

Abstract
Targeting immune checkpoints, such as PD‐L1 and its receptor PD‐1, has opened a new avenue for treating cancers. Understanding the regulatory mechanism of PD‐L1 and PD‐1 will improve the clinical response rate and efficacy of PD‐1/PD‐L1 blockade in cancer patients and the development of combinatorial strategies. VGLL4 inhibits YAP‐induced cell proliferation and tumorigenesis through competition with YAP for binding to TEADs. However, whether VGLL4 has a role in anti‐tumor immunity is largely unknown. Here, we found that disruption of Vgll4 results in potent T cell‐mediated tumor regression in murine syngeneic models. VGLL4 deficiency reduces PD‐L1 expression in tumor cells. VGLL4 interacts with IRF2BP2 and promotes its protein stability through inhibiting proteasome‐mediated protein degradation. Loss of IRF2BP2 results in persistent binding of IRF2, a transcriptional repressor, to PD‐L1 promoter. In addition, YAP inhibits IFNγ‐inducible PD‐L1 expression partially through suppressing the expression of VGLL4 and IRF1 by YAP target gene miR‐130a. Our study identifies VGLL4 as an important regulator of PD‐L1 expression and highlights a central role of VGLL4 and YAP in the regulation of tumor immunity.
Keywords: cancer immunity, Hippo, murine syngeneic tumor model, PD‐L1, VGLL4
Subject Categories: Cancer, Immunology
Introduction
Evasion of immune surveillance is a hallmark of cancer, which enables tumor cells to escape the attack from immune cells. Accumulating evidence has revealed that the interaction between the tumor cells and the host microenvironment, in particular the infiltrating immune cells, is critical for disease progression, metastasis, and relapse. The recent breakthroughs in the discovery of cancer immune checkpoints such as PD‐L1 (also known as B7‐H1 or CD274) and the success of checkpoint inhibitors in stimulating anti‐tumor immune response (Miller & Sadelain, 2015) have opened an avenue in the understanding of tumor immunology and the development of new strategy for cancer therapies.
Expression of PD‐L1 is an immunosuppressive mechanism by which cancer cells suppress the immune system. The binding of PD‐L1 to its receptor PD‐1 (Dong et al, 1999; Freeman et al, 2000) leads to the inhibition of T lymphocyte proliferation, cytokine production, cytolytic activity, and the suppression of the body's immune response. Many studies reported that the dysregulation of several oncogenic or tumor‐suppressive pathways constitutively activates the expression of PD‐L1, suggesting that this is a general mechanism of tumorigenesis. PTEN deletions, PI3K/AKT mutations (Parsa et al, 2007; Lastwika et al, 2016), EGFR mutations (Akbay et al, 2013), MYC overexpression (Casey et al, 2016), CDK5 disruption (Dorand et al, 2016), and YAP/TAZ activation (Feng et al, 2017; Lee et al, 2017; Miao et al, 2017; Janse van Rensburg et al, 2018; Kim et al, 2018) represent a rapidly growing list of genetic mechanisms of constitutive PD‐L1 expression. Expression of PD‐L1 can be induced by many cytokines, of which interferon‐gamma (IFNγ) is the most potent via JAK1/2‐STAT1/2/3‐IRF1 signaling axis, with IRF1 binding to PD‐L1 promoter (Dong et al, 2002; Loke & Allison, 2003; Lee et al, 2006; Garcia‐Diaz et al, 2017). The IFNγ‐inducible expression of PD‐L1 is more common than the constitutive expression in most cancers and can be detected as a patchy pattern of PD‐L1 expression in T cell‐rich areas of tumors, in particular at the invasive margin (Taube et al, 2012; Tumeh et al, 2014). Despite substantial clinical benefit, many patients with cancer fail to respond to therapies that target the PD‐1 and PD‐L1 interaction as a single agent (Zou et al, 2016). Thus, a better understanding of the mechanisms of regulation of PD‐1/PD‐L1 pathway in tumor microenvironment will improve the clinical response rate and efficacy of PD‐1/PD‐L1 blockade in cancer patients, and the development of combinatorial strategies for treating cancers.
Hippo pathway is first discovered in Drosophila and involved in organ‐size control, tissue homeostasis, and tumorigenesis. The conserved Hippo signaling is composed of a kinase cascade that controls the activity of the transcriptional coactivators, YAP and TAZ, by the kinases MST1/2 and LATS1/2 (Yu et al, 2015; Meng et al, 2016) to govern multiple aspects of cell physiology. When Hippo signaling is inactivated, YAP/TAZ enter the nucleus, bind to the transcription factor TEADs, and induce gene transcription. Recently, several studies identified VGLL4 as a transcriptional suppressor that inhibits YAP‐induced overgrowth and tumorigenesis through direct competition with YAP for binding to TEADs (Guo et al, 2013; Koontz et al, 2013; Jiao et al, 2014; Zhang et al, 2014). Previous studies have convincingly demonstrated the Hippo pathway as a suppressor signal for cellular transformation and tumorigenesis (Moroishi et al, 2015a). Recently, the effects of the Hippo pathway components on tumor initiation and growth in the context of reciprocal interactions between tumor cells and host anti‐tumor immune responses have emerged (Moroishi et al, 2016; Guo et al, 2017; Kim et al, 2017; Zhang et al, 2017b). However, the underlying mechanisms of how Hippo pathway cooperates with inflammatory signaling to regulate tumor microenvironment remain elusive.
In the present study, we investigated the role of VGLL4 in the context of anti‐tumor immunity. Surprisingly, inactivation of VGLL4 in murine tumor cells strongly suppresses tumor growth in immune‐competent mice due to the attenuation of tumor immune evasion. Our data indicate a new paradigm for how tumor immunogenicity is regulated through VGLL4 and YAP in tumor cells.
Results
Loss of Vgll4 inhibits murine tumor growth in syngeneic mice
To elucidate the role of Vgll4 in anti‐tumor immunity, we took advantage of murine syngeneic tumor models which comprise intact immune system. The syngeneic tumor model has been widely used to study reciprocal interactions between tumor cells and anti‐tumor immune responses. First, we assayed VGLL4 protein levels in a number of murine cancer cells. We found that VGLL4 protein levels were high in LLC lung cancer and MB49 bladder cancer cells, and low in 4T‐1 breast cancer and B16F10 melanoma cells (Fig 1A). Therefore, we inactivated Vgll4 in LLC and MB49 cells using shRNA‐mediated gene knockdown. The efficiency of knockdown was verified by immunoblot (Fig 1B). Knockdown of Vgll4 did not affect cell proliferation in cell cultures (Fig EV1A and B). To investigate the role of Vgll4 in tumor growth in vivo, we subcutaneously transplanted equal numbers of WT‐ or Vgll4‐knockdown LLC cells into the back flanks of C57BL/6 mice and monitored their growth. Surprisingly, downregulation of Vgll4 in LLC cells strongly inhibited tumor growth in vivo. All mice developed large tumors in the control group 21 days after inoculation, whereas injection of Vgll4‐knockdown LLC cells resulted in much smaller tumors (Figs 1C and D, and EV1C). However, Vgll4‐knockdown LLC tumors grew similar as control cells in immune‐compromised nude mice (Figs 1E and F, and EV1D). Next, we examined the tumor growth of Vgll4‐knockdown MB49 cells in C57BL/6 mice and nude mice. Consistently, control MB49 cells developed tumors in both C57BL/6 mice and nude mice (Figs 1G–J, and EV1E and F). However, Vgll4‐knockdown MB49 cells only developed tumors in nude mice, but not in C57BL/6 mice (Figs 1G–J, and EV1E and F). Thus, collectively, our observations indicate that loss of Vgll4 in tumor cells significantly inhibits the tumor growth in murine syngeneic tumor models.
Figure 1. Loss of Vgll4 in tumor cells inhibits tumor growth in syngeneic mouse tumor models.
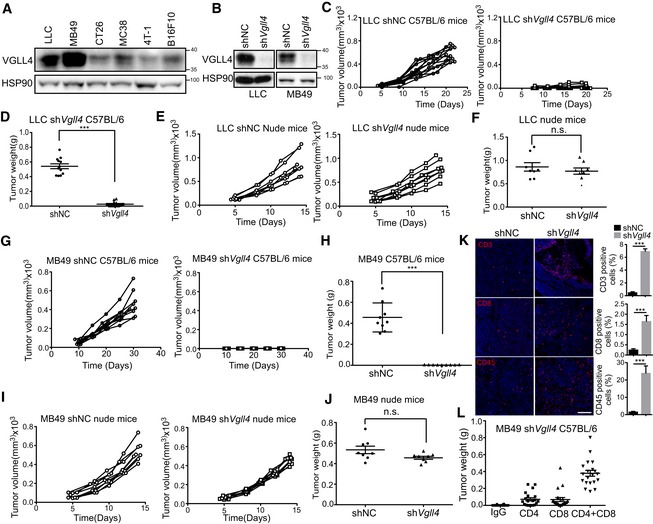
- VGLL4 protein expression levels in six murine tumor cell lines. Total cell lysates were analyzed by Western blot.
- VGLL4 protein levels were reduced in Vgll4‐depleted cells. Total cell lysates of Vgll4‐knockdown LLC and MB49 cells were analyzed by Western blot.
- Control or shVgll4 LLC cells were transplanted into C57BL/6 mice, and tumor‐growth kinetics was measured at the indicated times. n = 12 tumors for each group.
- Quantification of tumor weights 21 days after transplantation of control or shVgll4 LLC cells into C57BL/6 mice from (C). n = 12 tumors for each group. ***P < 0.001, two‐tailed Student's t‐test. The solid line represents the average weight ± SEM.
- Control or shVgll4 LLC cells were transplanted into nude mice, and tumor‐growth kinetics was measured at the indicated times. n = 8 tumors for each group.
- Quantification of tumor weights 15 days after transplantation of control or shVgll4 LLC cells into nude mice from (E). n = 8 tumors for each group. n.s. P > 0.05, two‐tailed Student's t‐test. The solid line represents the average weight ± SEM.
- Control or shVgll4 MB49 cells were transplanted into C57BL/6 mice, and tumor‐growth kinetics was measured at the indicated times. n = 9 tumors for each group.
- Quantification of tumor weights 30 days after transplantation of control or shVgll4 MB49 cells into C57BL/6 mice. n = 9 tumors for each group. ***P < 0.001, two‐tailed Student's t‐test. The solid line represents the average weight ± SEM.
- Control or shVgll4 MB49 cells were transplanted into nude mice, and tumor‐growth kinetics was measured at the indicated times. n = 8 tumors for each group.
- Quantification of tumor weights 15 days after transplantation of control or shVgll4 MB49 cells into nude mice from (I). n = 8 tumors for each group. n.s. P > 0.05; two‐tailed Student's t‐test. The solid line represents the average weight ± SEM.
- Immunofluorescent staining of CD3, CD8, and CD45 (red) counterstained with DAPI for DNA (blue) using control or shVgll4 LLC tumors from C57BL/6 mice. Statistical analysis of the percentage of CD3‐, CD8‐, and CD45‐positive cells in the tumors is shown in the right panel, respectively. n = 3 tumors for each group. ***P < 0.001, two‐tailed Student's t‐test, mean ± SEM. Scale bar 100 μm.
- Quantification of tumor weights 30 days after transplantation of shVgll4 MB49 cells into C57BL/6 mice receiving anti‐CD4 alone, anti‐CD8 alone, or both anti‐CD4 and anti‐CD8 blocking antibodies. n = 20 tumors for each group. The solid line represents the average weight ± SEM.
Source data are available online for this figure.
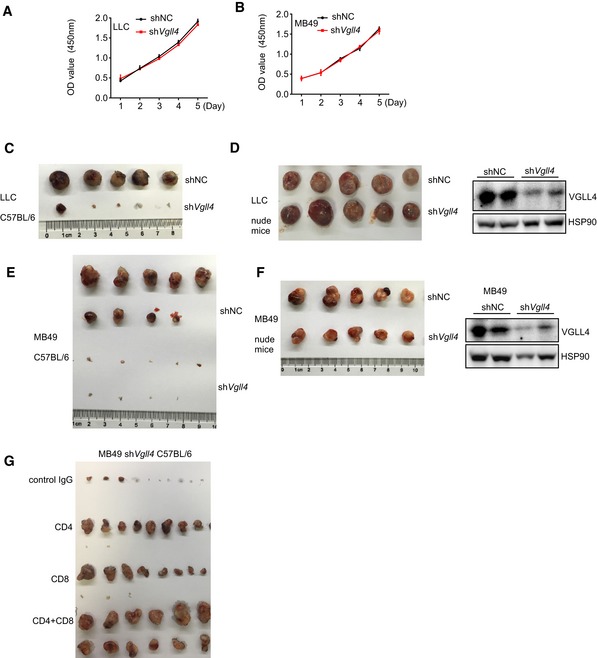
Figure EV1. Loss of Vgll4 in tumor cells does not affect cell proliferation, but inhibits tumor growth in syngeneic mouse model.
-
A, BshVgll4 LLC (A) and MB49 (B) cells grew similar as control cells in regular cell cultures using CCK8 assay. n = 3, mean ± SEM.
-
CThe gross appearance of the primary tumors of control or shVgll4 LLC from C57BL/6 mice.
-
DThe gross appearance of the primary tumors of control or shVgll4 LLC from nude mice. VGLL4 protein expression levels in the tumor lysates from nude mice were analyzed by Western blot and shown in the right panel.
-
EThe gross appearance of the primary tumors of control or shVgll4 MB49 from C57BL/6 mice.
-
FThe gross appearance of the primary tumors of control or shVgll4 MB49 from nude mice. VGLL4 protein expression levels in the tumor lysates from nude mice were analyzed by Western blot and shown in the right panel.
-
GThe gross appearance of the primary shVgll4 MB49 tumors from C57BL/6 mice receiving anti‐CD4 alone, anti‐CD8 alone, or both anti‐CD4 and anti‐CD8 blocking antibodies.
Source data are available online for this figure.
Since Vgll4 loss displays completely opposite effects on tumor growth in immune‐compromised vs. immune‐competent mice, we speculated that host immune factors may contribute to the apparent discrepancy of Vgll4‐knockdown tumor cells. Therefore, we examined the histopathology of control and Vgll4‐knockdown LLC tumors from C57BL/6 mice. We found massive infiltration of inflammatory cells in Vgll4‐knockdown LLC tumors, which were shown by staining with the leukocyte marker CD45 and the T‐cell markers CD3 and CD8 (Fig 1K). To identify T‐cell populations mediating this potent rejection, we depleted CD8+ T cells, CD4+ T cells, or both subsets in mice bearing with Vgll4‐knockdown MB49 cells. By day 31, 100% of mice injected with Vgll4‐knockdown MB49 cells had much smaller tumors, and mice receiving CD4 or CD8 blocking antibodies developed measurable but smaller tumors, whereas depletion of both CD8+ and CD4+ T cells yielded much larger tumor formation (Figs 1L and EV1G). Together, these studies suggest a T cell‐dependent rejection and robust anti‐tumor immunity of Vgll4‐deficient tumors.
Loss of VGLL4 decreases the expression of PD‐L1 in tumor cells
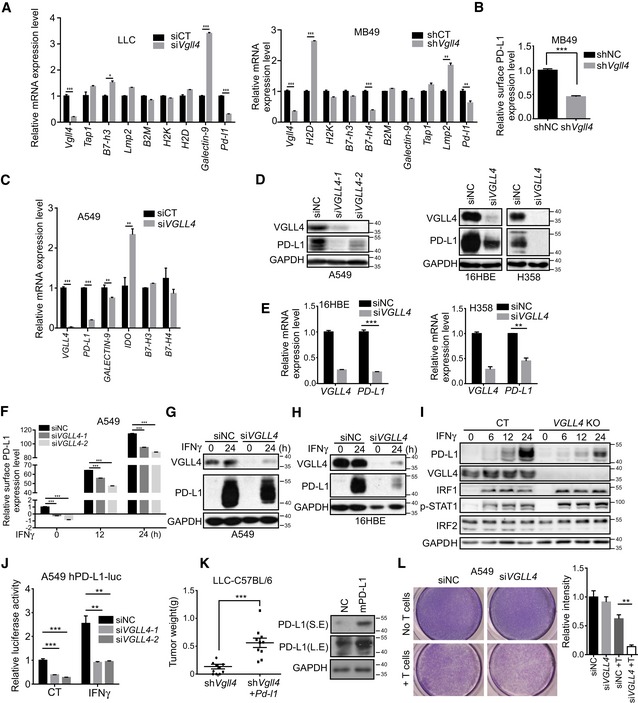
A major feature of immune evasion of cancer cells is the expression of multiple immune modulators, such as PD‐L1. We sought to test whether disruption of VGLL4 expression in cells impaired PD‐L1 expression. We examined the expression of PD‐L1 and other immune modulators in Vgll4‐knockdown LLC and MB49 cells. We observed that knockdown of Vgll4 significantly attenuated both PD‐L1 mRNA and cell surface expression using qRT–PCR and flow cytometry analysis, respectively (Figs 2A and B, and EV2A). Since VGLL4 has been shown to play a role in the tumorigenesis of lung cancer, we examined the PD‐L1 expression in several human lung cancer cell lines and human bronchial epithelial 16HBE cells when VGLL4 expression was inhibited by siRNA‐mediated gene knockdown. Notably, the mRNA levels of PD‐L1 were decreased in parallel with its protein levels after loss of VGLL4 expression in these cell lines (Fig 2C–E), indicating a transcriptional control of PD‐L1 expression by VGLL4. This finding was further supported by the reduced cell surface PD‐L1 expression through flow cytometry analysis in A549 cells (Figs 2F and EV2B). In addition, knockdown of VGLL4 increased the expression of YAP/TEAD target genes in several lung cancer cells (Fig EV2C), which is consistent with previous studies (Zhang et al, 2014). Notably, removal of VGLL3, which is highly expressed in A549 cells, had no effect on the expression of PD‐L1 in A549 cells (Fig EV2D). IFNγ expressed by infiltrating immune cells is known to potently induce PD‐L1 expression. We examined whether disruption of VGLL4 impaired PD‐L1 induction in response to IFNγ stimulation. We found that IFNγ induced less PD‐L1 expression in VGLL4‐knockdown cells compared to control A549 and 16HBE cells by Western blot analysis (Fig 2G and H). Moreover, we generated knockout cells for VGLL4 using CRISPR/Cas9. Consistent with siRNA‐mediated VGLL4 knockdown in A549, VGLL4‐knockout A549 cells also produced less PD‐L1 upon IFNγ stimulation (Fig 2I). To further analyze the effect of VGLL4 on PD‐L1 transcription, a 340‐bp fragment of the PD‐L1 promoter was cloned into a luciferase reporter plasmid. Consistently, the PD‐L1 promoter activity was significantly reduced in both INFγ‐untreated and INFγ‐treated VGLL4‐knockdown A549 cells compared to control cells (Fig 2J), indicating that VGLL4 expression levels correlate with PD‐L1 expression levels.
Figure 2. Disruption of VGLL4 expression suppresses PD‐L1 expression.
-
ARelative mRNA levels of immune‐related genes in Vgll4‐knockdown LLC or MB49 cells by qRT–PCR analysis. n = 3, mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, two‐tailed Student's t‐test.
-
BKnockdown of Vgll4 in MB49 cells decreased the surface expression of PD‐L1 protein on control or shVgll4 MB49 cells measured by flow cytometry analysis with anti‐mPD‐L1 antibody. n = 3, mean ± SEM. ***P < 0.001, two‐tailed Student's t‐test.
-
CRelative mRNA levels of immune‐related genes in Vgll4‐knockdown A549 cells by qRT–PCR analysis. n = 3, mean ± SEM. **P < 0.01, ***P < 0.001, two‐tailed Student's t‐test.
-
DImmunoblot analysis revealed the reduced expression of PD‐L1 protein in VGLL4‐knockdown A549, 16HBE, and H358 cells compared with control cells.
-
EqRT–PCR analysis revealed a reduction in PD‐L1 mRNA levels in VGLL4‐knockdown 16HBE and H358 cells by siRNA. n = 3, mean ± SEM. **P < 0.01, ***P < 0.001, two‐tailed Student's t‐test.
-
FFlow cytometry analysis of surface PD‐L1 levels in control and VGLL4‐knockdown A549 cells treated with IFNγ for the indicated times. n = 3, mean ± SEM. ***P < 0.001, one‐way ANOVA followed by Dunnett's test.
-
G, HImmunoblot analysis revealed the reduced IFNγ‐inducible PD‐L1 expression in VGLL4‐knockdown A549 (G) and 16HBE (H) cells.
-
IImmunoblot analysis of total cell lysates with the indicated antibodies revealed the attenuated IFNγ‐inducible PD‐L1 expression in VGLL4‐knockout (KO) A549 cells generated by CRISPR/Cas9.
-
JLoss of VGLL4 dampened the activity of PD‐L1 promoter reporter in a luciferase assay. A549 cells were transfected with human PD‐L1‐promoter luciferase plasmid and indicated siRNA, treated with IFNγ for 12 h, and subjected to a luciferase assay. n = 3, mean ± SEM. **P < 0.01, ***P < 0.001, one‐way ANOVA followed by Dunnett's test.
-
KOverexpression of mouse PD‐L1 in Vgll4‐knockdown LLC cells restored the tumor growth in C57BL/6 mice. Tumor weights of shVgll4 or shVgll4‐lenti‐mPD‐L1 LLC cells were determined 21 days after tumor cell transplantation into C57BL/6 mice (left panel). Expression of mPD‐L1 is shown in the right panel by immunoblot analysis with anti‐mPD‐L1 antibody. n = 10 tumors for each group. The solid line represents the average weight ± SEM. ***P < 0.001, two‐tailed Student's t‐test.
-
LKnockdown of VGLL4 in A549 cells enhances T cell‐mediated tumor cell killing. Activated T cells and A549 cells were co‐cultured in 24‐well plates for 4 days, and then, surviving tumor cells were visualized by crystal violet staining. Relative fold ratios of surviving cell intensities are shown in right panel. n = 3, mean ± SEM, **P < 0.01, two‐tailed Student's t‐test.
Source data are available online for this figure.
Figure EV2. Disruption of VGLL4 expression suppresses PD‐L1 expression.
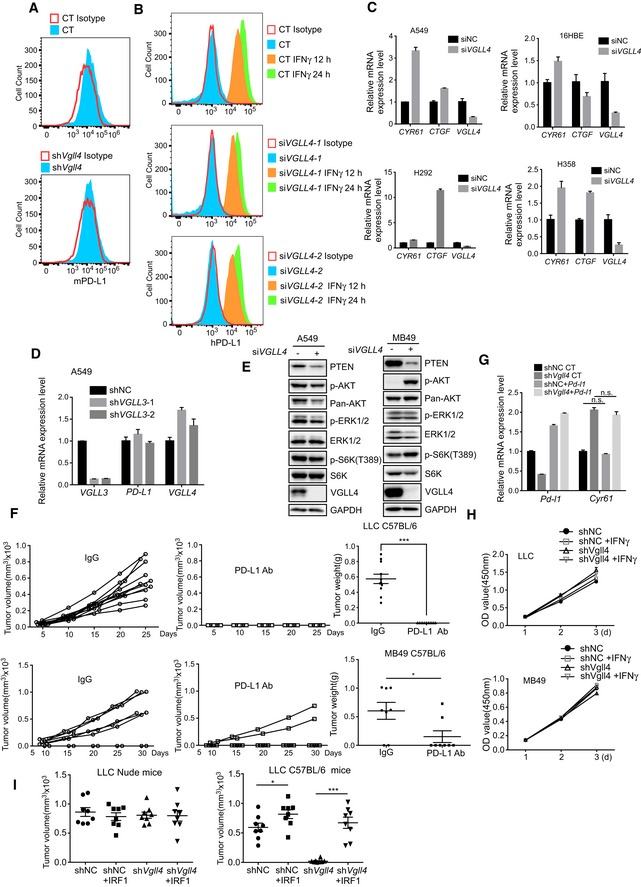
- Cell surface PD‐L1 expression is analyzed by flow cytometry in MB49 cells transfected with control or siVgll4 siRNA and stained with PE‐conjugated anti‐mouse PD‐L1 antibody or isotype control antibody.
- Cell surface PD‐L1 expression is analyzed by flow cytometry on the IFNγ‐stimulated A549 cells transfected with control or siVgll4 siRNA and stained with APC‐conjugated anti‐human PD‐L1 antibody or isotype control antibody.
- Knockdown of VGLL4 by siRNA increases the expression of YAP target genes. The levels of CTGF and CYR61 were determined by qRT–PCR. n = 3, mean ± SEM.
- Knockdown of VGLL3 does not affect PD‐L1 expression in A549 cells. PD‐L1 mRNA levels were analyzed by qRT–PCR in A549 cells transfected with control or siVGLL3 siRNA. n = 3, mean ± SEM.
- Depletion of VGLL4 results in reduced PTEN protein levels. A549 or MB 49 cells transfected with control or siVGLL4 siRNA were analyzed with indicated antibodies by Western blot.
- LLC and MB49 tumors respond to PD‐L1 blocking antibody treatment. C57BL/6 mice inoculated with LLC (n = 11 tumors) or MB49 (n = 8 tumors) tumor cells were administrated with anti‐mouse PD‐L1 antibody every 3 days. Tumor‐growth kinetics was measured at the indicated times. mean ± SEM. *P < 0.05, ***P < 0.001, two‐tailed Student's t‐test.
- Expression of PD‐L1 in Vgll4‐knockdown LLC tumors does not affect YAP target gene expression. Cyr61 mRNA expression levels were analyzed in control or Vgll4‐knockdown PD‐L1‐overexpression LLC tumors by qRT–PCR. n = 3 tumors, mean ± SEM, n.s. P > 0.05. One‐way ANOVA followed by Tukey's test.
- Analysis of proliferation rate of LLC and MB49 cells treated with IFNγ. Control, and Vgll4‐knockdown LLC or MB49 cells were treated with IFNγ, and the proliferation kinetics was measured by CCK8 assay. n = 3, mean ± SEM.
- Expression of IRF1 in Vgll4‐knockdown LLC tumors restored the tumor growth in C57BL/6 mice. Tumor volumes were measured 15 days for nude mice and 21 days for C57BL/6 mice after LLC tumor cell inoculation. n = 8 tumors for each group. *P < 0.05, ***P < 0.001, two‐tailed Student's t‐test. The solid line represents the average volume ± SEM.
Source data are available online for this figure.
In addition to the PD‐L1 expression, we also examined several pathways related to the regulation of PD‐L1 expression in VGLL4‐knockdown tumor cells. Of note, we found that PTEN protein levels were reduced in VGLL4‐knockdown A549 and MB49 cells (Fig EV2E). The significance of this finding was not addressed in this study. To establish whether there is a functional link between PD‐L1 expression and rejection of Vgll4‐deficient tumor cells in murine syngeneic tumor models, we tested whether expression of mouse PD‐L1 could rescue the tumor growth of Vgll4‐knockdown LLC cells in immune‐competent mice. We first showed that mice bearing WT LLC or MB49 tumor cells responded effectively to anti‐PD‐L1 antibody treatment (Fig EV2F). Next, we stably overexpressed mouse PD‐L1 in Vgll4‐knockdown LLC cells and transplanted them into C57BL/6 mice. As expected, expression of PD‐L1 restored the tumorigenesis potential of Vgll4‐knockdown LLC cells (Fig 2K). We also showed that the expression of PD‐L1 did not affect YAP target gene expression (Fig EV2G) and IFNγ treatment had no effect on the proliferation of control and Vgll4‐knockdown LLC and MB49 cells in vitro (Fig EV2H). Furthermore, the expression of IRF1, the major transcriptional factor for PD‐L1 expression, also restored the growth of Vgll4‐knockdown LLC tumors in C57BL/6 mice (Fig EV2I). In addition, knockdown of VGLL4 in A549 cells enhanced the T cell‐mediated cancer cell killing in vitro (Fig 2L). Together, these data suggest that loss of VGLL4 suppresses PD‐L1 expression in tumor cells, leading to the establishment of anti‐tumor immunity.
VGLL4 interacts with IRF2BP2 independent of TDU domains
IFNγ is known to be the major cytokine to induce PD‐L1 expression through JAK1/2‐STAT1/2/3‐IRF1 axis (Garcia‐Diaz et al, 2017). IRF2 is generally considered as a repressor to suppress IRF1 activity (Harada et al, 1989). Interestingly, one study showed that IRF2BP2, an IRF2‐binding protein, interacts with VGLL4 (Teng et al, 2010). This promoted us to investigate the possibility that VGLL4 might regulate PD‐L1 expression through modulating IRF2/IRF2BP2 complex. First, we confirmed physical interaction between VGLL4 and IRF2BP2 by immunoprecipitation of endogenous VGLL4 proteins in A549 cells (Fig 3A). Furthermore, epitope‐tagged VGLL4 expressed in HEK293T cells efficiently immunoprecipitated endogenous IRF2BP2 proteins (Fig 3B). Moreover, the interaction between IRF2BP1 (a homolog of IRF2BP2) and VGLL4 was also observed (Fig 3C), suggesting that VGLL4 interacts with the conserved motif shared by IRF2BP2 and IRF2BP1. Immunoprecipitation assay using serial IRF2BP2 truncations revealed that C‐terminus of IRF2BP2 is responsible for the interaction between VGLL4 and IRF2BP2 (Fig 3D).
Figure 3. VGLL4 interacts with IRF2BP2 independent of TDU domains.
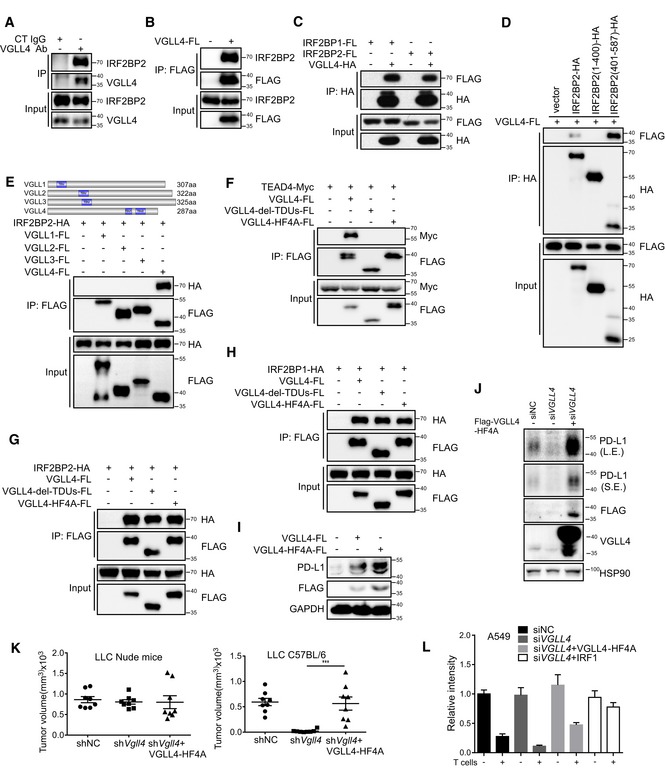
-
AThe endogenous complex of VGLL4 and IRF2BP2 was detected by immunoprecipitation using anti‐VGLL4 antibody and visualized by anti‐IRF2BP2 antibody in A549 cells.
-
BThe endogenous IRF2BP2 was immunoprecipitated by VGLL4‐FLAG proteins in HEK293T cells.
-
CInteraction between VGLL4 and IRF2BP1 or IRF2BP2 was revealed by co‐immunoprecipitation of differentially tagged proteins in HEK293T cells.
-
DC‐terminus of IRF2BP2 interacted with VGLL4 by co‐immunoprecipitation assay in HEK293T cells.
-
EInteraction of IRF2BP2 with VGLL1‐4 was revealed by co‐immunoprecipitation of differentially tagged proteins in HEK293T cells.
-
FTDU domains in VGLL4 are required for the interaction with TEADs by co‐immunoprecipitation assay in HEK293T cells.
-
G, HTDU‐deleted or HF4A VGLL4 mutations interacted with IRF2BP2 (G) and IRF2BP1 (H) by co‐immunoprecipitation analysis in HEK293T cells.
-
IExpression of VGLL4‐WT or VGLL4‐HF4A enhances PD‐L1 expression in A549 cells by immunoblot analysis. A549 cells were transfected with indicated plasmids and subjected to immunoblot with indicated antibodies.
-
JVGLL4‐HF4A rescues PD‐L1 expression in VGLL4‐knockdown A549 cells. A549 cells were transfected with siRNA targeting to the 3′UTR of VGLL4 mRNA and followed by transduction with lenti‐VGLL4‐HF4A virus. Cell lysates were analyzed by Western blot with indicated antibodies.
-
LExpression of VGLL4‐HF4A rescues tumor growth of VGLL4‐knockdown LLC cells in C57BL/6 mice. Control, Vgll4 knockdown, or Vgll4 knockdown together with VGLL4‐HF4A‐overexpressing LLC cells were transplanted into nude mice or C57BL/6 mice. Tumor volumes were measured 15 days for nude mice and 21 days for C57BL/6 mice after tumor cell inoculation. n = 8 tumors for each group. ***P < 0.001, two‐tailed Student's t‐test. The solid line represents the average volume ± SEM.
-
MExpression of VGLL4‐HF4A attenuates the T cell‐mediated tumor cell killing. Activated T cells and A549 cells were co‐cultured in 24‐well plates for 4 days, and relative fold ratios of surviving cells are shown by measuring the intensities of surviving cells stained with crystal violet. n = 3, mean ± SEM.
Source data are available online for this figure.
The mammalian genome encodes four Vestigial‐like proteins, VGLL1‐4, all of them containing Tondu (TDU) domain. Among them, VGLL4 is the only protein which can interact with IRF2BP2 in immunoprecipitation assay (Fig 3E). This result suggested that TDU domains in VGLL4 may not be required for the interaction with IRF2BP2. To test this, we generated two VGLL4 mutants, ΔTDUs and HF4A (H212A/F213A/H240A/F241A; Jiao et al, 2017), both of which have been shown to lose the ability to interact with the YAP‐binding domain of TEADs. Consistent with previous findings, deletion of TDU domains or HF4A mutation in VGLL4 abolished the interaction between VGLL4 and TEAD4 (Fig 3F). Importantly, both of the VGLL4 mutants still interacted with IRF2BP2 and IRF2BP1 (Fig 3G and H), suggesting the non‐overlap interaction domains in VGLL4 required for interaction with TEADs and IRF2BP2. To follow this observation, we tested whether TEADs are involved in VGLL4‐mediated PD‐L1 regulation. As shown in Fig 3I, both VGLL4‐WT and VGLL4‐HF4A promoted the expression of PD‐L1 in A549 cells and VGLL4‐HF4A displayed a higher activity. Because VGLL4‐WT, but not VGLL4‐HF4A, significantly suppressed the proliferation of A549 cells (Fig EV3A), we sought to test whether VGLL4‐HF4A could restore the expression of PD‐L1 in VGLL4‐knockdown A549 cells, and the growth of Vgll4‐knockdown mouse tumors. We found that the expression of VGLL4‐HF4A markedly promoted the PD‐L1 expression in VGLL4‐knockdown A549 cells (Fig 3J) and the growth of Vgll4‐knockdown LLC tumors in C57BL/6 mice (Fig 3K). In addition, the expression of VGLL4‐HF4A promoted the growth of B16F10 tumors in C57BL/6 mice (Fig EV3B) and attenuated the T cell‐mediated A549 tumor cell killing in vitro (Figs 3L and EV3C). Together, these results indicate that VGLL4 interacts with IRF2BP2, and that TDU domains in VGLL4 are not required for the interaction with IRF2BP2 and the regulation of PD‐L1 expression.
Figure EV3. VGLL4‐HF4A rescues the defects of VGLL4‐knockdown tumor cells.
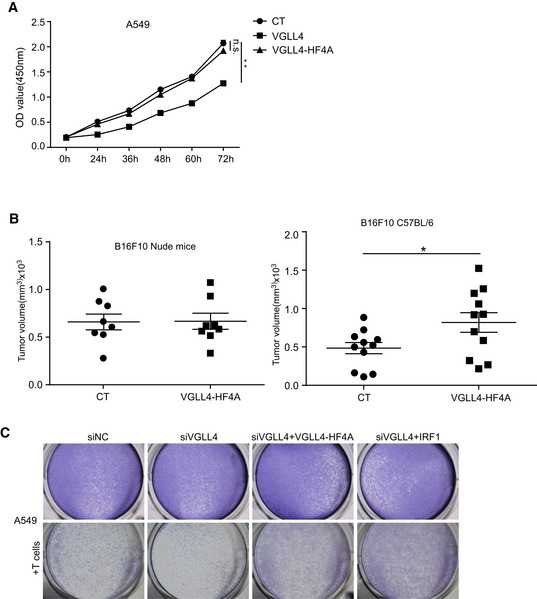
- VGLL4 suppresses A549 cell growth in vitro. The proliferation of A549 cells with VGLL4 or VGLL4‐HF4A expression was detected by CCK8 assay. n = 3, mean ± SEM, **P < 0.01, one‐way ANOVA followed by Dunnett's test.
- Overexpression of VGLL4‐HF4A promotes B16F10 tumor growth in C57BL/6 mice. Control or VGLL4‐HF4A‐overexpressing B16F10 cells were transplanted into nude mice (n = 8 tumors each group) or C57BL/6 (n = 11 tumors each group) mice, and tumor volumes were measured 21 days after tumor cell inoculation. *P < 0.05, two‐tailed Student's t‐test. The solid line represents the average weight ± SEM.
- Expression of VGLL4‐HF4A attenuates the T cell‐mediated tumor cell killing. Activated T cells and A549 cells were co‐cultured in 24‐well plates for 4 days and surviving cells stained with crystal violet.
VGLL4 stabilizes IRF2BP2 through inhibiting K48‐linked polyubiquitination of IRF2BP2
While investigating the abundance of VGLL4 and IRF2BP2 in a battery of cell lines, we noticed that there is a positive correlation between the protein levels of VGLL4 and IRF2BP2 (Fig 4A and B). To expand this observation, we tested a hypothesis that VGLL4 may regulate IRF2BP2 protein stability. First, we overexpressed VGLL4 in A549 cells and found that the endogenous IRF2BP2 protein level was upregulated (Fig 4C). Since VGLL4 can directly bind to TEADs, we took advantage of the VGLL4‐HF4A mutant to avoid the involvement of YAP/TEAD complex in the regulation of IRF2BP2. We found that overexpression of VGLL4‐HF4A still enhanced the expression of IRF2BP2 (Fig 4C). Conversely, IRF2BP2 protein levels were downregulated in several cell lines when VGLL4 was eliminated by siRNA‐mediated knockdown (Fig 4D and E). Notably, loss of VGLL4 did not reduce the mRNA level of IRF2BP2 in A549 cells (Fig 4F), indicating that the effect of VGLL4 on IRF2BP2 protein level was post‐transcriptional.
Figure 4. VGLL4 protects proteasome‐mediated IRF2BP2 degradation.
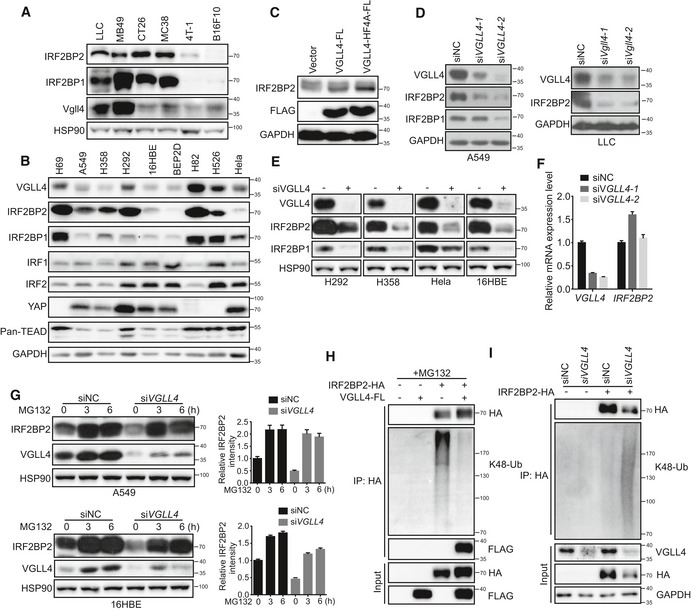
-
A, BCorrelation between protein expression of VGLL4 and IRF2BP2 in six murine tumor cell lines (A) and human cell lines (B) with the indicated antibodies.
-
CImmunoblot analysis of A549 cells transfected with VGLL4‐FLAG or VGLL4‐HF4A plasmids revealed increased IRF2BP2 protein levels compared with control cells.
-
DDeficiency of VGLL4 reduces IRF2BP2 protein levels. Immunoblot analysis of A549 and LLC cell lysates transfected with siRNA for control and VGLL4.
-
EThe indicated cell lines were transfected with control or VGLL4 siRNA and subjected to immunoblot analysis with the indicated antibodies.
-
FqRT–PCR analysis of IRF2BP2 mRNA levels in control and siVGLL4 A549 cells. n = 3, mean ± SEM.
-
GA549 or 16HBE cells were transfected with control or VGLL4 siRNA and treated with MG132 for the indicated times and subjected to immunoblot analysis with the indicated antibodies. Relative IRF2BP2 expression levels were quantified in the right panel. n = 3, mean ± SEM.
-
HLys48‐linked ubiquitylation of IRF2BP2 is strongly inhibited when VGLL4 was overexpressed in HEK293T cells. HEK293T cells were transfected with indicated plasmids, treated with MG132 for 3 h before harvest, and subjected to immunoprecipitation and immunoblot analysis as indicated.
-
ILys48‐linked ubiquitylation of IRF2BP2 is increased in the absence of VGLL4 in HEK293T cells. HEK293T cells were transfected with indicated siRNA and plasmids, and subjected to immunoprecipitation and immunoblot analysis as indicated.
Source data are available online for this figure.
Proteasome is the major pathway for protein degradation. To determine whether proteasome pathway is involved in IRF2BP2 degradation, we studied the effect of MG132 (a universal proteasome inhibitor) on VGLL4‐mediated IRF2BP2 stability. MG132 efficiently restored the protein level of IRF2BP2 in VGLL4‐knockdown A549 and 16HBE cells (Fig 4G), suggesting that IRF2BP2 degradation is through proteasome. K48‐linked polyubiquitination generally targets proteins for proteasomal degradation (Passmore & Barford, 2004). We investigated whether VGLL4 regulates IRF2BP2 protein stability through K48‐linked polyubiquitination and degradation. As expected, overexpression of VGLL4 decreased K48‐linked polyubiquitination of IRF2BP2 (Fig 4H), while knockdown of VGLL4 increased K48‐linked polyubiquitination of IRF2BP2 and reduced IRF2BP2 protein level (Fig 4I). Taken together, these results suggest that VGLL4 protects IRF2BP2 from polyubiquitination and proteasome‐dependent degradation.
Since VGLL4 interacts with both IRF2BP2 and TEADs, we sought to test whether TEADs are involved in the regulation of PD‐L1 expression and IRF2BP2 protein level. We found that depletion of TEADs in VGLL4‐knockdown A549 cells did not affect the expression of PD‐L1 compared to VGLL4 single‐knockdown A549 cells (Fig EV4A). Intriguingly, IRF2BP2 protein levels were upregulated in TEAD‐knockdown or VGLL4‐TEAD double‐knockdown A549 cells. We speculated that the removal of TEADs leads to increased interaction between VGLL4 and IRF2BP2, which will result in the stabilization of IRF2BP2. To test this, we examined the interaction of VGLL4 and IRF2BP2 in TEAD‐knockdown HEK293T cells. We found that VGLL4‐FLAG proteins bound more endogenous IRF2BP2 in TEAD‐knockdown HEK293T cells compared to the control cells (Fig EV4B). These results further support the role of VGLL4 in regulating PD‐L1 expression and IRF2BP2 protein stability.
Figure EV4. Knockdown of TEADs increases the association of VGLL4 with IRF2BP2.
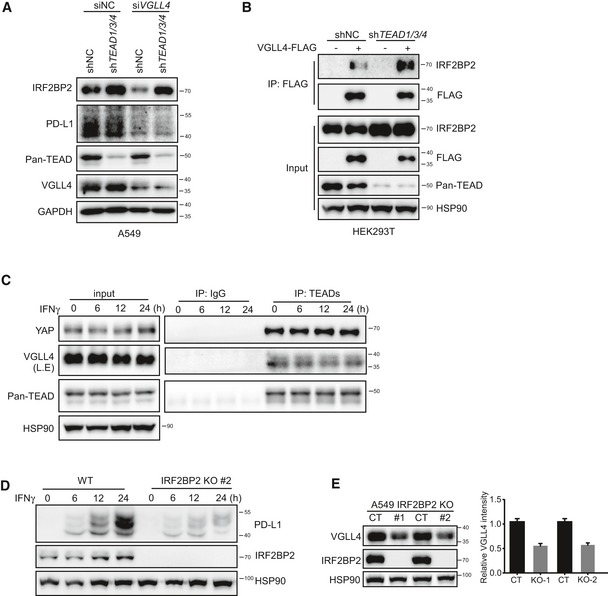
- A549 cells were treated with lenti‐shTEAD virus or together with siRNA against VGLL4, and cell lysates were analyzed by Western blot with indicated antibodies.
- Knockdown TEADs increase the association of VGLL4 with IRF2BP2. Immunoprecipitation analysis using anti‐FLAG antibody was performed in HEK293T cells transduced with lenti‐shTEAD virus and followed by transfection with VGLL4‐FLAG plasmid.
- The interaction between TEADs with YAP or VGLL4 is not affected in response to IFNγ stimulation. Immunoprecipitation analysis using anti‐pan TEAD antibody was performed in A549 cells treated with IFNγ.
- Loss of IRF2BP2 attenuated IFNγ‐inducible expression of PD‐L1. An independent IRF2BP2 KO A549 clone was treated with IFNγ for the indicated time, and cell lysates were analyzed by Western blot.
- VGLL4 protein levels were reduced in IRF2BP2 KO A549 cells. Immunoblot analysis of control and IRF2BP2 KO A549 cells with indicated antibodies. Quantification of VGLL4 protein levels in control and IRF2BP2 KO A549 cells is shown in the right panel. n = 3, mean ± SEM.
Source data are available online for this figure.
IFNγ stimulation triggers the release of IRF2 from PD‐L1 promoter and the dynamic association of IRF2 with IRF2BP2
Previous studies revealed that IFNγ signaling plays the key role in regulating PD‐L1 expression through inducing IRF1 expression and its binding to PD‐L1 promoter to activate PD‐L1 expression (Garcia‐Diaz et al, 2017). IRF2 may act as a repressor that competes with IRF1 for binding to the same PD‐L1 promoter element. We first showed that the expression of IRF2 indeed inhibited IRF1 transcriptional ability using PD‐L1 promoter luciferase assay (Fig 5A). In addition, knockdown of IRF2 led to the slightly increased PD‐L1 expression (Fig 5B). Importantly, knockdown of IRF2 restored the PD‐L1 expression in VGLL4‐knockdown A549 cells (Fig 5B). Furthermore, the expression of IRF1 significantly stimulated the expression of PD‐L1 in VGLL4‐knockdown A549 cells (Fig 5C). These results suggest that IRF2 and IRF1 act downstream of VGLL4 to regulate the expression of PD‐L1.
Figure 5. IFNγ stimulation elicits the release of IRF2 from PD‐L1 promoter and the dynamic interaction of IRF2 with IRF2BP2.
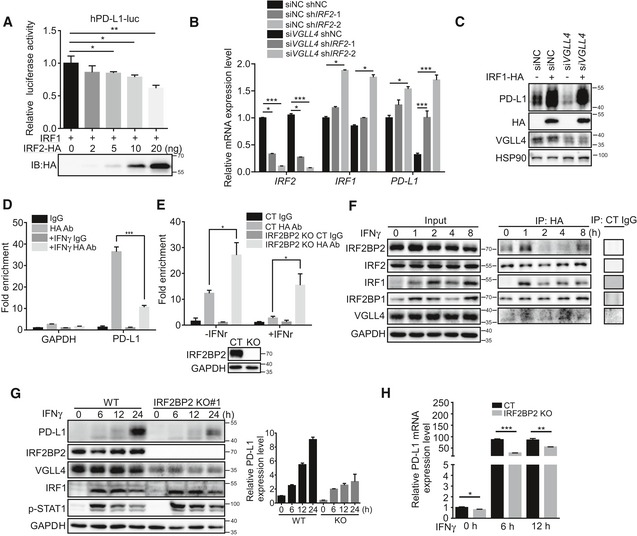
-
AIRF2 inhibits IRF1 transcriptional activity. A549 cells transfected with human PD‐L1 promoter luciferase reporter, and indicated plasmids were analyzed using a luciferase assay. n = 3, mean ± SEM. *P < 0.05, **P < 0.01, one‐way ANOVA followed by Dunnett's test.
-
BKnockdown IRF2 restores PD‐L1 expression in VGLL4‐knockdown A549 cells. Gene expression levels were analyzed by qRT–PCR in A549 cells transfected or transduced as indicated. n = 3, mean ± SEM. *P < 0.05, ***P < 0.001, one‐way ANOVA followed by Dunnett's test.
-
COverexpression of IRF1 increases PD‐L1 expression in VGLL4‐knockdown A549 cells. A549 cells were transfected with VGLL4 siRNA or together with HA‐IRF1 plasmid, and total cell lysates were analyzed by Western blot.
-
DIRF2 is released from PD‐L1 promoter upon IFNγ stimulation. IRF2‐3 × HA knockin A549 cells were stimulated with IFNγ and subjected to ChIP‐qPCR analysis in the PD‐L1 promoter using control and HA antibodies. Normal mouse IgG was used as control. n = 3, mean ± SEM. ***P < 0.001, one‐way ANOVA followed by Tukey's test.
-
EIncreased association of IRF2 with PD‐L1 promoter in IRF2BP2‐knockout (KO) A549 cells revealed by ChIP‐qPCR analysis. Immunoblot showed the loss of IRF2BP2 in IRF2BP2 KO A549 cells in the lower panel. n = 3, mean ± SEM. *P < 0.05, one‐way ANOVA followed by Tukey's test.
-
FDynamic association of IRF2 with IRF2BP2 during IFNγ stimulation by immunoprecipitation analysis using anti‐HA antibody in IRF2‐3 × HA knockin A549 cells.
-
G, HLoss of IRF2BP2 attenuated IFNγ‐inducible PD‐L1 protein (G) and mRNA (H) expression revealed by immunoblot and qRT–PCR analysis, respectively, in A549 control and IRF2BP2‐knockout cells. Quantification of PD‐L1 protein levels in (G) is shown in the right panel. n = 3, mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, two‐tailed Student's t‐test.
Source data are available online for this figure.
To test the binding of IRF2 to PD‐L1 promoter region, we carried out chromatin immunoprecipitation (ChIP) assays in the PD‐L1 promoter region. To facilitate the ChIP assay, we used the CRISPR‐Cas9 system to create an insertion of 3 × HA tag at the C‐terminus of IRF2 locus in A549 cells. As shown in Fig 5D, IFNγ stimulation induced the release of IRF2 from PD‐L1 promoter 2 h after treatment. Interestingly, deletion of IRF2BP2 by CRISPR/Cas9 in A549 cells enhanced the IRF2 binding to PD‐L1 promoter (Fig 5E). Furthermore, we found that the association of IRF2BP2 with IRF2 was dynamically regulated during IFNγ stimulation (Fig 5F). During the early stage, there was an enhanced interaction between IRF2 and IRF2BP2 (Fig 5F). Later, IRF2BP2 gradually dissociated and re‐associated with IRF2 (Fig 5F). Interestingly, the interaction between IRF2 and IRF2BP1 was not affected by IFNγ stimulation (Fig 5F). Moreover, the interaction of TEADs with YAP or VGLL4 was not affected by IFNγ stimulation (Fig EV4C). IRF2BP2 hyperphosphorylation has been indicated with the reduced PD‐L1 expression (Dorand et al, 2016). To investigate the role of IRF2BP2 in PD‐L1 regulation, we created IRF2BP2‐knockout A549 cells. Interestingly, deletion of IRF2BP2 hampered the IFNγ‐inducible PD‐L1 expression at both mRNA and protein levels (Figs 5G and H, and EV4D). Notably, VGLL4 protein levels were also reduced in IRF2BP2‐knockout A549 cells (Figs 5G and EV4E). Taken together, our results suggest that IFNγ stimulation triggers the release of IRF2 from PD‐L1 promoter and dynamic interaction between IRF2 and IRF2BP2.
YAP inhibits IFNγ‐inducible PD‐L1 expression
Recently, several studies suggest that YAP/TAZ promote PD‐L1 expression through TEAD‐mediated transcriptional regulation (Feng et al, 2017; Lee et al, 2017; Miao et al, 2017; Janse van Rensburg et al, 2018; Kim et al, 2018). Interestingly, one study showed that VGLL4 expression is repressed by YAP/TEAD transcriptional complex through miR‐130a (Shen et al, 2015), which promotes us to test whether miR‐130a and YAP could suppress IFNγ‐inducible PD‐L1 expression. Indeed, the expression of miR‐130a mimic or miR‐130a sponge suppressed or enhanced VGLL4 and PD‐L1 levels, respectively, in A549 cells (Fig 6A–C). Furthermore, expression of YAP target genes CTGF and CYR61 was slightly decreased by inhibition of miR‐130a (Fig 6D), while knockdown of VGLL4 slightly increased miR‐130a expression (Fig EV5A), confirming the positive feedback loop of YAP‐miR‐130a‐VGLL4 axis. Consistent with previous reports (Feng et al, 2017; Lee et al, 2017; Miao et al, 2017; Janse van Rensburg et al, 2018; Kim et al, 2018), we observed an increased PD‐L1 mRNA level when YAP5SA, a transcriptionally active and Hippo pathway‐resistant YAP mutant, was expressed in A549 cells (Fig EV5B). However, we detected a strong inhibition on PD‐L1 expression at both mRNA and protein levels induced by IFNγ treatment when YAP5SA or WT‐YAP was expressed (Figs 6E and F, and EV5C). Interestingly, disruption of YAP‐TEAD‐mediated transcription by YAPS94A displayed little inhibitory effect (Fig 6E and F). This result suggested that YAP target genes mediate the suppression on IFNγ‐inducible PD‐L1 expression. Furthermore, we used chemical approach to manipulate YAP activity. Serum‐borne lysophosphatidic acid (LPA) has been shown to active YAP activity through G protein‐coupled receptor (GPCR; Yu et al, 2012). LPA treatment induced YAP target gene expression (Fig EV5D). Next, we examined its effect on PD‐L1 expression under IFNγ stimulation. We found that co‐treatment of IFNγ with LPA decreased PD‐L1 expression (Fig EV5D). In addition, IFNγ did not affect the localization and phosphorylation of YAP and its target gene expression (Fig EV5E–G). Thus, our data indicate that activation of YAP inhibits IFNγ‐inducible PD‐L1 expression through its target genes. To examine whether YAP suppresses the tumor growth in mouse syngeneic model, we generated stably overexpressing YAP LLC cells. Similar to the previous study (Moroishi et al, 2016), overexpression of YAP significantly inhibited LLC tumor growth in C57BL/6 mice (Fig 6G), indicating the importance of YAP in regulating anti‐cancer immunity.
Figure 6. YAP inhibits IFNγ‐inducible PD‐L1 and IRF1 expression.
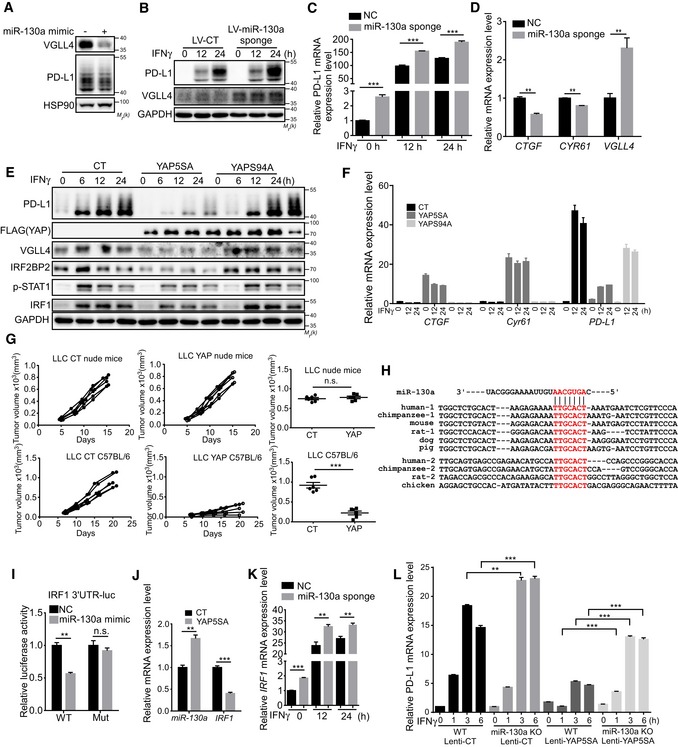
-
AmiR‐130a represses VGLL4 and PD‐L1 protein levels. A549 cells were transfected with control and miR‐130a mimic and subjected to immunoblot analysis.
-
B, CInhibition of miR‐130a by its sponge enhances IFNγ‐inducible PD‐L1 expression. A549 cells were transduced with control or miR‐130a sponge lentivirus and subjected to immunoblot (B) or qRT–PCR (C) analysis. n = 3, mean ± SEM, ***P < 0.001, two‐tailed Student's t‐test.
-
DDecreased YAP target gene expression by inhibition of miR‐130a using its sponge in A549 cells. n = 3, mean ± SEM, **P < 0.01, two‐tailed Student's t‐test.
-
EYAP5SA expression attenuates IFNγ‐inducible PD‐L1 expression. A549 cells were transduced with control, and YAP‐5SA or YAP‐S94A lentivirus and treated with IFNγ for the indicated times and subjected to immunoblot analysis.
-
FqRT–PCR analysis of cell lysates from control, and YAP‐5SA‐ or YAP‐S94A‐expressing A549 cells treated with IFNγ for the indicated times. n = 3, mean ± SEM.
-
GControl or YAP‐overexpressing LLC cells were transplanted into nude mice (upper panel) or C57BL/6 (lower panel) mice, and tumor‐growth kinetics was measured at the indicated times. Tumor volumes at the end time points were analyzed and shown at the right panel. n = 6 tumors for each group. The solid line represents the average weight ± SEM. n.s. P > 0.05, ***P < 0.001, two‐tailed Student's t‐test.
-
HConservation of miR‐130a sites in IRF1 3′UTR in vertebrates. miR‐130a binding sites from IRF1 3′UTR of different species are aligned with human miR‐130a.
-
ImiR‐130a regulates IRF1 3′UTR sensor activity. A549 cells were transfected with WT or mutant IRF1 3′UTR sensor plasmids together with miR‐130a mimic. Sensor activities were determined by dual‐luciferase assay. n = 3, mean ± SEM. n.s. P > 0.05, **P < 0.01, two‐tailed Student's t‐test.
-
JYAP5SA induces miR‐130a expression and represses IRF1 expression. A549 cells were transduced with control or YAP‐5SA lentivirus and subjected to qRT–PCR analysis. n = 3, mean ± SEM. **P < 0.01, ***P < 0.001, two‐tailed Student's t‐test.
-
KInhibition of miR‐130a by its sponge enhances IFNγ‐inducible IRF1 expression. A549 cells were transduced with control or miR‐130a sponge lentivirus and subjected to qRT–PCR analysis. n = 3, mean ± SEM. **P < 0.01, ***P < 0.001, two‐tailed Student's t‐test.
-
LSuppression of IFNγ‐inducible PD‐L1 expression by YAP5SA is compromised in miR‐130a KO A549 cells. PD‐L1 expression levels were analyzed by qRT–PCR in IFNγ‐treated control or miR‐130a KO A549 cells pre‐transduced with lenti‐YAP5SA virus. n = 3, mean ± SEM, **P < 0.01, ***P < 0.001, One‐way ANOVA followed by Tukey's test.
Source data are available online for this figure.
Figure EV5. YAP inhibits IFNγ and TNFα‐inducible PD‐L1 expression.
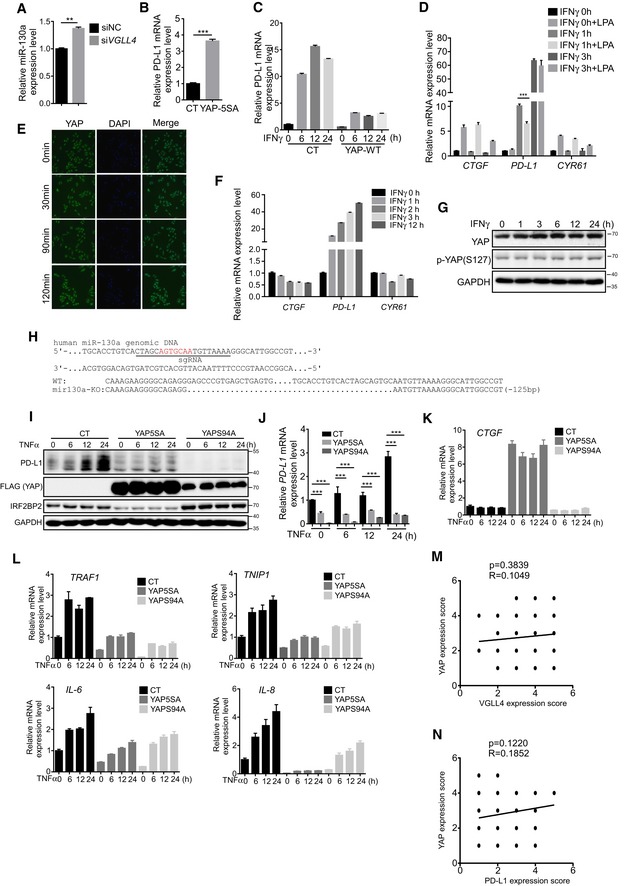
-
AmiR‐130a expression level was analyzed in VGLL4‐knockdown A549 cells by qRT–PCR. n = 3, mean ± SEM, **P < 0.01, two‐tailed Student's t‐test.
-
BYAP5SA expression increases PD‐L1 mRNA levels in A549 cells. A549 cells were transduced with control or YAP‐5SA lentivirus and subjected to qRT–PCR analysis. n = 3, mean ± SEM, ***P < 0.001, two‐tailed Student's t‐test.
-
CWT‐YAP inhibits IFNγ‐inducible PD‐L1 expression. A549 cells were transduced with control or WT‐YAP lentivirus and stimulated with IFNγ for the indicated time. PD‐L1 mRNA expression was analyzed by qRT–PCR. n = 3, mean ± SEM.
-
DLPA treatment attenuates IFNγ‐inducible PD‐L1 expression. A549 cells were treated with LPA together with IFNγ and subjected to qPCR analysis. n = 3, mean ± SEM, ***P < 0.001, one‐way ANOVA followed by Tukey's test.
-
EImmunofluorescent staining of YAP (green) counterstained with DAPI for DNA (blue) in A549 cells treated with IFNγ for the indicated time.
-
FThe expression of YAP target genes was analyzed in IFNγ‐treated A549 cells for the indicated time. n = 3, mean ± SEM.
-
GYAP‐S127 phosphorylation was analyzed in IFNγ‐treated A549 cells for the indicated time by Western blot.
-
HDNA sequence targeted by sgRNA is indicated by the solid line. The seed region is in red. miR‐130a KO A549 cells were generated by CRISPR‐/Cas9‐mediated gene editing, and deleted sequence is shown.
-
I, JYAP inhibits TNFα‐inducible PD‐L1 expression. A549 cells were transduced with control, and YAP‐5SA or YAPS94A lentivirus and treated with TNFα and subjected to immunoblot (I) and qRT–PCR (J) analysis. n = 3, mean ± SEM, ***P < 0.001, one‐way ANOVA followed by Dunnett's test.
-
KA549 cells were transduced with YAP5SA or YAPS94A lentivirus, treated with TNFα and subjected to qRT–PCR analysis for the expression of YAP target gene CTGF. n = 3, mean ± SEM.
-
lYAP inhibits TNFα target gene expression. A549 cells were transduced with control, YAP‐5SA or YAPS94A lentivirus, treated with TNFα and subjected to qRT–PCR analysis. n = 3, mean ± SEM.
-
MCorrelation of VGLL4 and YAP expressions was no significant on human non‐small‐cell lung cancer tissue arrays containing 71 samples (Pearson correlation test; R = 0.1049, P = 0.1049).
-
NCorrelation of PD‐L1 and YAP expressions was no significant on human non‐small‐cell lung cancer tissue arrays containing 71 samples (Pearson correlation test; R = 0.1852, P = 0.1220).
Source data are available online for this figure.
miR‐130a induced by YAP inhibits IRF1 expression
While investigating the regulation of PD‐L1 by YAP, we noticed that YAP5SA overexpression not only suppressed VGLL4 and PD‐L1 expression, but also the induction of IRF1 protein by IFNγ (Fig 6E). Interestingly, one of the miR‐130a targets was IRF1, a highly potent transcriptional factor for PD‐L1 expression using the TargetScan algorithm (www.targetscan.org) to predict potential miRNA targets. On the basis of these findings, we explored whether miR‐130a regulates IRF1 expression. Two miR‐130a seed‐binding sites are found in human IRF1 3′UTR, and one site is in mouse Irf1 3′UTR (Fig 6H). To determine the functionality of these predicted sites, we constructed a human IRF1 3′UTR luciferase sensor. Despite substantial repression of the WT sensor by miR‐130a mimic, the seed‐matching region mutant sensor remained unresponsive (Fig 6I). Therefore, miR‐130a could specifically bind to IRF1 3′UTR to regulate its expression. Furthermore, we showed that YAP5SA stimulated the expression of miR‐130a and inhibited IRF1 transcription simultaneously in A549 cells (Fig 6J). Consistently, inhibition of miR‐130a by microRNA sponge enhanced IFNγ‐inducible IRF1 expression (Fig 6K). Thus, IRF1 is a miR‐130a target gene. To further examine the miR‐130a‐mediated suppression of IFNγ‐inducible PD‐L1 expression, we generated a miR‐130a‐knockout A549 cell line by CRISPR/Cas9 (Fig EV5H). We found that the inhibition of IFNγ‐inducible PD‐L1 expression by YAP‐5SA was compromised in miR‐130a‐knockout A549 cells (Fig 6L). Together, these results indicate that miR‐130a significantly, though may not entirely, mediates the suppression of IFNγ‐inducible PD‐L1 expression by YAP.
Since TNFα/NF‐κB pathway induced the PD‐L1 expression (Donia et al, 2015; Lim et al, 2016). We also tested whether YAP could suppress the TNFα‐inducible PD‐L1 expression. Surprisingly, we unexpectedly observed a profound inhibition of TNFα‐inducible PD‐L1 expression and TNFα target gene expression by YAP5SA or YAPS94A (Fig EV5I–L). This observation suggests that YAP inhibits the activity of TNFα/NF‐κB pathway, which might be a direct effect rather than through its transcriptional targets.
Lower VGLL4 correlates with better clinical outcomes in human cancers
To examine the potential clinical association between VGLL4 and IRF2BP2, we evaluated the protein levels of VGLL4 and IRF2BP2 on human non‐small‐cell lung cancer tissue microarrays containing 71 samples by immunohistochemistry (Fig 7A). Expression of VGLL4 and IRF2BP2 proteins was found in most cases. Immunostaining quantification and statistical analyses revealed the correlations to be significant (Fig 7B). We also analyzed the correlation between the expression of VGLL4 with YAP or PD‐L1. We found that there is a weak correlation of VGLl4 and PD‐L1 (Fig 7C). However, we did not find a significant correlation of the expression of YAP with VGLL4 or PD‐L1 (Fig EV5M and N), indicating a complex regulation in clinical tumors. Together, these data further support the notion that VGLL4 regulates IRF2BP2 protein stability.
Figure 7. VGLL4 expression predicts cancer patient outcome and correlates with IRF2BP2 levels in non‐small‐cell lung cancer.
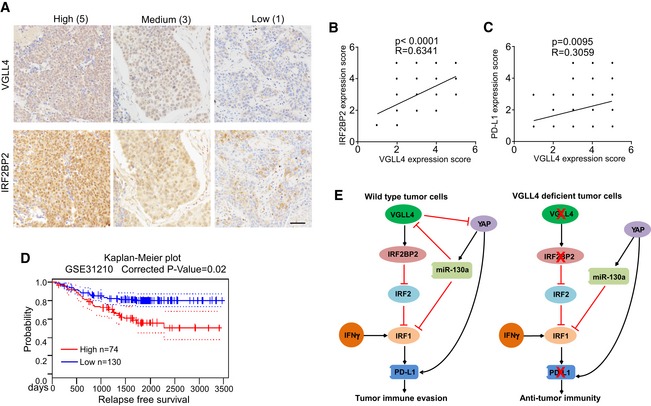
- Representative immunohistochemical staining for VGLL4 and IRF2BP2 in human non‐small‐cell lung cancer tissues. IHC score is indicated. Scale bar: 50 μm.
- Staining of VGLL4 and IRF2BP2 on human non‐small‐cell lung cancer tissue arrays containing 71 samples was quantified as described in the Materials and Methods section. Correlation of VGLL4 and IRF2BP2 expressions was significant (Pearson correlation test; R = 0.6341, P < 0.001).
- Correlation analysis of VGLL4 and PD‐L1 expression on human non‐small‐cell lung cancer tissue arrays containing 71 samples.
- Kaplan–Meier survival analysis of patients with high (n = 74) or low (n = 130) VGLL4 mRNA levels from GSE31210, a lung cancer data set. Survival curves are calculated according to the Kaplan–Meier method.
- Model for the regulation of PD‐L1 expression by VGLL4 and YAP. VGLL4 regulates PD‐L1 expression through controlling IRF2BP2 protein stability. YAP represses IFNγ‐inducible PD‐L1 expression partially through miR‐130a‐mediated inhibition of VGLL4 and IRF1 expression.
We next asked whether the expression levels of VGLL4 are relevant to human cancers. We first searched a database PrognoScan (http://www.prognoscan.org; Mizuno et al, 2009) to find any correlation between VGLL4 mRNA expression levels and patient outcome in different types of human cancer. Among 165 epidemiological data available, 22 studies show significant (P < 0.05) correlation between VGLL4 mRNA levels and patient outcome, in which 18 studies show better patient survival with low VGLL4 expression (Table EV1 and Fig 7D). Human epidemiological data strongly suggest that the expression levels of VGLL4 are clinically relevant and that lower expression of VGLL4 correlates with better patient outcome.
Collectively, our results suggest that YAP activation inhibits IFNγ‐inducible PD‐L1 expression partially through miR‐130a‐mediated suppression of VGLL4 and IRF1 expression, and loss of VGLL4 leads to the suppression of PD‐L1 expression by destabilizing IRF2BP2, thereby reducing PD‐L1 interaction with PD‐1 to escape T‐cell immune surveillance (Fig 7E).
Discussion
As a tumor suppressor pathway, the dysregulation of Hippo pathway has been linked to various cancers. VGLL4 was identified as an antagonist of YAP/TEAD complex. Several reports demonstrated that VGLL4 is a tumor suppressor in lung, gastric, and colorectal cancers by negatively regulating the YAP‐TEAD transcriptional complex and TCF4‐TEAD4 transactivation (Jiao et al, 2014, 2017; Zhang et al, 2014; Jiang et al, 2015). However, although its suppressive tumor‐growth activity has been well documented, little attention has been directed at the possible regulation for VGLL4 in cancer immunity. In this study, we uncovered that VGLL4 loss restrains tumor growth due to the suppression of tumor PD‐L1 expression and immune evasion. In the murine syngeneic tumor models, the deficiency of VGLL4 dramatically reduced the tumor growth of LLC and MB49 cells in C57BL/6 mice. Mechanistically, loss of VGLL4 promotes the degradation of IRF2BP2 through proteasome, which leads to the enhanced binding of IRF2 to PD‐L1 promoter. In addition, we found that YAP inhibits IFNγ‐inducible PD‐L1 expression partially through miRNA‐130a‐mediated suppression of VGLL4 and IRF1 expression. Our work herein implies the dual roles of VGLL4 and YAP in regulating tumor cell growth and immunogenicity.
PD‐L1 is a critical mediator in the interaction between T lymphocytes in the tumor microenvironment and tumor cells. It is not surprising that the expression of PD‐L1 is commonly elevated in cancer cells and the inhibition of PD‐L1 results in potent anti‐tumor immunity. Anti‐PD‐L1 and anti‐PD‐1 antibodies have been used for the treatment of cancer, showing promising outcomes. However, only a proportion of patients respond to the treatments. Therefore, the understanding of the PD‐L1 regulation could be helpful for the improvement of anti‐PD‐L1/PD‐1 treatments and may reveal superior biomarkers and therapeutic targets for cancer treatment. Our findings provide new insights into how PD‐L1 is regulated by VGLL4‐IRF2BP2‐IRF2 axis. In our IHC scoring analysis based on the staining intensity, the correlation of VGLL4 and IRF2BP2 expression is significant. However, the correlation of PD‐L1 expression with VGLL4 or YAP is not significant. PD‐L1 expression may be clustered rather than uniformly diffuse in tumor tissues and is often localized to the area where IFNγ+ T cells infiltrate (Ribas & Hu‐Lieskovan, 2016; Zou et al, 2016). Thus, human tumor tissue array may miss the PD‐L1‐positive area and give false‐negative results, or contain a high T‐cell infiltrate area and give a false‐positive result. Alternatively,quantitative or semiquantitative measures, such as Western blot of fresh clinical tumor samples will give a more precise result. Nevertheless, it will be interesting to dissect the mechanisms by which the balance between IFNγ‐inducible and constitutive PD‐L1 expression when YAP is activated in an immunocompetent environment.
Previous work identified IRF2BP2 as a VGLL4‐interacting protein (Teng et al, 2010). Our data support this finding and uncovered an additional unrecognized mechanism by which VGLL4 regulates IRF2BP2 protein stability and further affects IRF2 DNA binding. Although IRF2BP2 has not been extensively studied, it has been implicated as an IRF2 co‐repressor to inhibit IRF1‐induced transcriptional activity (Childs & Goodbourn, 2003). IRF2BP2 hyperphosphorylation is associated with reduced IFNγ signaling activity and PD‐L1 expression (Dorand et al, 2016). Our study indicates that the association of IRF2BP2 with IRF2 is dynamically regulated by IFNγ signaling, in which post‐translational modification might be involved. VGLL4 has been found to be in multiple protein complexes, which are involved in several signaling pathways, such as YAP/TEAD, TEAD4/TCF4, and IRF2/IRF2BP2. In the different complexes, VGLL4 plays distinct roles. VGLL4 targets TEAD4/TCF4 or TEAD/YAP complex to interfere the functional interplay between TEAD with TCF4 or YAP. Interestingly, VGLL4 stimulates TEAD1 degradation to suppress YAP activity during mouse heart development (Lin et al, 2016). On the contrary, VGLL4 promotes IRF2BP2 protein stability through inhibiting ubiquitination of IRF2BP2. Ubiquitin‐specific protease 11 (USP11) has been shown to control VGLL4 protein stability through deubiquitinating and stabilizing VGLL4 protein (Zhang et al, 2016). Future work is required to define the mechanism by which VGLL4 enhances IRF2BP2 stability, and the potential E3 ligase or deubiquitinase that regulates IRF2BP2 stability, such as USP11.
YAP has a well‐established cell autonomous oncogenic function. However, a few studies indicated that oncogenic function of YAP might be context dependent. For example, YAP expression levels positively correlate with patient survival in colorectal cancer, and multiple myeloma (Barry et al, 2013; Cottini et al, 2014), indicating YAP might play distinct roles in tumor initiation and progression. Several recent studies showed that human YAP or TAZ activates PD‐L1 expression through TEAD‐mediated transcriptional regulation (Feng et al, 2017; Lee et al, 2017; Miao et al, 2017; Janse van Rensburg et al, 2018; Kim et al, 2018). Signaling pathways have built‐in feedback mechanisms that contribute to the steadiness and robustness of cell signaling. For example, YAP/TEAD directly induces LATS2 and miR‐130a expression to form negative and positive feedback loops, respectively (Chen et al, 2015; Dai et al, 2015; Moroishi et al, 2015b; Shen et al, 2015). In this study, we also observed increased basal level of PD‐L1 expression upon YAP activation. However, IFNγ‐inducible PD‐L1 expression was significantly inhibited by YAP, which forms a negative regulation loop. Our results indicate that deletion of miR‐130a partially rescued the suppression of IFNγ‐inducible PD‐L1 expression by YAP. Given the profound effects of YAP activation on transcription (Meng et al, 2016), global miRNA biogenesis (Mori et al, 2014), and crosstalk with other major signaling, we speculate that there may be other factors contributing to the YAP‐mediated PD‐L1 regulation and anti‐tumor immunity. Interestingly, one study reported that IFNγ‐inducible expression of PD‐L1 was dependent on NF‐κB signaling (Gowrishankar et al, 2015). In this study, we showed that YAP also suppressed TNFα‐inducible PD‐L1 expression through inhibiting NF‐κB signaling. Alternatively, YAP may regulate IFNγ‐inducible expression of PD‐L1 through NF‐κB signaling. Recent reports demonstrate critical roles of the Hippo pathway in modulation of the tumor immune microenvironment. Loss of LATS1/2 inhibits immune evasion partially through activation of YAP in syngeneic mouse tumor models by regulating the secretion of nucleic acid‐containing extracellular vesicles (Moroishi et al, 2016). So the composition of immune cells and cytokines in the tumor microenvironment should be considered in the Hippo‐YAP‐regulated anti‐tumor immunity. Nevertheless, the Hippo‐YAP‐mediated crosstalk between tumor cells and immune cells needs further investigation.
Except for the function in cancer immunity, recent studies have demonstrated novel roles of Hippo signaling in regulating inflammatory signals (Liu et al, 2016; Wang et al, 2017; Zhang et al, 2017a). Our study indicates that YAP inhibits TNFα/NF‐κB signaling‐mediated PD‐L1 expression in a transcription‐independent manner. More speculatively, YAP/TAZ may differentially contribute to tumorigenesis by binding to various partners, depending on the cellular context. This flexibility adds an extra level of complexity to the regulation of cancer by YAP and TAZ in different tissues and contexts. Nevertheless, our study may also help to reconcile notable discrepancies between earlier publications that argue that the Hippo pathway both promotes and antagonizes cancer immune evasion, and underscore the need to consider model systems chosen for studying tumor immunology.
Successful treatment of cancers requires a multidisciplinary approach in which different strategies such as surgery, chemotherapy, and immunotherapy are combined. It is critical to improve our understandings into the cancer cell‐intrinsic and cell‐extrinsic mechanisms underlying tumor development, metastasis, and therapy responsiveness. Syngeneic mouse tumor models, which retain intact immune systems, are particularly relevant for studies of immune‐based targeted therapies, either used alone or in combination with other drugs that modulate the immune system's ability to seek and destroy cancer cells. It should be evident that the tumor genetics and immune system of mouse models obviously do not entirely reflect the complexity of human cancers. We observed interesting phenotype in syngeneic mouse tumor models when Vgll4 was depleted in mouse tumor cells. We selected human lung cancer cells to perform the mechanistic studies in our study, because VGLL4 has been implicated to play an important role in the carcinogenesis of human lung cancer (Zhang et al, 2014). Importantly, the major findings, such as the regulation of PD‐L1 expression and IRF2BP2 protein stability by VGLL4, are consistent in human lung cancer cells and murine cancer cell lines. More comprehensive and thorough investigations of VGLL4 function are needed to delineate its role in the modulation of immune response in human cancers.
In summary, our current study identifies VGLL4 as an important regulator in controlling PD‐L1 expression and directing anti‐cancer immune evasion, and further provides a mechanism for the PD‐L1 expression regulated by VGLL4 and YAP. Our study suggests that VGLL4 may act as a hub through which Hippo signaling controls cell proliferation and immune response, and that manipulation of the balance between IRF2/IRF2BP2 and TEAD/YAP complexes through VGLL4 may be of use for cancer treatment by suppressing cancer immune evasion and cancer cell growth.
Materials and Methods
Cell culture and transfection
The A549, H292, 16HBE, Hela, H358, and MB49 cells were cultured in DMEM (GIBCO) supplemented with 10% fetal bovine serum (GIBCO) and 1% penicillin/streptomycin. LLC, 4T‐1, and CT26 cells were cultured in RPMI 1640 (GIBCO) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were treated with IFNγ at 50 ng/ml and TNFα at 20 ng/ml.
Plasmids, shRNA siRNA, and CRISPR/Cas9 genome editing
Expression plasmids for VGLL1‐4, IRF1, IRF2, IRF2BP1, and IRF2BP2 were generated by standard molecular biology techniques using cDNAs from HEK293T cells as template. YAP‐lentivirus expression plasmids were generated by PCR using YAP2‐5SA (Addgene # 27371), YAP‐S94A (Addgene #33102) and YAP (Addgene #33091) as templates. Gene editing was performed by CRISPR/Cas9 system. Cells were transiently transfected with a Cas9 and guide RNA (gRNA) expression plasmid encoding puromycin resistance (PX459; Addgene #48139). Following transfection and transient selection with puromycin for 3 days, cells were cultured without puromycin. Knockout clones were selected by immunoblot analysis. Two independent clones were analyzed, and the parental WT cells (not transfected with PX459) were used as control. A549 IRF2‐HA knockin cells were created with the CRISPR‐Cas9 system. Targeting donor contains 800‐bp left arm and 800‐bp right arm with 3XHA at the C‐terminus of IRF2 locus. Correct knockin clones were verified by sequencing and immunoblot analysis. Guide RNA, shRNA, and siRNA sequences are listed in Table EV2.
Immunoprecipitation, immunoblot, immunofluorescence, and immunohistochemistry
Cells were lysed in lysis buffer A (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 0.5% NP‐40, 1 mM DTT, 1 mM PMSF). The lysates were incubated with anti‐HA or anti‐Flag magnetic beads overnight at 4°C. Beads were washed three times with lysis buffer A, and immunoprecipitates were eluted with SDS‐loading buffer (50 mM Tris–HCl, pH 6.8, 10% glycerol, 1% SDS, 1% beta‐mercaptoethanol). The eluates were separated by SDS–PAGE and transferred to PVDF membranes (EMD Millipore). Western blotting images were captured by ChemiScope5600 (Clinx, Shanghai) with ECL substrate.
Tumors were fixed in 4% PFA overnight and frozen in OCT compounds. The sections were cut at 10 μm and followed by regular immunofluorescence staining. Human lung cancer tissue arrays were purchased from Fanpu Biotech, Inc. (Guilin, China). For the immunohistochemistry, tissue arrays were treated with pH 6.0 sodium citrate for heat‐induced epitope retrieval and stained with VGLL4, IRF2BP2, YAP, or PD‐L1 antibody followed with biotinylated secondary antibodies. The signal was detected using HRP‐conjugated streptavidin with the chromogenic substrate DAB. Slides were counterstained in Mayer's hematoxylin. The staining intensity score was defined as follows: 1 for weak staining, 3 for moderate staining, and 5 for strong staining. Two individuals, who were both blinded to the slides examined, scored each sample and agreed the final scores. Antibody information is described in Table EV3. IHC scores of tissue arrays are in Table EV4.
Reverse transcription (RT) and quantitative PCR (qPCR) analysis
Total RNA was extracted using the RNAiso Plus reagent (Takara). cDNA was generated using PrimeScript™ RT Master Mix (Takara) according to the manufacturer's protocol. The cDNA was subjected to quantification by real‐time PCR using a Biorad CFX96 connect real‐time PCR system with SYBR Green PCR Master Mix (Takara). Relative quantification was expressed as 2‐▵ C t, where ▵ C t is the difference between the main C t value of triplicates of the sample and that of GAPDH mRNA control. Quantitative PCR primers were listed in Table EV5.
Dual‐luciferase reporter analysis
For luciferase assays, cells were transiently transfected with the indicated plasmids or siRNA with Renilla luciferase as the internal control. Cells were lysed by passive lysis buffer (Promega) 24 or 72 h after transfection and with indicated treatment. Luciferase assays were performed using a dual luciferase assay kit (Promega) and normalized to the internal Renilla luciferase control.
ChIP (chromatin immunoprecipitation) assay
ChIP assays were performed using Simple ChIP Plus Enzymatic Chromatin IP Kits (CST, #9003) according to the manufacturer's instructions. Briefly, 5 × 106 A549 IRF2‐HA knockin cells were cross‐linked, lysed, and digested to generate DNA fragments to length of approximately 150–900 bp. ChIP was performed using control IgG or antibodies against HA. Two percent of the chromatin extract was set aside for input. After the immunoprecipitation, crosslink reversal was carried out and the precipitated DNA was purified. All ChIP signals were normalized to the input, and relative fold‐change was compared with IgG controls. The resultant DNA was analyzed with qPCR, and the IP efficiency was calculated using the equation shown below.
C[T] = C T = Threshold cycle of PCR. With this method, signals obtained from each immunoprecipitation are expressed as a percent of the total input chromatin.
Flow cytometry analysis
For flow cytometry analysis, fluorochrome‐labeled antibodies were used as recommended by the manufacturer. Briefly, 3.5 × 105 to 1 × 106 cells in 100 μl FACS buffer were incubated with indicated or isotype control antibodies on ice in the dark for 30 min. After washing, cells were resuspended in 300 μl FACS buffer. Analysis was performed using Beckman CytoFlex, and results were analyzed using FlowJo software.
T cell‐mediated tumor cell killing assay
T cells were activated and expanded from peripheral blood mononuclear cells (PBMCs) of healthy donors using Dynabeads® Human T‐Activator CD3/CD28 (Gibco, 11161D) and IL‐2 (10 ng/ml; Peprotech). To analyze the killing of tumor cells by T cells, we co‐cultured tumor cells with activated primary human T cells in 24‐well plates for 4 days. To visualize the survived tumor cells at the end point, wells were washed with PBS twice to remove T cells, and then, the survived tumor cells were fixed and stained with crystal violet solution. The pictures of the dried plates were taken, and the intensity was quantified.
In vivo mouse studies
C57BL/6 and nude mice were purchased from Shanghai SLAC Laboratory Animal Company. Five‐ to 10‐week‐old mice were used in all animal experiments. No statistical method was used to predetermine sample size in the animal studies. Animal studies were approved by the Zhejiang University Animal Care and Use Committee. 5 × 105 tumor cells were subcutaneously inoculated into both back flanks of C57BL/6 or nude mice. Mice were observed regularly for tumor presence by visual inspection and manual palpation. Tumors were measured in the long and short dimensions, and tumor volumes were estimated using the equation:
Tumor tissues were harvested for weight measurement and further analyses. Antibodies used for in vivo immune checkpoint blockade experiments were given intraperitoneally at a dose of 200 μg per mouse PD‐L1 (10F.9G2) and rat IgG (LTF‐2; BioXCell). Blocking antibodies were given on day 3 after tumor cell inoculation and every 3 days for the duration of the study.
In vivo depletion of T cells was performed following VGLL4‐knockdown inoculation. Four groups of mice were injected with 100 μg of IgG, anti‐CD4 (GK1.5) antibody, anti‐CD8 (2.43) antibody or both antibodies 3 days and 1 day prior to tumor inoculation and then twice 1 week thereafter to ensure sustained depletion of T‐cell subset depletion during the experimental period. The mice were sacrificed and analyzed at day 30.
Statistical analysis
Statistical analyses were performed with a two‐tailed, unpaired Student's t‐test. When multiple comparisons were performed, one‐way ANOVA followed by Tukey's test or one‐way ANOVA with Dunnett's test was performed. P‐values < 0.05 were considered significant. *P < 0.05, **P < 0.01, ***P < 0.001. Except where otherwise indicated, experiments were repeated three times. Quantitative data were presented as mean ± SEM. All images shown were representative. Epidemiological data are obtained using the PrognoScan database (Mizuno et al, 2009). First, patients are ordered by expression value of the gene. Next, patients are divided into two (high and low) expression groups at all potential cutpoint, and the risk differences of the two groups are estimated by log‐rank test. Then, optimal cutpoint that gives the most pronounced P‐value (P min) is selected, and a value of P < 0.05 is considered statistically significant.
Author contributions
HS and AW designed the experiments. AW and QW performed all the experiments with assistance from YD, YL, JL, LL, and XL. BZ provided with reagents. CL helped in T cell‐mediated tumor cell killing assay. HS wrote the manuscript. All authors provided editorial comments.
Conflict of interest
Cheng Liao is a full‐time employee of Jiangsu Hengrui Medicine CO., LTD. All other authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank members of the Song laboratory for discussion. This work was supported by the National Natural Science Foundation of China (grant number 31471368), the Zhejiang Provincial Natural Science Foundation of China (grant number LR16C120001) to H.S., and the National Natural Science Foundation of China (grant number 81700002) to Y.D. H.S. is a scholar in the National 1000 Young Talents Program.
The EMBO Journal (2019) 38: e99506 30396996
References
- Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, Reibel JB, Soucheray M, Cohoon TJ, Janne PA, Meyerson M, Hayes DN, Shapiro GI et al (2013) Activation of the PD‐1 pathway contributes to immune escape in EGFR‐driven lung tumors. Cancer Discov 3: 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, Kuo CJ, Camargo FD (2013) Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 493: 106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gutgemann I, Eilers M, Felsher DW (2016) MYC regulates the antitumor immune response through CD47 and PD‐L1. Science 352: 227–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhang N, Xie R, Wang W, Cai J, Choi KS, David KK, Huang B, Yabuta N, Nojima H, Anders RA, Pan D (2015) Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev 29: 1285–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs KS, Goodbourn S (2003) Identification of novel co‐repressor molecules for Interferon Regulatory Factor‐2. Nucleic Acids Res 31: 3016–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L, ten Hacken E, Bertilaccio MT, Antonini E, Neri A, Ponzoni M, Marcatti M, Richardson PG, Carrasco R, Kimmelman AC, Wong KK, Caligaris‐Cappio F, Blandino G, Kuehl WM, Anderson KC et al (2014) Rescue of Hippo coactivator YAP1 triggers DNA damage‐induced apoptosis in hematological cancers. Nat Med 20: 599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Liu H, Shen S, Guo X, Yan H, Ji X, Li L, Huang J, Feng XH, Zhao B (2015) YAP activates the Hippo pathway in a negative feedback loop. Cell Res 25: 1175–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, Chen L (1999) B7‐H1, a third member of the B7 family, co‐stimulates T‐cell proliferation and interleukin‐10 secretion. Nat Med 5: 1365–1369 [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L (2002) Tumor‐associated B7‐H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med 8: 793–800 [DOI] [PubMed] [Google Scholar]
- Donia M, Andersen R, Kjeldsen JW, Fagone P, Munir S, Nicoletti F, Andersen MH, Thor Straten P, Svane IM (2015) Aberrant expression of MHC class II in melanoma attracts inflammatory tumor‐specific CD4+ T‐ cells, which Dampen CD8+ T‐cell antitumor reactivity. Can Res 75: 3747–3759 [DOI] [PubMed] [Google Scholar]
- Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, Chirieleison SM, Pareek TK, Abbott DW, Stearns DS, Letterio JJ, Huang AY, Petrosiute A (2016) Cdk5 disruption attenuates tumor PD‐L1 expression and promotes antitumor immunity. Science 353: 399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B, Yang M, Cao W, Wang L, Wu Z (2017) Tumor cell‐derived lactate induces TAZ‐dependent upregulation of PD‐L1 through GPR81 in human lung cancer cells. Oncogene 36: 5829–5839 [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T (2000) Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192: 1027–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Diaz A, Shin DS, Moreno BH, Saco J, Escuin‐Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, Parisi G, Saus CP, Torrejon DY, Graeber TG, Comin‐Anduix B, Hu‐Lieskovan S, Damoiseaux R, Lo RS, Ribas A (2017) Interferon receptor signaling pathways regulating PD‐L1 and PD‐L2 expression. Cell Rep 19: 1189–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P (2015) Inducible but not constitutive expression of PD‐L1 in human melanoma cells is dependent on activation of NF‐kappaB. PLoS One 10: e0123410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Lu Y, Li P, Yin MX, Lv D, Zhang W, Wang H, Zhou Z, Ji H, Zhao Y, Zhang L (2013) A novel partner of Scalloped regulates Hippo signaling via antagonizing Scalloped‐Yorkie activity. Cell Res 23: 1201–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai X, Ji X, Ji F, Gong XG, Li L, Bai X, Feng XH, Liang T, Ji J, Chen L, Wang H, Zhao B (2017) Single tumor‐initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev 31: 247–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, Miyata T, Taniguchi T (1989) Structurally similar but functionally distinct factors, IRF‐1 and IRF‐2, bind to the same regulatory elements of IFN and IFN‐inducible genes. Cell 58: 729–739 [DOI] [PubMed] [Google Scholar]
- Janse van Rensburg HJ, Azad T, Ling M, Hao Y, Snetsinger B, Khanal P, Minassian LM, Graham CH, Rauh MJ, Yang X (2018) The Hippo pathway component TAZ promotes immune evasion in human cancer through PD‐L1. Cancer Res 78: 1457–1470 [DOI] [PubMed] [Google Scholar]
- Jiang W, Yao F, He J, Lv B, Fang W, Zhu W, He G, Chen J, He J (2015) Downregulation of VGLL4 in the progression of esophageal squamous cell carcinoma. Tumour Biol 36: 1289–1297 [DOI] [PubMed] [Google Scholar]
- Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, Wang X, Guo T, Li P, Zhao Y, Ji H, Zhang L, Zhou Z (2014) A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 25: 166–180 [DOI] [PubMed] [Google Scholar]
- Jiao S, Li C, Hao Q, Miao H, Zhang L, Li L, Zhou Z (2017) VGLL4 targets a TCF4‐TEAD4 complex to coregulate Wnt and Hippo signalling in colorectal cancer. Nat Commun 8: 14058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Khan SK, Liu Y, Xu R, Park O, He Y, Cha B, Gao B, Yang Y (2017) Hepatic Hippo signaling inhibits protumoural microenvironment to suppress hepatocellular carcinoma. Gut 67: 1692–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Kim CG, Kim SK, Shin SJ, Choe EA, Park SH, Shin EC, Kim J (2018) YAP‐induced PD‐L1 expression drives immune evasion in BRAFi‐resistant melanoma. Cancer Immunol Res 6: 255–266 [DOI] [PubMed] [Google Scholar]
- Koontz LM, Liu‐Chittenden Y, Yin F, Zheng Y, Yu J, Huang B, Chen Q, Wu S, Pan D (2013) The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped‐mediated default repression. Dev Cell 25: 388–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastwika KJ, Wilson W III, Li QK, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S, Liu LN, Gills JJ, Dennis PA (2016) Control of PD‐L1 expression by oncogenic activation of the AKT‐mTOR pathway in non‐small cell lung cancer. Can Res 76: 227–238 [DOI] [PubMed] [Google Scholar]
- Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, Oh S, Shin JG, Yao S, Chen L, Choi IH (2006) Interferon regulatory factor‐1 is prerequisite to the constitutive expression and IFN‐gamma‐induced upregulation of B7‐H1 (CD274). FEBS Lett 580: 755–762 [DOI] [PubMed] [Google Scholar]
- Lee BS, Park DI, Lee DH, Lee JE, Yeo MK, Park YH, Lim DS, Choi W, Lee DH, Yoo G, Kim HB, Kang D, Moon JY, Jung SS, Kim JO, Cho SY, Park HS, Chung C (2017) Hippo effector YAP directly regulates the expression of PD‐L1 transcripts in EGFR‐TKI‐resistant lung adenocarcinoma. Biochem Biophys Res Comm 491: 493–499 [DOI] [PubMed] [Google Scholar]
- Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, Kim T, Chang WC, Hsu JL, Yamaguchi H, Ding Q, Wang Y, Yang Y, Chen CH, Sahin AA, Yu D et al (2016) Deubiquitination and stabilization of PD‐L1 by CSN5. Cancer Cell 30: 925–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Guo H, Cao Y, Zohrabian S, Zhou P, Ma Q, VanDusen N, Guo Y, Zhang J, Stevens SM, Liang F, Quan Q, van Gorp PR, Li A, Dos Remedios C, He A, Bezzerides VJ, Pu WT (2016) Acetylation of VGLL4 regulates Hippo‐YAP signaling and postnatal cardiac growth. Dev Cell 39: 466–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zheng Y, Yin F, Yu J, Silverman N, Pan D (2016) Toll receptor‐mediated Hippo signaling controls innate immunity in Drosophila . Cell 164: 406–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loke P, Allison JP (2003) PD‐L1 and PD‐L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA 100: 5336–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, Guan KL (2016) Mechanisms of Hippo pathway regulation. Genes Dev 30: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Hsu PC, Yang YL, Xu Z, Dai Y, Wang Y, Chan G, Huang Z, Hu B, Li H, Jablons DM, You L (2017) YAP regulates PD‐L1 expression in human NSCLC cells. Oncotarget 8: 114576–114587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JF, Sadelain M (2015) The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 27: 439–449 [DOI] [PubMed] [Google Scholar]
- Mizuno H, Kitada K, Nakai K, Sarai A (2009) PrognoScan: a new database for meta‐analysis of the prognostic value of genes. BMC Med Genomics 2: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, Gregory RI (2014) Hippo signaling regulates microprocessor and links cell‐density‐dependent miRNA biogenesis to cancer. Cell 156: 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, Guan KL (2015a) The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, Taniguchi K, Yu FX, Karin M, Pan D, Guan KL (2015b) A YAP/TAZ‐induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev 29: 1271–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Hayashi T, Pan WW, Fujita Y, Holt MV, Qin J, Carson DA, Guan KL (2016) The Hippo pathway kinases LATS1/2 suppress cancer immunity. Cell 167: 1525–1539 e1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO (2007) Loss of tumor suppressor PTEN function increases B7‐H1 expression and immunoresistance in glioma. Nat Med 13: 84–88 [DOI] [PubMed] [Google Scholar]
- Passmore LA, Barford D (2004) Getting into position: the catalytic mechanisms of protein ubiquitylation. Biochem J 379: 513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Hu‐Lieskovan S (2016) What does PD‐L1 positive or negative mean? J Exp Med 213: 2835–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Guo X, Yan H, Lu Y, Ji X, Li L, Liang T, Zhou D, Feng XH, Zhao JC, Yu J, Gong XG, Zhang L, Zhao B (2015) A miR‐130a‐YAP positive feedback loop promotes organ size and tumorigenesis. Cell Res 25: 997–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, Chen L (2012) Colocalization of inflammatory response with B7‐h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 4: 127ra137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng AC, Kuraitis D, Deeke SA, Ahmadi A, Dugan SG, Cheng BL, Crowson MG, Burgon PG, Suuronen EJ, Chen HH, Stewart AF (2010) IRF2BP2 is a skeletal and cardiac muscle‐enriched ischemia‐inducible activator of VEGFA expression. FASEB J 24: 4825–4834 [DOI] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA et al (2014) PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515: 568–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xie F, Chu F, Zhang Z, Yang B, Dai T, Gao L, Wang L, Ling L, Jia J, van Dam H, Jin J, Zhang L, Zhou F (2017) YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKvarepsilon‐mediated phosphorylation. Nat Immunol 18: 733–743 [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, Fu XD, Mills GB, Guan KL (2012) Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell 150: 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Gao Y, Li P, Shi Z, Guo T, Li F, Han X, Feng Y, Zheng C, Wang Z, Li F, Chen H, Zhou Z, Zhang L, Ji H (2014) VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP‐TEAD transcriptional complex. Cell Res 24: 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang E, Shen B, Mu X, Qin Y, Zhang F, Liu Y, Xiao J, Zhang P, Wang C, Tan M, Fan Y (2016) Ubiquitin‐specific protease 11 (USP11) functions as a tumor suppressor through deubiquitinating and stabilizing VGLL4 protein. Am J Cancer Res 6: 2901–2909 [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Meng F, Chen S, Plouffe SW, Wu S, Liu S, Li X, Zhou R, Wang J, Zhao B, Liu J, Qin J, Zou J, Feng XH, Guan KL, Xu P (2017a) Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ‐mediated TBK1 blockade. Nat Cell Biol 19: 362–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang H, Zhao B (2017b) Hippo signaling in the immune system. Trends Biochem Sci 43: 77–80 [DOI] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, Chen L (2016) PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med 8: 328rv324 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6