

Abstract
Urodele newts have unique biological properties, notably including prominent regeneration ability. The Iberian ribbed newt, Pleurodeles waltl, is a promising model amphibian distinguished by ease of breeding and efficient transgenic and genome editing methods. However, limited genetic information is available for P. waltl. We conducted an intensive transcriptome analysis of P. waltl using RNA-sequencing to build and annotate gene models. We generated 1.2 billion Illumina reads from a wide variety of samples across 12 different tissues/organs, unfertilized egg, and embryos at eight different developmental stages. These reads were assembled into 1,395,387 contigs, from which 202,788 non-redundant ORF models were constructed. The set is expected to cover a large fraction of P. waltl protein-coding genes, as confirmed by BUSCO analysis, where 98% of universal single-copy orthologs were identified. Ortholog analyses revealed the gene repertoire evolution of urodele amphibians. Using the gene set as a reference, gene network analysis identified regeneration-, developmental-stage-, and tissue-specific co-expressed gene modules. Our transcriptome resource is expected to enhance future research employing this emerging model animal for regeneration research as well as for investigations in other areas including developmental biology, stem cell biology, and cancer research. These data are available via our portal website, iNewt (http://www.nibb.ac.jp/imori/main/).
Keywords: transcriptome, Iberian ribbed newt, NGS, model organism
1. Introduction
Urodele amphibian newts have an outstanding history as model organisms in experimental biology. The ‘Spemann organizer’ was discovered using European newts, Triturus cristatus and T. taeniatus.1 ‘Wolffian lens regeneration’ was discovered using these species,2 and Eguchi et al., subsequently demonstrated transdifferentiation of pigment epithelial cells to lens cells by clonal cell culture using the Japanese common newt, Cynops pyrrhogaster.3 Newts have provided the clearest examples of natural reprogramming events in the process of lens regeneration, facilitating the study of mechanisms of cellular reprogramming.4–8 Additionally, experiments with newts have yielded a great deal of knowledge about the regeneration of various tissues and organs, including limb,9 joint,10 heart,11 jaw,12 retina,13,14 brain,15–17 spinal cord,18 intestine,19 testis,20,21 and lung.22 Among vertebrates, only newts are known to be capable of regenerating all of the above-mentioned organs and body parts. Furthermore, comparative studies of regeneration ability in newts and frogs have provided new insights for future regenerative medicine, given that frogs (like mammals) lose the ability to regenerate various tissues and organs after metamorphosis.10,12,23,24
Newts also have been employed in research other than that on regeneration, reflecting these animals’ unique biological properties. The genome sizes of newt species are 8–10 times larger (e.g. Axolotl Ambystoma mexicanum: 32 Gb) than the human genome (3 Gb).24–26 Newts are tumour-resistant, despite having a long lifetime.27,28 Newt eggs are fertilized via physiological polyspermy.29 Male newts can form new testes even after sexual maturation.30 Moreover, the mating behaviour of newts is mediated by sexual pheromones.31,32 Furthermore, several groups have shown the utility of newts for the toxicity testing of chemical compounds.33–35 Indeed, aquatic tetrapods like newts can serve as important indicators of the influence of chemical compounds on the environment.36 Therefore, newts are versatile model animals that can be used in various fields of research, including regeneration, stem cell biology, cancer research, developmental biology, reproductive biology, evolution, ethology, and toxico-genomics.
Although these properties make newts attractive model animals, the newt species that have been used (e.g. the American common newt, Notophthalmus viridescens, and the Japanese common newt, C. pyrrhogaster) in classic experiments are not suitable for reverse or molecular genetics because of the difficulty of breeding these species in captivity. For example, Japanese common newts spawn seasonally, and each female spawns only a small number of eggs per cycle.37,38 Under typical conditions, three or more years can be required for sexual maturation. Several different newt species have been used for studies performed across different laboratories, countries, and continents, making it difficult for members of this research community to share resources and expertise. In fact, it is estimated that the American common newt and the Japanese common newt diverged 150 million years ago,39 an evolutionary distance that results in substantial differences between the two species.24
The Iberian ribbed newt (Pleurodeles waltl) is an emerging model newt.24,40 In contrast to conventional newt species, P. waltl has high fecundity and is easy to maintain in the laboratory. P. waltl newts have a relatively short generation time, and females of this species spawn a large number of eggs all year around in the laboratory compared with the conventional newts (e.g. C. pyrrhogaster). Using P. waltl newts, we have established a model experimental system that is amenable to molecular genetics.40 Notably, efficient genome editing was recently demonstrated in P. waltl.41,42 Despite the availability of these tools, limited genetic information is available for this species. Recently, Elewa et al. reported a draft sequence for the P. waltl genome, which is 20 Gb in size, along with the corresponding transcriptome.24 These data provided pioneering resources for the P. waltl newt research field. However, assembly data or gene models are not publicly available for this species; there remains a popular demand for a reference gene catalogue that can be openly shared among researchers who are interested in this emerging model newt.
In the present study, we sought to create a reference set of comprehensive gene models for P. waltl. To that end, we prepared 29 libraries of mRNA from various tissues and embryonic stages of P. waltl and subjected the libraries to RNA-seq. We combined the resulting >1.2 billion reads and assembled these reads. This assembly yielded 1,395,387 contigs and permitted the annotation of 202,788 predicted ORFs. Using these data, we built a reference set of gene models of P. waltl providing good coverage of P. waltl protein-coding genes. Moreover, we demonstrated that the expression patterns of regeneration-, developmental stage-, and tissue-specific genes could be analysed using our gene model and transcriptome data sets. Furthermore, we have established a portal website that provides the research community with access to our data sets.
2. Materials and Methods
2.1. Animals
The P. waltl newts used in this study originated from a breeding colony at Tottori University. The animals were maintained as described previously.40 The developmental stages (St) were categorized according to criteria described by Shi and Boucaut.43 To isolate organs or perform surgical operations for limb and heart regeneration, embryos and adults were anaesthetized/euthanized by immersion in 0.01–0.2% MS-222 (tricaine; Sigma-Aldrich, MO). All procedures were carried out in accordance with Institutional Animal Care and Use Committee of the respective institutes and with the national guidelines of the Ministry of Education, Culture, Sports, Science & Technology of Japan.
2.2. RNA preparation, library construction, and RNA sequencing
Sequence data collection was performed in five laboratories. We here describe a representative workflow used in preparing the libraries from gonadal tissues and unfertilized eggs. Minor modifications were made for the other libraries (see Supplementary Table S1 for details). Total RNA was extracted using the TRIzol reagent (ThermoFisher, Waltham, MA.) and purified by NucleoSpin kits (Takara Bio, Siga, Japan), following the respective manufacturer’s instructions. Sequencing libraries were generated using TruSeq Stranded mRNA Sample Preparation kit (Illumina) starting from 1.0 μg total RNA for each sample, according to manufacturer’s protocol (Low Throughput Protocol) with minor modifications: RNA fragmentation was conducted for 4 min instead of 8 min at 94 °C and the number of PCR cycles was reduced to seven to minimize PCR biases. In total, eight multiplexed libraries were sequenced per lane using a Hiseq1500 system (Illumina) with 106-bp paired-end readings in the RapidRun mode.
2.3. Assembly and ORF prediction
All sequenced reads were employed for de novo assembly using the Trinity program ver. 2.4.044 under default parameter settings; the trimming option was performed using the trimmomatic software.45 Assembled contigs were processed using the TransDecoder program ver. 3.0.146 to predict open reading frames (ORFs) and amino acid sequences; the parameter of ‘minimum protein length’ was set to 50 amino acids (default: 100) to preclude overlooking short-length proteins such as neuropeptides and antimicrobial peptides. We also used the BLASTP and Pfam options in the TransDecoder ORF prediction. Redundant ORFs were filtered using the CD-HIT program.47 The quality of the assembly was evaluated using the BUSCO program ver. 248 to compare the newt transcriptome assembly against a core-vertebrate gene (CVG) data set49 and a vertebrate data set (Vertebrata_odb9).
2.4. Gene annotation and ortholog analysis
We searched predicted amino acid sequences derived from the newt transcriptome for homologs of previously published protein sequences using a BLASTP search against the NCBI non-redundant database (nr DB) (parameters: BLAST+ ver. 2.6.0; the nr DB was the latest version as of 23 November 2017). Gene ontology (GO) terms for each sequence were also annotated using the BLAST2GO program (Version 4.1.9) with the NCBI nr DB.50 To identify orthologs of each P. waltl protein-coding gene, orthologous groups within vertebrates were inferred using the OrthoFinder2 program (version 2.0.0).51 We carried out two different orthologous clustering analyses. First, we compared P. waltl proteomes with those of nine other vertebrates: green anole (Anolis carolinensis), zebrafish (Danio rerio), chicken (Gallus gallus), human (Homo sapiens), coelacanth (Latimeria chalumnae), mouse (Mus musculus), Chinese softshell turtle (Pelodiscus sinensis), African clawed frog (Xenopus laevis), and western clawed frog (X. tropicalis). All of the protein sequences were downloaded from OrthoDB ver. 9.1, except that the X. laevis sequences were obtained from Xenbase ver. 9.1 (http://www.xenbase.org/other/static/ftpDatafiles.jsp). Second, we performed a pairwise comparison between the P. waltl and axolotl (Ambystoma mexicanum) transcriptomes.52
2.5. Expression and network analysis
We quantified expression of each gene in each sample by mapping to the reference transcript database that was created by the de novo assembly (see above). The kallisto program v0.43.1 with 100 bootstrap replicates was used for quantifying abundance of the transcripts.53 The read count data were normalized by the trimmed mean M values (TMM) method available in the edgeR software package of R language software (version 3.12.1).54 After TMM normalization, we estimated the Reads Per Kilobase of exon per Million mapped reads (RPKM) value of each gene. We used the RPKM values for the gene network analysis. To visualize profiles of gene expressions, a multi-dimensional scaling (MDS) plot was generated using the edgeR software package.
To detect modules of co-expressed genes from the transcriptome data, Weighted Gene Correlation Network Analysis (WGCNA) was applied. Normalized RPKM data were used for this analysis, implemented in the WGCNA library (version 1.66)55 of R language. Genes expressed at low levels, and genes with low expression variance across the libraries, were filtered out; the 16,367 surviving gene models were used in the WGCNA analysis. A signed network was constructed in WGCNA with specific parameter settings of power = 8, networkType = ‘signed’, TOMType = ‘unsigned’, and minModuleSize = 30.
2.6. Identification of bmp2/4/16
To identify P. waltl bmp genes and infer phylogeny of this gene family among vertebrates, the corresponding predicted protein sequences were used to search the genome database as described below. We searched the Ensembl database version 9156 to identify bmp2/4/16 homologs in eight vertebrate species, including A. carolinensis, D. rerio, G. gallus, H. sapiens, L. chalumnae, M. musculus, Oryzias latipes, and Takifugu rubripes; the X. tropicalis ver. 9.1 gene model in the Xenbase also was searched.57 Additionally, we searched three independent amphibian-specific data sets that were described in previous reports, including those for Nanorana parkeri,58A. mexicanum,52 and C. pyrrhogaster.59 Homologous sequences encoded by bmp2/4/16 genes were aligned using the MUSCLE algorithm with default settings in the MEGA7 software (see Supplementary Data S1).60,61 A phylogenetic tree was constructed using the maximum-likelihood (ML) analysis implemented in MEGA7 with the JTT model and gamma distribution. Bootstrap probabilities were computed using 1,000 replicates.
3. Results and Discussion
3.1. Collection and preparation of material
We sought to create a comprehensive transcriptome reference covering the P. waltl gene repertoire, in the hope that the resulting database will be useful for various subsequent studies. Therefore, we collected RNA samples from a wide variety of tissues and developmental stages (and Table 1). The 29 resulting libraries were derived from 12 different normal tissues (heart, limb, tail, brain, kidney, liver, pancreas, intestine, testicular connective tissue, testicular gland, testis, and ovary) and two regenerating tissues (heart and limb) of adult newts, and from whole embryos at each of nine time points from early to late developmental stages (unfertilized egg and stages 7–7.5, 8b, 11, 12, 15, 18, 25, and 30).
Table 1.
Summary of the sample preparation and sequence profiles
| Labela | Tissue/organ | Age/dev. stageb | Sex | Description | Platform (Hiseq) | Read length# (bp) | Total reads |
|---|---|---|---|---|---|---|---|
| HtN | Heart | Adult | Mixed | Ventricle, normal | 2,000 | 101 | 55,882,914 |
| HtR | Heart | Adult | Mixed | Ventricle, regenerating | 2,000 | 101 | 50,516,110 |
| Lb0 | Limb | Adult | Mixed | Limb, normal | 2,000 | 101 | 55,246,408 |
| Lb3 | Limb | Adult | Mixed | Limb, regenerating day3 | 2,000 | 101 | 47,286,228 |
| Lb19 | Limb | Adult | Mixed | Limb, regenerating day19 | 2,000 | 101 | 64,373,580 |
| Tl | Tail | Adult | Female | Tail | 2,500 | 125 | 27,630,846 |
| Br | Brain | Adult | Female | Brain | 2,500 | 125 | 27,626,778 |
| Kn | Kidney | Adult | Female | Kidney | 2,500 | 125 | 27,441,344 |
| Lv | Liver | Adult | Female | Liver | 2,500 | 125 | 30,427,600 |
| Pc | Pancreas | Adult | Female | Pancreas | 2,500 | 125 | 24,594,822 |
| It | Intestine | Adult | Female | Intestine | 2,500 | 125 | 28,214,656 |
| Cn | Connective tissue | 3 months juvenile | Male | Connective tissue adjacent to testis | 2,000 | 106 | 34,403,376 |
| Tg | Testicular gland | Adult | Male | Testicular gland, matured | 2,000 | 106 | 33,182,908 |
| Tt3 | Testis | 3 months juvenile | Male | Testis, not matured | 2,000 | 106 | 36,052,648 |
| TtA | Testis | Adult | Male | Testis, matured | 2,000 | 106 | 35,305,096 |
| Ov3 | Ovary | 3 months juvenile | Female | Ovary, not matured | 2,000 | 106 | 37,990,436 |
| Ov7 | Ovary | 7 months juvenile | Female | Ovary, not matured | 2,000 | 106 | 39,617,046 |
| Eg-1 | Whole | Unfertilized egg | Unknown | Biological replicate 1 | 2,000 | 106 | 35,758,440 |
| Eg-2 | Whole | Unfertilized egg | Unknown | Biological replicate 2 | 2,000 | 106 | 38,172,320 |
| St7 | Whole embryo | Stage 7–7.5 | Unknown | Early gastrula | 2,500 | 125 | 29,361,580 |
| St8b | Whole embryo | Stage 8b | Unknown | Slightly advanced early gastrula | 1,500 | 100 | 65,423,479 |
| St11 | Whole embryo | Stage 11 | Unknown | Middle-late gastrula | 2,500 | 125 | 31,400,058 |
| St12 | Whole embryo | Stage 12 | Unknown | Late gastrula | 1,500 | 100 | 55,684,175 |
| St15 | Whole embryo | Stage 15 | Unknown | Neural plate stage | 1,500 | 100 | 48,604,306 |
| St18 | Whole embryo | Stage 18 | Unknown | Late neural fold stage | 1,500 | 100 | 55,446,723 |
| St25-1 | Whole embryo | Stage 25 | Unknown | Elongated tail bud stage | 1,500 | 100 | 49,345,987 |
| St25-2 | Whole embryo | Stage 25 | Unknown | Tail bud stages (st25-28) were mixed. | 2,000 | 101 | 51,692,566 |
| St30-1 | Whole embryo | Stage 30 | Unknown | Gill protrusion stage | 1,500 | 100 | 49,079,133 |
| St30-2 | Whole embryo | Stage 30 | Unknown | st30 and st31were mixed. | 2,000 | 101 | 49,920,498 |
3.2. Sequencing and de novo assembly of transcriptome
We sequenced the 29 libraries, each of which yielded 24 million to 65 million paired-end reads of 100 to 125 bases each, totalling >1.2 billion reads. To build a reference P. waltl transcriptome, cleaned reads from all of these libraries were assembled together using Trinity, yielding 1,395,387 contigs with an average length and N50 of 700.56 and 1,490 bp, respectively (Table 2 and Supplementary Table S2). From these contigs, we predicted 202,788 non-redundant ORFs, ranging from 147 to 73,080 bp with an N50 of 591 bp (Table 2 and Supplementary Table S2). The ORF set was designated PLEWA04_ORF and used as a reference P. waltl coding-sequence catalogue for downstream analysis.
Table 2.
Overview of de novo assembly and ORF prediction
| Number of samples | 29 |
|---|---|
| De novo assembly | |
| Number of contigs | 1,395,387 |
| N50 | 1,490 bp |
| Total length | 977,554,621 bp |
| ORF prediction | |
| Number of proteins | 202,788 |
| N50 | 591 bp |
| Total length | 113,316,939 bp |
| BUSCO completeness a | 99.0%b |
Analysed using Vertebrata_odb9.
Including 1% of fragmented.
We evaluated the completeness of our transcriptome by comparison (via the BUSCO program) with two different datasets (CVG and Vertebrata_odb9). The CVG data consists of 233 genes that are shared as one-to-one orthologs among 29 representative vertebrate genomes and are widely used for phylogenomic studies.39 We found that our P. waltl transcriptome covered all 233 CVG genes, indicating that we successfully reconstructed most of the protein-coding gene sequences in this species. In addition, our P. waltl transcriptome corresponded to 98% of the Vertebrata_odb9 gene set. We compared our result with earlier urodele transcriptome studies. Previous P. waltl and A. mexicanum transcriptomes covered 82% and 88% of the Vertebrata_odb9 data, respectively.24,52 Thus, our P. waltl transcriptome data significantly enhanced the gene space of urodeles, attaining a near-complete gene repertoire.
3.3. Gene annotation and ortholog analysis
All translated sequences of PLEWA04_ORF were compared with the NCBI non-redundant protein database (nr DB) using BLASTP. Among the 202,788 ORFs identified in our DB, 121,837 genes (60.1%) encoded proteins exhibiting sequence similarity to proteins in the NCBI nr DB (Supplementary Data S2). The two most-frequent BLASTP top-hit species corresponded to clawed frogs (X. tropicalis and X. laevis), followed by coelacanth (L. chalumnae) and turtles (C. picta, P. sinensis, and C. mydas) (Table 3), which reasonably reflects the phylogenetic position of P. waltl among these species.
Table 3.
Species of top BLAST hits in NCBI nr databases for the P. waltl transcriptome
| Species | Common name | # Top hits |
|---|---|---|
| X. tropicalis | Western clawed frog | 12,199 |
| X. laevis | African clawed frog | 8,604 |
| L. chalumnae | Coelacanth | 7,941 |
| C. picta | Western painted turtle | 7,631 |
| P. sinensis | Chinese soft-shelled turtle | 5,200 |
| C. mydas | Green sea turtle | 4,229 |
| Nanorana parkeri | Nanorana parkeri | 3,347 |
| Larimichthys crocea | Large yellow croaker | 2,827 |
| Shewanella frigidimarina | Shewanella frigidimarina | 1,960 |
| A. carolinensis | Green anole | 1,937 |
| Alligator mississippiensis | American alligator | 1,632 |
| Cyprinus carpio | Common carp | 1,584 |
| Oncorhynchus mykiss | Rainbow trout | 1,443 |
| Cordyceps militaris | Cordyceps militaris CM01 | 1,395 |
| Gekko japonicus | Gekko japonicus | 1,360 |
| Plasmodium malariae | Plasmodium malariae | 1,350 |
| Stylophora pistillata | Stylophora pistillata | 1,323 |
| Crassostrea virginica | Eastern oyster | 1,286 |
| Strongylocentrotus purpuratus | Purple sea urchin | 1,013 |
| Austrofundulus limnaeus | Austrofundulus limnaeus | 978 |
| Oreochromis niloticus | Nile tilapia | 923 |
| Pogona vitticeps | Central bearded dragon | 857 |
| Daphnia magna | Daphnia magna | 850 |
| H. sapiens | Human | 791 |
| Arabidopsis thaliana | Thale cress | 735 |
| M. musculus | House mouse | 722 |
| Acropora digitifera | Acropora digitifera | 710 |
| Trichuris suis | Pig whipworm | 679 |
| Crassostrea gigas | Pacific oyster | 668 |
We used InterProScan to query the predicted coding regions for known functional domains. We identified 90,471 Pfam motifs (Supplementary Data S3) in the products of 55,075 P. waltl gene models. In addition, 814,803 GO terms were assigned to 86,516 genes (42.7%) (Supplementary Data S4).
Figure 1.

Organs and embryos used for RNA preparation. Panel (A) provides a picture of a whole adult female. (B–N) Examples of organs, tissues, and embryos used for RNA extraction. dpa, days post amputation. Scale bars: 1 mm.
To understand gene repertoire evolution in the P. waltl proteome, we generated clusters of orthologous and paralogous gene families comparing the P. waltl proteome with those of nine other vertebrates (Table 4). The OrthoFinder program identified 18,559 orthogroups consisting of 215,304 genes. The P. waltl gene models were clustered into 15,923 orthogroups, among which 13,283 and 14,183 groups were shared with H. sapiens and X. laevis, respectively (Table 4 and Fig. 2). We found 660 orthologous groups, consisting of 2,958 gene models that are unique to P. waltl; these loci presumably represent evolutionarily young genes that have undergone considerable divergence following gene duplications. These lineage-specific genes might account for the traits unique to P. waltl. Additionally, we found 784 orthologous groups that are shared only among amphibians (P. waltl, and X. tropicalis).
Table 4.
Orthogroup overlaps
| H. sap | M. mus | G. gal | P. sin | A. car | P. waltl | X. lae | X. tro | L. cha | D. rer | |
|---|---|---|---|---|---|---|---|---|---|---|
| H. sap | 14,745 | 14,479 | 11,648 | 12,095 | 12,313 | 13,283 | 12,903 | 11,904 | 12,320 | 12,250 |
| M. mus | 14,656 | 11,603 | 12,049 | 12,259 | 13,203 | 12,839 | 11,858 | 12,263 | 12,199 | |
| G. gal | 12,162 | 11,240 | 11,129 | 11,748 | 11,536 | 10,787 | 11,054 | 11,010 | ||
| P. sin | 12,936 | 11,664 | 12,372 | 11,977 | 11,149 | 11,569 | 11,370 | |||
| A. car | 13,218 | 12,685 | 12,237 | 11,387 | 11,775 | 11,570 | ||||
| P. waltl | 15,923 | 14,183 | 12,499 | 13,091 | 12,844 | |||||
| X. lae | 15,185 | 13,043 | 12,606 | 12,556 | ||||||
| X. tro | 13,255 | 11,686 | 11,583 | |||||||
| L. cha | 13,664 | 12,078 | ||||||||
| D. rer | 13,340 | |||||||||
H. sap, H. sapiens; M. mus, M. musculus; G. gal, G. gallus; P. sin, P. sinensis; A. car, A. carolinensis; X. lae, X. laevis; X. tro, X. tropicalis; L. cha, L. chalumnae; D. rer, D. rerio.
Figure 2.
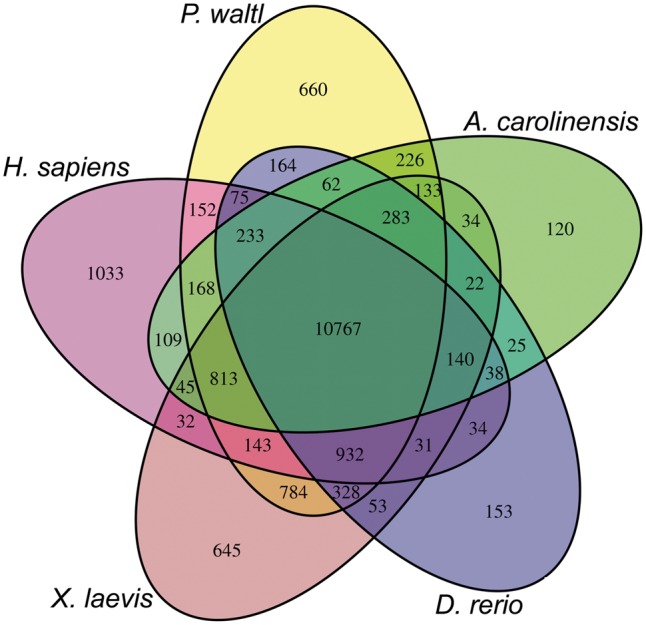
Venn diagram of shared and unique orthogroups in five vertebrates. Orthogroups were identified by clustering of orthologous groups using OrthoFinder.
Axolotl is another model salamander that belongs to a different family (within the order Urodeles) from that of P. waltl. We compared our P. waltl transcriptome with that of axolotl and identified 22,907 orthologous groups from the pairwise comparison. These two species shared 22,307 orthologous groups, while retaining 321 and 279 species-specific groups, respectively (Supplementary Fig. S1). In both organisms, these species-specific groups often contained LINE elements. Previous reports have shown that LINE elements are abundant in urodele amphibian genomes.24,26,62 Genes containing LINE elements may have evolved more rapidly, accumulating lineage-specific mutations as a result of retrotransposition events.
3.4. Gene co-expression pattern analysis
We quantified gene expression and profiled the expression patterns across all of the samples examined. A MDS plot of the 29 samples was used to depict the transcriptome similarities among the samples (Fig. 3). Samples derived from differentiated tissues/organs of adults (red dots in Fig. 3) yielded transcriptomes that were clearly distinct from those of samples from developing embryos (blue dots in Fig. 3). Samples at similar developmental stages clustered closer to each other than to those of differentiated tissues/organs; samples derived from the amputation experiments clustered on the MDS plot based on the amputated tissue. Notably, directional distances on the dimension-2 axis indicated a continuum in the direction of changes that was consistent with developmental progression. Specifically, the embryonic samples were clearly ordered along the dimension-2 axis from unfertilized egg to gastrula (St 7–12), neurula (St 15–18), and tailbud stage (St 25–28), implying that gene expression gradually changes with progression during embryogenesis.
Figure 3.
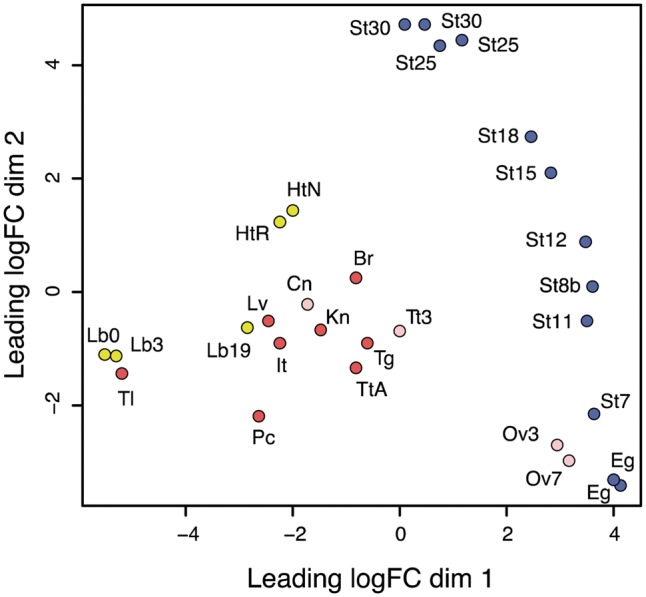
MDS plot for RNA-Seq gene expression of P. waltl tissues, organs, and embryogenesis samples. Multi-dimensional scaling (MDS) plot showing relatedness between transcript expression profiles of organs, tissues, and embryos of P. waltl at different developmental stages. Red dots represent the expression profiles of adult tissues/organs and pink dots represent those of juveniles (3 or 7 months). The labels indicating the tissues and sources are defined in Table 1. Yellow dots represent the expression profiles in samples undergoing regeneration after amputation, where the labels Lb0, Lb3, and Lb19 indicate limb or limb blastema expression profiles at 0, 3, and 19 dpa, respectively; HtR and HtN indicate expression profiles of the hearts regenerating after amputation and in unamputated controls, respectively.
To understand the co-expression relationships between genes at a systems level, we performed WGCNA. This unsupervised and unbiased analysis identified distinct co-expression modules corresponding to clusters of correlated transcripts (Fig. 4). WGCNA identified 19 co-expressed modules from the expression data spanning 29 samples; each module contained 58 to 3,585 co-expressed genes (Fig. 5). Each module represents genes with highly correlated expression profiles, either in a single tissue or in a narrow window of developmental stages. Out of 19 modules, 11 represent a tissue-specific pattern in the adult tissues: the modules indicated by different colours represent different tissues (green, lightgreen, tan, light yellow, black, pink, red, grey60, purple, blue, and brown for heart-, tail-, brain-, kidney-, liver-, pancreas-, intestine-, testicular connective tissue-, testicular gland-, adult testis-, and ovary-specific expression patterns, respectively) (Fig. 5A). Four modules were embryonic (Fig. 5B). The turquoise module was composed of 3,585 genes whose expression was observed only in unfertilized eggs, representing maternal transcripts that functioned in the early stages during P. waltl embryogenesis. Indeed, some genes such as those encoding daz-like protein (M0089929_PLEWA04), ddx25 (Dead south) (M0209664_PLEWA04), and nanos1 (M0208351_PLEWA04), which are known to be expressed maternally in Xenopus or other amphibians, are included in the turquoise module.63,64 On the other hand, modules indicated in cyan, yellow, and midnightblue exhibit zygotic expression after the mid-blastula transition (MBT; St 6–7), representing a progressive pattern showing peaks at St 8b–12 (early), St 15–18 (middle), and St 30 (late), respectively. In the regeneration experiments, limb-enriched genes were clustered into three modules based on a pattern corresponding to the responsiveness to amputation treatment: genes designated in greenyellow, magenta, and lightcyan showed peaks at 0-, 3-, and 19-day post-amputation (dpa), respectively (Fig. 5C).
Figure 4.

Gene co-expression analysis of the P. waltl transcriptome. Hierarchical cluster tree of the P. waltl genes showing co-expression modules identified using WGCNA. Modules correspond to branches are labelled by colours as indicated by the colour band underneath the tree.
Figure 5.

Co-expression gene modules. The co-expression gene modules identified using WGCNA are shown. Each grey dot represents the value of the respective module’s Eigengene. The number at the top left in each panel indicates the number of genes belonging to that module that exhibit unique expression. The modules are classified into four categories based on the expression pattern: modules associated with (A) specific tissues/organs, (B) embryogenesis, (C) regeneration processes, and (D) others. The sample abbreviations indicated by labels at the bottom of each panel are defined in Fig. 3 legend.
3.5. Major signalling pathways
Cell signalling pathways are essential for embryogenesis and organogenesis and are highly conserved in vertebrates. We inspected the gene repertoire of major signal-factor encoding genes and analysed the corresponding expression patterns at the various developmental stages. Specifically, the following genes (or gene families) were surveyed: wingless-type MMTV integration site (wnt) family (Supplementary Fig. S2A), fibroblast growth factor (fgf) (Supplementary Fig. S2B), hedgehog (hh) (Supplementary Fig. S2C), transforming growth factor-beta (tgf-beta) (Supplementary Fig. S2D), and insulin-like growth factor binding protein (igfbp) (Supplementary Fig. S2E). This analysis demonstrated that the repertoire of signalling genes of P. waltl is typical for vertebrates, but we found a few cases of urodele amphibian-specific gene duplications and potential losses.
The wnt gene family set is conserved in P. waltl as in other vertebrates. However, we detected two additional paralogous genes encoding Wnt ligands, wnt11b and wnt7-like (Supplementary Fig. S2A); we postulate that these additional loci were generated by duplication in the lineage leading to P. waltl. In vertebrates, six highly conserved igfbp family genes are typically observed, and P. waltl has all six igfbp orthologous genes (Supplementary Fig. S2E), while Xenopus lacks igfbp3 and igfbp6 orthologs.65
Orthologs of bmp2 and bmp16 were identified in the transcriptome of P. waltl, while the bmp4 ortholog was not found. Furthermore, no orthologs of bmp4 were identified in the genome and/or transcriptomes of two other urodeles, A. mexicanum, and C. pyrrhogaster (Fig. 6), suggesting a possible loss of bmp4 in the urodele lineage, although further genome-based investigation is required. The bmp16 gene was thought to be confined solely to teleost fish species.66 However, we found bmp16 in this urodele, consistent with results recently reported in coelacanths and reptiles.67 Thus, our phylogenetic analysis suggested that urodeles and anurans have lost bmp4 and bmp16, respectively, in each lineage.
Figure 6.

Phylogenetic tree of bmp2/4/16 genes among vertebrates. This phylogenetic tree was reconstructed using 34 vertebrate orthologs, including 12 bmp4, 16 bmp2, and 7 bmp16 genes; 2 ascidian bmp2/4 genes were used as the outgroup. The number at each node represents the bootstrap probability.
In summary, most of the orthologous genes for major signalling molecules were identified in P. waltl, which therefore harbours a gene repertoire typical of vertebrates, with a few exceptions. The expression patterns of the signalling molecule-encoding genes of P. waltl in some instances differed from those of Xenopus species. Further research on these differences is expected to expand our understanding of the evolution and development of amphibians.
3.6. Hox genes and their expression dynamics during embryogenesis
In tetrapod genomes, approximately 40 hox genes are present and organized into four hox clusters. hoxd12 has not been identified in any amphibians, while the hoxc3 gene is retained.68 No hoxc1 ortholog has been identified in amphibians, with the exception of caecilians.69 The genomes of the diploid X. tropicalis and the allotetraploid X. laevis harbour 38 and 75 functional hox genes, respectively.70 In this study, we identified 37 hox gene orthologs in the P. waltl transcriptome data (Fig. 7).
Figure 7.
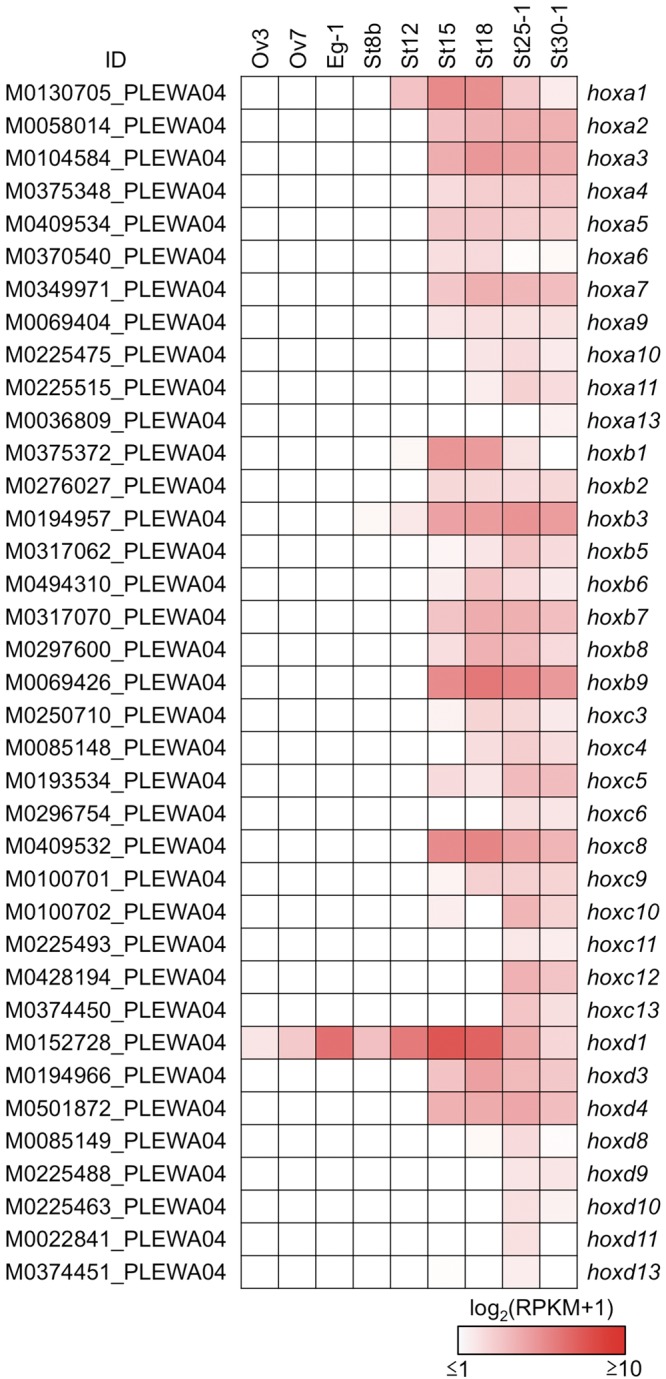
Expression profile of hox genes during oogenesis and embryogenesis. A total of 37 hox genes are listed from the assembly data of PLEWA04. The sample abbreviations indicated by the labels at the top of each panel are defined in Table 1. Ovaries were sampled at three and seven months after metamorphosis (Ov3 and Ov7, respectively). Note that P. waltl hoxc1, and hoxd12 orthologs were not identified from our transcriptome data. A hoxb13 ortholog was not confidently identified; the candidate contig (M1173232_PLEWA04) showed low homology compared with other vertebrates (Supplementary Data S5 and S6https://doi.org/10.6084/m9.figshare.7550768.v1). Most of the hox genes were zygotically activated; only the hoxd1 mRNA was synthesized during oogenesis and stored at the one-cell stage. RPKM values of each gene are indicated as a colour gradient on a log2 scale, ranging from red (maximum) to white (minimum).
The expression profile of the P. waltl hox genes during embryogenesis was similar to those of axolotl and Xenopus, suggesting that the regulation of this gene family is conserved among amphibians (Fig. 7).68,70 Anterior hox genes were activated starting around the time of the MBT; posterior hox genes were gradually up-regulated at the late embryonic stage, reflecting their spatio-temporal collinearity during embryogenesis. Interestingly, hoxd1 of P. waltl was found to be stored as a maternal mRNA at the oocyte and one-cell stage (Fig. 7), whereas the orthologous genes are expressed after MBT in axolotl and Xenopus.68,70,71
Newt genomic gigantism has been interpreted to suggest that the genome of a prototypical newt underwent species-specific whole genome duplication. Because hox genes are maintained as highly conserved gene clusters in vertebrate genomes, the number of hox clusters usually reflects the number of whole-genome duplications each genome experienced during evolution.72 In our P. waltl gene repertoire, we only found one-to-one orthologs of hox genes when comparing among available amphibian genes. This result suggested that the newt genome did not undergo additional whole-genome duplication. Similarly, the recently published axolotl giant genome showed no evidence for additional whole-genome duplication; instead, the axolotl genome has a correspondingly enlarged genic component, primarily due to the presence of especially long introns.24,26,73 In the salamander genome, expansion of LTR retrotransposons also contributes to genome gigantism.74 Such mechanisms also may have contributed to newt genome gigantism and may be related to the incredible regenerative ability of this species.
3.7. Transcriptomic features of regenerating limbs
Numerous developmental pathway genes have been reported to be reactivated during limb regeneration. For example, hox13-paralogous genes are expressed in distal regions of developing and regenerating amphibian limbs.75–78 Indeed, the present work confirmed the reactivation of the hoxa/c/d13 genes in regenerating P. waltl forelimb at 19 dpa (Fig. 8A), when the blastema is fully formed. Other transcription factor-encoding genes known to be involved in limb development and regeneration (msx1, msx2, prrx1, prrx2, tbx5, and hand2) were also drastically up-regulated at 19 dpa (Fig. 8A), indicating that the transcriptome defined here successfully recaptured the expression pattern of limb regeneration. Consistent with the results reported for axolotl,52 genes encoding RNA-binding proteins (rbmx, hnrnpa1, and sfrs1), matrix metalloproteinases (mmp3 and mmp11), and a novel proteinase inhibitor (kazald1) were up-regulated in regenerating limb in P. waltl at 19 dpa (Supplementary Fig. S3). These results suggested that these genes are commonly involved in regeneration in the two urodele amphibians.
Figure 8.
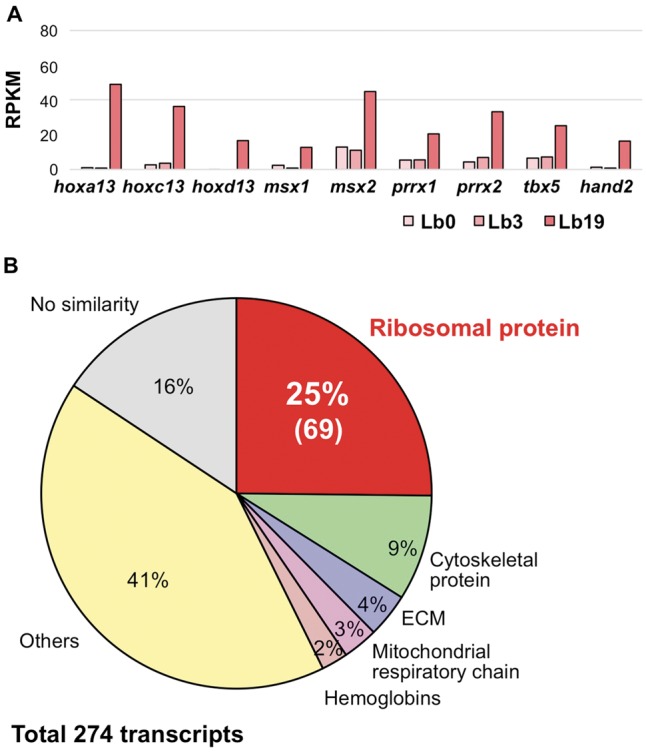
Expression profile of regenerating limb-enriched genes. (A) Expression (during regeneration) of transcription factor-encoding genes involved in limb development. The hoxa13, hoxc13, hoxd13, msx1 and 2, prrx1 and 2, tbx5, and hand2 genes were significantly up-regulated in the forelimb at 19 dpa. RPKM values of each gene were determined from the assembly data of PLEWA04. (B) Details of co-expressed genes in regenerating limb at 19 dpa. A total of 274 genes in this WGCNA module (indicated by lightcyan symbol in Fig. 5) were identified. Notably, genes encoding proteins of the large and small ribosomal subunits accounted for 25% (69 out of 274) of the genes in this module.
Intriguingly, WGCNA revealed a unique transcriptomic feature of regeneration (Fig. 5C). The lightcyan module contained 274 genes that are co-regulated in regenerating limb at 19 dpa (Fig. 8B). Notably, this module included 69 ribosomal protein-encoding genes (Fig. 8B). These transcripts were not detected in other tissues and organs transcriptome data, suggesting that these ribosomal protein-encoding genes are likely to have limb- or regeneration-specific roles. These ribosomal proteins may contribute to organ remodelling via regeneration-specific protein synthesis. Consistent with this inference, the expression of ribosomal proteins is down-regulated in Xenopus spinal cord regeneration at the non-regenerative stage after metamorphosis.79
The axolotl salamander is another model organism used for the study of regeneration, and the 32-Gb axolotl genome was recently reported.26 Despite being closely related in evolutionary terms, axolotl and P. waltl exhibit distinct regenerative properties. First, axolotl is a neotenic animal, that is, one that retains aspects of the larval state even after sexual maturation, unlike P. waltl. Second, axolotl shows restricted regenerative capacity compared with newts.24 What is the mechanism underlying the differences in metamorphosis and regenerative capacity between the two urodeles? Tanaka argued that the origin of regenerating limb muscle progenitors accompanying metamorphosis is switched when comparing axolotl and N. viridescens.80 Comparative study of the two model urodeles should enhance our understanding of the mechanisms used during regeneration in these animals. Our P. waltl reference transcriptome, together with the recently reported P. waltl draft genome sequence,24 is expected to facilitate genome-wide comparisons between these two model urodele amphibians.
3.8. Conclusions
In the present study, we built a reference gene catalogue of P. waltl using transcriptome data sets generated from a wide variety of samples. The reference gene set is expected to cover a large fraction of P. waltl protein-coding genes, as confirmed by BUSCO analysis, where 98% of universal single-copy orthologs were identified. The gene catalogue and the associated information will be valuable resources for any researchers studying P. waltl. To share these resources within the community, we have established a portal website, designated iNewt (http://www.nibb.ac.jp/imori/main/), from which these transcriptome data (including gene models, annotations, and expression profiles) can be obtained. The portal site also permits BLAST searches against these data sets. With these resources, P. waltl promises to serve as a good model to expand our understanding of the molecular mechanisms underlying regeneration. Given the newts’ unique biological properties, we further expect that our reference gene catalogue, together with the technique of highly efficient CRISPR/Cas9 genome editing,42 will open new avenues for the use of P. waltl in research besides the topic of regeneration, including developmental biology, stem cell biology, cancer research, reproductive biology, evolutionary biology, ethology, and toxico-genomics.
Supplementary Material
Acknowledgements
Computations were performed in part on the NIG supercomputer at ROIS. The Kyorin Corporation (Hyogo, Japan) kindly provided the feed for the newts.
Accession numbers
DRA006957, DRA006956, PRJDB6598, and PRJDB7442
Funding
This work was supported by MEXT/JSPS KAKENHI Grant Numbers, No.JP16H06376 to K.A., No. JP16H0125 and No. JP16K08467 to T.H., No. JP17J04796 to M.S., No. JP16K18613 to M.M., No. JP17K14980 to Y.H., No. JP16H04794 to T.T., and No. JP15K06802 to K.T.S., a Grant for Basic Science Research Projects of the Sumitomo Foundation (No. 170845 to M.M.), and a Chuo University Personal Research Grant (to A.F.). This work also was supported by the NIBB Collaborative Research Program (17-431, 18-204).
Conflict of interest
None declared.
References
- 1. Spemann H., Mangold O.. 1924, Über Induktion von Embryonalanlagen durch Implantation artfremder Organisatoren, Archiv Mikr. Anat. u Entwicklungsmechanik, 100, 599–638. [Google Scholar]
- 2. Wolff G. 1895, Entwicklungsphysiologische Studien 1. Die regeneration der Urodelenlinse, Wilhelm Roux’s Arch, Entwined Meghan Org., 1, 380–90. [Google Scholar]
- 3. Eguchi G., Abe S. I., Watanabe K.. 1974, Differentiation of lens-like structures from newt iris epithelial cells in vitro, Proc. Natl. Acad. Sci. U S A., 71, 5052–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agata K., Inoue T.. 2012, Survey of the differences between regenerative and non-regenerative animals, Dev. Growth Differ., 54, 143–52. [DOI] [PubMed] [Google Scholar]
- 5. Hayashi T., Mizuno N., Kondoh H.. 2008, Determinative roles of FGF and Wnt signals in iris-derived lens regeneration in newt eye, Dev. Growth Differ., 50, 279–87. [DOI] [PubMed] [Google Scholar]
- 6. Inoue T., Inoue R., Tsutsumi R., et al. 2012, Lens regenerates by means of similar processes and timeline in adults and larvae of the newt Cynops pyrrhogaster, Dev. Dyn., 241, 1575–83. [DOI] [PubMed] [Google Scholar]
- 7. Maki N., Takechi K., Sano S., Tarui H., Sasai Y., Agata K.. 2007, Rapid accumulation of nucleostemin in nucleolus during newt regeneration, Dev. Dyn., 236, 941–50. [DOI] [PubMed] [Google Scholar]
- 8. Maki N., Tsonis P. A., Agata K.. 2010, Changes in global histone modifications during dedifferentiation in newt lens regeneration, Mol. Vis., 16, 1893–7. [PMC free article] [PubMed] [Google Scholar]
- 9. Brockes J. P. 1997, Amphibian limb regeneration: rebuilding a complex structure’, Science, 276, 81–7. [DOI] [PubMed] [Google Scholar]
- 10. Tsutsumi R., Inoue T., Yamada S., Agata K.. 2015, Reintegration of the regenerated and the remaining tissues during joint regeneration in the newt Cynops pyrrhogaster, Regeneration (Oxf), 2, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mercer S. E., Odelberg S. J., Simon H. G.. 2013, A dynamic spatiotemporal extracellular matrix facilitates epicardial-mediated vertebrate heart regeneration, Dev. Biol., 382, 457–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kurosaka H., Takano-Yamamoto T., Yamashiro T., Agata K.. 2008, Comparison of molecular and cellular events during lower jaw regeneration of newt (Cynops pyrrhogaster) and West African clawed frog (Xenopus tropicalis), Dev. Dyn., 237, 354–65. [DOI] [PubMed] [Google Scholar]
- 13. Ikegami Y., Mitsuda S., Araki M.. 2002, Neural cell differentiation from retinal pigment epithelial cells of the newt: an organ culture model for the urodele retinal regeneration, J. Neurobiol., 50, 209–20. [DOI] [PubMed] [Google Scholar]
- 14. Chiba C., Oi H., Saito T.. 2005, Changes in somatic sodium currents of ganglion cells during retinal regeneration in the adult newt, Brain Res. Dev. Brain Res., 154, 25–34. [DOI] [PubMed] [Google Scholar]
- 15. Okamoto M., Ohsawa H., Hayashi T., Owaribe K., Tsonis P. A.. 2007, Regeneration of retinotectal projections after optic tectum removal in adult newts, Mol. Vis., 13, 2112–8. [PubMed] [Google Scholar]
- 16. Berg D. A., Kirkham M., Beljajeva A.,et al. 2010, Efficient regeneration by activation of neurogenesis in homeostatically quiescent regions of the adult vertebrate brain, Development, 137, 4127–34. [DOI] [PubMed] [Google Scholar]
- 17. Urata Y., Yamashita W., Inoue T., Agata K.. 2018, Spatio-temporal neural stem cell behavior leads to both perfect and imperfect structural brain regeneration in adult newts, Biol. Open, 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang F., Clarke J. D., Santos-Ruiz L., Ferretti P.. 2002, Differential regulation of fibroblast growth factor receptors in the regenerating amphibian spinal cord in vivo, Neuroscience, 114, 837–48. [DOI] [PubMed] [Google Scholar]
- 19. Grubb R. B. 1975, An autoradiographic study of the origin of intestinal blastemal cells in the newt, Notophthalmus viridescens, Dev. Biol., 47, 185–95. [DOI] [PubMed] [Google Scholar]
- 20. Uchida T., Hanaoka K. I.. 1949, The occurrence of oviform cells by hormonal injection in the regenerated testes of a newt, Cytologia, 15, 109–30. [Google Scholar]
- 21. Flament S., Dumond H., Chardard D., Chesnel A.. 2009, Lifelong testicular differentiation in Pleurodeles waltl (Amphibia, Caudata), Reprod. Biol. Endocrinol., 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romanova, 1959, Regenerating potential of a hypertrophied lung in Triturus cristatus, Biull. Eksp. Biol. Med., 47, 89–94. [PubMed] [Google Scholar]
- 23. Tsutsumi R., Yamada S., Agata K.. 2016, Functional joint regeneration is achieved using reintegration mechanism in Xenopus laevis, Regeneration (Oxf), 3, 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elewa A., Wang H., Talavera-López C., et al. 2017, Reading and editing the Pleurodeles waltl genome reveals novel features of tetrapod regeneration, Nat. Commun., 8, 2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gregory T. R. 2005, Synergy between sequence and size in large-scale genomics, Nat. Rev. Genet., 6, 699–708. [DOI] [PubMed] [Google Scholar]
- 26. Nowoshilow S., Schloissnig S., Fei J. F., et al. 2018, The axolotl genome and the evolution of key tissue formation regulators, Nature, 554, 50–5. [DOI] [PubMed] [Google Scholar]
- 27. Ingram A. J. 1972, The lethal and hepatocarcinogenic effects of dimethylnitrosamine injection in the newt Triturus helveticus, Br. J. Cancer, 26, 206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Okamoto M. 1997, Simultaneous demonstration of lens regeneration from dorsal iris and tumour production from ventral iris in the same newt eye after carcinogen administration, Differentiation, 61, 285–92. [DOI] [PubMed] [Google Scholar]
- 29. Ueno T., Ohgami T., Harada Y., Ueno S., Iwao Y.. 2014, Egg activation in physiologically polyspermic newt eggs: involvement of IP3 receptor, PLCγ, and microtubules in calcium wave induction, Int. J. Dev. Biol., 58, 315–23. [DOI] [PubMed] [Google Scholar]
- 30. Uribe M. C., Mejía-Roa V.. 2014, Testicular structure and germ cells morphology in salamanders, Spermatogenesis, 4, e988090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kikuyama S., Toyoda F., Ohmiya Y., Matsuda K., Tanaka S., Hayashi H.. 1995, Sodefrin: a female-attracting peptide pheromone in newt cloacal glands, Science, 267, 1643–5. [DOI] [PubMed] [Google Scholar]
- 32. Nakada T., Toyoda F., Matsuda K., et al. 2017, Imorin: a sexual attractiveness pheromone in female red-bellied newts (Cynops pyrrhogaster), Sci. Rep., 7, 41334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mouchet F., Gauthier L., Mailhes C., Ferrier V., Devaux A.. 2006, Comparative evaluation of genotoxicity of captan in amphibian larvae (Xenopus laevis and Pleurodeles waltl) using the comet assay and the micronucleus test, Environ. Toxicol., 21, 264–77. [DOI] [PubMed] [Google Scholar]
- 34. Mouchet F., Gauthier L., Baudrimont M., et al. 2007, Comparative evaluation of the toxicity and genotoxicity of cadmium in amphibian larvae (Xenopus laevis and Pleurodeles waltl) using the comet assay and the micronucleus test, Environ. Toxicol., 22, 422–35. [DOI] [PubMed] [Google Scholar]
- 35. Hirako A., Takeoka Y., Hayashi T., Takeuchi T., Furukawa S., Sugiyama A.. 2017, Effects of cadmium exposure on Iberian ribbed newt, J. Toxicol. Pathol., 30, 345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bour A., Mouchet F., Cadarsi S., et al. 2016, Toxicity of CeO2 nanoparticles on a freshwater experimental trophic chain: a study in environmentally relevant conditions through the use of mesocosms, Nanotoxicology, 10, 245–55. [DOI] [PubMed] [Google Scholar]
- 37. Makita R., Kondoh H., Okamoto M.. 1995, Transgenesis of newt with exogenous gene expression facilitated by satellite 2 repeats, Dev. Growth Differ., 37, 605–16. [DOI] [PubMed] [Google Scholar]
- 38. Ueda Y., Kondoh H., Mizuno N.. 2005, Generation of transgenic newt Cynops pyrrhogaster for regeneration study, Genesis, 41, 87–98. [DOI] [PubMed] [Google Scholar]
- 39. Irisarri I., Baurain D., Brinkmann H., et al. 2017, Phylotranscriptomic consolidation of the jawed vertebrate timetree, Nat. Ecol. Evol., 1, 1370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hayashi T., Yokotani N., Tane S., et al. 2013, Molecular genetic system for regenerative studies using newts, Dev. Growth Differ., 55, 229–36. [DOI] [PubMed] [Google Scholar]
- 41. Hayashi T., Sakamoto K., Sakuma T., et al. 2014, Transcription activator-like effector nucleases efficiently disrupt the target gene in Iberian ribbed newts (Pleurodeles waltl), an experimental model animal for regeneration, Dev. Growth Differ., 56, 115–21. [DOI] [PubMed] [Google Scholar]
- 42. Suzuki M., Hayashi T., Inoue T., et al. 2018, Cas9 ribonucleoprotein complex allows direct and rapid analysis of coding and noncoding regions of target genes in Pleurodeles waltl development and regeneration, Dev. Biol., 443, 127–36. [DOI] [PubMed] [Google Scholar]
- 43. Shi D. L., Boucaut J. C.. 1995, The chronological development of the urodele amphibian Pleurodeles waltl (Michah), Int. J. Dev. Biol., 39, 427–41. [PubMed] [Google Scholar]
- 44. Grabherr M. G., Haas B. J., Yassour M., et al. 2011, Full-length transcriptome assembly from RNA-Seq data without a reference genome, Nat. Biotechnol., 29, 644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bolger A. M., Lohse M., Usadel B.. 2014, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, 30, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haas B. J., Papanicolaou A., Yassour M., et al. 2013, De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis, Nat. Protoc., 8, 1494–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fu L., Niu B., Zhu Z., Wu S., Li W.. 2012, CD-HIT: accelerated for clustering the next-generation sequencing data, Bioinformatics, 28, 3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M.. 2015, BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs, Bioinformatics, 31, 3210–2. [DOI] [PubMed] [Google Scholar]
- 49. Hara Y., Tatsumi K., Yoshida M., Kajikawa E., Kiyonari H., Kuraku S.. 2015, Optimizing and benchmarking de novo transcriptome sequencing: from library preparation to assembly evaluation, BMC Genomics, 16, 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Götz S., García-Gómez J. M., Terol J., et al. 2008, High-throughput functional annotation and data mining with the Blast2GO suite, Nucleic Acids Res., 36, 3420–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emms D. M., Kelly S.. 2015, OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy, Genome Biol., 16, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bryant D. M., Johnson K., DiTommaso T., et al. 2017, A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors, Cell Rep., 18, 762–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bray N. L., Pimentel H., Melsted P., Pachter L.. 2016, Near-optimal probabilistic RNA-seq quantification, Nat. Biotechnol., 34, 525–7. [DOI] [PubMed] [Google Scholar]
- 54. Robinson M. D., McCarthy D. J., Smyth G. K.. 2010, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics, 26, 139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Langfelder P., Horvath S.. 2008, WGCNA: an R package for weighted correlation network analysis, BMC Bioinformatics., 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zerbino D. R., Achuthan P., Akanni W., et al. 2018, Ensembl 2018, Nucleic Acids Res., 46, D754–D61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Session A. M., Uno Y., Kwon T., et al. 2016, Genome evolution in the allotetraploid frog Xenopus laevis, Nature, 538, 336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sun Y. B., Xiong Z. J., Xiang X. Y., et al. 2015, Whole-genome sequence of the Tibetan frog Nanorana parkeri and the comparative evolution of tetrapod genomes, Proc. Natl. Acad. Sci. U S A., 112, E1257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nakamura K., Islam M. R., Takayanagi M., et al. 2014, A transcriptome for the study of early processes of retinal regeneration in the adult newt, Cynops pyrrhogaster, PLoS One, 9, e109831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Edgar R. C. 2004, MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Res., 32, 1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kumar S., Stecher G., Tamura K.. 2016, MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets, Mol. Biol. Evol., 33, 1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhu W., Kuo D., Nathanson J., et al. 2012, Retrotransposon long interspersed nucleotide element-1 (LINE-1) is activated during salamander limb regeneration, Dev. Growth Differ., 54, 673–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Owens D. A., Butler A. M., Aguero T. H., Newman K. M., Van Booven D., King M. L.. 2017, High-throughput analysis reveals novel maternal germline RNAs crucial for primordial germ cellpreservation and proper migration, Development, 144, 292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tamori Y., Iwai T., Mita K., Wakahara M.. 2004, Spatio-temporal expression of a DAZ-like gene in the Japanese newt Cynops pyrrhogaster that has no germ plasm, Dev. Genes Evol., 214, 615–27. [DOI] [PubMed] [Google Scholar]
- 65. Haramoto Y., Oshima T., Takahashi S., Ito Y.. 2014, Characterization of the insulin-like growth factor binding protein family in Xenopus tropicalis, Int. J. Dev. Biol., 58, 705–11. [DOI] [PubMed] [Google Scholar]
- 66. Feiner N., Begemann G., Renz A. J., Meyer A., Kuraku S.. 2009, The origin of bmp16, a novel Bmp2/4 relative, retained in teleost fish genomes, BMC Evol. Biol., 9, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Marques C. L., Fernández I., Viegas M. N., et al. 2016, Comparative analysis of zebrafish bone morphogenetic proteins 2, 4 and 16: molecular and evolutionary perspectives, Cell. Mol. Life Sci., 73, 841–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jiang P., Nelson J. D., Leng N., et al. 2017, Analysis of embryonic development in the unsequenced axolotl: waves of transcriptomic upheaval and stability, Dev. Biol., 426, 143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wu R., Liu Q., Meng S., Zhang P., Liang D.. 2015, Hox cluster characterization of Banna caecilian (Ichthyophis bannanicus) provides hints for slow evolution of its genome, BMC Genomics, 16, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kondo M., Kondo M., Yamamoto T., Takahashi S., Taira M.. 2017, Comprehensive analyses of hox gene expression in Xenopus laevis embryos and adult tissues, Dev. Growth Differ., 59, 526–39. [DOI] [PubMed] [Google Scholar]
- 71. Wacker S. A., Jansen H. J., McNulty C. L., Houtzager E., Durston A. J.. 2004, Timed interactions between the Hox expressing non-organiser mesoderm and the Spemann organiser generate positional information during vertebrate gastrulation, Dev. Biol., 268, 207–19. [DOI] [PubMed] [Google Scholar]
- 72. Kuraku S. 2011, Hox gene clusters of early vertebrates: do they serve as reliable markers for genome evolution? Genomics Proteomics Bioinformatics. 9, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Smith J. J., Putta S., Zhu W., et al. 2009, Genic regions of a large salamander genome contain long introns and novel genes, BMC Genomics, 10, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sun C., Shepard D. B., Chong R. A., et al. 2012, LTR retrotransposons contribute to genomic gigantism in plethodontid salamanders, Genome Biol. Evol., 4, 168–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gardiner D. M., Blumberg B., Komine Y., Bryant S. V.. 1995, Regulation of HoxA expression in developing and regenerating axolotl limbs, Development, 121, 1731–41. [DOI] [PubMed] [Google Scholar]
- 76. Endo T., Tamura K., Ide H.. 2000, Analysis of gene expressions during Xenopus forelimb regeneration, Dev. Biol., 220, 296–306. [DOI] [PubMed] [Google Scholar]
- 77. Beck C. W., Christen B., Slack J. M.. 2003, Molecular pathways needed for regeneration of spinal cord and muscle in a vertebrate, Dev. Cell, 5, 429–39. [DOI] [PubMed] [Google Scholar]
- 78. Satoh A., Endo T., Abe M., et al. 2006, Characterization of Xenopus digits and regenerated limbs of the froglet, Dev. Dyn., 235, 3316–26. [DOI] [PubMed] [Google Scholar]
- 79. Lee-Liu D., Sun L., Dovichi N. J., Larraín J.. 2018, Quantitative proteomics after spinal cord injury (SCI) in a regenerative and a nonregenerative stage in the frog, Mol. Cell. Proteomics, 17, 592–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tanaka E. M. 2016, The molecular and cellular choreography of appendage regeneration, Cell, 165, 1598–608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.