

Abstract
The target of rapamycin kinase complex 1 (TORC1) regulates cell growth and metabolism in eukaryotes. In Saccharomyces cerevisiae, TORC1 activity is known to be controlled by the conserved GTPases, Gtr1/2, and movement into and out of an inactive agglomerate/body. However, it is unclear whether/how these regulatory steps are coupled. Here we show that active Gtr1/2 is a potent inhibitor of TORC1-body formation, but cells missing Gtr1/2 still form TORC1-bodies in a glucose/nitrogen starvation-dependent manner. We also identify 13 new activators of TORC1-body formation and show that seven of these proteins regulate the Gtr1/2-dependent repression of TORC1-body formation, while the remaining proteins drive the subsequent steps in TORC1 agglomeration. Finally, we show that the conserved phosphatidylinositol-3-phosphate (PI(3)P) binding protein, Pib2, forms a complex with TORC1 and overrides the Gtr1/2-dependent repression of TORC1-body formation during starvation. These data provide a unified, systems-level model of TORC1 regulation in yeast.
INTRODUCTION
The target of rapamycin kinase complex I (TORC1) is a key regulator of cell growth and metabolism in eukaryotes (Loewith and Hall, 2011; Gonzalez and Hall, 2017; Saxton and Sabatini, 2017). In the presence of progrowth hormones and ample nutrients, TORC1 is active and drives protein, lipid, and nucleotide synthesis by phosphorylating a wide range of proteins (Bodenmiller et al., 2010; Hsu et al., 2011; Loewith and Hall, 2011; Robitaille et al., 2013; Gonzalez and Hall, 2017; Saxton and Sabatini, 2017). In contrast, when hormone, nutrient, or energy levels fall—or cells are exposed to noxious stress—TORC1 is inhibited, causing the cell to switch from anabolic to catabolic metabolism and eventually enter a quiescent state (Barbet et al., 1996; Noda and Ohsumi, 1998; Duvel et al., 2010; Loewith and Hall, 2011; Gonzalez and Hall, 2017; Saxton and Sabatini, 2017).
TORC1 is made up of three essential proteins: mTOR, Raptor, and mLst8 in humans, and Tor1, Kog1, and Lst8 in yeast (Kim et al., 2002; Loewith et al., 2002). Biochemical and structural studies show that these proteins form a stable, ring-like structure containing two copies of each subunit (Adami et al., 2007; Yip et al., 2010; Aylett et al., 2016; Yang et al., 2017). Kog1/Raptor recruits substrates to the TOR kinase (Tor1) and is required for the regulation of Tor1 activity (Hara et al., 2002; Kim et al., 2002; Aylett et al., 2016). Lst8, however, binds directly to Tor1 and may help stabilize the TOR complex (Kim et al., 2002). In yeast, TORC1 also includes the nonessential and poorly characterized subunit Tco89 (Reinke et al., 2004).
TORC1 activity is controlled by a variety of proteins and pathways (Loewith and Hall, 2011; Gonzalez and Hall, 2017; Nicastro et al., 2017; Saxton and Sabatini, 2017), but in the model organism Saccharomyces cerevisiae, two major modes of regulation have been identified.
First, nitrogen and amino acid signals are transmitted to TORC1 via a pair of small GTPases called Gtr1 and Gtr2 (Rag A/B and Rag C/D in humans) (Kim et al., 2008; Sancak et al., 2008; Binda et al., 2009). Gtr1 and 2 form a heterodimer that is tethered to the vacuolar/lysosomal membrane by a palmitoylated and myristoylated complex made up of Ego1, Ego2, and Ego3 (EGO-TC or Ragulator in humans) (Roth et al., 2006; Binda et al., 2009; Nadolski and Linder, 2009; Sancak et al., 2010; Powis et al., 2015). In the presence of abundant nitrogen/amino acids, Gtr1 and 2 are in their GTP and GDP bound forms, respectively, and bind tightly to Kog1/Raptor and Tco89 (Gao and Kaiser, 2006; Sancak et al., 2008; Binda et al., 2009). In contrast, when nitrogen/amino acid levels fall, Npr2, Npr3, and Iml1 (SEACIT in yeast and GATOR1 in humans) bind to Gtr1 and induce its GTPase activity (Neklesa and Davis, 2009; Bar-Peled et al., 2013; Panchaud et al., 2013; Su et al., 2017). The resulting switch—from GTP bound Gtr1, to GDP bound Gtr1—then causes a conformational change that weakens the interaction between Gtr1/2 and TORC1 and rapidly inhibits (τ = 1–2 min) TORC1 signaling (Sancak et al., 2008; Binda et al., 2009; Hughes Hallett et al., 2014).
TORC1 is also rapidly (τ = 1–2 min) inactivated in glucose starvation conditions, but this transition occurs normally in strains carrying mutations that lock Gtr1/2 in their active (GTP and GDP bound) conformations (Urban et al., 2007; Hughes Hallett et al., 2014), indicating either that glucose does not signal through Gtr1/2 (in contrast to the Rags) (Efeyan et al., 2013) or that other regulators dominate the response.
Second, glucose and nitrogen starvation cause Kog1, Tco89, and—as shown in recent studies—Tor1 (Kira et al., 2016; Prouteau et al., 2017) to move from their position spread across the vacuolar membrane to a single focus on the edge of the vacuole (Hughes Hallett et al., 2015). TORC1-body formation is sped-up ∼20-fold by Snf1/AMPK-dependent phosphorylation of Kog1 at Ser 491 and 494, and more than 100-fold by two glutamine-rich prion-like domains in Kog1 (Hughes Hallett et al., 2015). Analysis of strains with mutations that limit TORC1-body formation show that the TORC1-bodies are not required for rapid TORC1 inactivation (in contrast to Gtr1/2) but rather increase the threshold for TORC1 activation in cells that have been starved for a significant period of time (Hughes Hallett et al., 2015). This creates hysteresis in the TORC1 pathway to help ensure that cells remain committed to a starvation state until they are exposed to optimal conditions.
It is currently unclear, however, whether or how Gtr1/2 and Ego1-3 (EGOC) influence TORC1 localization or what signaling proteins/pathways besides Snf1 regulate TORC1-body formation. As a result, there is no unified model of TORC1 regulation in yeast.
Here, to address these questions, we examine the influence that Gtr1/2, Ego1-3, and 209 additional proteins (including nearly all nonessential kinases and phosphatases in yeast) have on TORC1 agglomeration using fluorescence microscopy. We show that the active (GTP bound) form of Gtr1 is a potent inhibitor of TORC1-body formation, but cells missing Gtr1 and Gtr2 still form TORC1-bodies in a glucose/nitrogen starvation-dependent manner. We also identify 13 new regulators of TORC1-body formation and show that they act in two steps: first, the protein kinase C (PKC) pathway, Sit4, Gcn2, and Cka1 work together with SEACIT to override the Gtr1/2-dependent inhibition of TORC1 agglomeration. Then Ypk1, Cmk1, Yak1, Sak1 (all stress/starvation signaling proteins) and the intrinsically disordered TORC1 subunit Tco89, help drive TORC1 into bodies. Finally, we show that the conserved phosphatidylinositol-3-phosphate (PI(3)P) binding protein, Pib2, forms a complex with TORC1 and overrides the Gtr1/2-dependent repression of TORC1-body formation during starvation.
Taken together, our data provide a comprehensive, network-level, view of TORC1 regulation and show that starvation signals have to be transmitted through Gtr1/2, before Pib2 and other stress/starvation signaling proteins can trigger TORC1-body formation and lock the TORC1 pathway in an inactive state.
RESULTS
EGOC-dependent control of Kog1-body formation
To measure the influence that EGOC has on TORC1-body formation, we followed the localization of Kog1 tagged with yellow fluorescent protein (Kog1-YFP) in a wild-type strain and a strain missing Gtr1/2 (and thus all interactions between EGOC and TORC1). Cells were kept in log phase growth for at least 12 h in synthetic medium with dextrose (SD medium) and loaded into chamber slides, and three-dimensional images were acquired using a fluorescence microscope. The cells were then washed with medium missing glucose, and additional images were acquired at regular time intervals.
The wild-type strain formed bodies in two phases; 8 ± 5% of cells had bodies at time zero, and this increased to ∼50% with a time constant of ∼10 min and then >90% with a time constant of 260 ± 100 min (Figure 1, A and B) (Hughes Hallett et al., 2015). In contrast, the gtr1Δgtr2Δ strain formed bodies in a single phase: 32 ± 4% of cells had bodies at time zero, and this increased to 73 ± 1% of cells with a time constant of 9 ± 2 min (Figure 1, A and B). These data show that deleting Gtr1/2 increases the fraction of cells containing TORC1-bodies in nutrient-replete conditions (from 8 ± 5% to 32 ± 4%) and speeds up body formation during starvation. Thus, EGOC acts to repress TORC1-body formation, especially in nutrient-replete conditions.
FIGURE 1:

EGOC regulates TORC1-body formation. (A) Kog1-YFP localization before (log growth) and 60 min after glucose starvation in the wild-type strain, and strains missing Gtr1/2 (gtr1Δgtr2Δ), Npr2 (npr2Δ), or carrying a constitutively active GTR1Q65L allele (Gtr1on). The dashed lines show the position of each cell in the bright-field image. (B) Time-course data showing the fraction of cells containing Kog1-YFP puncta in the wild-type strain and strains missing Npr2, Gtr1, Gtr2, Gtr1/2, Ego1, Ego3, or carrying a constitutively active Gtr1 allele (Gtr1on), during glucose starvation (as labeled). Each time point shows the average and SD from experiments carried out on three different days, with 75–300 cells per time point per replicate. The solid lines show the best fit to a single exponential for the gtr1Δgtr2Δ, ego1Δ, ego3Δ, gtr1Δ, and gtr2Δ strains, a double exponential for the wild-type strain, and a straight line for the npr2Δ and Gtr1on strains. The broken lines in the bottom panels show the best fit to the wild-type data (from the top panel) for comparison.
Next, to determine whether starvation signals transmitted through EGOC control TORC1-body formation, we followed Kog1-YFP localization in strains carrying mutations that lock Gtr1 in its active state (npr2Δ and GTR1Q65L) (Binda et al., 2009; Panchaud et al., 2013). These strains had gross defects in TORC1-body formation in both glucose and nitrogen starvation conditions (Figure 1, A and B, and Supplemental Figure S1), indicating 1) that the active form of EGOC—normally found in nutrient-replete conditions—is a potent inhibitor of TORC1-body formation and 2) that EGOC is converted into its inactive form during both glucose and nitrogen starvation, and this in turn allows TORC1 to form bodies.
Kog1 and EGOC colocalize during starvation
To learn more about the influence that EGOC has on TORC1-body formation, we followed EGOC localization during glucose and nitrogen starvation.
Recent studies have shown that EGOC localizes to both the vacuolar membrane, and distinct puncta associated with the vacuolar membrane, during growth in nutrient-replete (SD) medium (Binda et al., 2009; Kira et al., 2014, 2016). Nitrogen starvation then increases the fraction of cells with EGOC puncta, while mutants that disrupt EGOC decrease the fraction of cells with EGOC puncta (Kira et al., 2016).
To confirm and extend these results, we tagged Gtr1, Ego1, Ego2, and Ego3 with YFP and followed their movement in both glucose and nitrogen starvation conditions, using the same procedures described above for the Kog1-YFP experiments.
In line with previous results, we found that EGOC localizes to the vacuolar membrane in all cells, and to foci located on the vacuolar membrane in around 70% of cells, during log phase growth (Figure 2, A and B). EGOC then moves into foci in an additional 10 and 20% of cells during nitrogen and glucose starvation, respectively, but maintains vacuolar membrane localization in all cells (Figure 2, A and B).
FIGURE 2:

EGOC forms puncta in log growth and starvation conditions. (A) Gtr1-YFP and Ego1-YFP localization before (log growth), and 60 min after, glucose and nitrogen starvation. The dashed lines show the position of each cell in the bright-field image. (B) Time-course data showing the fraction of cells containing Ego1-YFP, Ego2-YFP, Ego3-YFP, and Gtr1-YFP puncta during glucose and nitrogen starvation (top and bottom panels, respectively). (C) Time-course data showing the fraction of cells containing Gtr1-YFP, Ego1-YFP, and Ego2-YFP puncta in strains missing Gtr1, Gtr2, Ego1, or Ego3 (as labeled). For B and C, each time point shows the average and SD from experiments carried out on two different days, with 100–300 cells per time point per replicate. (D) Localization of Gtr1-YFP and Kog1-DuDre (top panel) and Ego2-YFP and Kog1-DuDre (bottom panel) after 60 min of glucose starvation.
The finding that EGOC foci are formed during log phase growth—before TORC1 moves into bodies—led us to ask whether TORC1-bodies assemble at the EGOC foci. To do this, we examined the localization of Kog1 labeled with red fluorescent protein (RFP) (Kog1-DuDre) in strains carrying Gtr1-YFP and Ego2-YFP in glucose starvation conditions. These experiments showed that TORC1 and EGOC foci colocalize in the vast majority of cells containing TORC1 bodies (98% overlap between Ego2 and Kog1 foci (n = 97), and 95% overlap between Gtr1 and Kog1 foci (n = 103), in cells containing Kog1-foci) (Figure 2D). Thus, TORC1 interacts with the pool of EGOC located on the vacuolar membrane in log growth conditions and then moves to the pool of EGOC located in foci during starvation.
The role of EGOC foci in TORC1-body formation
The microscopy data described above suggest that EGOC foci act as nucleation sites for TORC1-body formation. If this is true, then eliminating the EGOC foci should slow down TORC1-body formation in strains that maintain interactions between Gtr1/2 and TORC1.
To test this prediction, we followed Kog1-YFP localization in strains missing Gtr1 alone and Gtr2 alone, since they do not form EGOC foci (Figure 2C) (Kira et al., 2016) but may maintain weak interactions between Gtr2 and TORC1 and then Gtr1 and TORC1, respectively. These strains form more TORC1-bodies than the wild-type strain in log growth conditions (indicating that the repression of TORC1-body formation by active EGOC requires an intact Gtr1/2 complex) but then form TORC1-bodies (7–11 times) slower than the wild-type strain (Figure 1B; τ = 107 ± 27 and 67 ± 37 min for the gtr1Δ and gtr2Δ strains vs. 11 ± 3 min for the wild-type strain).
We then followed Kog1-YFP localization in strains missing Ego1 alone and Ego3 alone, since they also fail to form EGOC foci (Figure 2C) but probably maintain some interaction between Gtr1/2 and TORC1. These strains form the same number of TORC1-bodies as the wild-type strain in log growth conditions (indicating that Gtr1/2 represses TORC1-body formation efficiently even when it is not tethered to the vacuolar membrane), and then—as predicted—form bodies (6–10 times) slower than the wild-type strain (Figure 1B; τ = 58 ± 19 and 99 ± 52 min for ego1Δ and ego3Δ strains vs. 11 ± 3 min for the wild-type strain).
Thus, while the main role of EGOC is to repress TORC1-body formation during log phase growth, it appears that interactions between TORC1 and the EGOC in foci (or TORC1 and other proteins in the EGOC foci) helps speed up TORC1-body formation during starvation (see the Supplemental Text and Supplemental Figure S2 for further discussion).
Activators of TORC1-body formation
To identify additional regulators of TORC1-body formation, we measured Kog1-YFP localization in 209 strains—each missing one of 139 nonessential kinases/phosphatases in yeast (Breitkreutz et al., 2010) or one of 70 genes of interest—many of which were found to interact with the TORC1 pathway in a previous screen (Worley et al., 2015). Cells were kept in log phase growth for at least 12 h and transferred into glucose-free medium for 60 min, and then three-dimensional images were acquired using a fluorescence microscope. Most of the strains formed more TORC1-bodies than the wild-type parental strain during starvation, but there were 40 outliers—all with significant (greater than twofold) defects in TORC1-body formation (Figure 3A). To follow this up, we grew the 45 strains that formed the fewest bodies in the screen (green bar, Figure 3A), and the 13 strains that formed the largest number of bodies in the screen (red bar, Figure 3A), and measured Kog1-YFP localization as a function of time in both glucose and nitrogen starvation conditions (Figure 3B). These experiments confirmed the findings from the initial screen and led to the identification of 13 strains that have dramatic defects in TORC1-body formation (<10% of cells with bodies after 2 h of glucose starvation vs. 78% of cells with bodies in the wild-type strain), namely pib2Δ, rom2Δ, tco89Δ, sit4Δ, yak1Δ, ypk1Δ, bck1Δ, sak1Δ, gcn2Δ, cmk1Δ, ypl150wΔ, cka1Δ, and slt2Δ (Figure 3, B and C). The defect in the pib2Δ strain was especially pronounced (blue line, Figure 3C), and in subsequent experiments we found that <5% of pib2Δ cells form TORC1-bodies after 24 h of starvation (vs. >90% in the wild-type strain).
FIGURE 3:

Screen for regulators of TORC1-body formation. (A) Histogram summarizing the influence that deleting 209 different genes (including all 139 nonessential kinases and phosphatases in yeast) has on Kog1-YFP foci formation. The score for each strain is based on the percentage of cells that form foci after 60 min of glucose starvation (based on data from at least 100 cells) and is normalized using wild-type data to calculate fold change. The raw data for each strain are included in Supplemental Table S1. (B) Heat map showing time-course data for the 45 strains that formed the fewest bodies in the initial screen (green/blue gene names), the 13 strains that formed the most bodies in the initial screen (red gene names), and the wild-type strain (wt), in glucose and nitrogen starvation conditions. Each colored square shows the fraction of cells with Kog1-puncta at a particular time point, based on images of at least 100 cells. (C) Time-course data for the 13 strains with the largest defects in body formation (green and blue lines; gene names shown in bold in B), and the wild-type strain (black line). (D, E) Quantification of band-shift data measuring Sch9 phosphorylation during glucose starvation in strains missing key regulators of TORC1-body formation. The data were normalized so that the level of Sch9 phosphorylation in the wild-type strain at time zero is set at 1.0. The raw bandshift data for each strain are shown in Supplemental Figure S3.
To determine whether the genes that regulate TORC1-body formation also control the rapid inactivation of TORC1, we measured TORC1 activity during glucose starvation in 14 different strains—each missing one gene that is important for TORC1-body formation (including all 13 genes listed above). To do this, we followed the phosphorylation of the key TORC1 substrate, Sch9, during glucose starvation (Urban et al., 2007; Hughes Hallett et al., 2014). These experiments showed that TORC1 was repressed as normal (or somewhat overrepressed) in 12 of these strains (Figure 3D and Supplemental Figure S3), and while the other two strains (rom2Δ and sak1Δ) do have defects in TORC1 repression (Figure 3E and Supplemental Figure S3), the defects are minor compared with those caused by the deletion of Snf1/AMPK—a gene we previously identified as regulating TORC1 activity in glucose starvation conditions (Figure 3E) (Hughes Hallett et al., 2014). Thus, the major regulators of TORC1-body formation identified in this study act downstream of, and/or in parallel with, the canonical TORC1 regulatory circuit to control TORC1 agglomeration and lock TORC1 in an inactive state.
Activators of TORC1-body formation acting upstream and downstream of Gtr1/2
Activators of TORC1-body formation could act 1) at, or above, the level of EGOC to promote release from the Gtr1/2-dependent repression of TORC1-body formation or 2) at a subsequent step to drive TORC1 agglomeration. To distinguish between these possibilities, we created a series of double mutant strains, each missing Gtr1 and one of the 13 regulators of TORC1-body formation identified in the screen. We also made a strain missing Gtr1 and Npr2 and a strain missing Gtr1 and carrying glutamine to alanine mutations in both prion-like domains of Kog1 (PrDm1+2) (Hughes Hallett et al., 2015) as controls for scenarios 1 and 2, respectively. All of these strains grew relatively well, except for bck1Δgtr1Δ (which we continued to study) and ypl150wΔgtr1Δ (which we dropped from the experiment), as described under Materials and Methods.
Measurement of TORC1-body formation in the double mutants revealed that the newly identified regulators fall into two distinct groups. Deletion of Gtr1 rescued the TORC1-body formation defects found in the bck1Δ, slt2Δ, and rom2Δ strains (all proteins in the PKC pathway) (Levin, 2011), as well as the defects found in the gcn2Δ, pib2Δ, cka1Δ, npr2Δ, and sit4Δ strains (Figure 4). In contrast, deletion of Gtr1 had very little impact on TORC1-body formation in the ypk1Δ, cmk1Δ, sak1Δ, tco89Δ, yak1Δ, and prDm1+2 strains (Figure 4). Thus, 7/13 genes we identified in the screen work together with Npr2 to promote release from the Gtr1/2-dependent repression of TORC1-body formation, while the remaining genes, including the intrinsically disordered TORC1 subunit Tco89, drive the subsequent steps in TORC1 agglomeration along with the prionlike domains in Kog1.
FIGURE 4:
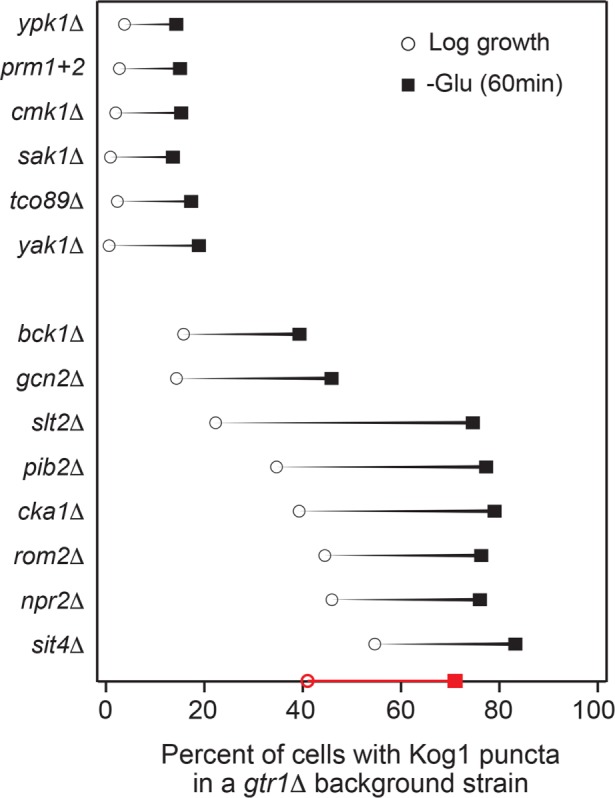
Cooperation between Gtr1 and key regulators of TORC1-body formation. Impact that deleting key regulators of TORC1-body formation, or mutating the prion domains in Kog1 (PrDm1+2), has on Kog1-YFP puncta formation in a gtr1Δ background. Black circles show the percentage of cells with bodies in log growth cultures, while black squares show the percentage of cells with bodies after 60 min of glucose starvation. The values shown are the average from experiments carried out on at least two different days with >100 cells per time point, per replicate. The SD is <5% for all mutants and time points except for sak1Δ (60 min), gcn2Δ (60 min), pib2Δ (60 min), cka1Δ (0 min), and sit4Δ (0 min), which have standard deviations of 5–10%; npr2Δ (0 min), sit4Δ (0 min), and sit4Δ (60 min), which have standard deviations of 10–15%; and slt2Δ (60 min) and npr2Δ (60 min), which have standard deviations of 15–20%. The red circle and square on the x-axis shows the data for the gtr1Δ single mutant for comparison.
Remarkably, most (six of seven) of the proteins that act on, or at the level of, Gtr1 to promote TORC1-body formation are repressed by, or in a pathway that is repressed by, TORC1, including the following: Sit4, a type 2A–related phosphatase repressed by TORC1 via Tap42, involved in regulating a wide range of processes, including cell-cycle regulation (Di Como and Arndt, 1996; Jacinto et al., 2001; Loewith and Hall, 2011); Slt2/Mpk1, a MAPK in the PKC pathway repressed by TORC1 and involved in the regulation of cell-wall integrity, cell-cycle progression, and proteasome activity (Krause and Gray, 2002; Torres et al., 2002; Moreno-Torres et al., 2015; Rousseau and Bertolotti, 2016); Rom2, an upstream activator in the PKC pathway (Ozaki et al., 1996; Levin, 2011); Bck1, a MAPKKK in the PKC pathway that interacts with TORC1 (Breitkreutz et al., 2010; Levin, 2011); Cka1, the alpha catalytic subunit of casein kinase 2 (CK2), recently found to be repressed by TORC1 and involved in regulating RNA Pol III activity and other proteins/pathways (Sanchez-Casalongue et al., 2015; Shekhar-Guturja et al., 2016); and Gcn2, a kinase repressed by TORC1 that has also been reported to act upstream of TORC1, involved in regulating amino acid biosynthesis (Cherkasova and Hinnebusch, 2003; Yuan et al., 2017).
These connections suggest that the inhibition of TORC1 signaling in starvation conditions helps drive the release of TORC1 from the EGOC-dependent repression of TORC1-body formation via feedback. This probably helps to ensure that all of the TORC1 molecules in a cell are primed to move into bodies during long-term starvation.
However, TORC1-body formation is also controlled by an additional group of proteins (Yak1, Ypk1, Cmk1, and Sak1) that activate TORC1-body formation once TORC1 is released from the EGOC-dependent repression of TORC1 agglomeration. With the exception of Yak1 (Martin et al., 2004), these proteins are not linked to the TORC1/EGOC signaling pathway and, instead, respond to membrane stress as part of the TORC2-Ypk1 pathway (Ypk1) (Roelants et al., 2011; Muir et al., 2014), calcium signals (Cmk1) (Cyert, 2001), and glucose/energy starvation (Sak1) (Elbing et al., 2006; Hedbacker and Carlson, 2008). It therefore appears that the second layer of regulation helps to ensure that multiple stress/starvation pathways are activated before a cell commits to the starvation state by forming TORC1-bodies.
Cooperation between Pib2 and EGOC
One of the most interesting findings from the double mutant analysis is that Pib2—a protein that is required for TORC1-body formation in wild-type cells (Figure 3) and known to interact with both EGOC and TORC1 (Tarassov et al., 2008; Kim and Cunningham, 2015; Michel et al., 2017)—becomes dispensable for TORC1-body formation in the absence of Gtr1 (Figure 4). But how? One possibility is that Pib2 is required for the starvation-dependent inactivation of Gtr1 and thus release of the Gtr1/2-dependent repression of TORC1-body formation, like Npr2. However, full-length Pib2 has been shown to activate TORC1 kinase activity (Kim and Cunningham, 2015; Michel et al., 2017; Tanigawa and Maeda, 2017; Varlakhanova et al., 2017), while Npr2 represses TORC1 kinase activity (Neklesa and Davis, 2009; Panchaud et al., 2013)—indicating that Pib2 drives TORC1-body formation via a different mechanism than Npr2. Therefore, to learn more about the role that Pib2 plays in TORC1-body formation, we examined the impact that each domain in Pib2 has on Kog1-YFP localization.
Previous studies have shown that Pib2 contains four distinct domains (Kim and Cunningham, 2015; Michel et al., 2017): 1) an N-terminal domain that inhibits TORC1 activity; 2) a central domain that binds to Kog1; 3) a FYVE domain that binds to PI(3)P and recruits Pib2 to the vacuolar membrane; and 4) a C-terminal domain, including a critical (and highly conserved) 15-amino-acid stretch from amino acids 620 to 635, that activates TORC1 (Figure 5A; see Supplemental Text and Supplemental Figures S4–S6 for further details).
FIGURE 5:
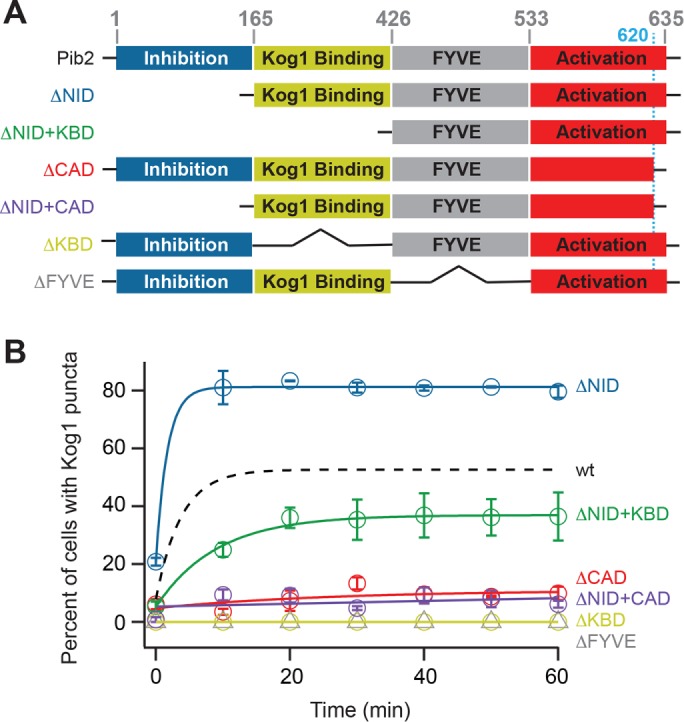
Impact of Pib2 domains on TORC1-body formation. (A) Map of the different domains in Pib2 (as described in the text), and the structure of the Pib2 truncation mutants we constructed. The black lines in the ΔKBD and ΔFYVE mutants show that the neighboring domains are connected and do not represent polypeptide. (B) Time-course data following Kog1-YFP localization in strains carrying truncated forms of Pib2 at the Pib2 locus. Each time point shows the average and SD from experiments carried out on two different days, with 70–200 cells per time point per replicate (except t = 0, ΔNID+CAD, which had >40 cells per replicate). The solid lines show the best fit to a single exponential for the ΔNID, ΔNID+KBD, ΔCAD, and ΔNID+CAD strains and a straight line for the ΔKBD and ΔFYVE strains. The broken line shows the best fit to the wild-type data (from Figure 2) for comparison. Overexpression of Pib2 had little impact on TORC1-body formation; see Supplemental Figure S6 and Supplemental Text for details.
Deletion of the N-terminal inactivation domain (NID) of Pib2 increased the fraction of cells that form TORC1-bodies in nutrient-replete conditions (from 8 ± 5% to 21 ± 1%) and on the 1-h timescale (from 56 ± 1% to 80 ± 2%)—indicating that this region of Pib2 inhibits TORC1-body formation (Figure 5B). In contrast, deletion of the C-terminal activating domain (CAD), Kog1-binding domain (KBD), and FYVE domain (FYVE) slowed or blocked TORC1-body formation, indicating that these domains promote TORC1-body formation (Figure 5B).
The data showing that the Kog1-binding domain in Pib2 is required for TORC1-body formation are especially interesting since previous studies have shown that this domain is dispensable for TORC1 activity (in SD medium). Moreover, they suggest that Pib2 drives TORC1-body formation via a direct interaction with Kog1/TORC1.
To test this idea, we created a strain carrying Pib2 tagged with green fluorescent protein (GFP-Pib2) and Kog1-DuDre and followed their localization during glucose starvation. This experiment revealed that 1) Pib2 is located on both the vacuolar membrane, and foci associated with the membrane, in nutrient-replete medium—just like EGOC—and 2) that Kog1 and Pib2 both reside in the TORC1-body (also occupied by EGOC) during starvation (93% overlap, n = 128 cells with Kog1 foci; Figure 6A). We also performed coimmunoprecipitation experiments (after cross-linking) to see whether Pib2, EGOC, and TORC1 bind to each other during log growth (when TORC1 is distributed across the vacuolar membrane) and/or in starvation conditions (when TORC1 is in a body). These experiments showed that Pib2 and Kog1, and Gtr1 and Kog1, interact at similar levels in both nutrient replete and starvation conditions (Figure 6B).
FIGURE 6:

Pib2, EGOC, and TORC1 interact in log growth and starvation conditions. (A) Localization of GFP-Pib2 and Kog1-DuDre during log growth (left panels) and after 60 min of glucose starvation (right panels). The dashed lines show the position of each cell in the bright-field image. (B) Coimmunoprecipitation experiments following interactions between Gtr1 and Kog1 (top panel) and Pib2 and Kog1 (bottom panel) before (0 min) and after 2 and 4 h of glucose starvation. The right-hand side of each blot shows the data for a mock IP (IP from cells missing the epitope tag on Kog1 or Pib2) used to measure the background levels of Gtr1 and Kog1 in the precipitate.
Thus, Pib2, EGOC, and TORC1 form a complex that blocks TORC1-body formation when EGOC is active (in nutrient-replete conditions) but permits TORC1 to form bodies when EGOC is inactive (during starvation). In this complex, EGOC constantly acts to inhibit TORC1-body formation, likely via direct binding to TORC1. However, when Gtr1/2 are in the inactive state, Pib2 overwhelms the repressive effect of EGOC so that TORC1-bodies can form.
DISCUSSION
Regulation of TORC1-body formation
In our original study of TORC1 localization (Hughes Hallett et al., 2015), we followed the movement of Kog1-YFP, Tco89-YFP, and Tor1 with a triple GFP insertion (Sturgill et al., 2008) in different stress and starvation conditions. Those experiments showed that Kog1-YFP and Tco89-YFP move into a body during glucose and nitrogen starvation (with a time constant of 10 min) while Tor1D330-3xGFP remains on the vacuolar membrane/cytoplasm—leading us to conclude that TORC1 dissociates in starvation conditions so that Kog1 and Tco89 can move into a “Kog1 body.” However, recent experiments examining the localization of GFP-Tor1 have shown that Tor1 also moves into a body (Kira et al., 2016; Prouteau et al., 2017), indicating that the internal 3xGFP tag disrupts Tor1 localization. We have confirmed these results (unpublished data) and therefore refer to TORC1-bodies, rather than Kog1-bodies, throughout this article.
In the same study, we showed that AMPK/Snf1 phosphorylates Kog1 during glucose starvation at two novel sites (Ser 491 and 494) and that these phosphorylation events help drive the formation of TORC1-bodies (TORC1-bodies form 20-fold slower in Kog1S491/494A and snf1Δ cells). We also showed that two glutamine-rich, prion-like domains in Kog1 help drive TORC1-body formation (TORC1-bodies form >100-fold slower in the strongest prion mutant; PrDm1+2). Then, by studying strains with mutations that limit TORC1-body formation (Kog1S491/494A, PrDm1+2, and others), we showed that TORC1-bodies are not required for the rapid inactivation of TORC1 but instead increase the threshold for TORC1 activation in cells that have been starved for a significant period of time (from around 0.02% glucose, to around 2% glucose). In other words, TORC1-body formation creates hysteresis (memory) in the TORC1 pathway to help ensure that cells remain committed to a starvation state until they are exposed to optimal conditions. In line with this, cells carrying Asp or Glu mutations at Ser 491 and 494 in Kog1 (phosphomimetics) fail to grow—even in rich medium.
Here, to build on our previous work, we set out to learn more about how TORC1-body formation is regulated and, in particular, whether/how the major TORC1 regulators Gtr1/2 (Rag A/B and C/D in humans) impact TORC1-body formation—with the goal of building an integrated model of TORC1 regulation in yeast.
Our new data show that TORC1-body formation and TORC1 inhibition are tightly coupled events. Specifically, we show that glucose and nitrogen starvation both trigger inhibition of the Gtr1/2 complex (i.e., a switch from the GTP/GDP to the GDP/GTP-bound state). This signaling event is well known to help drive inhibition of TORC1 kinase activity (Binda et al., 2009; Nicastro et al., 2017): In the case of nitrogen starvation, Gtr1/2 inhibition is responsible for ∼50% of the rapid and complete repression of TORC1 activity (Hughes Hallett et al., 2014). In the case of glucose starvation, Gtr1/2 inhibition is responsible for only ∼20% of the rapid and complete repression of TORC1 activity, since Snf1/AMPK and other unknown pathways play a dominant role in the response (Hughes Hallett et al., 2014). At the same time, Gtr1/2 inhibition also allows TORC1 to form bodies. Importantly, however, it is not TORC1 inhibition itself that promotes TORC1-body formation, since TORC1 is fully repressed in numerous mutant strains that have dramatic defects in TORC1-body formation (some of which can be rescued by deletion of Gtr1). Instead, it appears that Gtr1/2 has a dual role: 1) helping control TORC1 activity, particularly in response to nitrogen and amino acid signals, and 2) acting as a glucose and nitrogen starvation-dependent gate for TORC1-body formation. This dual role ensures that starvation signals are sent through the Gtr1/2—at least partially inactivating TORC1—before TORC1 can agglomerate and lock the pathway in a hyperrepressed state.
Our data also point to a second mechanism coupling TORC1 inhibition to TORC1-body formation: most (six of seven) of the proteins that are required for the release from Gtr1/2-dependent repression of TORC1-body formation are repressed by, or are in a pathway that is repressed by, TORC1 itself. It therefore appears that TORC1 inhibition promotes TORC1-body formation by activating several feedback loops.
While TORC1-body formation is tightly coupled to TORC1 inhibition via Gtr1/2 and feedback, it is important to note that the late steps in TORC1-body formation (those that occur after the release from the Gtr1/2-dependent repression of body formation) depend on stress and starvation signaling proteins that are not directly linked to the TORC1 pathway (Sak1, Ypk1, and Cmk1). Further work is needed to pick apart the influence that of these proteins have on TORC1 signaling, but it seems likely that they help limit TORC1-body formation until multiple stress/starvation pathways are activated.
Our data also provide important insight into the mechanism underlying the Gtr1/2-dependent gating of TORC1-body formation. We show that Gtr1/2 inhibits TORC1-body formation in nutrient replete conditions (when it is active) but becomes dispensable for TORC1-body formation during starvation (when it is inactive). We also show that the inactivation of the Gtr1/2-dependent repression of TORC1-body formation requires Pib2—a protein that forms a complex with TORC1 in both log growth and starvation conditions. These results suggest that that the tight interaction between Gtr1/2 and TORC1 in nutrient-replete conditions pins TORC1 in a conformation that has a low propensity to agglomerate and/or covers a surface of TORC1 that is needed for body formation (e.g., the prion-like domains in Kog1; Figure 7). Then, when the interaction between Gtr1/2 and TORC1 is weakened in starvation conditions (by a switch to the inactive GDP/GTP bound form of Gtr1/2), Pib2 pulls TORC1 into a new conformation that is primed to agglomerate (Figure 7).
FIGURE 7:
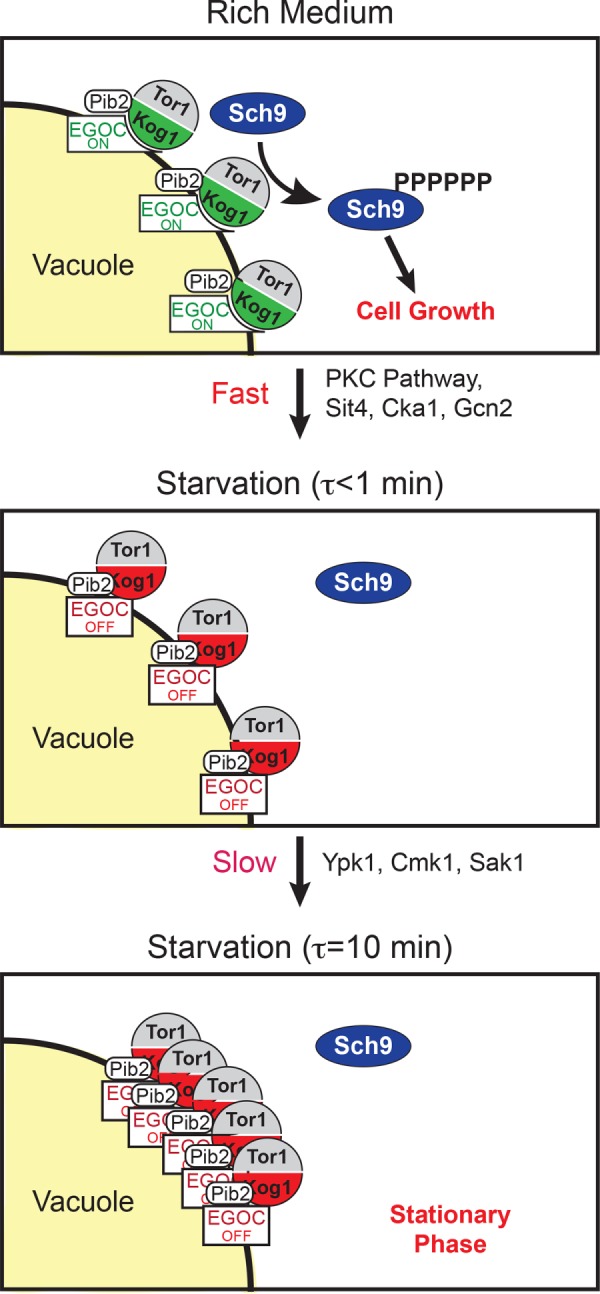
Working model of TORC1 regulation. See the text for details.
The discovery that Pib2 is required for the Gtr1/2-dependent regulation of TORC1-body formation builds on previous work showing that Pib2 and Gtr1/2 work together to control TORC1 activation (Kim and Cunningham, 2015; Worley et al., 2015; Michel et al., 2017; Tanigawa and Maeda, 2017; Varlakhanova et al., 2017). We therefore propose that EGOC and Pib2 act as a single complex machine—to control both Gtr1/2-dependent TORC1 activation and TORC1-body formation. In this context, it will be interesting to determine whether/how the related protein, LAPF/phafin-1, modulates Rag GTPase function in humans.
Comparison between TORC1-bodies and TOROIDS
While we were completing this work, Prouteau et al. (2017) published a new study of TORC1 localization. They report that TORC1 moves rapidly into and out of foci on a timescale that matches Sch9 phosphorylation and dephosphorylation (τ = 2 min). They also report that active Gtr1/2 limits TORC1-body formation and that deletion of Gtr1/2 leads to TORC1 agglomeration in 60% of cells, even in nutrient-replete medium—leading them to conclude that Gtr1/2 are the dominant regulators of TORC1 agglomeration.
The differences between their results and our results may be due to different experimental setups, since Prouteau et al. (2017) examined TORC1 localization in cells that were transitioning in and out of stationary phase (adding and subtracting nutrients where appropriate), while we followed TORC1 localization in cells that had been kept in log growth phase for >12 h. There may also be some strain-to-strain differences in the exact rates and levels of TORC1-body formation.
Prouteau et al. (2017) also analyzed the structure of TORC1 purified from cells in stationary phase using CryoEM. Their analysis shows that TORC1 molecules can pack in a helical array to form a hollow tube with a length of up to 1 μM. This packing occludes the active site of the complex. Using STORM microscopy, the authors then confirm that some (<20%) of the TORC1 agglomerates formed in vivo are elongated. To test the impact that these TORC1 fibers (called TOROIDS) have on TORC1 activity, Prouteau et al. then introduced the same Tor1D330-3XGFP construct that we used in our original study (and found does not move to bodies), arguing that the 3xGFP is in an ideal position to disrupt TOROID assembly. They find that Sch9 is phosphorylated at a high level in the Tor1D330-3XGFP strain, even during glucose starvation—leading them to conclude that TOROID formation is required for TORC1 inactivation in glucose starvation conditions.
The argument that TORC1 agglomeration is required for rapid TORC1 inactivation is incompatible with our data, showing that 14 different strains with major defects in TORC1-body formation all turn off TORC1 signaling normally (or nearly normally) in glucose-starvation conditions (GTR1Q65L and the 13 mutants identified in the screen; Figure 3 and Supplemental Figure S3). Instead, we believe there are two possible explanations for the TORC1 signaling defect found in the Tor1D330-3XGFP strain. The first is simply that the insertion of three GFPs into Tor1 blocks the access of a key regulatory protein to TORC1. The second is that interactions between individual TORC1 molecules (like those found in the TOROID) do help inactivate TORC1 in starvation conditions (leading to the signaling defect in the Tor1D330-3XGFP strain), but the assembly into a higher-order agglomerate—visible in the microscope and disrupted in our mutants—is only required to stabilize the off state of TORC1. A model of this type could also help resolve additional conflicts between our data and the data of Prouteau et al. (2017), namely, 1) that the TOROID structure does not include Tco89 (a key driver of TORC1 agglomeration) or explain why the prion-like domains in Kog1 help drive TORC1 agglomeration and 2) that in our microscopy experiments, we never see the kind of highly elongated fibers found in the structural studies of Prouteau et al. (2017) Therefore, it may be that the individual TORC1–TORC1 interactions identified in the CryoEM structure form in vivo, but the interactions formed with, and between, Tco89, the prion domains in Kog1, Pib2, and EGOC pull small TORC1 agglomerates into globular bodies and block the formation of the long fibers seen in vitro.
Further experiments are needed to resolve these issues and integrate the exciting findings of Prouteau et al. (2017) with our data and two-step model of TORC1 regulation (rapid TORC1 inhibition followed by TORC1-body formation; Figure 7). However, from the work completed to date, it is clear that the movement of TORC1 into and out of higher-order structures is a carefully controlled process that plays an important role in regulating TORC1 activity. Moving forward, it will therefore be interesting to determine precisely how TORC1-body formation tunes signaling through the TORC1 pathway and to see whether other kinases are regulated by reversible agglomeration.
MATERIALS AND METHODS
Saccharomyces cerevisiae strains
Most of the strains used in this study were generated in a haploid S. cerevisiae strain, W303 background (trp1, can1, leu2, his3, ura3), using standard methods. The exceptions were as follows: 1) To make the strain carrying a Gtr1Q65L allele at the native Gtr1 locus, we first knocked out GTR1 using the pCORE cassette, containing both a URA3 and Kan markers (Storici and Resnick, 2006). We then cloned the mutant form of GTR1 from a plasmid provided from the De Virgilio lab and introduced it into the gtr1Δ cell line. Finally, we selected for cells that had integrated the mutant GTR1 at the native locus via selection with 5FOA and by searching for colonies that die on kanamycin (G418) plates. 2) We also used the pCORE cassette to build the strains carrying truncated forms of Pib2 at the native locus. PCR was used to create truncated versions of the Pib2 gene, and the fragments were integrated at the native Pib2 locus by transforming them into strains that had the entire Pib2, or part of the Pib2 gene, replaced with the pCORE cassette. 3) GFP-Pib2 was created by cloning the tagged gene from a plasmid provided by the Cunningham lab and integrating it at the native locus—as described above for the truncation mutants. In cases 1–3, the integrity of the final gene product was confirmed by sequencing the Pib2 or Gtr1 gene and surrounding regions. 4) Strains carrying Kog1-DuDre and Ego1-YFP or Gtr1-YFP and the strains carrying Kog1-YFP and Ego1/Ego3/Gtr1-DuDre were made by mating and tetrad dissection.
The double mutant strains examined in Figure 4 were made by knocking each of the relevant Kog1-body regulators out in the gtr1Δ strain. In all but two cases (ypl150wΔ gtr1Δ and bck1Δgtr1Δ) the transformation led to a standard number of positive colonies and the resulting strain grew at a rate similar to that of the gtr1Δ parental strain. However, since previous reports indicated that the gtr1Δpib2Δ strain is inviable (Kim and Cunningham, 2015), we checked that we had not made a mistake by building it several times. In all cases, we identified a standard number of colonies and the strains grew at approximately the same rate as the gtr1Δ strain.
All strains used in this study are listed in Supplemental Table S2.
Fluorescence microscopy
Cells were taken from a fresh yeast extract peptone dextrose (YEPD) plate and grown for 5–6 h in SD medium in a 35-ml test tube on a rotator at 30°C, until they reached an OD600 of ∼0.1. The starter cultures were then used to inoculate 20 ml of SD medium in a 125-ml conical flask to an OD600 of ∼0.001 and allowed to grow shaking at 200 rpm and 30°C, until they reached mid–log phase. At this point, 400-μl samples were transferred to a chamber slide (Ibidi μ-slide, eight-well; 80827) that had been treated with 2 mg/ml concanavilin A and examined on the microscope. The slides were then washed three times with 400 μl starvation medium (either SD minus glucose or SD minus nitrogen [no amino acids or ammonium sulfate]) and loaded into a 30°C chamber on the microscope, and protein localization was followed over time.
Images were acquired using a DeltaVision Elite Microscope equipped with an Olympus 100×, 1.4NA, objective and a scientific complementary metal–oxide–semiconductor (sCMOS) camera. We collected a Z-series of 16 images with 0.4-μm spacing in the YFP (YFP filter; Ex. 496–528 nm, Em. 537–559 nm), GFP (GFP filter; Ex. 425–495 nm, Em. 500–550 nm), and/or RFP (RFP filter; Ex. 555–590 nm, Em. 600–675 nm) channels at each time point to ensure that all of the fluorescent foci in the cell were detected. Image files were then processed in ImageJ (Schneider et al., 2012) to create the maximum projection from the stack.
We calculated the fraction of cells containing one or more TORC1-bodies using a custom pipeline in CellProfiler (Carpenter et al., 2006). In all cases, the results were checked by manual inspection and adjusted if necessary. Where appropriate time-course data were fitted to a single exponential equation, A*(1-e-t/τ) + c, or double exponential equation, A1*(1-e-t/τ1) + A2*(1-e-t/τ2) + c, where A (or A1 + A2) is the fraction of cells that form bodies during the time course, τ is the apparent time constant, and c is the fraction of cells that have bodies at the start of the time course. In cases where there was no change in TORC1-body levels during the time course, the data were fitted to a line. All fitting was done in Igor Pro 6.3 (WaveMetrics), and the errors reported are the SD estimated from the fit.
Sch9 bandshift experiments
Bandshift measurements were performed as described previously (Urban et al., 2007; Hughes Hallett et al., 2014, 2015), except that the data were quantified using Image Studio software (LiCor) by comparing the total intensity of the upper (phosphorylated) bands to the total intensity of the upper and lower (phosphorylated and dephosphorylated) bands at each time point (to calculate the fraction Sch9 phosphorylated). The values for all time points and strains were then multiplied by a single constant so that the fraction of Sch9 phosphorylated at time zero in the wild-type strain is set to 1.0.
Coimmunoprecipitation experiments
Cells were grown in 750 ml of SD medium, shaking at 200 rpm and 30°C, until they reached mid–log phase (OD600 between 0.55 and 0.6). At that point, one-third of the culture was harvested by filtration and flash frozen. Approximately 1 min later, the other two-thirds of the culture was collected on a separate filter, washed with SD medium missing glucose, and added to a flask containing 500 ml of SD medium missing glucose and grown for an additional 2 or 4 h before it was collected using filtration.
Immunopurifications were performed using a modified version of the protocol by Murley and Nunnari (Murley et al., 2017) designed to identify protein–protein interactions on membranes. Cells were resuspended in 4 ml of lysis buffer (20 mM HEPES, 150 mM potassium acetate, 2 mM magnesium acetate, 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid [EGTA], and 0.6 M sorbitol at pH 7.4) containing Roche complete protease and phosphatase inhibitors and lysed by bead beating (6 × 1 min). The lysates were then cleared by centrifugation at 3000 rpm for 5 min on a benchtop centrifuge and treated with 1 mM dithiobis succinimidyl propionate (a reversible cross-linker; Thermo Fisher Scientific) for 30 min at 4°C. The reaction was then quenched by the addition of 100 mM Tris–HCl (pH 7.5) to the sample and incubation on ice for 30 min. Membranes were then solubilized by the addition of 1% digitonin (Cayman) and nutation at 50 rpm and 4°C for 60 min, and the lysates cleared by centrifugation at 12,000 × g for 10 min at 4°C.
In the GFP-Pib2 purifications, the clarified lysates were incubated with 50 μl of μMACs monoclonal mouse anti-GFP magnetic microbeads (Miltenyi Biotec) at 4°C for 30 min. μMAC columns were then equilibrated in the lysis buffer +1% digitonin and protease inhibitors; loaded with the beads; washed three times with 800 μl lysis buffer, 0.1% digitonin, and Roche protease inhibitors; and then washed two times with 800 μl lysis buffer. The proteins were then eluted in 50 mM Tris–HCl (pH 6.8), 50 mM dithiothreitol, 1% SDS, 1 mM EDTA at 95°C.
In the Kog1-FLAG purifications, the clarified lysates were incubated with 10 μg of anti-FLAG M2 antibody (Sigma) for 3 h, and then Protein A/G Ultralink resin (Pierce) for an additional 2 h, all at 4°C with slow rotation. The resin was then washed three times with 800 μl lysis buffer, 0.1% digitonin, and Roche protease inhibitors, and two times with 800 μl lysis buffer, before the resin was boiled in SDS–PAGE loading buffer to elute Kog1 and associated proteins.
The protein samples from each experiment (three time points for each IP and the matching controls) were run on a single 8% SDS–PAGE gel and transferred to a nitrocellulose membrane using standard procedures. The membrane was then cut in half to separate the higher- and lower-molecular-weight regions (using a prestained ladder as a guide). The high-molecular-weight portion of the membrane was then incubated with anti-FLAG antibody (M2; Sigma) to detect Kog1-FLAG, while the lower weight portion of the membrane was incubated with anti-GFP to detect GFP-Pib2 (4B10B2; Roche) or anti-myc (9E10; Roche) to detect Gtr1-myc.
Supplementary Material
Acknowledgments
We thank Claudio De Virgilio for sharing the GTR1 Q65L plasmid, Kyle Cunningham for sharing the GFP-Pib2 plasmid, and Takeshi Noda for sharing the GFP-Tor1 strain. This work was supported by National Institutes of Health grants R01GM097329 and T32GM008659.
Abbreviations used:
- PI(3)P
phosphatidylinositol-3-phosphate
- PKC
protein kinase C
- TORC1
target of rapamycin complex I.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-05-0297) on November 28, 2018.
REFERENCES
- Adami A, Garcia-Alvarez B, Arias-Palomo E, Barford D, Llorca O. (2007). Structure of TOR and its complex with KOG1. Mol Cell , 509–516. [DOI] [PubMed] [Google Scholar]
- Aylett CH, Sauer E, Imseng S, Boehringer D, Hall MN, Ban N, Maier T. (2016). Architecture of human mTOR complex 1. Science , 48–52. [DOI] [PubMed] [Google Scholar]
- Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. (1996). TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell , 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. (2013). A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science , 1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda M, Peli-Gulli MP, Bonfils G, Panchaud N, Urban J, Sturgill TW, Loewith R, De Virgilio C. (2009). The Vam6 GEF controls TORC1 by activating the EGO complex. Mol Cell , 563–573. [DOI] [PubMed] [Google Scholar]
- Bodenmiller B, Wanka S, Kraft C, Urban J, Campbell D, Pedrioli PG, Gerrits B, Picotti P, Lam H, Vitek O, et al (2010). Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci Signal , rs4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin ZY, Breitkreutz BJ, Stark C, Liu G, et al (2010). A global protein kinase and phosphatase interaction network in yeast. Science , 1043–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol , R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkasova VA, Hinnebusch AG. (2003). Translational control by TOR and TAP42 through dephosphorylation of eIF2alpha kinase GCN2. Genes Dev , 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyert MS. (2001). Genetic analysis of calmodulin and its targets in Saccharomyces cerevisiae. Annu Rev Genet , 647–672. [DOI] [PubMed] [Google Scholar]
- Di Como CJ, Arndt KT. (1996). Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases. Genes Dev , 1904–1916. [DOI] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell , 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM. (2013). Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature , 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbing K, McCartney RR, Schmidt MC. (2006). Purification and characterization of the three Snf1-activating kinases of Saccharomyces cerevisiae. Biochem J , 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Kaiser CA. (2006). A conserved GTPase-containing complex is required for intracellular sorting of the general amino-acid permease in yeast. Nat Cell Biol , 657–667. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Hall MN. (2017). Nutrient sensing and TOR signaling in yeast and mammals. EMBO J , 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell , 177–189. [DOI] [PubMed] [Google Scholar]
- Hedbacker K, Carlson M. (2008). SNF1/AMPK pathways in yeast. Front Biosci , 2408–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al (2011). The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science , 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes Hallett JE, Luo X, Capaldi AP. (2014). State transitions in the TORC1 signaling pathway and information processing in Saccharomyces cerevisiae. Genetics , 773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes Hallett JE, Luo X, Capaldi AP. (2015). Snf1/AMPK promotes the formation of Kog1/Raptor-bodies to increase the activation threshold of TORC1 in budding yeast. Elife , e09181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Guo B, Arndt KT, Schmelzle T, Hall MN. (2001). TIP41 interacts with TAP42 and negatively regulates the TOR signaling pathway. Mol Cell , 1017–1026. [DOI] [PubMed] [Google Scholar]
- Kim A, Cunningham KW. (2015). A LAPF/phafin1-like protein regulates TORC1 and lysosomal membrane permeabilization in response to endoplasmic reticulum membrane stress. Mol Biol Cell , 4631–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell , 163–175. [DOI] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol , 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kira S, Kumano Y, Ukai H, Takeda E, Matsuura A, Noda T. (2016). Dynamic relocation of the TORC1-Gtr1/2-Ego1/2/3 complex is regulated by Gtr1 and Gtr2. Mol Biol Cell , 382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kira S, Tabata K, Shirahama-Noda K, Nozoe A, Yoshimori T, Noda T. (2014). Reciprocal conversion of Gtr1 and Gtr2 nucleotide-binding states by Npr2-Npr3 inactivates TORC1 and induces autophagy. Autophagy , 1565–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause SA, Gray JV. (2002). The protein kinase C pathway is required for viability in quiescence in Saccharomyces cerevisiae. Curr Biol , 588–593. [DOI] [PubMed] [Google Scholar]
- Levin DE. (2011). Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics , 1145–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Hall MN. (2011). Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics , 1177–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. (2002). Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell , 457–468. [DOI] [PubMed] [Google Scholar]
- Martin DE, Soulard A, Hall MN. (2004). TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell , 969–979. [DOI] [PubMed] [Google Scholar]
- Michel AH, Hatakeyama R, Kimmig P, Arter M, Peter M, Matos J, De Virgilio C, Kornmann B. (2017). Functional mapping of yeast genomes by saturated transposition. Elife , e23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Torres M, Jaquenoud M, De Virgilio C. (2015). TORC1 controls G1-S cell cycle transition in yeast via Mpk1 and the greatwall kinase pathway. Nat Commun , 8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir A, Ramachandran S, Roelants FM, Timmons G, Thorner J. (2014). TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. Elife , e03779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A, Yamada J, Niles BJ, Toulmay A, Prinz WA, Powers T, Nunnari J. (2017). Sterol transporters at membrane contact sites regulate TORC1 and TORC2 signaling. J Cell Biol , 2679–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadolski MJ, Linder ME. (2009). Molecular recognition of the palmitoylation substrate Vac8 by its palmitoyltransferase Pfa3. J Biol Chem , 17720–17730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa TK, Davis RW. (2009). A genome-wide screen for regulators of TORC1 in response to amino acid starvation reveals a conserved Npr2/3 complex. PLoS Genet , e1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro R, Sardu A, Panchaud N, De Virgilio C. (2017). The architecture of the Rag GTPase signaling network. Biomolecules , 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y. (1998). Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem , 3963–3966. [DOI] [PubMed] [Google Scholar]
- Ozaki K, Tanaka K, Imamura H, Hihara T, Kameyama T, Nonaka H, Hirano H, Matsuura Y, Takai Y. (1996). Rom1p and Rom2p are GDP/GTP exchange proteins (GEPs) for the Rho1p small GTP binding protein in Saccharomyces cerevisiae. EMBO J , 2196–2207. [PMC free article] [PubMed] [Google Scholar]
- Panchaud N, Peli-Gulli MP, De Virgilio C. (2013). Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci Signal , ra42. [DOI] [PubMed] [Google Scholar]
- Powis K, Zhang T, Panchaud N, Wang R, De Virgilio C, Ding J. (2015). Crystal structure of the Ego1-Ego2-Ego3 complex and its role in promoting Rag GTPase-dependent TORC1 signaling. Cell Res , 1043–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prouteau M, Desfosses A, Sieben C, Bourgoint C, Lydia Mozaffari N, Demurtas D, Mitra AK, Guichard P, Manley S, Loewith R. (2017). TORC1 organized in inhibited domains (TOROIDs) regulate TORC1 activity. Nature , 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke A, Anderson S, McCaffery JM, Yates J, 3rd, Aronova S, Chu S, Fairclough S, Iverson C, Wedaman KP, Powers T. (2004). TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J Biol Chem , 14752–14762. [DOI] [PubMed] [Google Scholar]
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN. (2013). Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science , 1320–1323. [DOI] [PubMed] [Google Scholar]
- Roelants FM, Breslow DK, Muir A, Weissman JS, Thorner J. (2011). Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc Natl Acad Sci USA , 19222–19227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR, 3rd, Davis NG. (2006). Global analysis of protein palmitoylation in yeast. Cell , 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau A, Bertolotti A. (2016). An evolutionarily conserved pathway controls proteasome homeostasis. Nature , 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell , 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science , 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Casalongue ME, Lee J, Diamond A, Shuldiner S, Moir RD, Willis IM. (2015). Differential phosphorylation of a regulatory subunit of protein kinase CK2 by target of rapamycin complex 1 signaling and the Cdc-like kinase Kns1. J Biol Chem , 7221–7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM. (2017). mTOR signaling in growth, metabolism, and disease. Cell , 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods , 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar-Guturja T, Gunaherath GM, Wijeratne EM, Lambert JP, Averette AF, Lee SC, Kim T, Bahn YS, Tripodi F, Ammar R, et al (2016). Dual action antifungal small molecule modulates multidrug efflux and TOR signaling. Nat Chem Biol , 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Resnick MA. (2006). The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol , 329–345. [DOI] [PubMed] [Google Scholar]
- Sturgill TW, Cohen A, Diefenbacher M, Trautwein M, Martin DE, Hall MN. (2008). TOR1 and TOR2 have distinct locations in live cells. Eukaryot Cell , 1819–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su MY, Morris KL, Kim DJ, Fu Y, Lawrence R, Stjepanovic G, Zoncu R, Hurley JH. (2017). Hybrid structure of the RagA/C-Ragulator mTORC1 activation complex. Mol Cell , 835–846 e833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa M, Maeda T. (2017). An in vitro TORC1 kinase assay that recapitulates the Gtr-independent glutamine-responsive TORC1 activation mechanism on yeast vacuoles. Mol Cell Biol , e00075-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW. (2008). An in vivo map of the yeast protein interactome. Science , 1465–1470. [DOI] [PubMed] [Google Scholar]
- Torres J, Di Como CJ, Herrero E, De La Torre-Ruiz MA. (2002). Regulation of the cell integrity pathway by rapamycin-sensitive TOR function in budding yeast. J Biol Chem , 43495–43504. [DOI] [PubMed] [Google Scholar]
- Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, et al (2007). Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell , 663–674. [DOI] [PubMed] [Google Scholar]
- Varlakhanova NV, Mihalevic MJ, Bernstein KA, Ford MGJ. (2017). Pib2 and the EGO complex are both required for activation of TORC1. J Cell Sci , 3878–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley J, Sullivan A, Luo X, Kaplan ME, Capaldi AP. (2015). Genome-wide analysis of the TORC1 and osmotic stress signaling network in Saccharomyces cerevisiae. G3 (Bethesda) , 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, Dhar A, Pavletich NP. (2017). Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature , 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. (2010). Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell , 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Guo S, Gao J, Zhong M, Yan G, Wu W, Chao Y, Jiang Y. (2017). General control nonderepressible 2 (GCN2) kinase inhibits target of rapamycin complex 1 in response to amino acid starvation in Saccharomyces cerevisiae. J Biol Chem , 2660–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.