and lower bounds at low molecular weights, and it is shown above that these bounds can be reasonably explained in terms of the length of the terminal cilia associated with the molecules comprising the surface nuclei and growth strip. We now ask why the values for σe intermittently vary between the upper and lower bounds. The explanation suggested below implies that βi and σe actually alternate between their upper and lower bounds. Despite the large number of samples studied, the data are insufficient to show actual alternation over a wide range of molecular weights. However, there is some evidence for a regular variation over a restricted range of molecular weight. Accordingly, the explanation offered below for the variation of σe and βi between their upper and lower bounds must be regarded as somewhat tentative.
For a given value of v, say v=2, the terminal cilium length in the model shown in figure 13A increases uniformly as the molecular weight increases. Then the cilium reaches a length of l*g where v→ v+ 1, and the cilium length again becomes very small. This will lead βi to alternate between the lower and upper bounds as the molecular weight increases as shown in figure 13B. We emphasize again that this holds only if J*g does not vary strongly with molecular weight as the molecular weight changes. The details of the variation of βi with molecular weight (increasing cilium length) with a given v is a matter of considerable uncertainty. Equation (21) suggests a situation as shown by the dotted lines in figures 13B and 14. This assumes an extremely sharp fraction. If it is arbitrarily assumed that the variation of βi with increasing cilium length is as shown in the solid line in figure 13B, the estimates of the rather abrupt variation of σe with molecular weight seem appropriately predicted at v = l → v = 2 and v = 2 → v = 3, as shown in figure 14.
It is possible that the tendency for many of the experimental points that lie between v and v+l that imply a lower βi than that given by eq (21) [dotted lines in figures 13B and 14] is a result of the existence of the distribution of molecular weight in the samples. There should be a natural tendency in the presence of a distribution for the nucleus to select out chains with short cilia (low βi) leading to σe values near lower bound. Then when just the appropriate molecular weight corresponding to a long cilium is reached, the search for molecules leading to short cilia is difficult, and the system shows a high βi and enhanced σe. At a slightly higher molecular weight, the long cilia folds over, and a low βi and σe is rather abruptly recovered.
Our overall conclusions are as follows. The upper and lower bounds exhibited by σe and βi, at low molecular weights are a result of cilium length effects in the growth nucleus. A terminal cilium in or near the fold surface is associated with a low βi and σe, and a terminal cilium that approximates l*g in length leads to a high βi and a σe near the upper bound. The increase and decrease of cilium length shown in figure 13A with increasing molecular weight is probably the cause of the existence of a rather definite upper and lower bound to σe at low molecular weights. This model implies that βi and σe should alternate in a regular way between their respective upper and lower bounds. The data are insufficient to show this in detail, but abrupt changes in βi and σe occur in two regions of molecular weight that are consistent with the model. Many more good fractions — and possibly much sharperones — than were available to us would be required to obtain experimentally the shape of the βi curve within a given v.
Attention is now drawn to the question of the constancy of l*g, which approximation was used in the foregoing analysis. The value of the initial lamellar thickness l*g for single crystals of polyethylene of high molecular weight can be accurately calculated as a function of undercooling with the expression [1]
| (24) |
where C2 = 4.3 × 10−7 cm or 43 Å. This expression, which has a general theoretical basis, is quantitative for a thickness range of ~ 100 Å to near 200 Å for crystals formed from dilute solution [1]. We adapt this expression to the present case, and calculate l*g for each of the fractions using σe from figures 12 or 14, the T°m relevant to the fraction, and the average undercooling used in the growth rate runs (table 3)14. The results are shown in figure 15. It is seen that l*g is predicted to be in the range 200±20 Å from Mw ≅ 6000 upwards, which covers the cases v = 1 → 2. and all higher v. Also shown are the LAXR “L2” data for several of the fractions. In general, these lie within the same range as the l*g values calculated with eq (24). This confirms the validity of the assumption concerning the approximate constancy and magnitude of l*g that was used in the calculations cited above for Mw ≥ 6000. At very low molecular weights, the low value of σe causes l*g as calculated by eq (24) to fall well below 200 ±20 Å.
Figure 15. Experimental and theoretical values of the initial lamellar thickness l*g as a function of molecular weight.

It is of interest to comment on why l*g was so nearly constant in the particular case of polyethylene fractions of Mw ~ 6000 and higher. We may write the expression for the growth rate in the form
| (25) |
to a good approximation. Then if G were measured within the restricted range of ~ 10−5 cm/s to ~ 10−8 cm/s, it is easy to show that l*g will increase by only about 10 percent or so as G goes from 10−5 cm/s to 10−8 cm/s. What happens is that l*g increases with temperature according to eq (24) in accord with theory and experiment, but this increase for any given specimen is small compared to l*g (or the maximum cilium length) because of the restricted temperature range allowed by the practical matter of the slowest and fastest rates that are feasible to measure.
With the analysis given in this section, it is justified to say that we have gained some insight into the factors that govern the growth rate of polyethylene from the melt from a molecular weight corresponding to but two folds per molecule (circa Mw ~ 6000) up to a molecular weight in excess of 800,000.
7. Work of Chain Folding
The “best” value of σe(∞) given in table 6 (item C.3) is 90.5 erg/cm2. While there are various ways to estimate this quantity, this value represents a reasonable compromise and is consistent with the growth rate data within rather narrow limits. This can be used to calculate a “kinetic” value of the work of chain folding q as
| (26) |
where σe0, which is the surface free energy in the absence of any work of chain folding, may be taken as equal to σ=14.2 erg/cm2 (table 6, item C.1). This leads to the estimate
| (27) |
The corresponding estimate of q from equilibrium data on single crystals of polyethylene (i.e., σe(eq) = 93±9 erg/cm2 from a Tm versus 1/l plot) gives with σe0 = 14.2 erg/cm2 the value [1]
| (28) |
The agreement between the “kinetic” and “equilibrium” estimates of q in polyethylene is sufficient to support our particular application of nucleation theory to the problem of chain folding. The folds are kinetically formed continuously during the crystallization process, and exhibit the same properties whether their free energy of formation is measured by kinetic or thermodynamic means.
It is of interest in connection with the foregoing estimates that theoretical calculations by Corradini and co-workers using inter- and intra-potential energy functions have suggested that
| (29) |
for (110) type folds [42]. It may be recalled that our analysis in which we find q (kinetic) = 4.09±0.4 kcal/mol is based on the assumption that (110) type folds control the growth kinetics; we also observe that the experimental value of q (equilibrium) = 4.23±0.4 kcal/mol is based on melting data on single crystals with mostly (110) folds.
Corradini and co-workers estimate a q value closeto 3.1 kcal/mol for (200) folds. The accuracy of these calculations is difficult to assess. Nevertheless, in view of the agreement attained between q(kinetic), q(equilibrium), and the q(theoretical) estimated by Corradini et al., in the case of (110) type folding, it is reasonable to assume that their theoretical estimate of q from potential energy functions for the (200) folds is basically correct. This then raises the question of why the lower energy (200) folds, known to occur in ordinary polyethylene spherulites [25], do not make themselves more apparent in the kinetics.
If two types of folds of different q were present in a given morphology, it is by no means certain that the low one would be clearly observed in kinetic measurements. Circumstances occur where both (110) and (200) type folds appear on the same overall growth front. For example, Keith [43] has shown that the overall growth front of polyethylene spherulites forming from concentrated solutions of pure hydrocarbons are serrated, with alternate and intersecting traverses of (110) and (200) folds, the latter being in the direction of overall growth, which is along the b axis of the unit cell. The polymer lamella resembles a laurel leaf with serrated edges, with the growth direction corresponding to the long axis of the leaf. The (110) folds occur at the growth tip, and at each serration. In this case — which we consider to be reasonable in a spherulitic morphology in bulk polyethylene — the kinetics of radial growth would tend to be dominated by the higher energy (110) folds until the (200) runs became quite long in comparison with the (110) runs. Possibly the presence of (200) folds is the reason that σσe(∞) for the coarse-grained non-banded spherulites is slightly lower than it is for the axialites (table 6). However, it must be noted again that the difference between the σσe(∞) in these two cases has a somewhat doubtful statistical significance. In any case it appears that (110) type folds generally dominate the kinetics of overall growth in axialites and irregular spherulites, and have the largest effect in the case of the coarse-grained non-banded spherulites.
8. Discussion
8.1. Relationship to Earlier Work: Growth Rate and Morphology
The principal previous research on the rate of growth of crystals from the melt in polyethylene appears to be that of Lindenmeyer and Holland [4]. They used five specimens ranging from Mv = 3900 to 135,000 in molecular weight. They mention the presence of banded spherulites only, whereas we found axialites over most of this molecular weight range at low undercoolings, and coarse-grained non-banded spherulites at higher undercoolings. There is no evidence of a break in log G versus T in their data, but this is explained by the relatively low growth temperatures used in their investigation. The quantitative agreement in absolute growth rates between our work and theirs is not particularly good. This can be traced in part to the fact that some of their runs were made at temperatures low enough to produce crystallization that was apparently sufficiently rapid to cause some self-heating. Also we infer from their paper that they may have used very high T1 values which might have degraded the samples somewhat. Nevertheless, their data, when plotted as log G versus l/T(ΔT) (see their fig. 3), show changes in slope in the low molecular weight region very similar in broad aspect to that shown in our figure 8A. This implies that Kg increases and then levels off, as we found in the present research. No intermittent high and low values at low molecular weights were observed, but this may have resulted from the relatively few samples used.
It is of considerable interest to compare what we have found for polyethylene fractions with the results reported for a systematic study of TMPS fractions by Magill [44]. In this work, it was found that σσe rose rapidly at first and then tended to level off at high molecular weights, and Magill interpreted this as meaning that σe behaved with increasing molecular weight in the same manner. Moreover, Magill noted some variations of σe at low molecular weights (but still well within the region where folding occurs) that are strongly reminiscent of the intermittent high and low data for σe for polyethylene fractions near Mw = 6200 and Mw,= 8500 as seen in figures 12 and 14. In particular, one of the low TMPS fractions (M=8700) has a σe that is substantially higher than that of neighboring points. (Recall that in polyethylene we have tentatively attributed this type of behavior to the occasional appearance of a terminal cilium close to l*g in length on the growth nucleus.)
Most of the data points on TMPS were taken at rather high undercoolings, corresponding, in our estimation, to regime II or mixed regime I and regime II [1]. It is therefore not surprising that Magill did not observe a regime I → regime II transition. Our analysis of the growth data, given elsewhere [1], suggests that σe(∞) for TMPS is 34 ± 11 erg/cm2, which is in fair accord with the value σe(eq) = 39 erg/cm2 obtained from a Tm versus 1/l plot.
Devoy and Mandelkern [45] have discussed polymers where σσe rises and then levels off with increasing molecular weight. Their analysis differs in a number of significant respects from that given in this work.
8.2. Regime I and Regime II in Other Polymers: Morphological Implications
Definite evidence for the existence of regime I and regime II crystallization was found for polyethylene fractions in the intermediate molecular weight range. The regime tests gave clear-cut results in this region, and the Kg values differed by a factor of about two. Also, the ratio of the pre-exponential factors was very large and of approximately the magnitude expected.
We must now raise the question as to why no such effect was seen by previous investigators for polyethylene or any other polymer crystallizing from the melt. In the case of polyethylene the answer is fairly straightforward. Few measurements, if any, were previously made at undercoolings sufficiently low to enter regime I. For example, Lindenmeyer and Holland carried out the main body of their investigations at temperatures below 127 °C, and the transition between regimes I and II occurs near or above this temperature. This same point probably accounts in part for the failure to observe the transition in other polymers — at high growth temperatures where regime I is entered, the growth rate tends to be extremely slow and the region is therefore quite inconvenient to survey from an experimental standpoint.
It has already been pointed out that the regime I → regime II transition is smeared out in polyethylene with a broad distribution of molecular weight. This is particularly clear in the case of the whole polymer SRM 1475 sample. On this basis, one might not expect to find the transition in other polymers where the distribution was broad. It will be a matter of interest to see if fractions of other polymers exhibit the transition.
It is of special interest to note that Cooper and Manley [46] have found some evidence for the existence of a regime I → regime II transition in polyethylene fractions crystallized from dilute solution. We observe that the transition, though not as well-defined in the dilute solution case as in the case of crystallization from the melt, appears to occur at approximately the same undercooling in both cases. We also observe that at a specified concentration, σσe as calculated with Kg = 3bσσeT°d/(Δhf)k, where T°d is the dissolution temperature, tends to rise and then level off with increasing molecular weight when the variation of T°d with molecular weight is taken into account (see their table 6). They find a σσe at high molecular weights comparable to that which we have found for melt-crystallized polyethylene.
In the case of polyethylene fractions in the low and intermediate molecular weight range, a good correlation exists between morphology and regime. Regime II kinetics could in general be identified with the existence of coarse-grained non-banded spherulites, (or spherulites with a few coarse bands at the lowest isothermal growth temperatures), and the appearance of axialites at high growth temperatures is, broadly speaking, associated with regime I kinetics. Also, the existence of irregular spherulites at high molecular weights seems to mostly involve mixed regime I and II kinetics. While these identifications seem reasonably definite for polyethylene fractions crystallized from the melt, it is not known how widely applicable the general idea of the relationship between regime and morphological characteristics really is for fractions in other polymers.
8.3. Overview
The following summarizes the basic picture of crystallization that has evolved.
The rate of crystallization of polyethylene fractions from the melt is governed by the rate of deposition of folded surface nuclei on the previously formed substrate. The nucleus is formed by a molecule (probably already associated with the substrate by physical adsorption [1]) that apparently arranges its first chain end in such a way as to keep the free energy near a minimum, i.e., the initial chain end is flush or nearly so with the fold surface. Then the rest of the molecule comes into crystallographic register with the substrate in a sequential process, leaving a dangling terminal cilium if the contour length of the molecule does not correspond to an integral multiple of the fold period l*g. On the average, the pendant cilium does not exceed the fold period; if it is longer than l*g a new fold is formed, and the long cilium goes down on the substrate. A sufficiently short terminal chain end has a low effective surface free energy σe(cilium) ≅ σ, and a long terminal cilium has a σe(cilium) that is proportional to the natural logarithm of the effective chain length z/z0, giving a σe(cilium) value that is many times larger than σ. The value of σe(cilium) for a long terminal cilium of length l*g is roughly comparable to, but probably not in excess of, σe. Since the average terminal cilium length is such that its local surface free energy σe(cilium) is less than that associated with a chain fold, the total average surface free energy (cilia plus folds) is rather low for material of low molecular weight where the fraction of cilia is rather high. As the molecular weight is increased, the fraction of folds likewise increases, and the average value of the surface free energy σe tends toward a limiting value, σe(∞).
For a given number of folds and a fixed substrate width l*g, a surface nucleus can have either a very short cilium, one of intermediate size, or one closely approaching l*g in length, depending on the contour length of the molecule forming the nucleus. Since the local surface free energy of a long cilium is greater than that of a short one, alternating values of σe are to be expected in very sharp fractions at low molecular weights as with increasing molecular weight a molecule goes through the sequence: short cilium → intermediate length cilium → cilium approaching l*g in length → formation of additional fold with short cilium. This effect is believed on a tentative basis to be the source of the intermittent low and high values of σe observed at low molecular weights in the particular fractions employed in the work reported here.
The surface nucleation rate ¿, which is proportional to exp [—4bσσeT°m/(Δhf)(ΔT)kTf], leads to overall observable growth of two types. At high growth temperatures (low undercoolings) the nucleation rate is low, and each nucleus causes the completion of the substrate of length L. Then a new nucleus forms, and the process is repeated forming a lamellar crystal. In this case, regime I, the growth rate G is proportional to the nucleation rate i. At lower growth temperatures, (high undercoolings) the nucleation rate becomes quite high, and many surface nuclei are formed on the substrate. Then regime II is entered, and G is proportional to i1/2, again with the formation of a lamellar crystal. The transition between regimes I and II is experimentally quite sharp in good fractions, and the undercooling where it occurs can be explained in terms of a value of L that is roughly 5 μm. The sharpness of the transition is fundamentally a result of the rapid change of i with undercooling.
At moderate to high molecular weight, the nucleation rate and the concomitant overall growth rate are not strongly dependent on changes of viscosity of the melt from which the various crystalline bodies are formed. Also, the nucleation rate, as measured by σσe, appearsto be nearly independent of such factors as the presence of interlamellar links at high molecular weights. The absolute value of the pre-exponential for regime I is much greater than that for regime II, principally because that for regime I contains the factor L, whereas that for regime II does not.
The observed value of the initial lamellar thickness l*g is in general close to that predicted from the under-cooling and the σe relevant to the molecular weight under consideration. Because the growth rate G is related to l*g according to G ∝ exp (−2bσl*g/kT), and because the experimental range of G is limited, l*g is itself restricted in polyethylene crystallizing isothermally from the melt to a rather narrow range close to 200 Å.
The work of chain folding q can be deduced from strictly kinetic measurements. The value so obtained is in close agreement with that from equilibrium measurements on polyethylene single crystals, and with certain theoretical calculations. There appear to be no inconsistencies of an energetic character in the picture of crystallization presented here.
The authors thank Esmund Drott of the Monsanto Chemical Company for supplying some of the fractions, and J.J. Weeks, Jr., for his considerable assistance in carrying out the WAXR and LAXR measurements. We also gratefully acknowledge a number of discussions with E. A. DiMarzio and F. Khoury. We are indebted to H. D. Keith for pointing out the significance of his work on crystallization of polyethylene from pure hydrocarbons, and for a number of timely discussions.
Footnotes
Figures in brackets indicate the literature references at the end of this paper.
See also Price [5], and Lauritzen and Hoffman [6] for early derivations applicable to crystallization with chain folding corresponding to the case f=1.
Note that an upright K is used to mean “thousand” in denoting the molecular weight. In accordance with international convention, an italic K is used later in this paper to indicate temperature in kelvins.
Certain company names and commercial products are identified in order to adequately specify the experimental procedure. In no case does such identification imply recommendation or endorsement by the National Bureau of Standards, nor does it imply that the products identified are necessarily the best available for the purpose.
One angstrom unit (Å) is equal to 10−10 m.
In its strictest definition, the term “axialite” refers to a crystalline object that is highly asymmetrical. The large rather open and sometimes roughly spherical structures that we have loosely called “axialites” in this paper are true axialites in their early stages of growth. In many cases the strictly axialitic mode persists up to rather large values of x (see for example, specimen 42.6 K in fig. 3).
The following customary units are used in this paper: G and G0 in cm/s, U* in cal/mol, with R=1.987 cal mol−1K−1, T and T∞ in kelvins (K), Kg in kelvins squared (K2). The quantity Kg in customary usage contains surface free energies σ and σe in units of erg/cm2, the heat of fusion in erg/cm3, a molecular layer thickness b in cm, and Boltzmann’s constant in erg K−1. The conversion factors to SI units are 1 erg/cm2= 1 mJ/m2,1 erg/cm3 = 0.1 J/m3,1 cal/mol = 4.184J/mol, and R = 8.314J mol−1K−1.
The factor z in eq (4) was omitted in our previous formulations [2], but its presence is noted in a review [1]
It is assumed in making the calculations here and elsewhere in this paper that (110) type folds prevail at the leading growth front no matter which particular morphology is under consideration. This does not necessarily imply that other types of folds, e.g., (200), may not be present at other parts of the lamellae in some cases. Points related to this issue are mentioned later in the paper, particularly in section 7.
Note that a and b refer to the width and thickness respectively of the polymer molecule on the substrate as shown in figure 10, and not to the dimensions of the unit cell. The dimensions of the unit cell mentioned later in this paper are denoted with the bold face letters a and b.
Note that a, b, and L are in cm in eqs (3) through (14) even though for convenience the> are often quoted elsewhere in the text in other units such as Å or μm.
The value L ~ 5 μm refers to polymer crystallized at an undercooling of ~ 17.5 °C. Actually, L is probably a function of temperature, and may vary in such a way as to cause a further sharpening of the regime I → regime II transition.
Keith has devised a scheme whereby he explains the shape of the overall growth front in polyethylene lamellae in spherulitic structures crystallizing from concentrated solution in long chain hydrocarbons as alternate and intersecting (200) and (110) growth fronts, the latter occurring at the leading tip. The subject of (110) and (200) growth is again taken up in section 7.
It is worth noting that the value of l*g calculated with eq (24) does not depend on the regime in which the crystallization occurs [1, 2).
9. References
- [1].Hoffman J. D., Davis G. T., and Lauritzen J. I. Jr., in Treatise on Solid State Chemistry, Hannay N. B., Ed. (Plenum Press, New York, Vol. 3, 1976). [Google Scholar]
- [2].Lauritzen J. I. Jr., and Hoffman J. D., J. Appl. Phys. 44, 4340 (1973). [Google Scholar]
- [3].J Hoffman J. D., Soc. Plastics Eng. Trans. 4, 315 (1964). [Google Scholar]
- [4].Lindenmeyer P. H., and Holland V. F., J. Appl. Phys. 35, 55 (1964). [Google Scholar]
- [5].Price F. P., J. Polymer Sci. 42, 49 (1960). [Google Scholar]
- [6].Lauritzen J. I. Jr., and Hoffman J. D., J. Res. Nat. Bur. Stand. (U.S.), 64A (Phys. and Chem.), No. 1, 73–102 (January-February 1960). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sanchez I. D. and DiMarzio E. A., J. Res. Nat. Bur. Stand. (U.S.), 76A (Phys. and Chem.), No. 3, 213–223 (May-June 1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lauritzen J. I. Jr., J. Appl. Phys. 44,4353 (1973). [Google Scholar]
- [9].NBS Special Publication 260–42, The Characterization of Linear Polyethylene SRM 1475, Wagner H. L. and Verdier P. H., Eds. (1972). Reprinted from J. Res. Nat. Bur. Stand. (U.S.), 76A (Phys. and Chem.), No. 2, 137–170 (March-April 1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hoffman J. D., and Weeks J. J.,J. Chem. Phys. 37, 1723 (1962). [Google Scholar]
- [11].Ross G. S., and Frolen L. J., to be published. [Google Scholar]
- [12].Broadhurst M. G., J. Res. Nat. Bur. Stand. (U.S.), 70A (Phys. and Chem.), No. 4, 481–486 (November-December 1966). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Flory P. J. and Vrij A., J. Am. Chem. Soc. 85, 3548 (1963). [Google Scholar]
- [14].Gieniewski C. and Moore R. S., Macromolecules 2,385 (1969). [Google Scholar]
- [15].Tearè P. W., and Holmes D. R.,J. Polymer Sci. 24,496(1957). [Google Scholar]
- [16].Niegisch W. D., and Swan P. R., J. Appl. Phys. 31, 1906 (1960). [Google Scholar]
- [17].Geil P. H., Polymer Single Crystals, (Interscience Publishers, New York, 1963). [Google Scholar]
- [18].Bunn C. W., and Alcock T. C., Trans. Faraday Soc. 41, 317 (1945). [Google Scholar]
- [19].Point J. J., Bull. Acad. Roy. Belg. 41, 982 (1955). [Google Scholar]
- [20].Point J. J., and Homès G. A., Compt. Rend. 242, 2557 (1956). [Google Scholar]
- [21].Fischer E. W., Z. Naturforsch. 12a, 753 (1957). [Google Scholar]
- [22].Anderson F. R., J. Appl. Phys. 35, 64 (1964). [Google Scholar]
- [23].Palmer R. P., and Cobbold A. j., Macromol. Chem. 74, 174 (1964). [Google Scholar]
- [24].Keller A., and Sawada S., ibid., 190 (1964). [Google Scholar]
- [25].Bank M. I., and Krimm S., J. Polymer Sci. A27, 1785 (1969). [Google Scholar]
- [26].Khoury F., and Passaglia E., in Treatise on Solid State Chemistry, Hannay N. B., Ed. (Plenum Press, New York, Vol. 3, 1976). [Google Scholar]
- [27].Suzuki T., and Kovacs A., Polymer J. 1, 82 (1970). [Google Scholar]
- [28].Davis G. T., and Eby R. K., J. Appl. Phys. 44, 4274 (1973). [Google Scholar]
- [29].Huseby T. W., and Bair H. E., J. Appl. Phys. 39, 4969 (1968). [Google Scholar]
- [30].Weeks J. J., J. Res. Nat. Bur. Stand. (U.S.), 67A, (Phys. and Chem.), No. 5, 441–451 (September-October 1963). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rijke A. M., and Mandelkern L., J. Polymer Sci. A-2, 8, 225 (1970). [Google Scholar]
- [32].Private communication from Keith H. D., Bell Telephone Laboratories. [Google Scholar]
- [33].Ergöz E., Fatou J. G., and Mandelkern L., Macromolecules 5, 147(1972). [Google Scholar]
- [34].Elban W., and Hoffman J. D., manuscript in preparation.
- [35].Ross G. S., and Frolen L. J., J. Res. Nat. Bur. Stand. (U.S.), 79A, (Phys. and Chem.), No. 6 (November-December 1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Keller A., and Priest D. J., J. Macromol. Sci. B2, No. 3, 479 (1969). [Google Scholar]
- [37].Lauritzen J. I. Jr., and DiMarzio E. A., manuscript in preparation. [Google Scholar]
- [38].Flory P. J., in Statistical Mechanics of Chain Molecules, (Interscience, New York, 1969). [Google Scholar]
- [39].DiMarzio E. A., XXIII International Congress of Pure and Applied Chemistry, 8, 239 (1971). [Google Scholar]
- [40].Sanchez I. C., and DiMarzio E. A., Macromolecules, 4, 677 (1971). [Google Scholar]
- [41].Sanchez I. C., and DiMarzio E. A., J. Chem. Phys. 55, 893 (1971). [Google Scholar]
- [42].Corradini P., Petraccone V., and Allegra G., Macromolecules 4, 770(1971). [Google Scholar]
- [43].Keith H. D., J. Appl. Phys. 35, 3115 (1964). [Google Scholar]
- [44].Magill J. H., Polymer Sci J.. Part A-2, 5, 89 (1967). [Google Scholar]
- [45].Devoy C., and Mandelkern L., J. Polymer Sci. Part A-2, 7, 1883 (1969). [Google Scholar]
- [46].Cooper M., and Manley R. St. J., Macromolecules 8, No. 2, 219 (1975). [Google Scholar]