

Abstract
A key remit of the NSF‐funded “Arabidopsis Research and Training for the 21st Century” (ART‐21) Research Coordination Network has been to convene a series of workshops with community members to explore issues concerning research and training in plant biology, including the role that research using Arabidopsis thaliana can play in addressing those issues. A first workshop focused on training needs for bioinformatic and computational approaches in plant biology was held in 2016, and recommendations from that workshop have been published (Friesner et al., Plant Physiology, 175, 2017, 1499). In this white paper, we provide a summary of the discussions and insights arising from the second ART‐21 workshop. The second workshop focused on experimental aspects of omics data acquisition and analysis and involved a broad spectrum of participants from academics and industry, ranging from graduate students through post‐doctorates, early career and established investigators. Our hope is that this article will inspire beginning and established scientists, corporations, and funding agencies to pursue directions in research and training identified by this workshop, capitalizing on the reference species Arabidopsis thaliana and other valuable plant systems.
Keywords: genomics, metabolomics, proteomics, training, transcriptomics
1.
…plant physiologists (collectively) have two, somewhat linked, responsibilities. One is to make profound discoveries about the behaviour of plants; the other is to make useful ones. (Passioura, 1979)
2. BIG QUESTIONS IN PLANT BIOLOGY
Colloquially, the study of the collective behavior of an ensemble of one category of biomolecules within a system is referred to as omics research. Familiar examples of omes, including those that are a major focus of this report, are the genome (the collection of all genetic material), the transcriptome (the collection of all RNAs), the proteome (the collection of all proteins), and the metabolome (the collection of all metabolites); with the ultimate phenotypic outcome being referred to as the phenome (Figure 1). Before addressing current strengths and needs in plant omics research specifically, it is illuminating to take a broader look at important directions in plant biology research. Workshop participants identified a non‐exhaustive set of “Big Questions” that could coalesce future research in plant biology. The Big Questions identified are described here as strictly biologically motivated, but have obvious applications to plant improvement, the bioeconomy, and human and environmental health. Similarly, these questions cannot be addressed adequately without technical and training advances described elsewhere in this article. Such Big Questions motivate interest in plant science, describe significant unknowns and thus opportunities, and suggest big‐picture research initiatives for the 21st century.
Figure 1.
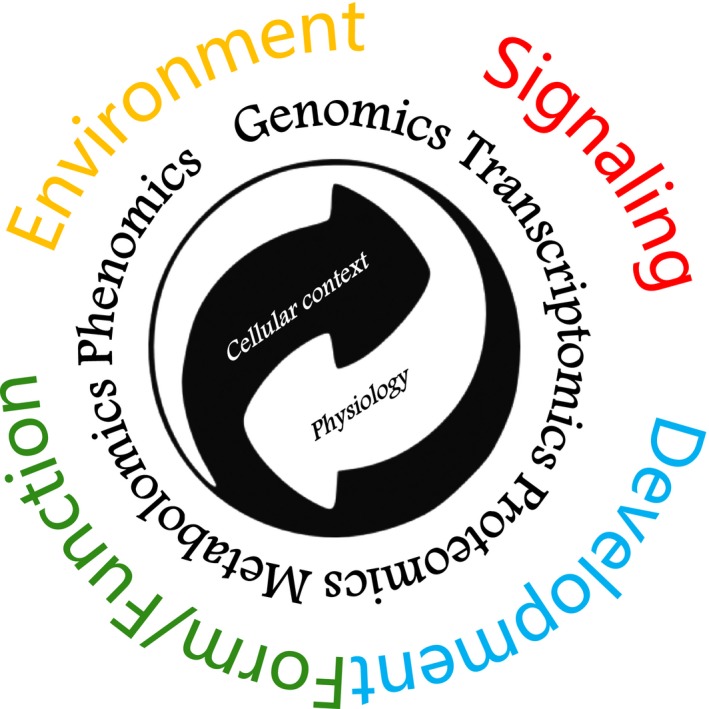
Diverse omics approaches provide insights into cell biology and physiology and inform our knowledge of plant functional development and environmental interaction
A primary question is “how does the whole organism assemble itself”? We now have sets of molecular markers that identify many specific plant cell types, but we fall short in understanding the mechanisms that drive the eventual expression of such markers, and their interplay. Research to date has focused to a large extent on plant transcription factors (Moreno‐Risueno, Van Norman, & Benfey, 2012) as master switches that control organization and differentiation of the plant body. However, it is becoming increasingly evident that upstream of these regulatory nodes are receptor interactions with cognate ligands (e.g., in stomatal patterning (Rowe & Bergmann, 2010; Torii, 2012)), and many of these upstream signaling elements remain to be discovered. Downstream of transcription, metabolites and enzymes that impose translational modulation and post‐translational regulation are equally important. In some systems, of which the Drosophila melanogaster embryo is a prime example, elegant research has identified transcription‐factor defined tissue and organ boundaries along a developmental timeline (Sandler & Stathopoulos, 2016), but the determinate growth of metazoans provides a level of simplicity that is absent from the plastic body plan of plants, which is shaped through indeterminate growth and environmentally‐responsive developmental programs.
Extending from the above, a second big picture question is “how do plants manifest plasticity” given their programmed modular patterning. To give a simple concrete example: how is it that we can readily distinguish oaks from maples, yet each individual oak tree is different from any other oak tree? What are the underlying bases for both the “sameness” and “differentness” of individual plants within the same species? Such “differentness” arises from individual genetic variation, local environmental differences, and their interplay. This interplay involves processes at the levels of the genome, epigenome, and “post‐genome” (including the –omes of the transcriptome, RNA structurome, proteome, post‐translationally modified proteome, interactome, and metabolome). Related to this phenomenon are questions of how plants maintain responsive capacity to the environment, and how the developmental program is modulated or modified in response to external stimuli, and how domestication has resulted in anatomical or metabolic tradeoffs that plants uniquely tolerate. While genome‐wide association study (GWAS) approaches in particular are providing large‐scale insights into the genetic bases, and whole‐plant phenomics approaches are helping to resolve macroscopic phenotypes arising from plant‐environment interactions, we are still distant from quantitative phenomics at the tissue, cellular, and subcellular levels. In this realm, omics approaches can accelerate progress.
A corollary to the above question stems from the observation that responses to environmental cues vary from cell to cell, yet the plant necessarily responds as a single organism. How does each individual plant generate a cohesive and evolutionarily successful emergent response to its environment from these varied signals, especially without a coordinating nervous system? A related question is: how do plants remember? Plants respond to the environment on time scales ranging from the microsecond (e.g., photosynthetic electron transport) to the minute (e.g., rapid gene expression), to the century (e.g., morphology of long‐lived deciduous trees). How much of this memory is potentially subject to modulation, for example by cell signaling or by coding and re‐coding of the epigenome, and how much is irretrievably fixed, as in anatomical form. In short, how do plants “learn” and to what extent do plants “forget”?
A third big picture question of particular relevance to the workshop's context is “how can we improve the ‘hit rate’ in translating knowledge and discoveries from research on Arabidopsis thaliana (henceforth referred to as Arabidopsis) into real‐world solutions for improving crop yield, quality, and resilience”? When selection of cultivars for desirable agronomic traits and yield is performed under optimal conditions rather than by assessing productivity across dynamic environments, this incurs potential costs, such as loss of environmental response resilience, specific metabolic pathways, and design principles present in non‐domesticated species, including Arabidopsis. As we accrue comprehensive and detailed ‐omics data integration on Arabidopsis grown under diverse conditions, how can we best leverage this information to inform breeding decisions? While some specialized aspects of crop development and physiology, e.g., nitrogen fixation by leguminous crops, are obviously absent from the biology of our favorite weedy annual, many of the processes underlying plant growth and development are conserved across species. For these, we need a better understanding of the basis for inconsistencies encountered in translating Arabidopsis research to agronomically‐relevant species and environments. If limitations in translation arise from the current ways in which Arabidopsis research is practiced, can this be surmounted with changes in federal or private funding strategies, or in research conception, design, and execution that improve the applicability of Arabidopsis as a “crop model”? If limitations reflect convergent evolution, in which Arabidopsis has evolved to solve the same problem in a different way than crop species (due either to natural selection or domestication), can various omics approaches be harnessed to identify the different pathways that then lead to a similar phenotypic outcome? This could then lead to an informed decision when choosing an experimental approach, e.g., whether to build the desired trait into the crop species by engineering in a whole pathway versus tweaking an existing one. Finally, to the extent that Arabidopsis employs truly unique mechanisms, advances in synthetic biology, including the CRISPR revolution, offer the potential to introduce those mechanisms to improve crops that lack them. For example, perhaps defenses against pathogens and herbivores that arise from Brassicaceae‐specific glucosinolate metabolism could be advantageously introduced into non‐cruciferous crops.
3. BIG BIOLOGY: BIG ADVANCES AND BIG CHALLENGES IN PLANT OMICS RESEARCH
This workshop had a deliberate focus on plant omics. Participants identified many exciting advances in technique development that are occurring in omics research, and also identified significant challenges. In many cases, advances and challenges are both agnostic to the biological system under study, but in some cases, particularly in metabolomics, plant systems offer specific opportunities and difficulties.
3.1. Nucleic acids
Research on the most widely studied omes, the genome and transcriptome, has been revolutionized by next generation high‐throughput sequencing methodologies. Research involving large‐scale nucleic acid sequencing, more than research on any other ome, has been democratized by decreases in cost, such that whole genome sequencing and resequencing is conceivable as an individual laboratory effort. There are now hundreds of sequenced plant species (Michael & VanBuren, 2015) and for some species, including Arabidopsis, there are full genome sequences for thousands of accessions or cultivars (Alonso‐Blanco et al., 2016; Li, Wang, & Zeigler, 2014). Genomic research continues to be advanced by technologies such as single molecule sequencing (e.g., Nanopore (Michael et al., 2018)). At the same time, plant genomes offer particular challenges, e.g., the spectrum of genome sizes in plants is wider than that of animals (Pellicer, Hidalgo, Dodsworth, & Leitch, 2018), and polyploidy and ancient genome duplications increase the complexity of many crop genomes (Michael & VanBuren, 2015).
Sequencing technologies also have expanded far beyond simple sequence identification and quantitation of nucleic acids. Accessible and affordable sequencing has enabled numerous applications at a genome‐wide scale. These include identification of causative mutations from forward genetic screens (Cuperus et al., 2010), determination of copy number variants (reviewed in Zmienko, Samelak, Kozlowski, & Figlerowicz (2014)), and elucidation of chromosomal architecture (Grob, Schmid, Luedtke, Wicker, & Grossniklaus, 2013), methylation status (Cokus et al., 2008; Lister et al., 2008), cis‐regulatory regions and motifs (Lu, Hofmeister, Vollmers, DuBois, & Schmitz, 2017; O'Malley et al., 2016; Zhang, Zhang, Wu, & Jiang, 2012), and sites of transcription factor‐DNA interaction (Kaufmann et al., 2009). With regard to the transcriptome, nucleic acid sequencing is being used to determine translation efficiency (reviewed in Mazzoni‐Putman & Stepanova, 2018), the RNA interactome of RNA‐binding proteins (Meyer et al., 2017; Zhang et al., 2015), and dynamics of RNA structure and stability (Ding et al., 2014; Li et al., 2012; Su et al., 2018). Low cost sequencing of transcriptomes along with reaction miniaturization is also powering large‐scale data collection of single‐cell transcriptomes (Birnbaum, 2018).
3.2. Proteins
At the level of the proteome, continuing advances in mass spectrometry (MS) are allowing a more complete picture of the qualitative proteome. However, our ability to characterize the proteome in a comprehensive and quantitative manner still lags far behind what is achievable for the genome and transcriptome. While the current generation of mass spectrometers has impressive sensitivity and data acquisition rates, limitations in the rate and extent of chromatographic separation (Shishkova, Hebert, & Coon, 2016) and in methods for enrichment of low abundance proteoforms prior to sample introduction into the mass spectrometer often restrict throughput and resolution in proteomics as well as metabolomics experiments. Thus, in a number of situations, the limitation in sensitivity is not the mass spectrometer itself but the systems for liquid chromatographic separations. Development of nanofluidic systems to separate small volumes could provide a solution for making, e.g., single cell analyses, more feasible.
It is increasingly recognized that a particular post‐translational modification (PTM) of a protein may be important for one biological phenotype but not another phenotype governed by the same protein; in these instances, genetic knockout analysis provides only a coarse‐grained tool, as it results in the loss of all PTM forms. The added layer of complexity imposed by PTMs also poses a challenge to comprehensive analysis of the proteome. First, because such modifications alter peptide mass and other properties, they increase the difficulty of peptide identification. Second, because such modifications occur on a probabilistic, rather than on an all‐or‐none basis, accurate quantitation of the extent of PTM of any given protein species remains challenging. Third, the universe of known PTMs that need to be accounted for is still expanding. For example, while protein phosphorylation has long been known as a PTM, new advances in redox proteomics (Rinalducci, Murgiano, & Zolla, 2008; Yang, Carroll, & Liebler, 2016) are revealing the diversity, ubiquity, and importance of protein redox status. A related issue that currently has no high throughput solution is that the same protein may have different functions in different cellular locations. For instance, an activated kinase may regulate unrelated processes at the plasma membrane versus the nucleus. Developing robust methods to monitor protein function at the subcellular level would provide important additional information, particularly valuable for building networks of activities. An exciting development in this area is in vivo proximity labeling, which holds promise for spatial analysis of protein‐protein interactions at subcellular resolution (Khan, Youn, Gingras, Subramaniam, & Desveaux, 2018; Lin et al., 2017; Lobingier et al., 2017), and for high resolution identification of interacting peptide domains (Kao et al., 2011; Stanton, Chory, & Crabtree, 2018).
Another area of recent advancement in the study of proteins and macromolecules, although not yet at the –omic scale, is cryo‐electron microscopy (cryoEM). Protein structures form the basis for modeling of enzyme kinetics and macromolecular interactions, yet large‐molecule structure analysis often depends on X‐ray crystallography, NMR and/or MS. X‐ray crystallography requires biomolecules to form uniform crystals, which is often a challenging task, especially with membrane proteins. NMR is a gold standard for structural analysis, but it is relatively insensitive and requires large amounts of purified sample. Mass spectrometry is highly sensitive, but provides limited structural information. Although MS excels in determining primary amino acid sequences, 3D structural analysis by MS requires tedious isotope exchange assays and complicated data analysis/interpretation. Recent advances in cryoEM with direct electron detectors have revolutionized structure determination of biological macromolecules, and cryoEM can be applied to analyzing large complexes at a super‐high resolution that rivals X‐ray crystallography. Newly developed electron detectors can achieve images with unprecedented quality with details to deduce the atomic structure of a range of large biomolecules. The combination of cryoEM and cross‐linking coupled mass spectrometry (CX‐MS) holds promise for deep interrogation of protein structures and their interactors (Schmidt & Urlaub, 2017). Continued advances in cryoEM imaging will allow plant scientists to gain unprecedented insights into many biomolecules and their interactions, while advances in cryoEM tomography (Pfeffer & Mahamid, 2018) and super‐resolution microscopy (Komis, Samajova, Ovecka, & Samaj, 2015) are facilitating visualization of molecules in a cellular context, spanning the gap between atomic level resolution and cell biology.
3.3. Metabolites
Metabolomics is a particularly exciting frontier in plant omics. Plants are both sessile and silent, and a vast chemical repertoire helps plants to self‐sustain and communicate. Plant primary metabolism stores energy through photosynthesis and produces the building blocks of life, including nucleic acids, amino acids, carbohydrates, and lipids. Specialized plant metabolism creates a plethora of chemicals that facilitate stress responses and communication. For instance, the cuticle and flavonoids are plants’ sunscreen to reduce damage by UV radiation (Jansen, Gaba, & Greenberg, 1998; Yeats & Rose, 2013) while osmolytes such as proline, glycine betaine, and sugar alcohols help plants to retain water in the presence of drought and high salinity (Deinlein et al., 2014). Terpenes, phenolics and alkaloids are all plant inventions that fend off pathogens and predators (Constabel, Yoshida, & Walker, 2014; Gershenzon & Dudareva, 2007; Mithofer & Boland, 2012), whereas nectar and caffeine attract and retain pollinators (Wright et al., 2013). Indeed, plants are unique in the number and diversity of small molecules that they produce, with an estimated over 1 million distinct metabolites produced by the plant domain of life (Afendi et al., 2012; D'Auria & Gershenzon, 2005; Fernie, 2007). Many of these small molecules are not only central to plant metabolism and physiology, but are also essential drugs for human health, or form the basis of scent and flavor in our foods.
In‐depth functional characterization of even a single metabolite can be a time‐consuming task, and phenomic outcomes may arise from crosstalk among multiple metabolites (Jin et al., 2013; Mundim & Pringle, 2018; Zhou & Wang, 2018). Researchers thus need to prioritize based on relevance and importance, identifying those metabolites that are more likely to have a major biological impact. This pre‐selection of targets requires knowledge of which hubs and branch points are implicated in specific metabolic pathways. We also need to identify the relevant impact of each metabolite. Is a given metabolite primarily for internal biological processes, or for intra‐ or inter‐species communication? Reciprocally, how many plant proteins and other macromolecules sense and interact with metabolites?
Another major challenge is to identify and functionally characterize the proteins involved in generating and regulating the metabolome. Despite profound progress in the last century, characterization of the enzymes responsible for the synthesis of many plant small molecules remains incomplete. Moreover, metabolite diversity is partially attributable to the promiscuous function of biogenesis and modifying enzymes. Arguably, the most famous example is RuBisCo, which produces different products by reacting with either CO2 or O2 for photosynthesis or photorespiration, respectively. Enzyme promiscuity is particularly common in specialized metabolism (Weng & Noel, 2012). In addition, little is known regarding the receptors and transporters for most metabolites, in part because of genetic redundancy and a lack of feasible read‐out assays.
Unlike the polymeric nature of nucleic acids and proteins that has facilitated development of technologies for their large‐scale identification and quantitation, even the building blocks that make up metabolites are highly diverse, leading to a vast combinatorial complexity in the metabolome. On the one hand, this complexity levels the playing field among plant systems—because metabolomics analyses do not rely on a sequenced genome for molecular identification, metabolomics techniques can be applied to plant species where such genomic information is incomplete or absent. However, this combinatorial complexity also confers a disadvantage, since, unlike the genetic code, there is no simple genomic template for structural identification of a metabolite. Accordingly, the most confident identification and quantification of a given metabolite requires matching the metabolite's chromatographic profile and mass spectrum with that of an authentic standard. However, such standards are often expensive or are not even available commercially, requiring custom organic syntheses that are beyond the scope of many labs. Moreover, instrument‐specific differences in liquid chromatography‐mass spectrometry (LC‐MS) preclude easy use of the mass spectrum of a metabolite obtained on one LC‐MS machine as a “fingerprint” which can computationally identify that metabolite on other LC‐MS machines. For these reasons, many metabolites in plants (as well as other biological systems) are identified as “features” in mass spectra but lack explicit identification of their chemical structure. While NMR remains the gold standard for molecular structure elucidation, its low sensitivity and low throughput precludes its application in large‐scale investigations.
In short, creating an atlas of the plant metabolome alone is already a daunting task. Due to the above technical limitations, compounded by the diversity of the physicochemical properties of metabolites, and the condition/species‐dependency of many specialized metabolites, the “dark matter” of the plant metabolome far surpasses what can be currently profiled. Moreover, to track a metabolite from cradle to grave also requires information on its flux, reactivity with other macromolecules, and spatial distribution. Regarding this last point, one exciting area of recent advancement in metabolomics is micro‐sampling approaches, particularly those that harness laser‐based techniques of metabolite volatilization that allow metabolite imaging from living tissues (Misra, Assmann, & Chen, 2014). The ability to profile biological molecules, as well as inorganic elements (Shimotohno et al., 2015), in situ from one or a few cells is one arena in which MS‐based approaches are advancing rapidly. This area of research has significant potential, even though at present the majority of detected molecules are those that are most abundant. Enhancements in sensitivity through improvement of MS instrumentation, on‐target sample preparation, and sampling will greatly enhance the depth and breadth of these analyses. In addition, enabling tandem MS (MS2) and multiple MS (MSn) data acquisition for structural elucidation will expand capabilities, given that acquiring single MS (MS1) spectra alone may not be informative/useful except for well‐defined molecules. When such information can be combined with functional genomic and genetic data, plant science research will be poised for metabolic engineering (Anarat‐Cappillino & Sattely, 2014) and synthetic biology to optimize small molecule production and flux, to control metabolite localization and activity, to create new metabolites, and to engineer plants as factories for chemical production (Vickery, La Clair, Burkart, & Noel, 2016). Clearly, both major challenges and significant opportunities lie ahead in metabolomics research.
3.4. Integration
While the above discussion has separately considered challenges associated with each types of ome, another major challenge is achieving meaningful integration across all the inter‐related omes. Even within a single cell type, life scientists have yet to completely meet the challenge of trans‐omic integration in a meaningful way. Moreover, even if starting with single cell types, each individual cell will be in a unique state. Single‐cell RNA‐seq was highlighted by Science as the 2018 breakthrough of the year, with an emphasis on its role in illuminating metazoan development (Pennisi, 2018), but progress in plant systems is also accelerating (Brennecke et al., 2013; Efroni et al., 2016; Ryu, Huang, Kang, & Schiefelbein, 2019). Because single‐cell omics is inherently variable, distinguishing the signal from the noise in these datasets requires both measurements on many individual cells and advanced statistical/machine learning approaches for data analyses (Yuan et al., 2017). Similar challenges arise when scaling from the single cell to incorporating interactions that occur between cells, tissues and organs, including the complexity of integrating impacts of cell‐to‐cell and long distance signaling. Yet another layer of complexity arises from addition of the fourth dimension, integrating across time. Although the magnitude of this challenge is enormous, for systems biology to inform successful synthetic biology efforts (e.g., in precision engineering of agricultural crops), ongoing attention to the integration problem is needed. Fortunately, even partial solutions may have significant impact; for example, in mammalian cell lines important new insights have recently been achieved concerning mechanistic relationships governing the extent of correlation between the transcriptome and proteome (Schwanhausser et al., 2011).
4. BIG WISHES: A WISH LIST FOR PLANT RESEARCH
In this section, we discuss some of the promising research directions and possibilities for tool development that arose during the workshop. While certainly a non‐exhaustive list, development of capacity in these areas would greatly enhance the capability of plant scientists to meet the challenges described in the previous section and, ultimately, to address the “Big Questions” in plant biology. This wish list loosely follows the topic order of the previous section, from genomics/transcriptomes to proteomics, metabolomics, and integration. Over time, this wish list will surely evolve, as new technologies are developed and as solutions to some of these items lead to new goals. For example, prior to 2000, such a wish list would probably have included the goal of one sequenced plant genome, as compared to the present day when we now have many thousands of sequenced genomes for multiple plant species.
4.1. A panel of true cell‐type specific immortal plant cell lines distributed from a stock center
Utilization of immortalized specific cell lines has transformed research in animal models. The development of cell‐type specific plant cell lines has the potential to similarly revolutionize research in plant biology. Cell lines enable uniform and scalable experiments, and facilitate acquisition of cell‐type specific information on multiple cellular omes, ranging from the genome to the transcriptome, proteome, metabolome and more, thereby promoting integrative analysis. Cell lines are particularly relevant toward enabling omics experiments requiring large amounts of material, which can often be limiting for specialized and hard to access cell‐types. Availability of such lines through a stock center would democratize this essential resource.
While the benefits are apparent, the task of actually creating these cell lines is quite challenging. To enable production of cell‐type specific lines, genes that specify cell fate need to be identified and then employed to re‐differentiate stem cells or protoplasts to maintain the given cell type. Another major challenge to practical implementation may be the reduced cell proliferation potential of highly differentiated plant cells, which would prevent their propagation. An alternative approach would be a community effort to develop and distribute standardized protocols to allow individual labs to perform short‐term re‐differentiation of dedifferentiated cells into specified cell types, as exemplified by the tracheary element system (Endo et al., 2009; Iakimova & Woltering, 2017). Because of the already substantial knowledge on developmental regulators in Arabidopsis, Arabidopsis is the optimal system with which to work towards these goals.
4.2. A desktop mass spectrometer for proteomics and metabolomics
With mass analyzers becoming greatly reduced in size, a miniature, affordable mass spectrometer is envisioned as providing metabolomics and proteomics capabilities to individual labs in the near future. Ion trap and orbitrap based systems, which allow structural elucidation using MSn (3D ion trap) and high mass resolution and high mass accuracy (Orbitrap), have great potential in these applications. Ideally, mass spectrometers would become instruments found in every lab or collaborative team. Such easy access would allow hands‐on experiences for students in individual labs, and could accelerate progress.
4.3. Improved computational tools to extract data from proteomics analyses
A common observation in processing MS data is that only ~25% of good quality spectra can be assigned an identity (i.e., a protein ID is matched with the spectrum). The most commonly held explanation is that extensive, combinatorial PTMs alter the mass in unpredictable ways that preclude assigning a match, but it is not possible simply to add all conceivable modifications into the queried database because the risk of false positives increases greatly with each additional inclusion of mass‐altered amino acids. Therefore, computational solutions for (easily) assigning identities to these many orphan spectra could provide a tremendous wealth of additional information from every proteomic experiment, particularly with regard to the possibility of identifying potentially hundreds of dynamic PTM changes that may currently be missed during cellular responses. Recent promise in this area is described in a report that employed a large mass‐tolerance of 500 Da in a database search, wherein a large number of unmatched spectra were identified that arose from modifications of different amino acids (Chick et al., 2015).
A related issue in need of a computational solution is the identification of small peptides that contain a currently unknown N‐terminus. Particularly for secreted peptides for which defined proteolytic processing is not known, database searching is limited by the vast number of possible peptides that would have to be included in the database. With the growing number of bioactive peptides that have already been demonstrated to play important roles in plant responses, including the observation that such peptides are ligands for receptor‐like kinases, a family with some 600 members in Arabidopsis (Osakabe, Yamaguchi‐Shinozaki, Shinozaki, & Tran, 2013), a solution to this problem is likely to identify numerous new regulatory molecules involved in cell signaling.
4.4. Overcoming mass spectrometry machine specificity for metabolomics
Peptide mass spectra acquired on different types of instruments usually do not affect the peptide sequence assignment due to the richness of information within the spectra, despite variations in fragmentation. Similarly, metabolite mass spectra acquired on electron impact/chemical ionization based GC‐MS are extremely reproducible on different instruments. However, metabolite mass spectra acquired on electrospray ionization (soft ionization) based LC‐MS are variable depending on the instrument types. Such variation often requires spectral libraries that are created on the same type of instrument in order to achieve success in metabolite structural annotation. This variation is one reason why untargeted metabolomics has had limited success in metabolite identification. Another limitation in metabolite identification is that libraries are incomplete and do not include modified metabolites. Untargeted metabolomics can generate thousands of peaks/features, but only tens or low hundreds of them can be identified through library searching. A potentially promising approach is to generate combined spectral libraries containing spectra acquired on different instruments. In addition, generating theoretical spectra based on knowledge of metabolic pathways and allowing in silico inclusion of modifications to existing spectra (e.g., oxidation, hydroxylation and carboxylation) could enhance spectral matching success. Finally, application of machine learning for feature classification appears to be a promising future direction (Cuperlovic‐Culf, 2018).
Some of these issues could be addressed in the short term by increased federal funding to support core metabolomics facilities that invested time in services and technologies to serve the plant community. NIH‐supported cores are biased toward dealing with human metabolites—their libraries and their methods rarely encompass phytochemicals. Cores that focused on libraries and protocols for plant metabolomics could perform the more standard metabolomic analyses, analogous to the standard RNA‐seq that has become ubiquitously available for nucleic acid identification and quantification, while individual laboratories could develop and distribute the specialized methods required for specific groups of metabolites, facilitating collaboration and eventual incorporation of these advances into the offering of core facilities.
4.5. Omics scale field‐deployable sensors
Development of transgenic optical sensors that are able to report in real time under field conditions would provide essential information needed to address the “translation gap” between laboratory experiments and field results. Real time and non‐invasive monitoring of multiple types of molecules, ranging from nucleic acids to proteins to metabolites, in plant cells under field as well as laboratory conditions would not only reveal important biological processes that may be missed or misinterpreted using in vitro methods, but also enable high‐resolution dynamic studies that would revolutionize plant biology and biology in general. Plant scientists already employ transcriptional sensors for single metabolite monitoring. These include the auxin reporter DR5 (Ulmasov, Murfett, Hagen, & Guilfoyle, 1997) and the cytokinin reporter TCSn (Zurcher et al., 2013), which are driven by synthetic promoters engineered to contain binding sites of regulatory transcription factors, thus bypassing regulation by other stimuli to provide sensor specificity. More recently, the development of genetically‐encoded sensors that exploit native molecule recognition mechanisms has enabled quantitative measurement of plant hormones (Brunoud et al., 2012; Jones, Danielson, et al., 2014; Larrieu et al., 2015; Samodelov et al., 2016; Waadt et al., 2014; Wend et al., 2013), ionic conditions (Swanson, Choi, Chanoca, & Gilroy, 2011) and metabolites such as glucose (Chen et al., 2010) with extraordinary sensitivity. Technologies that allowed large‐scale implementation, including field deployment, and omics level application of sensors (e.g., to identify all proteins that bound a given substrate) would be challenging, innovative and of high impact. Such impact would be further extended by the development of deep tissue imaging techniques that would allow reporting from internal tissues.
4.6. Databases that inform rational data integration across laboratories, scales, and species
A priority raised in this and other workshops (Friesner et al., 2017; International Arabidopsis Informatics Consortium [IAIC], 2019) is the need to promote community‐driven science that can answer big questions and address grand challenges in the field, such as food security and crop adaptation to future climate scenarios. Innovative methods for biological data integration across temporal and spatial scales are needed to achieve these goals. It is the meta‐analysis across datasets and from the fine‐ to coarse‐scales that will most strongly enable predictive capability to address pressing issues in plant biology across topics as diverse as engineering crop ideotypes or designing conservation strategies for rare and endangered plant species.
Accurate data integration across laboratories requires community adoption of standards for both experimental protocols and annotation of datasets. Although some standards for data reporting have already been established by the larger biological community, for example MIAME (Brazma et al., 2001), MIQE (http://www.rdml.org/miqe.php) and HUPO Proteomics Standards (https://www.hupo.org/Proteomics-Standards-Initiative), critical experimental information is often poorly documented. Oft‐neglected details such as circadian time of sampling, tissue type and age, and detailed plant growth conditions are needed not only to facilitate reproducibility and comparability of results among laboratories, but also to identify the most biologically appropriate data for meaningful integration. Databases such as Gene Expression Omnibus and, to some extent, Proteomics IDEntifications, require specific and detailed information that is subject to approval upon submission. However, a single comparable database for metabolomics does not exist. NCBI or cross‐agency establishment of a centralized database for transcriptomics, proteomics, and metabolomics data would greatly strengthen the impact of omics research for the entire community of life scientists.
There is also a need for a centralized cyberinfrastructure for plant science. Many excellent and informative databases exist for multiple species; examples for Arabidopsis being TAIR (subscription‐based for full access) and Araport (open access). However, there is no singular database in which researchers can readily access, download, and integrate high‐quality data from genome, transcriptome, metabolome, proteome, physiological, and phenomic experiments, and do so across multiple species. The existence of scattered databases for specific species and experimental techniques has resulted in the time‐consuming creation of redundant tools and difficulty in consolidating knowledge across plant species. Unless resolved, we run the risk of reinventing the wheel for every new species for which omic tools become available. Many of these same points have been emphasized in a recent white paper from the International Arabidopsis Informatics Consortium (2019). Resources that integrate across plant species, e.g., to allow facile comparison of genomes and functional networks between model species and crops, also might improve the hit rate for translational research. An ultimate goal would be to develop mathematical approaches that could identify relationships across these data sets, formulate mathematical constructs that would characterize these relationships, and then use the mathematical constructs to predict how novel perturbations at any biological level would impact the resulting phenotype.
5. BIG VISIONS: TRAINING THE OMICS SCIENTIST
A recent report from another NSF‐funded RCN, the Plant Science Research Network, advocates for increased empowerment of trainees to personalize their Ph.D. training program, and proposes a modular approach that could, e.g., facilitate incorporation of training in both wet bench and analytical skills, and would be conducted under the guidance of a mentoring team (Henkhaus, Taylor, Greenlee, Sickler, & Stern, 2018). Such a shift in training paradigms might also help to create a more inclusive environment and promote workforce diversity.
Another recent NAASC report has focused on training needs for computational and quantitative plant biology (Friesner et al., 2017). Purely “in silico” biology offers new avenues for collaborations with, e.g., mathematicians and engineers, and can level the playing field for investigators with fewer experimental resources. A third NAASC workshop that focused on broadening the impact of plant science through effective outreach programs will make recommendations for how to innovate, evaluate and disseminate activities in this area.
This workshop focused on experimental training relative to omics science. Improving training in omics approaches has both conceptual and practical considerations. Among the conceptual considerations are the need for trainees to understand how to ask a good question before initiating an omics experiment. This understanding has three components: first, developing a hypothesis that is both testable and worth testing; second, identifying whether the specific omics technique under consideration is actually applicable to the question at hand; third, if an omics approach has been chosen, designing the experiment so as to ensure the collection of useful data. There may also be instances where a discovery‐based approach (as opposed to a hypothesis‐based approach) is justified, especially when the ensuing omics experiments will yield a data resource of widespread utility. Clearly, among the first things that a student needs to learn is what omics tools exist, what information each can provide, and what information each cannot provide. An additional point is that while some genomics techniques remain difficult to perform in crops due to barriers to transformation and long generation times, other genomics approaches, including many transcriptomic and epigenomic approaches, require only high‐quality genome sequence information. Proteomics methods are applicable to most species with reasonably good genome annotation, and metabolomics approaches do not even require a sequenced genome.
When field experiments are involved, additional skills that often must be acquired by omics‐centric researchers include the ability to identify a relevant set of environmental conditions and to learn what is involved in managing and sampling from a field site. Appreciation of the ecology or agro‐ecology of the organism can aid in the optimal design of field experiments.
Another issue for consideration is the extent to which trainees need to understand and be able to execute the wet bench aspects of an omics method. For example, for RNA‐seq, students often do not need to know more than how to isolate high quality RNA, as most sequencing facilities will construct the actual RNA‐seq libraries. For such highly standardized omics approaches, wet bench training primarily concerns sample preparation, and this training can often be obtained in the individual laboratory. On the other hand, failure to provide students with deeper knowledge, e.g., of how RNA‐seq libraries are generated, will obstruct the student's ability to either develop an innovative new variant of the method, or envision an entirely new approach. Students should also have an understanding of how the raw data are generated so that they can appreciate where issues of quality or reproducibility may arise, and how to implement quality control measures.
Therefore, we see a need for students to have an appreciation of how the equipment that generates omic data, e.g., next generation sequencers and mass spectrometers—work. This goal is achievable by several methods, including workshops held by core facilities and short internships with knowledgeable labs. One limitation to training in modern experimental omics is that not every college and university has a core facility where these technologies can be accessed. Distance learning opportunities spearheaded by core facilities, in both real and virtual time (in the latter category would be webinars and YouTube videos), can help to redress this issue. One innovation is emerging WebEx methods whereby students can actually control the equipment (at least for mass spectrometers) remotely. Such training would be further facilitated by the development of kits that would serve as technology training tools. A good analogy for this is the current use of Raspberry Pi as a teaching and learning tool that allows students to experiment with electronic technologies. Raspberry Pi is also a cost‐effective conduit between theoretical topics and practical applications of those topics, allowing students to play with and learn diverse applications with one accessible training module. Development of a similar tool for MS would help beginners understand both the concepts behind and the practicalities and limitations of these machines.
Another conceptual skill that can be difficult for students to develop is the ability to derive important new questions from large‐scale omics data, once those data have been generated. Typically, omics discovery is not the end, but the beginning of further hypothesis generation and testing. Because principles derived from meta‐analyses may or may not hold true when any one nucleic acid, protein or metabolite is evaluated, it is vital to develop skills in choosing the targets for any such experiments. In short, students need to learn how to design, test, and then refine their experiments, from beginning to end. A measured approach to experiment planning, as described above, will help avoid the situation in which an omics experiment is performed simply because the methodology is feasible and accessible.
Tantamount to achieving the above goal is the essentiality of obtaining computational skills to analyze the large amounts of data produced by omics methods. In this area as well, turn‐key applications such as those available on Galaxy (https://usegalaxy.org/) are excellent resources. However, without more sophisticated training, students will not appreciate the limitations of such approaches, and will not be able to go beyond them. A basic understanding of shell coding, a scripting language (increasingly, Python), and the R programming environment that empowers statistical analysis and data visualization, are becoming essential tools for plant biologists. The necessary core computational and quantitative skills for plant scientists, and approaches to empower training and access to such skills, have been described elsewhere, along with a useful enumeration of a number of curricula and vehicles for obtaining such training (Friesner et al., 2017). As synthetic biology gains in tools and resources (Church, Elowitz, Smolke, Voigt, & Weiss, 2014), knowledge of fundamental concepts from diverse engineering disciplines (e.g., modularity, sensitivity, reliability, and robustness) and quantitative modeling approaches will also benefit omics students.
A final comment has to do with the soft skills required for omics research. As both wet bench techniques and computational analyses increase in diversity and complexity, the ability to identify key collaborators in other disciplines and to work productively in a team setting is gaining in importance. Such team approaches have long been emphasized in industry, are increasingly essential in academe as well, and, when successful, can significantly decrease the time to publication, or to graduation for Ph.D. trainees. The report from the first workshop of the ART‐21 Research Coordination Network provides concrete suggestions on how to define and design collaborative efforts (Friesner et al., 2017).
6. BIG AND LITTLE PLANTS: ARABIDOPSIS RESEARCH AND BEYOND
The living laboratory of Arabidopsis provides plant researchers with an advantage over their counterparts in medical research in that manipulative research can readily be performed on the organism itself; thus, Arabidopsis is often referred to as a “reference species” rather than as a “model species”. The numerous contributions of Arabidopsis research to our understanding of both universal and plant‐specific biological mechanisms, and the importance of Arabidopsis in translational research for crop improvement are extensive (Provart et al., 2016). Through the omics lens, the well‐known advantages of Arabidopsis that originally made it a superior system for (now) classical molecular genetic approaches currently provide a wellspring of knowledge from which to interpret omics data. For example, the multiple iterations of sequencing and gene model identification that Arabidopsis has undergone provide, more completely and rigorously than for any other plant species, the genome annotation needed for accurate identification of transcriptomes and proteomes. Arabidopsis remains the most convenient testbed for the development of new experimental approaches (Holland & Jez, 2018), as witnessed by the many landmark omics publications on Arabidopsis, some of which are summarized in Table 1. While the number of sequenced rice genomes now exceeds that of Arabidopsis (Rellosa et al., 2014; Wang et al., 2018), “platinum level” (i.e., extremely high quality) genome sequence and gene annotation lags behind; and rice transformation is slow. The ease of Arabidopsis transformation—without a tissue culture step that may itself elicit epigenomic effects (Ong‐Abdullah et al., 2015)—allows rapid experimental testing of hypotheses that arise from omics data analyses. Moreover, CRISPR technologies are expected to facilitate gene knockout as well as transgene introduction in any Arabidopsis accession, enabling direct testing of the functional impacts of natural variants within the diverse, fully sequenced 1,135 genomes of Arabidopsis (Alonso‐Blanco et al., 2016).
Table 1.
Some landmark advances in plant omics facilitated by use of Arabidopsis as a model system
| Key reference | Title | |
|---|---|---|
| Advance: Genome | ||
| Plant genome sequence | Arabidopsis Initiative (2000) | Analysis of the genome sequence of the flowering plant Arabidopsis thaliana |
| Whole‐genome methylation analysis | Zhang et al. (2006) | Genome‐wide high‐resolution mapping and functional analysis of DNA methylation in Arabidopsis |
| Whole genome histone modification maps | Zhang et al. (2007) | Whole‐genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis |
| Whole‐genome methylome analysis at single nucleotide resolution | Cokus et al. (2008) | Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning |
| Whole‐genome methylome analysis at single nucleotide resolution | Lister et al. (2008) | Highly integrated single‐base resolution maps of the epigenome in Arabidopsis |
| 1001 genomes | Cao et al. (2011) | Whole‐genome sequencing of multiple Arabidopsis thaliana populations |
| Multi‐genome comparison | Long et al. (2013) | Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden |
| Parallel population‐wide sequencing of genomes, transcriptomes, and methylomes | Schmitz et al. (2013) | Patterns of population epigenomic diversity |
| Transcription factor‐wide analysis of cis‐element preferences across multiple eukaryotic clades including Arabidopsis | Weirauch et al., (2014) | Determination and inference of eukaryotic transcription factor sequence specificity |
| 1001 genomes | Alonso‐Blanco et al. (2016) | 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana |
| 1001 epigenomes | Kawakatsu et al. (2016) | Epigenomic diversity in a global collection of Arabidopsis thaliana accessions |
| DAP‐Seq analysis | O'Malley et al. (2016) | Cistrome and epicistrome features shape the regulatory DNA landscape |
| Size‐resolved chromatin‐seq | Pass et al. (2017) | Genome‐wide chromatin mapping with size resolution reveals a dynamic sub‐nucleosomal landscape in Arabidopsis |
| Advance: Transcriptome | ||
| Large scale EST sequencing | Yamada et al. (2003) | Empirical analysis of transcriptional activity in the Arabidopsis genome |
| Cell‐type gene expression atlas | Birnbaum et al. (2003) | A gene expression map of the Arabidopsis root |
| Developmental gene expression map | Schmid et al. (2005) | A gene expression map of Arabidopsis thaliana development |
| Deep sequencing of small RNAs | Lu et al. (2006) | MicroRNAs and other small RNAs enriched in the Arabidopsis RNA‐dependent RNA polymerase‐2 mutant |
| Deep sequencing of small RNAs | Henderson et al. (2006) | Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning |
| RNA‐seq analysis | Lister et al. (2008) | Highly integrated single‐base resolution maps of the epigenome in Arabidopsis |
| Cell‐type specific transcriptome profiling of an environmental response | Gifford, Dean, Gutierrez, Coruzzi, and Birnbaum (2008) | Cell‐specific nitrogen responses mediate developmental plasticity |
| Cell‐type specific transcriptome profiling of an environmental response | Dinneny et al. (2008) | Cell identity mediates the response of Arabidopsis roots to abiotic stress |
| Single‐cell RNA‐seq | Brennecke et al. (2013) | Accounting for technical noise in single‐cell RNA‐seq experiments |
| Translatome/ribosome footprinting analysis | Juntawong, Girke, Bazin, and Bailey‐Serres (2014) | Translational dynamics revealed by genome‐wide profiling of ribosome footprints in Arabidopsis |
| In vivo transcriptome‐wide analysis of RNA structure | Ding et al. (2014) | In vivo genome‐wide profiling of RNA secondary structure reveals novel regulatory features |
| Advance: Proteome | ||
| Cell‐type proteomics | Wienkoop et al. (2004) | Cell‐specific protein profiling in Arabidopsis thaliana trichomes: identification of trichome‐located proteins involved in sulfur metabolism and detoxification |
| Vacuolar proteomics analysis | Carter et al. (2004) | The vegetative vacuole proteome of Arabidopsis thaliana reveals predicted and unexpected proteins |
| Genome‐scale proteome map | Baerenfaller et al. (2008) | Genome‐scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics |
| Targeted interactomics application in basic cell cycle complex machinery. | Van Leene et al. (2010) | Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana |
| Large‐scale plant protein interactome | Braun et al. (2011) | Evidence for network evolution in an Arabidopsis interactome map |
| Large‐scale plant – microbe protein interactome | Mukhtar et al. (2011) | Independently evolved virulence effectors converge onto hubs in a plant immune system network |
| Membrane protein interactome | Jones, Xuan, et al. (2014) | Border control—A membrane‐linked interactome of Arabidopsis |
| Advance: Metabolome | ||
| Integration of transcriptomics and metabolomics | Hirai et al. (2004) | Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana |
| Genome‐scale metabolic model | Poolman, Miguet, Sweetlove, and Fell (2009) | A genome‐scale metabolic model of Arabidopsis and some of its properties |
| Single cell metabolomics | Holscher et al. (2009) | Matrix‐free UV‐laser desorption/ionization (LDI) mass spectrometric imaging at the single‐cell level: distribution of secondary metabolites of Arabidopsis thaliana and Hypericum species |
| Cell‐type metabolomics | Ebert et al. (2010) | Metabolic profiling of Arabidopsis thaliana epidermal cells |
| Advance: Phenome | ||
| Large scale insertional mutant collection | Sussman, Amasino, Young, Krysan, and Austin‐Phillips (2000) | The Arabidopsis knockout facility at the University of Wisconsin‐Madison |
| Large scale insertional mutant collection | Samson et al. (2002) | FLAGdb/FST: a database of mapped flanking insertion sites (FSTs) of Arabidopsis thaliana T‐DNA transformants |
| Large scale insertional mutant collection | Sessions et al. (2002) | A high‐throughput Arabidopsis reverse genetics system |
| Large scale insertional mutant collection | Alonso et al. (2003) | Genome‐wide insertional mutagenesis of Arabidopsis thaliana |
| Large scale insertional mutant collection | Rosso et al. (2003) | An Arabidopsis thaliana T‐DNA mutagenized population for flanking sequence tag‐based reverse genetics. |
| GWAS analysis | Aranzana et al. (2005) | Genome‐wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes |
| Automated phenome analyses | Granier et al. (2006) | PHENOPSIS, an automated platform for reproducible phenotyping of plant responses to soil water deficit in Arabidopsis thaliana permitted the identification of an accession with low sensitivity to soil water deficit |
| Large scale phenome analysis | Kuromori et al. (2006) | A trial of phenome analysis using 4000 Ds‐insertional mutants in gene‐coding regions of Arabidopsis |
| Large scale phenotyping GWAS study | Atwell et al. (2010) | Genome‐wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines |
GWAS: genome‐wide association study.
7. CONCLUDING REMARKS
In summary, with enhanced research and training in emerging omics technologies, a 4D –omics analysis of Arabidopsis, providing superior resolution in both spatial and temporal domains, is conceivable. From such information, we will be able not only to identify mechanisms that can be leveraged for agricultural solutions, but also, ideally, to mine these omics datasets to identify improvements that have not been favored by natural selection (Keren et al., 2016) but can nevertheless be engineered by synthetic biology approaches for the improvement of agricultural systems and global health.
AUTHOR CONTRIBUTIONS
All authors contributed substantively to the ideas presented in this report and participated in manuscript preparation.
ACKNOWLEDGMENTS
This work was supported by the U.S. National Science Foundation (award no. NSF‐RCN 1518280), which funded the workshop that led to generation of this white paper.
CONFLICT OF INTEREST
The authors declare no conflict of interest associated with the work described in this manuscript.
Argueso CT, Assmann SM, Birnbaum KD, et al. Directions for research and training in plant omics: Big Questions and Big Data. Plant Direct. 2019;3:1–16. 10.1002/pld3.133
REFERENCES
- Afendi, F. M. , Okada, T. , Yamazaki, M. , Hirai‐Morita, A. , Nakamura, Y. , Nakamura, K. , … Kanaya, S. (2012). KNApSAcK family databases: Integrated metabolite‐plant species databases for multifaceted plant research. Plant and Cell Physiology, 53(2), e1 10.1093/pcp/pcr165 [DOI] [PubMed] [Google Scholar]
- Alonso, J. M. , Stepanova, A. N. , Leisse, T. J. , Kim, C. J. , Chen, H. M. , Shinn, P. , … Ecker, J. R. (2003). Genome‐wide insertional mutagenesis of Arabidopsis thaliana . Science, 301(5633), 653–657. 10.1126/science.1086391 [DOI] [PubMed] [Google Scholar]
- Alonso‐Blanco, C. , Andrade, J. , Becker, C. , Bemm, F. , Bergelson, J. , Borgwardt, K. M. , … Consortium, G. (2016). 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana . Cell, 166(2), 481–491. 10.1016/j.cell.2016.05.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anarat‐Cappillino, G. , & Sattely, E. S. (2014). The chemical logic of plant natural product biosynthesis. Current Opinion in Plant Biology, 19, 51–58. 10.1016/j.pbi.2014.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Initiative, T. (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana . Nature, 408(6814), 796–815. 10.1038/35048692 [DOI] [PubMed] [Google Scholar]
- Aranzana, M. J. , Kim, S. , Zhao, K. Y. , Bakker, E. , Horton, M. , Jakob, K. , … Nordborg, M. (2005). Genome‐wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genetics, 1(5), 531–539. 10.1371/journal.pgen.0010060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwell, S. , Huang, Y. S. , Vilhjalmsson, B. J. , Willems, G. , Horton, M. , Li, Y. , … Nordborg, M. (2010). Genome‐wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature, 465(7298), 627–631. 10.1038/nature08800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenfaller, K. , Grossmann, J. , Grobei, M. A. , Hull, R. , Hirsch‐Hoffmann, M. , Yalovsky, S. , … Baginsky, S. (2008). Genome‐scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science, 320(5878), 938–941. 10.1126/science.1157956 [DOI] [PubMed] [Google Scholar]
- Birnbaum, K. D. (2018). Power in numbers: Single‐cell RNA‐seq strategies to dissect complex tissues. Annual Review of Genetics, 52(52), 203–221. 10.1146/annurev-genet-120417-031247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum, K. , Shasha, D. E. , Wang, J. Y. , Jung, J. W. , Lambert, G. M. , Galbraith, D. W. , & Benfey, P. N. (2003). A gene expression map of the Arabidopsis root. Science, 302(5652), 1956–1960. 10.1126/science.1090022 [DOI] [PubMed] [Google Scholar]
- Braun, P. , Carvunis, A. R. , Charloteaux, B. , Dreze, M. , Ecker, J. R. , Hill, D. E. , … Co, A. I. M. (2011). Evidence for network evolution in an Arabidopsis interactome map. Science, 333(6042), 601–607. 10.1126/science.1203877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma, A. , Hingamp, P. , Quackenbush, J. , Sherlock, G. , Spellman, P. , Stoeckert, C. , … Vingron, M. (2001). Minimum information about a microarray experiment (MIAME)‐toward standards for microarray data. Nature Genetics, 29(4), 365–371. 10.1038/ng1201-365 [DOI] [PubMed] [Google Scholar]
- Brennecke, P. , Anders, S. , Kim, J. K. , Kolodziejczyk, A. A. , Zhang, X. W. , Proserpio, V. , … Heisler, M. G. (2013). Accounting for technical noise in single‐cell RNA‐seq experiments. Nature Methods, 10(11), 1093–1095. 10.1038/Nmeth.2645 [DOI] [PubMed] [Google Scholar]
- Brunoud, G. , Wells, D. M. , Oliva, M. , Larrieu, A. , Mirabet, V. , Burrow, A. H. , … Vernoux, T. (2012). A novel sensor to map auxin response and distribution at high spatio‐temporal resolution. Nature, 482(7383), 103–106. 10.1038/nature10791 [DOI] [PubMed] [Google Scholar]
- Cao, J. , Schneeberger, K. , Ossowski, S. , Gunther, T. , Bender, S. , Fitz, J. , … Weigel, D. (2011). Whole‐genome sequencing of multiple Arabidopsis thaliana populations. Nature Genetics, 43(10), 956–963. 10.1038/ng.911 [DOI] [PubMed] [Google Scholar]
- Carter, C. , Pan, S. , Zouhar, J. , Avila, E. L. , Girke, T. , & Raikhel, N. V. (2004). The vegetative vacuole proteome of Arabidopsis thaliana reveals predicted and unexpected proteins. Plant Cell, 16(12), 3285–3303. 10.1105/tpc.104.027078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. Q. , Hou, B. H. , Lalonde, S. , Takanaga, H. , Hartung, M. L. , Qu, X. Q. , … Frommer, W. B. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature, 468(7323), 527–532. 10.1038/nature09606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick, J. M. , Kolippakkam, D. , Nusinow, D. P. , Zhai, B. , Rad, R. , Huttlin, E. L. , & Gygi, S. P. (2015). A mass‐tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nature Biotechnology, 33(7), 743–749. 10.1038/nbt.3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church, G. M. , Elowitz, M. B. , Smolke, C. D. , Voigt, C. A. , & Weiss, R. (2014). Realizing the potential of synthetic biology. Nature Reviews Molecular Cell Biology, 15(4), 289–294. 10.1038/nrm3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus, S. J. , Feng, S. , Zhang, X. , Chen, Z. , Merriman, B. , Haudenschild, C. D. , … Jacobsen, S. E. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature, 452(7184), 215–219. 10.1038/nature06745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constabel, C. P. , Yoshida, K. , & Walker, V. (2014). Diverse ecological roles of plant tannins: Plant defense and beyond. Recent Advances in Polyphenol Research, 4, 115–142. [Google Scholar]
- Cuperlovic‐Culf, M. (2018). Machine learning methods for analysis of metabolic data and metabolic pathway modeling. Metabolites, 8(1), pii: E4 10.3390/metabo8010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuperus, J. T. , Montgomery, T. A. , Fahlgren, N. , Burke, R. T. , Townsend, T. , Sullivan, C. M. , & Carrington, J. C. (2010). Identification of MIR390a precursor processing‐defective mutants in Arabidopsis by direct genome sequencing. Proceedings of the National Academy of Sciences of the United States of America, 107(1), 466–471. 10.1073/pnas.0913203107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Auria, J. C. , & Gershenzon, J. (2005). The secondary metabolism of Arabidopsis thaliana: Growing like a weed. Current Opinion in Plant Biology, 8(3), 308–316. 10.1016/j.pbi.2005.03.012 [DOI] [PubMed] [Google Scholar]
- Deinlein, U. , Stephan, A. B. , Horie, T. , Luo, W. , Xu, G. H. , & Schroeder, J. I. (2014). Plant salt‐tolerance mechanisms. Trends in Plant Science, 19(6), 371–379. 10.1016/j.tplants.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Y. , Tang, Y. , Kwok, C. K. , Zhang, Y. , Bevilacqua, P. C. , & Assmann, S. M. (2014). In vivo genome‐wide profiling of RNA secondary structure reveals novel regulatory features. Nature, 505(7485), 696–700. 10.1038/nature12756 [DOI] [PubMed] [Google Scholar]
- Dinneny, J. R. , Long, T. A. , Wang, J. Y. , Jung, J. W. , Mace, D. , Pointer, S. , … Benfey, P. N. (2008). Cell identity mediates the response of Arabidopsis roots to abiotic stress. Science, 320(5878), 942–945. 10.1126/science.1153795 [DOI] [PubMed] [Google Scholar]
- Ebert, B. , Zoller, D. , Erban, A. , Fehrle, I. , Hartmann, J. , Niehl, A. , … Fisahn, J. (2010). Metabolic profiling of Arabidopsis thaliana epidermal cells. Journal of Experimental Botany, 61(5), 1321–1335. 10.1093/jxb/erq002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni, I. , Mello, A. , Nawy, T. , Ip, P. L. , Rahni, R. , DelRose, N. , … Birnbaum, K. D. (2016). Root regeneration triggers an embryo‐like sequence guided by hormonal interactions. Cell, 165(7), 1721–1733. 10.1016/j.cell.2016.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo, S. , Pesquet, E. , Yamaguchi, M. , Tashiro, G. , Sato, M. , Toyooka, K. , … Demura, T. (2009). Identifying new components participating in the secondary cell wall formation of vessel elements in zinnia and Arabidopsis. Plant Cell, 21(4), 1155–1165. 10.1105/tpc.108.059154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernie, A. R. (2007). The future of metabolic phytochemistry: Larger numbers of metabolites, higher resolution, greater understanding. Phytochemistry, 68(22–24), 2861–2880. 10.1016/j.phytochem.2007.07.010 [DOI] [PubMed] [Google Scholar]
- Friesner, J. , Assmann, S. M. , Bastow, R. , Bailey‐Serres, J. , Beynon, J. , Brendel, V. , … Brady, S. M. (2017). The next generation of training for Arabidopsis researchers: Bioinformatics and quantitative biology. Plant Physiology, 175(4), 1499–1509. 10.1104/pp.17.01490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershenzon, J. , & Dudareva, N. (2007). The function of terpene natural products in the natural world. Nature Chemical Biology, 3(7), 408–414. 10.1038/nchembio.2007.5 [DOI] [PubMed] [Google Scholar]
- Gifford, M. L. , Dean, A. , Gutierrez, R. A. , Coruzzi, G. M. , & Birnbaum, K. D. (2008). Cell‐specific nitrogen responses mediate developmental plasticity. Proceedings of the National Academy of Sciences of the United States of America, 105(2), 803–808. 10.1073/pnas.0709559105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier, C. , Aguirrezabal, L. , Chenu, K. , Cookson, S. J. , Dauzat, M. , Hamard, P. , … Tardieu, F. (2006). PHENOPSIS, an automated platform for reproducible phenotyping of plant responses to soil water deficit in Arabidopsis thaliana permitted the identification of an accession with low sensitivity to soil water deficit. New Phytologist, 169(3), 623–635. 10.1111/j.1469-8137.2005.01609.x [DOI] [PubMed] [Google Scholar]
- Grob, S. , Schmid, M. W. , Luedtke, N. W. , Wicker, T. , & Grossniklaus, U. (2013). Characterization of chromosomal architecture in Arabidopsis by chromosome conformation capture. Genome Biology, 14(11), R129 10.1186/gb-2013-14-11-r129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, I. R. , Zhang, X. Y. , Lu, C. , Johnson, L. , Meyers, B. C. , Green, P. J. , & Jacobsen, S. E. (2006). Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nature Genetics, 38(6), 721–725. 10.1038/ng1804 [DOI] [PubMed] [Google Scholar]
- Henkhaus, N. A. , Taylor, C. B. , Greenlee, V. R. , Sickler, D. B. , & Stern, D. B. (2018). Reinventing postgraduate training in the plant sciences; T‐training defined through modularity, customization, and distributed mentorship. Plant Direct, 2(11), e00095 10.1002/pld3.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai, M. Y. , Yano, M. , Goodenowe, D. B. , Kanaya, S. , Kimura, T. , Awazuhara, M. , … Saito, K. (2004). Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana . Proceedings of the National Academy of Sciences of the United States of America, 101(27), 10205–10210. 10.1073/pnas.0403218101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, C. K. , & Jez, J. M. (2018). Arabidopsis: The original plant chassis organism. Plant Cell Reports, 37(10), 1359–1366. 10.1007/s00299-018-2286-5 [DOI] [PubMed] [Google Scholar]
- Holscher, D. , Shroff, R. , Knop, K. , Gottschaldt, M. , Crecelius, A. , Schneider, B. , … Svatos, A. (2009). Matrix‐free UV‐laser desorption/ionization (LDI) mass spectrometric imaging at the single‐cell level: Distribution of secondary metabolites of Arabidopsis thaliana and Hypericum species. The Plant Journal, 60(5), 907–918. 10.1111/j.1365-313X.2009.04012.x [DOI] [PubMed] [Google Scholar]
- Iakimova, E. T. , & Woltering, E. J. (2017). Xylogenesis in zinnia (Zinnia elegans) cell cultures: Unravelling the regulatory steps in a complex developmental programmed cell death event. Planta, 245(4), 681–705. 10.1007/s00425-017-2656-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Arabidopsis Informatics Consortium . (2019). Arabidopsis bioinformatics resources: The current state, challenges, and priorities for the future. Plant Direct. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, M. A. K. , Gaba, V. , & Greenberg, B. M. (1998). Higher plants and UV‐B radiation: Balancing damage, repair and acclimation. Trends in Plant Science, 3(4), 131–135. 10.1016/s1360-1385(98)01215-1 [DOI] [Google Scholar]
- Jin, X. , Wang, R. S. , Zhu, M. , Jeon, B. W. , Albert, R. , Chen, S. , & Assmann, S. M. (2013). Abscisic acid‐responsive guard cell metabolomes of Arabidopsis wild‐type and gpa1 G‐protein mutants. Plant Cell, 25(12), 4789–4811. 10.1105/tpc.113.119800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, A. M. , Danielson, J. A. H. , ManojKumar, S. N. , Lanquar, V. , Grossmann, G. , & Frommer, W. B. (2014). Abscisic acid dynamics in roots detected with genetically encoded FRET sensors. eLife, 3, e01741 10.7554/elife.01741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, A. M. , Xuan, Y. , Xu, M. , Wang, R. S. , Ho, C. H. , Lalonde, S. , … Frommer, W. B. (2014). Border control—A membrane‐linked interactome of Arabidopsis . Science, 344(6185), 711–716. 10.1126/science.1251358 [DOI] [PubMed] [Google Scholar]
- Juntawong, P. , Girke, T. , Bazin, J. , & Bailey‐Serres, J. (2014). Translational dynamics revealed by genome‐wide profiling of ribosome footprints in Arabidopsis . Proceedings of the National Academy of Sciences of the United States of America, 111(1), E203–E212. 10.1073/pnas.1317811111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, A. , Chiu, C. L. , Vellucci, D. , Yang, Y. , Patel, V. R. , Guan, S. , … Huang, L. (2011). Development of a novel cross‐linking strategy for fast and accurate identification of cross‐linked peptides of protein complexes. Molecular & Cellular Proteomics, 10 (1), M110 002212 10.1074/mcp.M110.002212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann, K. , Muino, J. M. , Jauregui, R. , Airoldi, C. A. , Smaczniak, C. , Krajewski, P. , & Angenent, G. C. (2009). Target genes of the MADS transcription factor SEPALLATA3: Integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biology, 7(4), e1000090 10.1371/journal.pbio.1000090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakatsu, T , Huang, SSC , Jupe, F , Sasaki, E , Schmitz, RJ , Urich, MA , … Consortium, G (2016). Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell, 166(2), 492–505. 10.1016/j.cell.2016.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren, L. , Hausser, J. , Lotan‐Pompan, M. , Slutskin, I. V. , Alisar, H. , Kaminski, S. , … Segal, E. (2016). Massively parallel interrogation of the effects of gene expression levels on fitness. Cell, 166(5), 1282–1294.e18. 10.1016/j.cell.2016.07.024 [DOI] [PubMed] [Google Scholar]
- Khan, M. , Youn, J. Y. , Gingras, A. C. , Subramaniam, R. , & Desveaux, D. (2018). In planta proximity dependent biotin identification (BioID). Scientific Reports, 8(1), 9212 10.1038/s41598-018-27500-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komis, G. , Samajova, O. , Ovecka, M. , & Samaj, J. (2015). Super‐resolution microscopy in plant cell imaging. Trends in Plant Science, 20(12), 834–843. 10.1016/j.tplants.2015.08.013 [DOI] [PubMed] [Google Scholar]
- Kuromori, T. , Wada, T. , Kamiya, A. , Yuguchi, M. , Yokouchi, T. , Imura, Y. , … Shinozaki, K. (2006). A trial of phenome analysis using 4000 Ds‐insertional mutants in gene‐coding regions of Arabidopsis. The Plant Journal, 47(4), 640–651. 10.1111/j.1365-313X.2006.02808.x [DOI] [PubMed] [Google Scholar]
- Larrieu, A. , Champion, A. , Legrand, J. , Lavenus, J. , Mast, D. , Brunoud, G. , … Laplaze, L. (2015). A fluorescent hormone biosensor reveals the dynamics of jasmonate signalling in plants. Nature Communications, 6, 6043 10.1038/ncomms7043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. Y. , Wang, J. , & Zeigler, R. S. (2014). The 3,000 rice genomes project: New opportunities and challenges for future rice research. Gigascience, 3, 8 10.1186/2047-217X-3-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Zheng, Q. , Vandivier, L. E. , Willmann, M. R. , Chen, Y. , & Gregory, B. D. (2012). Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome. Plant Cell, 24(11), 4346–4359. 10.1105/tpc.112.104232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Q. , Zhou, Z. , Luo, W. , Fang, M. , Li, M. , & Li, H. (2017). Screening of proximal and interacting proteins in rice protoplasts by proximity‐dependent biotinylation. Frontiers in Plant Science, 8, 749 10.3389/fpls.2017.00749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister, R. , O'Malley, R. C. , Tonti‐Filippini, J. , Gregory, B. D. , Berry, C. C. , Millar, A. H. , & Ecker, J. R. (2008). Highly integrated single‐base resolution maps of the epigenome in Arabidopsis . Cell, 133(3), 523–536. 10.1016/j.cell.2008.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobingier, B. T. , Huttenhain, R. , Eichel, K. , Miller, K. B. , Ting, A. Y. , von Zastrow, M. , & Krogan, N. J. (2017). An approach to spatiotemporally resolve protein interaction networks in living cells. Cell, 169(2), 350–360.e312. 10.1016/j.cell.2017.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, Q. , Rabanal, F. A. , Meng, D. Z. , Huber, C. D. , Farlow, A. , Platzer, A. , … Nordborg, M. (2013). Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nature Genetics, 45(8), 884–890. 10.1038/ng.2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Z. , Hofmeister, B. T. , Vollmers, C. , DuBois, R. M. , & Schmitz, R. J. (2017). Combining ATAC‐seq with nuclei sorting for discovery of cis‐regulatory regions in plant genomes. Nucleic Acids Research, 45(6), e41 10.1093/nar/gkw1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, C. , Kulkarni, K. , Souret, F. F. , MuthuValliappan, R. , Tej, S. S. , Poethig, R. S. , … Meyers, B. C. (2006). MicroRNAs and other small RNAs enriched in the Arabidopsis RNA‐dependent RNA polymerase‐2 mutant. Genome Research, 16(10), 1276–1288. 10.1101/gr.5530106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni‐Putman, S. M. , & Stepanova, A. N. (2018). A plant biologist's toolbox to study translation. Frontiers in Plant Science, 9, 873 10.3389/fpls.2018.00873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, K. , Koster, T. , Nolte, C. , Weinholdt, C. , Lewinski, M. , Grosse, I. , & Staiger, D. (2017). Adaptation of iCLIP to plants determines the binding landscape of the clock‐regulated RNA‐binding protein AtGRP7. Genome Biology, 18(1), 204 10.1186/s13059-017-1332-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael, T. P. , Jupe, F. , Bemm, F. , Motley, S. T. , Sandoval, J. P. , Lanz, C. , … Ecker, J. R. (2018). High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nature Communications, 9(1), 541 10.1038/s41467-018-03016-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael, T. P. , & VanBuren, R. (2015). Progress, challenges and the future of crop genomes. Current Opinion in Plant Biology, 24, 71–81. 10.1016/j.pbi.2015.02.002 [DOI] [PubMed] [Google Scholar]
- Misra, B. B. , Assmann, S. M. , & Chen, S. (2014). Plant single‐cell and single‐cell‐type metabolomics. Trends in Plant Science, 19(10), 637–646. 10.1016/j.tplants.2014.05.005 [DOI] [PubMed] [Google Scholar]
- Mithofer, A. , & Boland, W. (2012). Plant defense against herbivores: Chemical aspects. Annual Review of Plant Biology, 63, 431–450. 10.1146/annurev-arplant-042110-103854 [DOI] [PubMed] [Google Scholar]
- Moreno‐Risueno, M. A. , Van Norman, J. M. , & Benfey, P. N. (2012). Transcriptional switches direct plant organ formation and patterning. Transcriptional Switches during Development, 98, 229–257. 10.1016/B978-0-12-386499-4.00009-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtar, M. S. , Carvunis, A. R. , Dreze, M. , Epple, P. , Steinbrenner, J. , Moore, J. , … Conso, E. U. E. (2011). Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science, 333(6042), 596–601. 10.1126/science.1203659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundim, F. M. , & Pringle, E. G. (2018). Whole‐plant metabolic allocation under water stress. Frontiers in Plant Science, 9, 852 10.3389/fpls.2018.00852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley, R. C. , Huang, S. C. , Song, L. , Lewsey, M. G. , Bartlett, A. , Nery, J. R. , … Ecker, J. R. (2016). Cistrome and epicistrome features shape the regulatory DNA landscape. Cell, 165(5), 1280–1292. 10.1016/j.cell.2016.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong‐Abdullah, M. , Ordway, J. M. , Jiang, N. , Ooi, S. E. , Kok, S. Y. , Sarpan, N. , … Martienssen, R. A. (2015). Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature, 525(7570), 533–537. 10.1038/nature15365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakabe, Y. , Yamaguchi‐Shinozaki, K. , Shinozaki, K. , & Tran, L. S. (2013). Sensing the environment: Key roles of membrane‐localized kinases in plant perception and response to abiotic stress. Journal of Experimental Botany, 64(2), 445–458. 10.1093/jxb/ers354 [DOI] [PubMed] [Google Scholar]
- Pass, D. A. , Sornay, E. , Marchbank, A. , Crawford, M. R. , Paszkiewicz, K. , Kent, N. A. , & Murray, J. A. H. (2017). Genome‐wide chromatin mapping with size resolution reveals a dynamic sub‐nucleosomal landscape in Arabidopsis . PLoS Genetics, 13(9), e1006988 10.1371/journal.pgen.1006988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passioura, J. B. (1979). Accountability, philosophy and plant physiology. Search, 10, 347–350. [Google Scholar]
- Pellicer, J. , Hidalgo, O. , Dodsworth, S. , & Leitch, I. J. (2018). Genome size diversity and its impact on the evolution of land plants. Genes, 9(2), pii: E88 10.3390/genes9020088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi, E. (2018). Development cell by cell. Science, 362(6421), 1344–1345. 10.1126/science.362.6421.1344 [DOI] [PubMed] [Google Scholar]
- Pfeffer, S. , & Mahamid, J. (2018). Unravelling molecular complexity in structural cell biology. Current Opinion in Structural Biology, 52, 111–118. 10.1016/j.sbi.2018.08.009 [DOI] [PubMed] [Google Scholar]
- Poolman, M. G. , Miguet, L. , Sweetlove, L. J. , & Fell, D. A. (2009). A genome‐scale metabolic model of Arabidopsis and some of its properties. Plant Physiology, 151(3), 1570–1581. 10.1104/pp.109.141267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provart, N. J. , Alonso, J. , Assmann, S. M. , Bergmann, D. , Brady, S. M. , Brkljacic, J. , … McCourt, P. (2016). 50 years of Arabidopsis research: Highlights and future directions. New Phytologist, 209(3), 921–944. 10.1111/nph.13687 [DOI] [PubMed] [Google Scholar]
- Rellosa, M. C. , Reano, R. A. , Capilit, G. L. S. , de Guzman, F. C. , Ali, J. , Hamilton, N. R. S. , … Project, R. G. (2014). The 3,000 rice genomes project. Gigascience, 3, 7 10.1186/2047-217x-3-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinalducci, S. , Murgiano, L. , & Zolla, L. (2008). Redox proteomics: Basic principles and future perspectives for the detection of protein oxidation in plants. Journal of Experimental Botany, 59(14), 3781–3801. 10.1093/jxb/ern252 [DOI] [PubMed] [Google Scholar]
- Rosso, M. G. , Li, Y. , Strizhov, N. , Reiss, B. , Dekker, K. , & Weisshaar, B. (2003). An Arabidopsis thaliana T‐DNA mutagenized population (GABI‐Kat) for flanking sequence tag‐based reverse genetics. Plant Molecular Biology, 53(1–2), 247–259. 10.1023/B:PLAN.0000009297.37235.4a [DOI] [PubMed] [Google Scholar]
- Rowe, M. H. , & Bergmann, D. C. (2010). Complex signals for simple cells: The expanding ranks of signals and receptors guiding stomatal development. Current Opinion in Plant Biology, 13(5), 548–555. 10.1016/j.pbi.2010.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, K. H. , Huang, L. , Kang, H. M. , & Schiefelbein, J. (2019). Single‐cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiology. 179, 1444–1456. 10.1104/pp.18.01482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samodelov, S. L. , Beyer, H. M. , Guo, X. J. , Augustin, M. , Jia, K. P. , Baz, L. , … Zurbriggen, M. D. (2016). StrigoQuant: A genetically encoded biosensor for quantifying strigolactone activity and specificity. Science Advances, 2(11), e1601266 10.1126/sciadv.1601266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson, F. , Brunaud, V. , Balzergue, S. , Dubreucq, B. , Lepiniec, L. , Pelletier, G. , … Lecharny, A. (2002). FLAGdb/FST: A database of mapped flanking insertion sites (FSTs) of Arabidopsis thaliana T‐DNA transformants. Nucleic Acids Research, 30(1), 94–97. 10.1093/nar/30.1.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler, J. E. , & Stathopoulos, A. (2016). Stepwise progression of embryonic patterning. Trends in Genetics, 32(7), 432–443. 10.1016/j.tig.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, M. , Davison, T. S. , Henz, S. R. , Pape, U. J. , Demar, M. , Vingron, M. , … Lohmann, J. U. (2005). A gene expression map of Arabidopsis thaliana development. Nature Genetics, 37(5), 501–506. 10.1038/ng1543 [DOI] [PubMed] [Google Scholar]
- Schmidt, C. , & Urlaub, H. (2017). Combining cryo‐electron microscopy (cryo‐EM) and cross‐linking mass spectrometry (CX‐MS) for structural elucidation of large protein assemblies. Current Opinion in Structural Biology, 46, 157–168. 10.1016/j.sbi.2017.10.005 [DOI] [PubMed] [Google Scholar]
- Schmitz, R. J. , Schultz, M. D. , Urich, M. A. , Nery, J. R. , Pelizzola, M. , Libiger, O. , … Ecker, J. R. (2013). Patterns of population epigenomic diversity. Nature, 495(7440), 193–198. 10.1038/nature11968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser, B. , Busse, D. , Li, N. , Dittmar, G. , Schuchhardt, J. , Wolf, J. , … Selbach, M. (2011). Global quantification of mammalian gene expression control. Nature, 473(7347), 337–342. 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- Sessions, A. , Burke, E. , Presting, G. , Aux, G. , McElver, J. , Patton, D. , … Goff, S. A. (2002). A high‐throughput Arabidopsis reverse genetics system. Plant Cell, 14(12), 2985–2994. 10.1105/tpc.004630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimotohno, A. , Sotta, N. , Sato, T. , De Ruvo, M. , Maree, A. F. M. , Grieneisen, V. A. , & Fujiwara, T. (2015). Mathematical modeling and experimental validation of the spatial distribution of boron in the root of Arabidopsis thaliana identify high boron accumulation in the tip and predict a distinct root tip uptake function. Plant and Cell Physiology, 56(4), 620–630. 10.1093/pcp/pcv016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkova, E. , Hebert, A. S. , & Coon, J. J. (2016). Now, more than ever, proteomics needs better chromatography. Cell Systems, 3(4), 321–324. 10.1016/j.cels.2016.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton, B. Z. , Chory, E. J. , & Crabtree, G. R. (2018). Chemically induced proximity in biology and medicine. Science, 359(6380), pii: eaao5902 10.1126/science.aao5902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, Z. , Tang, Y. , Ritchey, L. E. , Tack, D. C. , Zhu, M. , Bevilacqua, P. C. , & Assmann, S. M. (2018). Genome‐wide RNA structurome reprogramming by acute heat shock globally regulates mRNA abundance. Proceedings of the National Academy of Sciences of the United States of America, 115(48), 12170–12175. 10.1073/pnas.1807988115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman, M. R. , Amasino, R. M. , Young, J. C. , Krysan, P. J. , & Austin‐Phillips, S. (2000). The Arabidopsis knockout facility at the University of Wisconsin‐Madison. Plant Physiology, 124(4), 1465–1467. 10.1104/pp.124.4.1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson, S. J. , Choi, W. G. , Chanoca, A. , & Gilroy, S. (2011). In vivo imaging of Ca2+, pH, and reactive oxygen species using fluorescent probes in plants. Annual Review of Plant Biology, 62(62), 273–297. 10.1146/annurev-arplant-042110-103832 [DOI] [PubMed] [Google Scholar]
- Torii, K. U. (2012). Mix‐and‐match: Ligand‐receptor pairs in stomatal development and beyond. Trends in Plant Science, 17(12), 711–719. 10.1016/j.tplants.2012.06.013 [DOI] [PubMed] [Google Scholar]
- Ulmasov, T. , Murfett, J. , Hagen, G. , & Guilfoyle, T. J. (1997). Aux/IAA proteins repress expression of reporter genes containing natural and highly active synthetic auxin response elements. Plant Cell, 9(11), 1963–1971. 10.1105/tpc.9.11.1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leene, J. , Hollunder, J. , Eeckhout, D. , Persiau, G. , Van de Slijke, E. , Stals, H. , … De Jaeger, G. (2010). Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana . Molecular Systems Biology, 6, 397 10.1038/msb.2010.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery, C. R. , La Clair, J. J. , Burkart, M. D. , & Noel, J. P. (2016). Harvesting the biosynthetic machineries that cultivate a variety of indispensable plant natural products. Current Opinion in Chemical Biology, 31, 66–73. 10.1016/j.cbpa.2016.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waadt, R. , Hitomi, K. , Nishimura, N. , Hitomi, C. , Adams, S. R. , Getzoff, E. D. , & Schroeder, J. I. (2014). FRET‐based reporters for the direct visualization of abscisic acid concentration changes and distribution in Arabidopsis. eLife, 3, e01739 10.7554/eLife.01739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. S. , Mauleon, R. , Hu, Z. Q. , Chebotarov, D. , Tai, S. S. , Wu, Z. C. , … Leung, H. (2018). Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature, 557(7703), 43–49. 10.1038/s41586-018-0063-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch, M. T. , Yang, A. , Albu, M. , Cote, A. G. , Montenegro‐Montero, A. , Drewe, P. , … Hughes, T. R. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell, 158(6), 1431–1443. 10.1016/j.cell.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wend, S. , Dal Bosco, C. , Kampf, M. M. , Ren, F. G. , Palme, K. , Weber, W. , … Zurbriggen, M. D. (2013). A quantitative ratiometric sensor for time‐resolved analysis of auxin dynamics. Scientific Reports, 3, 2052 10.1038/srep02052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng, J. K. , & Noel, J. P. (2012). The remarkable pliability and promiscuity of specialized metabolism. Cold Spring Harbor Symposia on Quantitative Biology, 77, 309–320. 10.1101/sqb.2012.77.014787 [DOI] [PubMed] [Google Scholar]
- Wienkoop, S. , Zoeller, D. , Ebert, B. , Simon‐Rosin, U. , Fisahn, J. , Glinski, M. , & Weckwerth, W. (2004). Cell‐specific protein profiling in Arabidopsis thaliana trichomes: Identification of trichome‐located proteins involved in sulfur metabolism and detoxification. Phytochemistry, 65(11), 1641–1649. 10.1016/j.phytochem.2004.03.026 [DOI] [PubMed] [Google Scholar]
- Wright, G. A. , Baker, D. D. , Palmer, M. J. , Stabler, D. , Mustard, J. A. , Power, E. F. , … Stevenson, P. C. (2013). Caffeine in floral nectar enhances a pollinator's memory of reward. Science, 339(6124), 1202–1204. 10.1126/science.1228806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, K. , Lim, J. , Dale, J. M. , Chen, H. M. , Shinn, P. , Palm, C. J. , … Ecker, J. R. (2003). Empirical analysis of transcriptional activity in the Arabidopsis genome. Science, 302(5646), 842–846. 10.1126/science.1088305 [DOI] [PubMed] [Google Scholar]
- Yang, J. , Carroll, K. S. , & Liebler, D. C. (2016). The expanding landscape of the thiol redox proteome. Molecular & Cellular Proteomics, 15(1), 1–11. 10.1074/mcp.O115.056051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeats, T. H. , & Rose, J. K. C. (2013). The formation and function of plant cuticles. Plant Physiology, 163(1), 5–20. 10.1104/pp.113.222737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, G. C. , Cai, L. , Elowitz, M. , Enver, T. , Fan, G. P. , Guo, G. J. , … Tirosh, I. (2017). Challenges and emerging directions in single‐cell analysis. Genome Biology, 18, 84 10.1186/s13059-017-1218-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. Y. , Clarenz, O. , Cokus, S. , Bernatavichute, Y. V. , Pellegrini, M. , Goodrich, J. , & Jacobsen, S. E. (2007). Whole‐genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis . PLoS Biology, 5(5), 1026–1035. 10.1371/journal.pbio.0050129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Gu, L. , Hou, Y. , Wang, L. , Deng, X. , Hang, R. , … Cao, X. (2015). Integrative genome‐wide analysis reveals HLP1, a novel RNA‐binding protein, regulates plant flowering by targeting alternative polyadenylation. Cell Research, 25(7), 864–876. 10.1038/cr.2015.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. Y. , Yazaki, J. , Sundaresan, A. , Cokus, S. , Chan, S. W. L. , Chen, H. M. , … Ecker, J. R. (2006). Genome‐wide high‐resolution mapping and functional analysis of DNA methylation in Arabidopsis . Cell, 126(6), 1189–1201. 10.1016/j.cell.2006.08.003 [DOI] [PubMed] [Google Scholar]
- Zhang, W. L. , Zhang, T. , Wu, Y. F. , & Jiang, J. M. (2012). Genome‐wide identification of regulatory DNA elements and protein‐binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell, 24(7), 2719–2731. 10.1105/tpc.112.098061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, M. , & Wang, W. (2018). Recent advances in synthetic chemical inducers of plant immunity. Frontiers in Plant Science, 9, 1613 . 10.3389/fpls.2018.01613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmienko, A. , Samelak, A. , Kozlowski, P. , & Figlerowicz, M. (2014). Copy number polymorphism in plant genomes. Theoretical and Applied Genetics, 127(1), 1–18. 10.1007/s00122-013-2177-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurcher, E. , Tavor‐Deslex, D. , Lituiev, D. , Enkerli, K. , Tarr, P. T. , & Muller, B. (2013). A robust and sensitive synthetic sensor to monitor the transcriptional output of the cytokinin signaling network in planta. Plant Physiology, 161(3), 1066–1075. 10.1104/pp.112.211763 [DOI] [PMC free article] [PubMed] [Google Scholar]