

Abstract
Eukaryotic cell division has been studied thoroughly and is understood in great mechanistic detail. Paradoxically, however, we lack an understanding of its core control process, in which the master regulator of the cell cycle, cyclin-dependent kinase (CDK), temporally coordinates an array of complex molecular events. The core elements of the CDK control system are conserved in eukaryotic cells, which contain multiple cyclin–CDK forms that have poorly defined and partially overlapping responsibilities in the cell cycle. However, a single CDK can drive all events of cell division in both mammalian and yeast cells, and in fission yeast a single mitotic cyclin can drive the cell cycle without major problems. But how can the same CDK induce different events when activated at different times during the cell cycle? This question, which has bewildered cell cycle researchers for decades, now has a sufficiently clear mechanistic answer. This Perspective aims to provide a synthesis of recent data to facilitate a better understanding of this central cellular control system.
CDK—the master regulator of the cell cycle—sends signals to control all major steps of cell division. CDK is activated at each stage of the cell cycle by binding of stage-specific cyclins. For example, in the late G1 phase, G1 cyclin–CDK complexes phosphorylate and inactivate transcriptional repressors to unleash the transcription of hundreds of genes required for cell cycle entry and S phase (Bertoli et al., 2013). Subsequently, DNA replication, centrosome duplication, spindle formation and other cell cycle processes are initiated by cyclin-CDK complexes that drive phosphorylation of multiple key regulators. Distinct cyclin-CDK complexes can be linked to specific processes, however, in most cases the responsibilities are shared among different cyclin–CDK complexes (Bloom and Cross, 2007). Importantly, often the later cyclin–CDK complexes can compensate for the absence of earlier complexes, the most extreme example being in fission yeast, where a single mitotic cyclin–CDK complex is sufficient to drive the cell cycle (Coudreuse and Nurse, 2010). To understand the scale of the networks formed by regulatory paths that emanate from the central CDK node, one can consider that in Saccharomyces cerevisiae, where the CDK system is best known, CDK is estimated to phosphorylate ∼500–700 proteins (Ubersax et al., 2003; Holt et al., 2009), which make up ∼10% of the proteome. Furthermore, the phosphoregulatory significance for roughly 100 of these targets has already been sufficiently well demonstrated both in vitro and in vivo (Enserink and Kolodner, 2010). Thus, each major cell cycle process is triggered and regulated by tens of different CDK phosphorylation events until cyclin accumulation culminates in metaphase. After the metaphase–anaphase transition, cyclin levels start to decline, and at least a fraction of CDK-controlled phosphorylations are reversed by phosphatases (Bremmer et al., 2012). These dephosphorylation events act as specific switches that drive mitotic exit (Bloom et al., 2011).
The ordering of cell cycle events is dependent on CDK activity (Coudreuse and Nurse, 2010; Swaffer et al., 2016). For example, if fission yeast cells arrested in G1 are directly released to mitotic cyclin–CDK activity, then S-phase and mitosis occur simultaneously (Swaffer et al., 2016). These findings have led to one of the core questions in cell cycle: how does activation of the same catalytic subunit induce different events at different times during the cell cycle?
A particular uniqueness of the CDK system among most of the eukaryotic protein kinases is that it is not a simple binary kinase-ON/kinase-OFF system, but rather, as elegantly proposed by Stern and Nurse in 1996, the accumulating CDK fires its downstream targets when reaching increasing thresholds at each subsequent cell cycle transition (Figure 1A; Stern and Nurse, 1996). The range of different thresholds and the complexity of the switching order could be further increased if CDK were sequentially activated by different cyclins, which might dynamically alter the functional specificity of the net mix of CDK kinase complexes (Figure 1B). Therefore, a multitude of discovered versions of cyclins (and CDKs in higher eukaryotes) have prompted researchers in past decades to pose the question of cyclin specificity in substrate recognition: are there cyclin-specific CDK substrates, and if so, what are the specificity mechanisms that the particular CDK complexes use to recognize them within the pool of all potential CDK targets?
FIGURE 1:
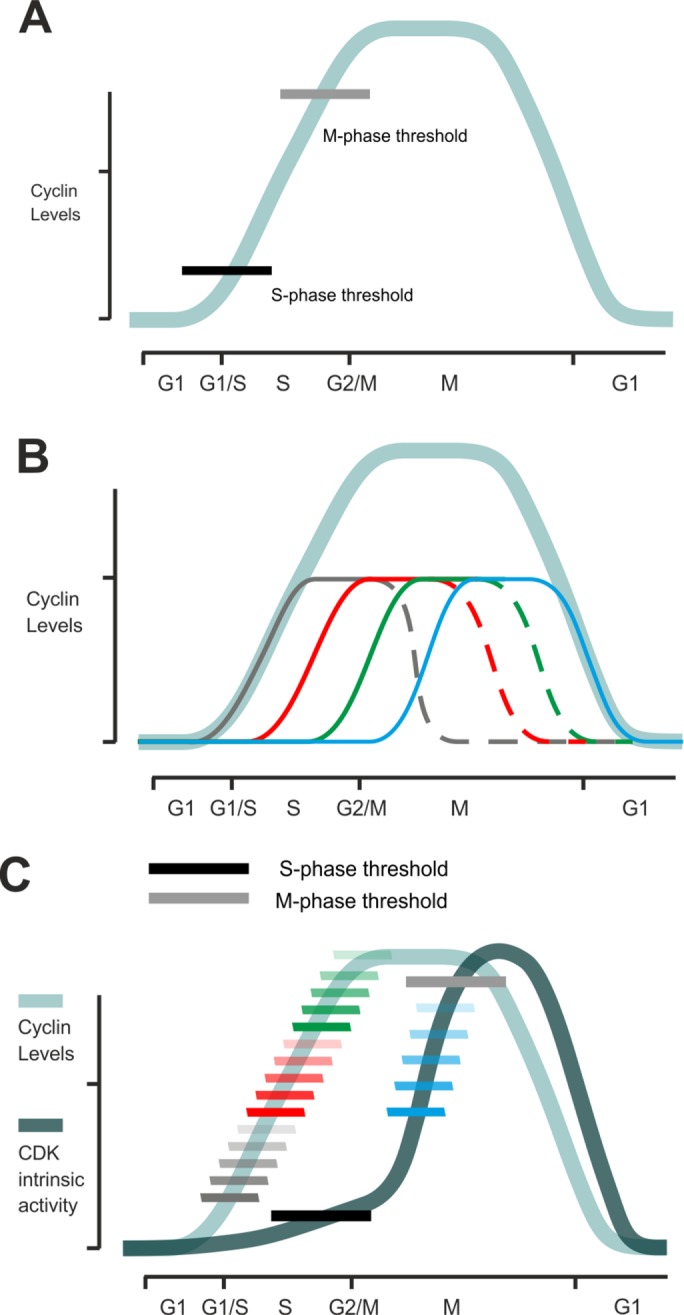
(A) The principle of the quantitative model of CDK function in the cell cycle (Stern and Nurse, 1996). Accumulating CDK activity triggers cell cycle stages at different activity thresholds. (B) The net profile of activated CDK complex (light blue line) is a sum of CDK complexes activated by cell cycle–specific cyclins. For example, in budding yeast, there are four major types of cyclins that drive the cell cycle, designated here by different colors. It is not yet clear how the quantitative model and the functional specificities of periodically expressed cyclins can be combined into a unified model. (C) When the increase in specificity of sequential CDK complexes is taken into account, the general activity profile of CDK (dark blue line) has a delayed response compared with the accumulation of cyclin–CDK complexes (light blue line). Patterns of linear docking motifs and cyclin-specific time windows can create a wide dynamic range of CDK thresholds in the cell cycle.
In fact, this question was often formulated without acknowledging the fact that protein kinases are generally very similar in their active site specificity. The whole kinome can roughly be divided into basophilic, acidophilic, and proline-directed kinases ,with large subfamilies of kinases having overlapping specificity (Miller and Turk, 2018). Although distant docking interactions provided by different cyclins could provide an additional level of specificity, the basal active site specificity of CDK kinase subunits, which recognize both full consensus motifs (S/TPXK/R) and minimal consensus motifs (S/TP), would apparently prevent absolute discrimination between targets. Because such extreme specificity among different CDK complexes was not conspicuous, the general mechanism of CDK function remained unexplained and presented several fundamental questions, including how different thresholds and cell cycle execution time are encoded into cyclin–CDK complexes and their targets.
The first possible solution to this problem was offered by early studies on cyclin specificity, which revealed that S-phase cyclins target the CDK complex to phosphorylate specific substrates via binding to a short linear motif—the RXL motif—on substrates (Schulman et al., 1998; Wilmes et al., 2004; Loog and Morgan, 2005). In S. cerevisiae, G1 cyclins have evolved a different linear motif for G1-specific CDK phosphorylation (Bhaduri and Pryciak, 2011; Kõivomägi et al., 2011b). In recent studies, we have also found that the G2- and M-phase specific cyclins can use specific docking sites of their own to enhance the phosphorylation of sites and fine tune the phosphorylation thresholds of different targets (unpublished data).
The second important finding was that the phosphorylation rate and specificity of the CDK complexes for a peptide containing a full consensus CDK phosphorylation site increase in the order of appearance of cyclins in the cell cycle (Figure 2 and Box 1; Loog and Morgan, 2005; Kõivomägi et al., 2011b). It is important to bear in mind that the kinetic analysis was performed using a short peptide substrate interacting only with the CDK active site and not with cyclins. A similar increasing specificity profile has been found in biochemical studies with mammalian CDKs (Jan Skotheim and Mardo Kõivomägi, personal communication). This mechanism ensures that early CDK complexes will not induce late cell cycle events and also makes it possible to keep the early substrates phosphorylated throughout the cell cycle, which is critical in the case of replication proteins, for example. The poor specificity of the early complexes can be compensated for by cyclin-specific docking interactions in G1- and S-phase specific CDK targets, which explains how, at very low CDK activity in the early stages of the cell cycle, efficient phosphorylation switches can be triggered (Box 1).
FIGURE 2:
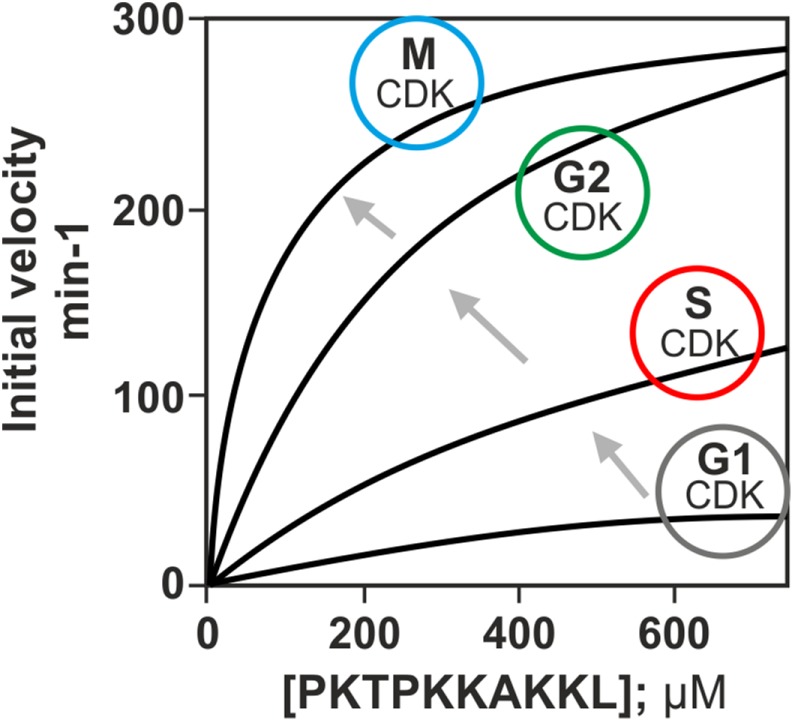
The activity of CDK complex toward a peptide containing a full consensus phosphorylation motif rises in the order of expression of cyclins in the cell cycle. The kinetic analysis was performed using a short peptide substrate that only interacts with the CDK active site and not with cyclins. Figure adapted from Kõivomägi et al., 2011b.
BOX 1: Cyclin specificity of CDK substrate phosphorylation.
Besides being activators of CDK kinase subunits, different cyclins can introduce different activation levels of cyclin–CDK complexes. Surprisingly, in both yeast and mammals, the intrinsic activity of cyclin–CDK complexes was found to increase in correlation with the appearance of the particular cyclin in the cell cycle. That is, the G1 and G1/S cyclins produce complexes with the lowest activity, which manifests in the highest KM and the lowest kcat values toward the model substrate, while the following S-, G2-, and M-phase complexes gradually lower the KM and have higher kcat values. These kinetic parameters were measured using a short model peptide containing a full consensus CDK site whose phosphorylation rate would be influenced only by the active site of CDK and not by any distant docking interaction with cyclin or Cks1. This cyclin-specific stepwise increase creates a delay in the CDK activity profile, if one considers only the active site. However, this delay can be used efficiently to create highly specific low-KM targets for early thresholds using distant docking interactions via pockets that only the early cyclins have. For example, the G1-specific cyclins Cln1/2 in budding yeast use so-called LLPP motifs in substrates for cyclin targeting, while the S-phase cyclins use the RXL motifs to lower the KM of specific substrates. This docking-induced potentiation can reach up to 100-fold when compared with the active site model peptide. In this way, the early cyclins do not prematurely trigger the later thresholds, but can phosphorylate specific substrates with cyclin-specific docking motifs at lower net CDK thresholds at early stages of the cell cycle.
Finally, in addition to cyclin-specific docking motifs, poor specificity of phosphorylation sites can be enhanced by the phosphoadaptor Cks1, which binds phosphorylated TP sites and potentiates the phosphorylation of secondary sites (Figure 3; Kõivomägi et al., 2011a, 2013; McGrath et al., 2013). Because the majority of CDK targets have multiple phosphorylation sites clustered in disordered regions, we set out to clarify the mechanistic logic of these multisite phosphorylation processes (Kõivomägi et al., 2013; Valk et al., 2014). First, we found that only phosphorylated TP sites, but not SP sites, bind to Cks1. Second, the effect of both Cks1-mediated docking and cyclin-specific docking on the phosphorylation rate is highly dependent on the relative positioning of docking sites and phosphorylation sites (Figure 3). Therefore, the docking interactions direct CDK to phosphorylate specific sites, which leads to an ordered multisite phosphorylation process. The net rate of multisite phosphorylation is governed by the distances between phosphorylation sites and docking motifs, the distribution of TP and SP sites, consensus motif elements around the phosphorylation sites, and other parameters (Kõivomägi et al., 2013). These multisite phosphorylation network parameters, or the CDK multisite phosphorylation code, could control the thresholds via the net rate of accumulation of a critical combination of phosphorylated sites required for the downstream signaling switch. For example, depending on the presence or absence, and also positioning of the Cks1-docking sites, the mechanism of multisite phosphorylation can have a high degree of processivity, or alternatively, be entirely distributive. The former means that every phosphate in the multisite cluster is added without the CDK complex dissociating between the subsequent phosphorylation events. In contrast, in the case of the distributive mechanism, the kinase complex dissociates between every pair of phosphorylation events. With respect to CDK thresholds, a more processive mechanism reaches the fully phosphorylated state—the output signal, at lower CDK activity. Taken together, the combination of cyclin-specific docking motifs and Cks1-dependent phosphorylation mechanism enables differential phosphorylation of a wide range of CDK substrates (Figures 1C and 3). Because CDK phosphorylation clusters are almost exclusively located in intrinsically disordered regions of the proteins (Holt et al., 2009), the coding can be entirely linear, using short linear motifs (SLiMs: CDK phosphorylation motifs, cyclin docking motifs, phosphodegrons, etc.), and linkers, where amino acids can be counted for necessary distances between the SLiMs.
FIGURE 3:
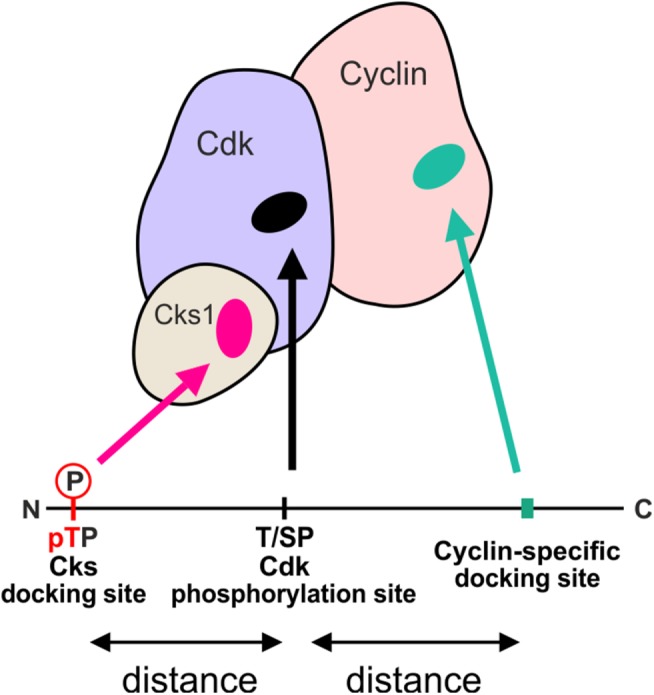
Schematic diagram showing the main interactions between substrate proteins and the CDK complex that determine the phosphorylation rate and specificity. The CDK active site phosphorylates full consensus motifs (S/TPXK/R) and minimal consensus (S/TP) motifs. The phosphorylation rate of a site can be increased by two docking interactions: Cks1 can bind to phosphorylated TP sites and cyclins can interact with substrates via specific short linear motifs. In both cases, the effect of docking is dependent on the relative positioning of docking sites and phosphorylation sites along the disordered substrate.
In a proteomics study aimed at analyzing the phosphorylation dynamics of CDK targets in Schizosaccharomyces pombe, it was found that CDK substrates can be divided into early, middle, and late targets (Swaffer et al., 2016). Importantly, phosphorylation of late targets required higher CDK activity than for earlier targets. Surprisingly, no correlation was found between the substrate’s sensitivity to CDK activity and the phosphorylation motif being either minimal or full consensus. This finding was unexpected, because early work on CDK specificity had shown a great increase in the phosphorylation rate of peptides that contained the full-consensus motif in comparison with ones that had a minimal-consensus motif (Songyang et al., 1994). This suggests that the protein context, including the linear patterns of helper docking motifs, may be even more important than the specificity of phosphorylation sites themselves.
Discrimination between early and late CDK substrates can also be driven by the differing phosphatase activity toward these sets of targets. For example, the S. cerevisiae phosphatase PP2ACdc55 specifically counteracts phosphorylation of threonine residues, and this leads to threonine-based CDK sites being phosphorylated at higher CDK activity and therefore later in the cell cycle than serine-based sites (Godfrey et al., 2017). Studies in S. pombe, however, have shown that while threonines are indeed dephosphorylated faster than serines, the dephosphorylation rates of early and late targets are similar (Swaffer et al., 2016). Therefore, while there is significant evidence that phosphatase activity denies early phosphorylation of some CDK targets (Ndd1, Net1; Queralt et al., 2006; Godfrey et al., 2017), it is not clear whether differential phosphatase activity leads to global ordering of CDK thresholds. In addition, phosphatase activity, in combination with cyclin-specific docking interactions, has been shown to play a role in ordering the thresholds during mitotic exit. Because the S-phase cyclin is degraded in metaphase before the M-phase cyclin, S-CDK-specific targets were found to be dephosphorylated earlier (Jin et al., 2008).
In conclusion, the system comprising a mix of CDK activities, mediated by different cyclin–CDK complexes, with changing but also a common baseline specificity, is apparently different from a hypothetical system of binary switches triggered by orthogonal kinase activities activated at each cell cycle stage. For example, in budding yeast, the changes in specificity are caused by different cyclins, while the common baseline activity occurs because these periodically expressed cyclins activate a common Cdk1 kinase. Most importantly, if there were an exclusively specific kinase pathway evolved for each stage of the cell cycle, then it would be difficult to maintain phosphorylation of the targets that should be phosphorylated during the whole span of S and M phases. These include the sets of targets that safeguard the mechanisms that prevent relicensing, rereplication, reduplication, and the like—the core principles of the once-per-cell-cycle. Thus, due to the special character of the cell cycle, a central control system with a set of ON–OFF-style branched kinase pathways with absolute specificity would not be an optimal solution. Instead, a fine ladder of thresholds encoded into the substrates by highly cyclin-specific docking motifs, different phosphorylation site patterns, and Cks1-binding sites would create a virtually unlimited set of CDK input–output functions and different mixtures of switching orders. However, because the mitotic cyclin, like a central administrator, has a key to every threshold, the system is safe and robust to withstand anomalies in cyclin accumulation waves. Simultaneous fine tuning and robustness of the CDK system are especially important for single-cell organisms such as yeasts that depend on competitive growth. Indeed, the extreme complexity of processive multisite phosphorylation schemes has been found in budding yeast to coordinate thresholds with the highest precision between Start and G1/S, a stage spanning just 10–15 min (Kõivomägi et al., 2011a; Repetto et al., 2018). In this respect, the single–mitotic cyclin system (Coudreuse and Nurse, 2010) is like a clock that shows hours, while the full cyclin-specific system ticks with minute or even second precision (Figure 1C).
Acknowledgments
We thank D. Kellogg for valuable comments on the manuscript. The work was supported by European Research Council (ERC) Consolidator Grant 649124, Phosphoprocessors (ML), and Estonian Science Agency Grant IUT2-21.
Abbreviations used:
- CDK
cyclin-dependent kinase
- PP2A
protein phosphatase 2A
Footnotes
REFERENCES
- Bertoli C, Skotheim JM, de Bruin RAM. (2013). Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol , 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaduri S, Pryciak PM. (2011). Cyclin-specific docking motifs promote phosphorylation of yeast signaling proteins by G1/S Cdk complexes. Curr Biol , 1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J, Cristea IM, Procko AL, Lubkov V, Chait BT, Snyder M, Cross FR. (2011). Global analysis of Cdc14 phosphatase reveals diverse roles in mitotic processes. J Biol Chem , 5434–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J, Cross FR. (2007). Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol , 149–160. [DOI] [PubMed] [Google Scholar]
- Bremmer SC, Hall H, Martinez JS, Eissler CL, Hinrichsen TH, Rossie S, Parker LL, Hall MC, Charbonneau H. (2012). Cdc14 phosphatases preferentially dephosphorylate a subset of cyclin-dependent kinase (Cdk) sites containing phosphoserine. J Biol Chem , 1662–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudreuse D, Nurse P. (2010). Driving the cell cycle with a minimal CDK control network. Nature , 1074–1079. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Kolodner RD. (2010). An overview of Cdk1-controlled targets and processes. Cell Div , 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey M, Touati SA, Kataria M, Jones A, Snijders AP, Uhlmann F. (2017). PP2ACdc55 phosphatase imposes ordered cell-cycle phosphorylation by opposing threonine phosphorylation. Mol Cell , 393–402.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO. (2009). Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science , 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Liu H, Liang F, Rizkallah R, Hurt MM, Wang Y. (2008). Temporal control of the dephosphorylation of Cdk substrates by mitotic exit pathways in budding yeast. Proc Natl Acad Sci USA , 16177–16182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kõivomägi M, Örd M, Iofik A, Valk E, Venta R, Faustova I, Kivi R, Balog ERM, Rubin SM, Loog M. (2013). Multisite phosphorylation networks as signal processors for Cdk1. Nat Struct Mol Biol , 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kõivomägi M, Valk E, Venta R, Iofik A, Lepiku M, Balog ERM, Rubin SM, Morgan DO, Loog M. (2011a). Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature , 128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kõivomägi M, Valk E, Venta R, Iofik A, Lepiku M, Morgan DO, Loog M. (2011b). Dynamics of Cdk1 substrate specificity during the cell cycle. Mol Cell , 610–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loog M, Morgan DO. (2005). Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature , 104–108. [DOI] [PubMed] [Google Scholar]
- McGrath DA, Balog ERM, Kõivomägi M, Lucena R, Mai M V, Hirschi A, Kellogg DR, Loog M, Rubin SM. (2013). Cks confers specificity to phosphorylation-dependent CDK signaling pathways. Nat Struct Mol Biol , 1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CJ, Turk BE. (2018). Homing in: mechanisms of substrate targeting by protein kinases. Trends Biochem Sci , 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queralt E, Lehane C, Novak B, Uhlmann F. (2006). Downregulation of PP2ACdc55 phosphatase by separase initiates mitotic exit in budding yeast. Cell , 719–732. [DOI] [PubMed] [Google Scholar]
- Repetto MV, Winters MJ, Bush A, Reiter W, Hollenstein DM, Ammerer G, Pryciak PM, Colman-Lerner A. (2018). CDK and MAPK synergistically regulate signaling dynamics via a shared multi-site phosphorylation region on the scaffold protein Ste5. Mol Cell , 938–952.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman BA, Lindstrom DL, Harlow ED. (1998). Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc Natl Acad Sci USA , 10453–10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, Cantley LC. (1994). Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr Biol , 973–982. [DOI] [PubMed] [Google Scholar]
- Stern B, Nurse P. (1996). A quantitative model for the Cdc2 control of S phase and mitosis in fission yeast. Trends Genet , 345–350. [PubMed] [Google Scholar]
- Swaffer MP, Jones AW, Flynn HR, Snijders AP, Nurse P. (2016). CDK substrate phosphorylation and ordering the cell cycle. Cell , 1750–1761. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. (2003). Targets of the cyclin-dependent kinase Cdk1. Nature , 859–864. [DOI] [PubMed] [Google Scholar]
- Valk E, Venta R, Örd M, Faustova I, Kõivomägi M, Loog M. (2014). Multistep phosphorylation systems: tunable components of biological signaling circuits. Mol Biol Cell , 3456–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmes GM, Archambault V, Austin RJ, Jacobson MD, Bell SP, Cross FR. (2004). Interaction of the S-phase cyclin Clb5 with an “RXL” docking sequence in the initiator protein Orc6 provides an origin-localized replication control switch. Genes Dev , 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]