

Abstract
During the asymmetric division of the Caenorhabditis elegans zygote, germ (P) granules are disassembled in the anterior cytoplasm and stabilized/assembled in the posterior cytoplasm, leading to their inheritance by the germline daughter cell. P granule segregation depends on MEG (maternal-effect germline defective)-3 and MEG-4, which are enriched in P granules and in the posterior cytoplasm surrounding P granules. Here we use single-molecule imaging and tracking to characterize the reaction/diffusion mechanisms that result in MEG-3::Halo segregation. We find that the anteriorly enriched RNA-binding proteins MEX (muscle excess)-5 and MEX-6 suppress the retention of MEG-3 in the anterior cytoplasm, leading to MEG-3 enrichment in the posterior. We provide evidence that MEX-5/6 may work in conjunction with PLK-1 kinase to suppress MEG-3 retention in the anterior. Surprisingly, we find that the retention of MEG-3::Halo in the posterior cytoplasm surrounding P granules does not appear to contribute significantly to the maintenance of P granule asymmetry. Rather, our findings suggest that the formation of the MEG-3 concentration gradient and the segregation of P granules are two parallel manifestations of MEG-3′s response to upstream polarity cues.
INTRODUCTION
RNA/protein (RNP) granules are hubs of RNA metabolism that can rapidly assemble and disassemble in response to cellular cues. Granules assemble through the interactions between RNA and proteins that cause them to demix from the surrounding cytoplasm through liquid–liquid phase separation (LLPS) (Shin and Brangwynne, 2017). Granules are not bounded by a membrane, which allows granule components to exchange freely with the surrounding cytoplasm. Interest in understanding how granule assembly and disassembly are controlled is motivated in part by the association between granule assembly defects and a number of pathologies (Ramaswami et al., 2013; Alberti and Carra, 2018; Purice and Taylor, 2018).
Germ (P) granules in Caenorhabditis elegans provide a striking example of the dynamic ways in which LLPS can be regulated in vivo. P granules are micron-sized RNP granules that are present in germ cells at all stages of their development (Voronina, 2012; Strome and Updike, 2015). Just before fertilization of the oocyte, P granule localization shifts from the cytoplasmic face of the nuclear envelope to the cytoplasm. Following fertilization, P granules are initially symmetrically distributed in the cytoplasm. As the zygote polarizes along the anterior/posterior axis, upstream polarity regulators drive P granule disassembly in the anterior cytoplasm and assembly/stabilization in the posterior cytoplasm. As a consequence, P granules become highly enriched in the posterior and are subsequently inherited by the posterior germline daughter cell (Brangwynne et al., 2009; Gallo, Wang, et al., 2010).
Dozens of proteins concentrate in P granules, including the constitutive P granule proteins PGL-1 and PGL-3 (Updike and Strome, 2010). MEG-3 and MEG-4 (MEG-3/4 hereafter) concentrate specifically in embryonic P granules and are particularly important for P granule segregation. MEG-3 and MEG-4 each contain a large intrinsically disordered region and MEG-3 has been shown to bind RNA with nanomolar affinity (Wang et al., 2014; Smith et al., 2016). In meg-3/4 mutant embryos, P granules are not disassembled in the anterior cytoplasm and therefore remain symmetrically distributed along the anterior/posterior axis (Wang et al., 2014). Intriguingly, MEG-3 concentration is higher in the posterior cytoplasm surrounding P granules than in the anterior cytoplasm (Wang et al., 2014), raising the possibility that the gradient in MEG-3 concentration along the anterior/posterior (A/P) axis might contribute to the establishment or maintenance of P granule asymmetry. This idea is supported by the observation that LLPS is a concentration-dependent process in vitro (Elbaum-Garfinkle et al., 2015; Zhang et al., 2015; Saha et al., 2016; Smith et al., 2016) and that increased expression of RNA granule proteins can drive LLPS in vivo (Molliex et al., 2015; Bolognesi et al., 2016).
Both the formation of the MEG-3 gradient (Wang et al., 2014) and the segregation of P granules depend on the cytoplasmic polarity regulators MEX-5 and MEX-6 (MEX-5/6 hereafter), two highly similar RNA-binding proteins that concentrate in the anterior cytoplasm (Schubert et al., 2000). In vitro, MEX-5 competes with MEG-3 and the P granule protein PGL-3 for RNA, thereby inhibiting MEG-3 and PGL-3 from undergoing RNA-stimulated LLPS (Saha et al., 2016; Smith et al., 2016). P granule disassembly in the anterior cytoplasm depends on MEX-5′s ability to bind RNA (Smith et al., 2016), consistent with the idea that MEX-5/6 destabilizes P granules by competing with P granule proteins for RNA. A second mechanism controlling P granule stability depends on MBK-2 (Pellettieri et al., 2003; Quintin et al., 2003; Pang et al., 2004), a dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) that phosphorylates MEG-3 and promotes the P granule disassembly in the anterior cytoplasm (Wang et al., 2014). The P granule PP2A phosphatase subunits PPTR-1 and PPTR-2 counteract MBK-2 by dephosphorylating MEG-3, thereby stabilizing P granules (Wang et al., 2014). MBK-2 also phosphorylates MEX-5 and MEX-6 (residue T186 in MEX-5), generating a binding site for the polo-like kinase PLK-1 (Chase et al., 2000; Nishi et al., 2008). In turn, PLK-1 drives the RNA-binding protein POS-1 to form a posterior-rich cytoplasmic gradient (Han et al., 2018). Whether PLK-1 also regulates MEG-3 gradient formation or P granule segregation is not known.
In this study, we address two questions raised by the above findings. First, what are the reaction/diffusion dynamics that maintain the posterior-rich MEG-3 concentration gradient? Second, what is the relationship between the MEG-3 gradient and MEG-3’s role in controlling P granule segregation? Imaging approaches such as spinning disk confocal microscopy have revealed a great deal about how the ensemble behaviors of P granule components contribute to P granule dynamics. However, these approaches obscure the behaviors of individual molecules as they move through the cytoplasm and in and out of P granules. To access these dynamics, we analyzed the single-molecule behaviors of MEG-3. Our findings suggest that the formation of the posterior-rich MEG-3 gradient does not promote MEG-3 association with P granules. Rather, our data suggest that the formation of the MEG-3 gradient and the incorporation of MEG-3 into P granules in the posterior are two parallel outputs of upstream polarity cues.
RESULTS
Near-TIRF imaging of PGL-1::GFP and MEG-3::Halo
We first used spinning disk confocal microscopy to image the localization of PGL-1::GFP and MEG-3::meGFP in the polarized C. elegans zygote. Both PGL-1::GFP and MEG-3::meGFP were present in P granules and in the cytoplasm outside of P granules. Whereas PGL-1::GFP was uniformly distributed in the cytoplasm outside of P granules, cytoplasmic MEG-3::meGFP levels were higher in the posterior cytoplasm than in the anterior cytoplasm (Wang et al., 2014) (Figure 1A). To access the behavior of individual molecules as they move through the cytoplasm and in and out of P granules, we used near-TIRF (total internal reflection fluorescence) microscopy. Embryos were imaged at ∼20 frames per second with the focal plane ∼0.5 μm from the cell cortex. For simplicity, we will refer to this region as the cytoplasm in the following sections, although we note that our near-TIRF imaging studies specifically report on this “subcortical” region of the cytoplasm and do not necessarily reflect dynamics deeper in the cytoplasm. The expression levels of transgenic PGL-1::GFP were reduced to the single-particle regime through partial GFP RNA interference (RNAi) (Robin et al., 2014). Under these conditions, PGL-1::GFP appeared highly dynamic and homogeneously distributed throughout the cytoplasm outside of P granules (Supplemental Movie S1). These findings are consistent with a model in which PGL-1::GFP exchanges between P granules and a well-mixed cytoplasmic pool.
FIGURE 1:

Single-molecule imaging of MEG-3::Halo. (A) Top, spinning disk confocal images of MEG-3::meGFP and PGL-1::GFP embryos at NEBD. Scale bar = 10 μm. Anterior is to the left, and posterior is to the right in this and all subsequent images. Bottom, intensity profile along the anterior/posterior axis of the embryos shown above (normalized to the anterior pole). (B) Near-TIRF imaging of MEG-3::Halo; PGL-1::GFP embryos. MEG-3::Halo molecules (cyan) are present both within PGL-1::GFP granules (magenta) and in the surrounding cytoplasm. PGL-1::GFP granules are segmented. Scale bar = 5 μm. (C) Enlargement of the region in B indicated by the dotted box. Note the presence of MEG-3::Halo molecules both within (PG molecule, marked by arrow) and outside (SD molecules) of the segmented PGL-1::GFP granules. Under these imaging conditions, fast-diffusing MEG-3::Halo molecules are blurred and cannot be tracked. Scale bar = 1 μm. (D, E) Examples of the appearance and disappearance of single MEG-3::Halo PG (D) and SD (E) molecules. Time relative to the initial appearance of the molecule is indicated above the panels. Right, the trajectories of the corresponding molecules with the black and the gray dot showing the appearance and disappearance events, respectively. Scale bar = 1 μm. (F) Violin plot (log-scale) of estimated Dc of PG and SD MEG-3::Halo molecules in the posterior. White dots = median, box = 25th and 75th percentiles, whiskers = 1.5X the interquartile range from the 25th and 75th percentiles. Statistical significance in F, H, and I were determined with Student’s t tests (two-tailed). Gray lines indicate Dc estimates from simulated Brownian motion of molecules with Dc = 0.01 μm2/s (left) and Dc = 0.11 μm2/s (right). (G) FCS autocorrelation curves of MEG-3::meGFP in the anterior and posterior cytoplasm (anterior, n = 28; posterior, n = 15). Error bars indicate SEM. (H) Ratio of fast and slow-diffusing components estimated by fitting the FCS curves (G) to a two-component model. For H and I, error bars represent standard deviation. (I) Dc of the fast-diffusing component estimated from FCS curves (G). (J) Schematic of an embryo with distinct FD, SD, and PG MEG-3 molecules and their median apparent Dc. P granules are in magenta.
Movie S1.
near-TIRF imaging of PGL-1::GFP.
We next used near-TIRF microscopy to image endogenously tagged MEG-3::Halo labeled with JF646-HaloTag ligand (Grimm et al., 2015). The dynamics of MEG-3::Halo molecules appeared qualitatively similar to those of MEG-3::meGFP molecules (Supplemental Movies S2 and S3). We chose to perform further analysis on MEG-3::Halo rather than MEG-3::meGFP for two reasons. First, MEG-3::Halo molecules could be imaged simultaneously with endogenously tagged PGL-1::GFP (Andralojc et al., 2017), which was used to mark P granules to distinguish MEG-3::Halo molecules that were associated with P granules and those that were present in the surrounding cytoplasm (Figure 1, B–E, and Supplemental Movies S2 and S4). Second, we found that using relatively low concentrations of JF646-HaloTag ligand resulted in sparse labeling of MEG-3::Halo, which facilitated single-molecule detection and tracking.
Movie S2.
near-TIRF imaging of MEG-3::Halo and PGL-1::GFP.
Movie S3.
near-TIRF imaging of Meg-3::meGFP.
Movie S4.
Spinning disk imaging of MEG-3::Halo and PGL-1::GFP.
We observed MEG-3::Halo molecules in three apparent diffusive states, which we refer to as P granule (PG), slow-diffusing (SD), and fast-diffusing (FD) MEG-3::Halo molecules. To estimate the short-term diffusion coefficient (Dc) of PG and SD MEG-3::Halo molecules, we analyzed the mean-squared displacement of trajectories longer than 20 frames and fitted it to MSD = 4Dcτα (Robin et al., 2014; described under Materials and Methods). PG MEG-3::Halo molecules were associated with P granules (as defined by their overlap with PGL-1::GFP granules) (Figure 1, C and D) and exhibited relatively slow, subdiffusive movements, with a median apparent diffusion coefficient (Dc) of 0.01 μm2/s (mean Dc = 0.05 μm2/s; anomaly coefficient α = 0.63) (Figure 1F). Slow-diffusing MEG-3::Halo molecules were dispersed in the cytoplasm outside of P granules (Figure 1, C and E) and had a median apparent Dc of 0.11 μm2/s (mean Dc = 0.16 μm2/s; α = 0.95) (Figure 1F). The range in the estimated diffusivities of SD and PG molecules (Figure 1F) could reflect heterogeneity in the diffusivities of these molecules or could result from error inherent in estimating diffusivities from short particle tracks (Robin et al., 2014). To distinguish between these possibilities, we estimated the Dc of simulated molecules with diffusion coefficients of 0.11 or 0.01 μm2/s and the same track lengths as those of the SD or PG molecules, respectively (Robin et al., 2014). The ranges in the estimated diffusivities of simulated 0.11 μm2/s molecules and SD molecules are similar (Figure 1F), indicating that this diffusion coefficient reasonably describes the diffusivity of SD molecules. In contrast, the range in the estimated diffusivities of simulated 0.01 μm2/s molecules is smaller than that of PG molecules (Figure 1F), suggesting heterogeneity in the diffusivity of P granule–associated MEG-3::Halo molecules.
In addition to the PG and SD MEG-3::Halo molecules, we could readily detect a population of highly dynamic MEG-3::Halo molecules that appeared to be homogeneously distributed throughout the zygotic cytoplasm (Supplemental Movie S2). We refer to these molecules as FD MEG-3::Halo molecules. Because they were highly dynamic, we could not reliably track FD MEG-3::Halo molecules and therefore did not include them in our single-particle tracking analysis. To estimate the diffusivity of FD MEG-3 molecules, we performed fluorescence correlation spectroscopy (FCS) on MEG-3::meGFP in the anterior cytoplasm and in the posterior cytoplasm outside of P granules. FCS autocorrelation curves (Figure 1G) were fitted with a two-component model in which the Dc of the slow component was constrained to 0.16 μm2/s and α = 0.95 (the average diffusivity of SD MEG-3::Halo particles) (Figure 1F). We estimate a Dc of ∼5 μm2/s (4.6 μm2/s in the anterior and 5.1 μm2/s in the posterior) for FD MEG-3::meGFP molecules (Figure 1I). We estimate that MEG-3::meGFP is predominantly in the FD state in the anterior cytoplasm (∼72% FD and 28% SD molecules in the anterior) and predominantly in the SD state in the posterior cytoplasm (∼44% FD and 56% SD molecules in the posterior) (Figure 1H).
During near-TIRF imaging, MEG-3::Halo molecules appeared and subsequently disappeared either within a P granule (PG molecules) or outside of P granules (SD molecules), which presumably reflects the association and subsequent dissociation of MEG-3::Halo with binding partners in P granules or distributed in the surrounding cytoplasm. On the basis of these initial observations, we conclude that MEG-3 exists in three diffusive states and that MEG-3 frequently transitions between diffusive states (Figure 1J).
MEG-3 gradient formation
Although we did not directly measure their distribution, the relatively fast diffusion of FD MEG-3::Halo molecules suggests that they were likely to be symmetrically distributed throughout the cytoplasm. In contrast, the concentration of SD MEG-3::Halo molecules was approximately threefold higher in the posterior cytoplasm than in the anterior cytoplasm of wild-type embryos (Figure 2A). We propose that the asymmetric distribution of SD molecules gives rise to the cytoplasmic MEG-3 gradient. To begin to understand how the MEG-3 gradient forms, we therefore characterized the single-molecule dynamics that enrich SD MEG-3::Halo molecules in the posterior. The displacement of MEG-3::Halo molecules in the SD state was small (<1 μm) and was symmetric along the A/P axis (Figure 2, B and D), suggesting that the gradient in SD MEG-3::Halo molecules does not reflect directional transport of SD molecules toward the posterior. Rather, we hypothesized that the kinetics of MEG-3::Halo switching between the FD and SD states might vary along the A/P axis such that the SD molecules become enriched in the posterior. To monitor the conversion of FD molecules into SD molecules, we quantified the rate at which new SD MEG-3::Halo molecules appeared along the A/P axis (Figure 2C). We found that new SD MEG-3::Halo molecules appeared twofold more frequently in the posterior cytoplasm relative to the anterior cytoplasm (Figure 2, C and E). SD MEG-3::Halo appearance events appeared to be broadly distributed in the cytoplasm outside of P granules, suggesting that the binding reactions that generate SD molecules occur at dispersed sites. Additionally, SD MEG-3::Halo molecules were significantly more persistent in the posterior cytoplasm than in the anterior cytoplasm (Figure 2F), which further contributed to their accumulation in the posterior.
FIGURE 2:

Single-molecule behaviors underlying MEG-3::Halo gradient formation. (A) The number of SD MEG-3::Halo particles across the A/P axis of embryos with the indicated genotype. Particles numbers were normalized to the anterior end and averaged among the indicated number (n) of embryos. Error bars indicate SEM. (B) Tracks of SD MEG-3::Halo molecules in representative embryos of the indicated genotype. Trajectories longer than 250 ms during 10 s acquisitions are shown. (C) Appearance events of SD MEG-3::Halo molecules in embryos of the indicated genotypes. The same embryos were analyzed in B and C. (D) Top, displacement of SD MEG-3::Halo molecules in the anterior and posterior cytoplasm from the embryo in B and C. For each molecule, the appearance position is normalized to the center of the graph and the disappearance position is indicated by a blue dot. Only the displacements of tracks >250 ms are shown. Bottom, frequency of displacements of tracks >250 ms along the A/P axis. Tracks are pooled from 37 embryos (n = total number of tracks analyzed). (E) Average appearance rate of SD MEG-3::Halo particles along the A/P axis. Appearance rate is normalized to the anterior region for each embryo. Note that we cannot directly compare appearance rates between embryos due to variability in Halo labeling and in illumination. The same embryos were analyzed in A, E, and F. n = number of embryos analyzed. Error bars indicate SEM. (F) Cumulative frequency of track durations for the indicated genotypes. Track duration values are pooled from the indicated number embryos in A. n = number of particle tracks analyzed. Note that genotypes within the brackets show similar cumulative frequency distributions.
The formation of the MEG-3 gradient depends on MEX-5/6 and on MBK-2 kinase (Wang et al., 2014; Smith et al., 2016). In mbk-2(RNAi) and mex-5/6(RNAi) embryos, the distribution (Figure 2, A and B), appearance rate (Figure 2, C and E), and persistence (Figure 2F) of SD MEG-3::Halo molecules were symmetric along the A/P axis (Supplemental Movies S5 and S6), indicating that MEX-5/6 and MBK-2 drive MEG-3 gradient formation by establishing asymmetries in the kinetics of SD molecule appearance and disappearance. MBK-2 phosphorylates MEX-5 on T186, priming MEX-5 to interact with PLK-1 kinase (Nishi et al., 2008). To test whether MEX-5 phosphorylation on T186 is required for MEX-5 to control MEG-3 dynamics, we analyzed mex-5(T186A) embryos in which the redundant MEX-5 homologue MEX-6 had been depleted by RNAi. The distribution, appearance rate and persistence of SD MEG-3::Halo molecules in mex-5(T186A);mex-6(RNAi) embryos were identical to mex-5/6(RNAi) and mbk-2(RNAi) embryos (Figure 2, A–C, E, and F, Supplemental Movies S5–S7). These results are consistent with a model in which the priming of the interaction between MEX-5 and PLK-1 by MBK-2 is required for MEG-3 segregation.
Movie S5.
near-TIRF imaging of Meg-3::meGFP and PGL-1::GFP in an mbk-2(RNAi) embryo.
Movie S6.
near-TIRF imaging of Meg-3::meGFP and PGL-1::GFP in an mex-5/6(RNAi) embryo.
Movie S7.
near-TIRF imaging of Meg-3::meGFP and PGL-1::GFP in an mex-5(T186A);mex-6(RNAi) embryo.
The uniform distribution of SD MEG-3::Halo molecules in mex-5(T186A);mex-6(RNAi) embryos suggests a potential role for PLK-1 in controlling the distribution of SD MEG-3::Halo molecules. Consistent with this possibility, in plk-1(RNAi) embryos, the posterior enrichment of SD MEG-3::Halo molecules was reduced (Figure 2, A and B) due to reductions in the differences in the appearance rate (Figure 2, C and E) and the persistence (Figure 2F) of SD MEG-3::Halo molecules between the anterior and posterior (Supplemental Movie S8). The plk-1(RNAi) embryos analyzed in these experiments likely retained some PLK activity because strong RNAi depletion of PLK-1 and its homologue PLK-2 results in sterility (Nishi et al., 2008). Therefore, we do not know whether the intermediate SD MEG-3::Halo molecule segregation phenotype in plk-1(RNAi) embryos was due to residual PLK activity or to PLK-independent function of MEX-5/6 and MBK-2.
Movie S8.
near-TIRF imaging of Meg-3::meGFP and PGL-1::GFP in an plk-1(RNAi) embryo.
Taken together, these data are consistent with a model in which MBK-2 and MEX-5/6 (potentially in conjunction with PLK-1) suppress the MEG-3::Halo SD state in the anterior cytoplasm, leading to the accumulation of SD MEG-3::Halo molecules in the posterior cytoplasm. Having analyzed the single-molecule behaviors that give rise to the MEG-3::Halo gradient, we next sought to analyze the connection between the MEG-3 gradient and the segregation of P granules to the posterior cytoplasm.
Estimation of the oligomeric state of SD MEG-3::meGFP
The observation that recombinant MEG-3 can phase separate in vitro indicates that MEG-3 is capable of homotypic interactions (Smith et al., 2016). Therefore, we considered the possibility that SD MEG-3 molecules reflect the formation of small MEG-3 assemblages that might be intermediates in the formation/growth of P granules. Such a model would predict the presence of multiple MEG-3 molecules within SD particles. To test this possibility, we assessed whether endogenously tagged MEG-3::meGFP was oligomeric within SD particles. Because of the transience of MEG-3::meGFP in the SD state, we could not reliably use the bleaching profile of individual SD particles to estimate the number of MEG-3::meGFP molecules present. Instead, we reasoned that if SD particles contain multiple MEG-3::meGFP molecules, then the brightness of SD particles would be reduced after extensive photobleaching of the embryo. Alternatively, if SD particles contain one MEG-3::meGFP molecule, then photobleaching would reduce the number of SD MEG-3::meGFP particles but not their brightness. We analyzed the brightness of SD MEG-3::meGFP particles before and after extensive photobleaching in mex-5/6(RNAi) embryos, in which the SD MEG-3::meGFP particles were dispersed throughout the cytoplasm and therefore more amenable to single-particle detection and intensity analysis. After 100 s of photobleaching, the total number of SD MEG-3::meGFP particles in the focal plane decreased by ∼80% (Figure 3, A and B). Strikingly, however, the intensity of SD MEG-3::meGFP particles did not change (Figure 3C). These data suggest that MEG-3::meGFP is predominantly monomeric in the SD state. We therefore propose that SD particles do not reflect the formation of small MEG-3::meGFP oligomers or granules. It is important to note that we cannot rule out the possibility that other factors may be associated with MEG-3::meGFP SD molecules.
FIGURE 3:

Brightness analysis of MEG-3::meGFP SD particles. (A) Representative near-TIRF images of MEG-3::meGFP in mex-5/6(RNAi) embryos before and after bleaching. The overall intensity of the embryo is brighter before bleaching because of higher particle density and out-of-focus signal. Scale bar = 5 μm. (B) Relative number of MEG-3::meGFP particles normalized to the average number between 0 and 1 s and plotted over time. After 100 s of photobleaching, the number of detectable MEG-3::meGFP particles dropped by an average of ∼80%. Average of five embryos. Error bars indicate SEM. (C) Brightness analysis of endogenously tagged MEG-3::meGFP particles in mex-5/6(RNAi) embryos before and after bleaching. Frequency of normalized intensity of all detectable MEG-3::meGFP particles pooled from the beginning of movies (0–1 s, the solid line) or from the end of the movies (95–100 s, the dashed line). Intensity is normalized to the peak value of 95–100 s for individual embryos as described in the methods. n = the total number of particle brightness estimates from five embryos. Note that because we did not track individual particles between frames, the brightness of each particle was estimated in each frame in which it was detected.
Single-molecule analysis of MEG-3::Halo association with P granules
We next considered the possibility that the higher concentration of MEG-3 in the posterior cytoplasm might increase the rate at which cytoplasmic MEG-3 encounters P granules, thereby promoting P granule stability/growth. The arrival rate Rarrive of a cytoplasmic protein at the surface of a granule with the radius Rg is described by
where ccyt is the cytoplasmic concentration and Dc is the cytoplasmic diffusion coefficient of the protein (Berg and Purcell, 1977). Therefore, the arrival rate of a cytoplasmic protein at the surface of an existing P granule (for example, during P granule maintenance) is a function of both the protein’s cytoplasmic concentration and its cytoplasmic diffusivity. To estimate the concentration of MEG-3::meGFP and PGL-1::GFP, embryos were bathed in either 300 nM recombinant GFP or 75 nM recombinant meGFP, which were used as standards to calibrate the intracellular protein concentration (Figure 4A). Background fluorescence intensity was estimated by imaging N2 embryos under the same conditions (Figure 4, B and D). The mean apparent concentration of PGL-1::GFP was 2281 nM in P granules and 530 nM in the cytoplasm outside of P granules (Figure 4, B and C), which is in reasonable agreement with previous mass spectrometry-based estimate of ∼1200 nM concentration of PGL-1 in mixed stage embryo lysates (Saha et al., 2016). The mean apparent concentration of MEG-3::meGFP was 126 nM in P granules and 22.6 nM in the posterior cytoplasm surrounding P granules (Figure 4, D and E). On the basis of our estimate that the ratio of FD:SD molecules in the posterior cytoplasm is 44:56 (Figure 1H), we estimate that the concentration of FD molecules is roughly 9.9 nM (0.44 * 22.6 nM) and the concentration of SD molecules is roughly 12.7 nM (0.56 * 22.6 nM) (Table 1). We could not reliably estimate MEG-3 concentration in the anterior cytoplasm because the intensity values were close to the background values. This analysis suggests that due to its higher cytoplasmic levels, cytoplasmic PGL-1 will encounter P granules at a significantly higher rate than cytoplasmic MEG-3. Furthermore, even though the concentration of FD molecules is lower than SD molecules in the posterior (44 and 56%, respectively; Figure 1H), FD molecules likely encounter P granules at a significantly higher rate than SD MEG-3 molecules because of the ∼30-fold difference in their diffusivity (Figure 1, F and I).
FIGURE 4:

Quantification of MEG-3::meGFP and PGL-1::GFP concentration. (A) Coomassie-stained SDS–PAGE gel of recombinant meGFP and GFP at the indicated concentrations. BSA was used as a loading standard. (B) PGL-1::GFP (right) and N2 (left, no GFP expression) embryos were bathed in 300 nM recombinant GFP. The top images (Bright) were normalized to show the GFP bath and cytoplasmic GFP and include saturated P granule signals. The bottom images (Dim) are normalized so that there are no saturated pixels. (C) Quantification of estimated PGL-1::GFP concentration in the cytoplasm outside of P granules (16 embryos) and in P granules (325 P granules from 16 embryos). Error bars = SD. Inset, fluorescence intensity of different concentration GFP baths in arbitrary units (AU). R2 value for the trendline is indicated. (D) MEG-3::meGFP (right) and N2 (left, no GFP expression) embryos were bathed in 75 nM recombinant meGFP. (E) Quantification of estimated MEG-3::meGFP concentration in the posterior cytoplasm outside of P granules (16 embryos) and in P granules (378 P granules from 16 embryos). Error bars = SD. Inset, fluorescence intensity of different concentration meGFP baths in arbitrary units (AU). R2 value for the trendline is indicated.
TABLE 1:
Summary of findings.
| MEG-3 diffusive states | |||||
|---|---|---|---|---|---|
| Diffusive state | FD (fast-diffusing) | SD (slow-diffusing) | PG (P granule) | ||
| Diffusion coefficient | 5 μm2/s (Figure 1I) | 0.11 μm2/s (Figure 1F) | 0.01 μm2/s (Figure 1F) | ||
| Distribution | Symmetric (assumed) | ∼3-fold posterior-rich gradient (Figure 2A) | Posterior (in P granules) | ||
To determine whether the predicted difference in the encounter rate of FD and SD MEG-3::HALO molecules with P granules was reflected in the rate at which FD and SD molecules incorporate into P granules, we assessed the relative contribution of FD and SD molecules to the recruitment of MEG-3::HALO into P granules. We first examined MEG-3::Halo particles that appeared within a 3-μm radius of individual P granules and observed that most SD MEG-3::Halo particles did not enter P granules (Figure 5A). To quantify the proportion of MEG-3::Halo molecules that entered P granules from the FD or the SD state, we very sparsely labeled MEG-3::Halo such that only ∼10 labeled molecules were present in the focal plane at a given time. To minimize potential tracking errors, we manually analyzed the association of individual MEG-3::Halo molecules with P granules that 1) initially had no labeled MEG-3::Halo within them, 2) remained in focus throughout the imaging, and 3) never contained more than 1 labeled MEG-3::Halo molecule at any given time. From 23 embryos imaged, we tracked a total of 43 MEG-3::Halo molecules associated with P granules that met these criteria.
FIGURE 5:

Recruitment of MEG-3::Halo molecules into P granules. (A) Top, trajectories of MEG-3::Halo molecules that appeared within a 3-μm radius from the center of a P granule. Because P granules move, the XY positions of each trajectory were registered to the P granule center for every time point. Black dots represent appearance events, and the dotted gray circle represents the P granule. Bottom, PGL-1::GFP (raw, unsegmented image on the left and segmented image on the right) and MEG-3::Halo from a single representative frame of the movie used for tracking. The dotted circle indicates the region analyzed. (B) Representative trajectories of MEG-3::Halo molecules that associate with P granules. Black dots represent appearance events and gray dots represent disappearance events. XY positions were registered to the P granule center for every time point. Percentage of each subgroup of PG MEG-3::Halo particles is indicated. The corresponding particles are shown in Supplemental Movies S9–S12. (C) Cumulative frequency of track durations for SD and PG MEG-3::Halo from the 23 very sparsely labeled embryos. n = the total numbers of particles analyzed.
MEG-3::Halo molecules that first appeared as SD molecules in the cytoplasm surrounding granules and subsequently moved into granules were counted as SD→PG transitions. PG MEG-3::Halo molecules that first appeared within the P granule were assumed to have transitioned from the FD to the PG state directly and were counted as FD→PG transitions. Of 43 molecules, 34 underwent apparent FD→PG transitions (Figure 5B and Supplemental Movie S9). Three of these 34 molecules subsequently left the P granule as SD molecules (FD→PG→SD; Figure 5B and Supplemental Movie S10). The remaining 31 molecules suddenly disappeared from the P granules, either due to photobleaching or due to dissociation into the FD state. Nine molecules underwent SD→PG transitions (Figure 5B and Supplemental Movie S11), two of which subsequently left the granules as SD molecules (SD→PG→SD; Figure 5B and Supplemental Movie S12). The persistence of MEG-3::Halo molecules in the PG state was significantly longer than in the SD state, which contributes to the enrichment of MEG-3 in P granules (Figure 5C). These results suggest that the majority of MEG-3::Halo molecules enter P granules from the pool of FD MEG-3::Halo molecules, even though the local concentration of MEG-3::Halo SD molecules is higher.
Movie S9.
near-TIRF imaging of Meg-3::meGFP and PGL-1::GFP embryos. FD to PG transition.
Movie S10.
near-TIRF imaging of Meg-3::meGFP and PGL-1::GFP embryos. FD to PG to SD transition.
Movie S11.
near-TIRF imaging of Meg-3::meGFP and PGL-1::GFP embryos. SD to PG transition.
Movie S12.
near-TIRF imaging of Meg-3::meGFP and PGL-1::GFP embryos. SD to PG to SD transition.
PLK-1 contributes to P granule segregation
On the basis of our observations suggesting that MEX-5/6 work in conjunction with PLK-1 to control MEG-3 gradient formation, we considered the possibility that PLK-1 might also contribute to the segregation of P granules. To test this possibility, we analyzed the distribution of MEG-3::meGFP and PGL-1::GFP granules in mex-5(T186A);mex-6(RNAi) and plk-1(RNAi) embryos. We found that similar to mex-5/6(RNAi) and mbk-2(RNAi) embryos, P granules were uniformly distributed in mex-5(T186A);mex-6(RNAi) embryos (Figure 6, A–D). In plk-1(RNAi) embryos, the P granule domain was significantly shifted toward the anterior (Figure 6, A–D). These data suggest that PLK-1, likely acting with MEX-5, contributes to the suppression of P granules in the anterior cytoplasm.
FIGURE 6:

PLK-1 regulates P granule segregation. (A) Spinning disk confocal images of MEG-3::meGFP in embryos of different genotypes at NEBD. Images were taken at the cell midplane. Scale bar = 10 μm. (B, D) Quantification of P granule distribution at NEBD. For embryos in which P granules are asymmetrically distributed, the positions of the anterior-most granules are plotted. Only granules >0.7 μm in diameter were counted to exclude small granules that are sometimes present in the anterior of wild-type embryos. The distribution of P granules in plk-1(RNAi) embryos was significantly shifted toward the anterior compared with the Control (p < 0.0001; Student’s t test). n = number of embryo analyzed. Error bars indicate SD. (C) Maximum projections of spinning disk confocal images of PGL-1::GFP at NEBD. Images were taken at the cell midplane. Scale bar = 10 μm.
DISCUSSION
Taking together the findings described in this study (summarized in Table 1), we propose the following model for the single-molecule behaviors underlying MEG-3 dynamics during polarity maintenance phase. We propose that outside of P granules, MEG-3 transitions between fast-diffusing states (Dc = 5 μm2/s; Figure 1I) and slow diffusing states (median Dc = 0.11 μm2/s, α = 0.95; Figure 1F), presumably through binding/dissociation with an unknown cytoplasmic binding partner(s). The threefold enrichment of MEG-3::Halo SD molecules in the posterior (Figure 2A) reflects both a twofold higher FD→SD transition rate (Figure 2, C and E) and an increased persistence in the SD state in the posterior cytoplasm relative to the anterior cytoplasm (Figure 2F). MEX-5/6, MBK-2 and PLK-1 act to suppress the MEG-3::Halo SD state in the anterior by regulating the kinetics with which MEG-3::Halo switches into and out of the SD state (Figure 2, C, E, and F). Although we have not quantified the distribution of FD MEG-3 molecules, their rapid diffusion suggests they are likely to be approximately symmetrically distributed along the A/P axis. MEG-3 is most highly concentrated in P granules, where MEG-3::Halo is relatively persistent (Figure 5C) and its diffusion is relatively slow (median Dc = 0.01 μm2/s, α = 0.63; Figure 1F). Even though the majority of MEG-3::meGFP molecules in the posterior cytoplasm outside of P granules are in the SD state (44:56 ratio of FD:SD; Figure 1H), the majority of MEG-3::Halo molecules appear to transition into P granules from the FD state (Figure 5B). We speculate that the disproportionate incorporation of FD molecules into P granules may, at least partly, be because the higher diffusivity of FD molecules results in FD molecules encountering P granules at a higher rate than SD molecules. We do not know whether there is a difference in the incorporation rates of FD and SD molecules when they encounter a P granule.
Importantly, our findings argue against a model in which the MEG-3 gradient contributes significantly to the maintenance of P granules in the posterior. Rather, we favor a model in which the maintenance of the MEG-3 gradient and P granule asymmetry are two separable outputs of the upstream polarity system. Consistent with the idea that gradient formation and P granule formation are separable, Smith et al. (2016) previously showed that the intrinsically disordered region of MEG-3 forms a gradient but does not form granules. In the future, it will be important to analyze MEG-3 dynamics early in the process of P granule segregation as it remains a possibility that the MEG-3 gradient plays a more instructive role during the establishment of P granule asymmetries. Additionally, such studies could address whether changes in the kinetics of MEG-3 association/dissociation from P granules drives the disassembly of P granules in the anterior cytoplasm and growth/stabilization of P granules in the posterior cytoplasm.
MEG-3 gradient formation
The dynamics underlying the MEG-3 gradient are similar in some respects to those that underlie the posterior-rich PIE-1 and POS-1 gradients. MEX-5/6 locally control the kinetics with which MEG-3, PIE-1, and POS-1 transition between FD and SD states such that the SD state accumulates in the posterior cytoplasm (Wu et al., 2015, 2018; Han et al., 2018; this study). In the case of PIE-1 and POS-1, the switch to the SD state appears to depend, at least in part, on their association with RNA (Han et al., 2018). While we do not know how MEG-3 switches to the SD state, MEG-3 binds RNA with high affinity in vitro (Smith et al., 2016), raising the possibility that the transition to the SD state reflects MEG-3 binding to RNA distributed in the cytoplasm surrounding P granules. Interestingly, the concentration of available MEG-3 target RNAs is higher in the posterior than in the anterior (Smith et al., 2016), which could explain the enrichment of MEG-3 SD molecules in the posterior. Another, nonexclusive possibility is that the association between MEG-3 and its binding partner(s) is inhibited in the anterior and/or stimulated in the posterior. MEX-5/6 appear to drive POS-1 segregation by recruiting PLK-1 to the anterior cytoplasm (Nishi et al., 2008), which phosphorylates POS-1 to locally inhibit POS-1 retention in SD molecules (Han et al., 2018). Whether an analogous mechanism, perhaps mediated by PLK-1, inhibits MEG-3’s switch to the SD in the anterior cytoplasm is not known. An important step in testing these different models will be to identify the MEG-3 binding partner(s) that mediate formation of SD molecules.
P granule segregation
MEG-3 can assemble into granules in vitro (Smith et al., 2016), suggesting homotypic MEG-3 interactions contribute to the recruitment of MEG-3 into P granules in vivo. In contrast, SD MEG-3::meGFP molecules appear to be monomeric, suggesting the MEG-3 aggregation/oligomerization is unlikely to underlie the transition into the SD state (Figure 3C). In addition to homotypic interactions, MEG-3 interactions with RNA (Smith et al., 2016) and potential interactions with other P granule proteins, some of which have been shown to phase separate on their own (Elbaum-Garfinkle et al., 2015; Saha et al., 2016), are likely to stabilize MEG-3 in P granules. The potential for multiple interactions within P granules may account for the observation that MEG-3 is less mobile and more persistent in P granules than in the SD state outside of P granules.
Likely because MBK-2 kinase stimulates P granule disassembly (Pellettieri et al., 2003; Quintin et al., 2003; Pang et al., 2004) both through phosphorylation of MEG-3/4 (Wang et al., 2014) and through phosphorylation of MEX-5 T186A (Nishi et al., 2008; this study), the P granule disassembly defect is stronger in mbk-2 mutant embryos than in mex-5/6 mutant embryos (Gallo, Wang, et al., 2010). Interestingly, we find similar defects in the distribution, appearance rate and persistence of SD MEG-3::Halo molecules in mbk-2 and mex-5/6 mutant embryos. One possible explanation for the difference in the severity of the P granule and SD MEG-3 molecule phenotypes in these mutant embryos is that MBK-2 phosphorylation might specifically inhibit MEG-3’s ability to scaffold P granules but not its ability to form SD molecules in the cytoplasm outside of P granules. As a consequence, MBK-2 activity would destabilize P granule in mex-5/6 mutant embryos but would not affect SD MEG-3 molecule dynamics. Our finding that P granule segregation is defective in mex-5(T186A);mex-6(RNAi) and plk-1(RNAi) embryos supports a role for PLK-1 kinase in P granule disassembly. We consider it unlikely that the P granule segregation defect in mex-5(T186A);mex-6(RNAi) and plk-1(RNAi) embryos is a consequence of the defect in SD MEG-3::Halo molecule segregation, which suggests PLK-1 may have a more direct role in regulating P granule disassembly, either through modification of a P granule component such as MEG-3 or through a regulator of P granule segregation.
Our findings highlight the importance of diffusivity in considering the propensity of a protein to undergo LLPS. Mechanisms that modulate the diffusivity of a protein outside of granules will modulate the rate at which the protein reaches the surface of the granule and therefore the rate at which it can incorporate into the granule. While both protein and RNA concentration are recognized as critical parameters that regulate phase-separation behavior, it will be interesting to learn the extent to which modulation of protein mobility is used to regulate LLPS in vivo.
MATERIALS AND METHODS
Caenorhabditis elegans strains and culturing
Caenorhabditis elegans strains were maintained on Nematode Growth Medium (NGM) plates with OP50 Escherichia coli as a source of food (Brenner 1974). The following strains were used in this study: JJ2101: zuIs242(nmy-2::PGL-1::GFP), unc119(+) II; unc-119(ed3) III (Sheth et al., 2010); DUP75: pgl-1(sam33[pgl-1::GFP::3XFLAG]) IV (Andralojc et al., 2017); EGD364: meg-3(egx4[meg-3::Halo]) X; EGD369: meg-3(egx4[meg-3::Halo]) X; pgl-1(sam33[pgl-1::GFP::3XFLAG]) IV; JH3441: mex-5(axls4301[T186A]) IV, JH3503 meg-3(ax3054[meg-3::meGFP]) X (Smith et al., 2016); EGD320: mex-5(axls4301[T186A]) IV; meg-3(ax3054[meg-3::meGFP]) X; EGD430: pgl-1(sam33[pgl-1::GFP::3XFLAG]) IV; mex-5(axls4301[T186A]) IV; EGD426: pgl-1(sam33[pgl-1::GFP::3XFLAG]), mex-5(axls4301[T186A]) IV; meg-3(egx4[meg-3::Halo]) X. The wild-type strain was N2. All strains were maintained at 20°C except for JJ2101, which was maintained at 25°C to prevent transgene silencing.
Feeding RNAi was performed as described previously (Timmons and Fire, 1998). mex-5/6(RNAi), mex-6(RNAi) and mbk-2(RNAi) were performed by culturing L4 worms on RNAi plates for 24 h at 25 °C and plk-1(RNAi) was performed by culturing L3 worms on RNAi plates for 26–28 h at 25°C. mex-5/6(RNAi) was performed by mixing equal volumes of mex-5(RNAi) and mex-6(RNAi) bacteria prior to seeding on RNAi culture plates. mex-6(RNAi) targets the 3′UTR of MEX-6 and specifically depletes MEX-6 but not MEX-5 (as demonstrated in Han et al., 2018). For near-TIRF imaging of PGL-1::GFP transgene (JJ2101), expression levels were reduced using GFP RNAi bacteria diluted 1:4 with control L4440 RNAi bacteria, which reduced the strength of the RNAi and resulted in a partial depletion of PGL-1::GFP levels.
MEG-3::Halo labeling
MEG-3::Halo labeling for spinning disk imaging (Supplemental Movie S4) was performed in liquid culture in a 96-well plate in 25 μl S medium containing concentrated OP50 and 2.5 μM JF646-HaloTag ligand (Grimm et al., 2015). L4s (30–40) were picked into each well, and 150 μl H2O was added into the surrounding wells to reduce evaporation. Plates were covered with aluminum foil and cultured for 24 h at room temperature at 150 rpm on a benchtop shaker.
MEG-3::Halo labeling for near-TIRF imaging was performed on solid medium in a 24-well plate. RNAi culture (50 μl) containing 0.6 μM of JF646-HaloTag ligand was spotted per well, which contained 500 μl solid medium with 25 μg/ml Carb and 1 mM IPTG (isopropyl b-d-1-thiogalactopyranoside). dsRNA expression was induced overnight. The next day, 40–50 worms were picked onto the plate and incubated for 22–26 h, depending on the RNAi (see above). Worms were then transferred to the appropriate RNAi plate without JF646-HaloTag ligand and cultured for either 2 h (Supplemental Movies S2 and S5–S8 and Figures 1, 2, and 5A) or 3 h (Figure 5, B and C, and Supplemental Movies S9–S12) to “wash out” excess free dye and to reduce the concentration of labeled MEG-3::Halo molecules.
Gene editing
Except for the T186A mutation, gene editing was performed with CRISPR/Cas9 using dpy-10 as the co-conversion marker as described previously (Arribere et al., 2014). To introduce halo into the meg-3 locus (between amino acids 121 and 122), we used a single guide RNA (sgRNA) sequence located internally in the MEG-3 intrinsically disordered region as described in Smith et al. (2016). Oligos containing the sgRNA sequence (gaagtctgcagagaagacgg) YW174 and YW175 were annealed and ligated into the sgRNA expression vector pRB1017 (Arribere et al., 2014). The repair template was a PCR product amplified from pDD378 (kind gift from Dan Dickinson) using the primers YW176 (gcatgcagctcaaggaaacggcagcaacgcctttaacagtattcctcctacggcccccgctgaaattggcacaggattcc) and YW177 (gctggagctattgcgtgtttgcaagttgcggcggaagtctgcagagaagactccggagatctcgagggtggag). The injection mix contained 40 ng/μl meg-3 sgRNA vector, 30 ng/μl repair template, 15 ng/μl dpy-10 sgRNA vector (pJA58) (Arriberel et al., 2014), 300 nM dpy-10 ssODN repair template (BH0271, IDT) and 50 ng/μl Cas9 expression vector (pDD162) (Dickinson et al., 2013). The mix was injected into N2 hermaphrodites, and F1s rollers were genotyped by duplex PCR using oligos YW180 (cctcaaaaccttacccaagcggtct), YW182 (ccaagacattgctggtgaacattgg), and YW156 (ctggcttgtcggactttcccattc). The ∼300 base pairs flanking the insertion site in the genome was verified by sequencing.
mex-5(T186A) was introduced into the N2 strain using preassembled CRISPR-Cas9 ribonucleoprotein complexes, as described (Paix et al., 2015). Crispr RNA (crRNA) JScr7 containing sgRNA sequence tgttggaagaggagtagacg and a single-stranded DNA repair template with the sequence gtggagattagtagcacacgcacagctccattgacgagctctgctcctcttccaacaagtctcgaatacgagactgttc were used. Genotyping was performed by PCR with oligos JS198 (ggagctcagcaagtcatgc) and JS199 (ccgaacttctcgccacg) followed by SacI digestion. The mutation was sequence verified.
Near-TIRF imaging
Near-TIRF imaging was conducted on a Nikon Ti-E inverted microscope equipped with a TI-TIRF illuminator and an 100×/1.49NA APO TIRF objective with the temperature correction collar set to 23°C. Both 488- and 640-nm laser illumination was delivered from the Andor Laser Combiner. Image collection was performed on two Andor iXon 897 cameras (17-MHz readout speed, gain = 3, intensification = 300) mounted on an Andor Tucam. Image collection was controlled by NIS Elements software (Advanced Research 4.50.00 Build 1117 Patch 03). The laser illumination angle was determined empirically to maximize signal to noise.
To image either MEG-3::meGFP or transgenic PGL-1::GFP embryos, embryos were dissected into M9 on a coverslip, to which approximately one hundred 20-μm polysterene beads were added (Bangs Laboratories). The coverslip was mounted on a slide and sealed with Vaseline. To image MEG-3::Halo; PGL-1::GFP embryos, embryos were dissected and then mouth pipetted into M9 on a new coverslip to minimize the amount of the free dye in the dissection media. Embryos were then washed twice with M9 before polysterene beads were added.
PGL-1::GFP transgene expression levels were depleted as described above. Endogenously tagged MEG-3::meGFP does not require partial depletion because it is expressed at relatively low levels; however, a short period of photobleaching (∼5 s) was conducted to reduce the density of fluorescent MEG-3::meGFP to facilitate single-particle tracking. For brightness analysis of MEG-3::meGFP SD particles, no prebleaching was performed. To image either PGL-1::GFP or MEG-3::meGFP, a 488/561 dual-band set was used, and movies were taken under 100% laser power for 50-ms exposures with continuous imaging (Fast Acquisition mode) in NIS-Elements (Nikon).
Spinning disk confocal microscopy
Embryos were dissected into M9 buffer and mounted on a 3% agarose pad and imaged on a spinning disk confocal microscope built around a Zeiss Axio Observer Z.1 equipped with a Zeiss Plan-Apochromat 63×/1.4NA oil immersion objective, a CSU-X1 spinning disk (Yokogawa), an Evolve 512 × 512 electron-multiplying charge-coupled device (EMCCD) camera (Photometrics) and a 50-mW 488-nm solid state laser and controlled by the Slidebook software package (3I, Inc). The still images of MEG-3::meGFP in Figures 1 and 6 were taken at the midplane with 60% laser power, 1-s exposures and camera intensification of 300 and gain of 1. Images of PGL-1::GFP in Figures 1 and 6 are from Z stacks (0.5-μm step size) covering the half of the embryo closest to the coverslip (the middle plane is shown in Figure 1, and maximum intensity projections are shown in Figure 6). Z stacks were collected using 20% laser power, 100-ms exposures, a gain setting of 1. To quantify the distribution of granules in Figure 6, granules with a diameter >0.7 μm were counted. For Supplemental Movie S4, images were captured at the middle plane at 15 s intervals with 20% laser power, 100-ms exposures, camera intensification of 300 and gain of 1 for the 488-nm channel and 60% laser power, 300-ms exposures, camera intensification of 300 and gain of 2 for the 640-nm channel.
Single-particle tracking and brightness analysis
Single-particle tracking of MEG-3::Halo was performed according to the methods described in Robin et al. (2014) and Sailer et al. (2015). Particles were detected using the Kilfoil (http://people.umass.edu/kilfoil/downloads.html) implementation of the Crocker–Grier algorithm (Crocker and Grier, 1996). Feature size, minimum intensity, and integrated intensity threshold were empirically chosen so that >90% of visible particles were detected. Particles were linked to trajectories with a gap size of three frames in μTrack using the Browning motion model (Jaqaman et al., 2008). No splitting or merging was allowed.
To segment P granules, our two-channel near-TIRF movies were segmented based on the PGL-1::GFP channel in Imaris 9 (Bitplane) with the Surface function using the Background Subtraction (Local Contrast) thresholding method. In the initial segmentation step, a maximum P granule size was set to 1 micron. This size limit was then removed, and we manually adjusted the segmentation intensity threshold. Decreasing the segmentation intensity threshold caused the segmentation domain of large, bright P granules to swell outside of the actual P granule boundary, whereas increasing the threshold excluded small, dim PGL-1::GFP granules. We chose thresholds that included ∼80% of the visible PGL-1::GFP granules and primarily excluded very small, dim PGL-1::GFP granules. The diameter of the P granule segmentation regions ranged from 0.3 to 2.5 μm (mean = 0.9 μm). Masks were then created for the segmented P granules and a pseudobinary image was generated by setting the intensity inside and outside the masks to constant values (0 for inside and 255 for outside). The pseudobinary image was exported from Imaris and then imported to Matlab (version R2016b; MathWorks) for further analyses. To separate slow-diffusing MEG-3 from P granular MEG-3, a custom Matlab script was used to identify tracks that are associated with P granules at any time point. These tracks are termed PG tracks. Tracks that are not associated with P granules at any given time are termed SD tracks.
To analyze the appearance rate and SD particle number, all tracks (longer than two frames) are divided into five equally spaced regions (regions 1–5 from the anterior to the posterior) along the anterior/posterior axis, depending on where they first appeared. To calculate the relative appearance rate, the number of new tracks per unit area in each region was normalized to that of region 1 (the anterior). Note that the absolute appearance rate cannot be compared between embryos due to variability in labeling density in different embryos. To analyze the distribution of SD particles, the total number of SD particles per unit area in each region was normalized to that of region 1. Cumulative frequency curves of track durations were plotted in Prism6 (GraphPad Software, La Jolla, CA). To minimize tracking errors in the PG particle analysis (Figure 5B), we report particles in P granules only where no other particles were observed within the last 20 frames. Particles that lasted for at least 20 frames were used for this analysis.
To estimate the diffusion coefficient of SD and PG particles, the first five lag times of trajectories longer than 20 frames were fitted to the equation MSD = 4Dctα (Robin et al., 2014). Estimated diffusion coefficients of SD and PG particles in regions 4 and 5 are graphed as violin plots, which were generated using an online graphing tool (http://shiny.chemgrid.org/boxplotr/). The same analysis was performed on simulated diffusion tracks that have the same lengths as the experimental tracks, with diffusion coefficients of 0.11 and 0.01 μm2/s.
For brightness analysis, the total intensity of each particle (without tracking) was subtracted by the integrated intensity within two-pixel radius around the particle (background signal). Histograms were plotted for particle intensity at 95–100 s in Matlab and then all the intensity values (including 0–1 s and 95–100 s) were normalized to the peak intensity value of 95–100 s for individual embryos. Normalized values from five embryos were then pooled to plot the distribution using ksdensity function in Matlab. We did not exclude P granule MEG-3 from our brightness analysis. However, given the relatively smaller number of P granules compared with the slow-diffusing particles, they do not significantly contribute to the overall distribution of the brightness values.
Fluorescence correlation spectroscopy
FCS was performed on a Nikon A1 laser scanning confocal microscope using a 60×/1.27NA water immersion objective (Plan Apo IR; Nikon) and a pulsed 485-nm laser at 20-MHz repetition frequency (PDL800-D; PicoQuant) with pinhole set to one Airy unit. Autocorrelation curves were obtained with afterpulsing suppression in SymPhoTime (PicoQuant). The effective confocal volume approximated by a Gaussian profile 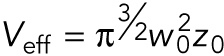 was calibrated by fitting the autocorrelation curve of 1 nM fluorescence dye ATTO488 (ATTO-TEC GmbH) to a three-dimensional triplet state model with one free diffusing species:
was calibrated by fitting the autocorrelation curve of 1 nM fluorescence dye ATTO488 (ATTO-TEC GmbH) to a three-dimensional triplet state model with one free diffusing species:
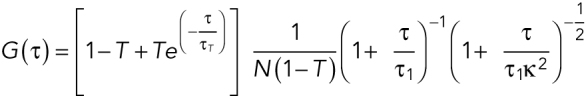 |
where w0 is the effective lateral focal radius at 1/e2 intensity, z0 is the effective focal radius along the optical axis at 1/e2 intensity, T is the triplet fraction of molecules, τT is the lifetime of the triplet state, N is the average number of molecules in the focal volume, τ1 is the diffusion time, k = z0 / w0 is the length to diameter ratio of the focal volume. The diffusion coefficient Dc is given by Dc = 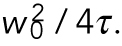 Given the diffusion coefficient of ATTO488 (360 µm2/s), mean k values of 5.2 (SEM = 0.0005), and mean effective confocal volume Veff = 0.27 fl (SEM = 0.005) were acquired. Embryos were dissected from young adults and imaged in M9. FCS data were collected for 60 s at ∼15% embryo length (anterior cytoplasm) or ∼85% embryo length (posterior cytoplasm) at nuclear envelope breakdown (NEBD). Autocorrelation curves were fitted to a three dimensional, two-component anomalous diffusion model with one triplet species,
Given the diffusion coefficient of ATTO488 (360 µm2/s), mean k values of 5.2 (SEM = 0.0005), and mean effective confocal volume Veff = 0.27 fl (SEM = 0.005) were acquired. Embryos were dissected from young adults and imaged in M9. FCS data were collected for 60 s at ∼15% embryo length (anterior cytoplasm) or ∼85% embryo length (posterior cytoplasm) at nuclear envelope breakdown (NEBD). Autocorrelation curves were fitted to a three dimensional, two-component anomalous diffusion model with one triplet species,
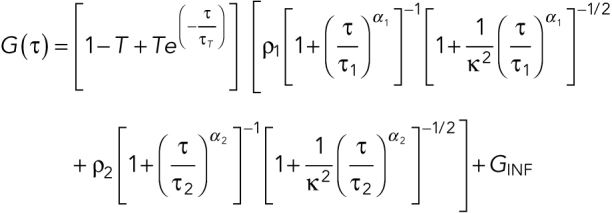 |
where α1 and α2 are the anomaly parameters, GINF is the correlation offset. We constrained one component to have a Dc = 0.16 μm2/s and α = 0.95 (the SD MEG-3::Halo molecules from our single-particle tracking data) and α of another component to be 1 (assumes FD molecules are free diffusing).
Partial purification of meGFP and GFP
The following amino acids differ between meGFP/GFP: 81Q/R, 85L/F, 86T/C, 207K/A, and 288L/H. meGFP was amplified from JH3503 genomic DNA (Smith et al., 2016) and GFP was synthesized as a gBlock gene fragment (IDT). Both open reading frames were cloned into the BamH1 and Xho1 sites of pGEX-KG (Guan and Dixon, 1991), which was modified to include a 6xHis-TEV cleavage site (gift from Henry Higgs, Dartmouth College). The resulting vectors, pTG91 (pGEX-KG-TEV-GFP) and pTG93 (pGEX-KG-TEV-meGFP), also included the linker GlySerGlyArgSer between TEV and meGFP/GFP.
pTG91 and pTG93 were transformed into BL21(DE3) pLysS (New England Biolabs), grown in 250 ml Terrific Broth at 37°C to OD600 = 0.8 and induced with 1 mM IPTG overnight at 16°C. Pellets were resuspended in 20 ml extraction buffer (50 mM Tris–HCl, pH 8, 500 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol [DTT], Roche complete protease inhibitors) and sonicated twice for 60 s. Extracts were pelleted at 17,000 × g for 10 min at 4°C, and supernatants were loaded onto column with glutathione resin (G Biosciences) equilibrated in glutathione S-transferase (GST) buffer (10 mM Tris–HCl, pH 8, 250 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.1 % NP40). Columns were washed two times with 3 ml GST buffer. The bound protein was cleaved with 10 μl GST-tagged AcTEV protease (Thermo Fisher Scientific) overnight at 4°C. Supernatants were loaded onto 1 ml Q Sepharose Fastflow resin (GE Lifesciences) equilibrated in 10 mM Tris–HCl, pH 8, 80 mM NaCl, 1 mM EDTA, 1 mM DTT and eluted with 4 ml 10 mM Tris–HCl, pH 8, 250 mM NaCl, 1 mM EDTA, 1 mM DTT. Elution fractions containing meGFP or GFP were pooled and dialyzed overnight at 4°C into 10 mM Tris–HCl, pH 8, 140 mM NaCl, 1 mM EDTA, 1 mM DTT using SepctraPor7 MWCO 8000 dialysis tubing. Protein concentrations were estimated by Bradford Assay (BioRad) and were confirmed by BCA Assay (Pierce) and on SDS–PAGE gels using a bovine serum albumin (BSA) standard.
Estimation of MEG-3::meGFP and PGL-1::GFP concentrations
N2, MEG-3::meGFP, and PGL-1::GFP gravid adults were dissected on a coverslip in 3 μl of M9 buffer. The buffer was exchanged at least four times with 10 μl M9 supplemented with 100 μg/ml BSA and either 75 nM meGFP or 300 nM GFP. Roughly one hundred 20 μm polystyrene beads (Bangs laboratory) in either M9/BSA/meGFP or M9/BSA/GFP were added, and the coverslip was sealed to a slide with Vaseline. The mean intensity within N2 embryos bathed in either 75 nM meGFP or 300 nM GFP was used to estimate the background signal within the embryo. meGFP and GFP fluorescence intensities were corrected for camera background, which was estimated from slides prepared with M9 only. The change in fluorescence intensity as a function of GFP or meGFP concentration was determined by measuring at least five images from at least two independent slides. The data points in the Figure 4, C and E, insets are the mean intensities from each slide.
PGL-1::GFP and MEG-3::meGFP concentrations were estimated in a second independent experiment with independent GFP and meGFP purifications. In the second experiment (which was not included in Figure 4), BSA was not added to the recombinant protein and MEG-3::meGFP embryos were bathed in 150 nM meGFP. In this experiment, we estimated MEG-3::meGFP concentration in the posterior cytoplasm to be 29.4 nM (n = 23 embryos; SEM = 7.1 nM) and in P granules to be 148 nM (n = 114 P granules from 14 embryos; SEM = 5.0 nM). We estimated PGL-1::GFP concentration in the cytoplasm to be 426 nM (n = 8 embryos; SEM = 23.1) and 1361 nM (n = 119 P granules from eight embryos; SEM = 72.3 nM). We report the estimates from the first experiment in Figure 4 because the addition of BSA resulted in a more uniform distribution of recombinant GFP and meGFP and because the fluorescence of 75 nM meGFP more closely matches the fluorescence of MEG-3::meGFP zygotes than 150 nM meGFP.
Supplementary Material
Acknowledgments
We thank Bing He, Geraldine Seydoux, and members of the Griffin lab for helpful discussions and for comments on the manuscript. We thank Luke Lavis (Janelia Research Campus) for generously providing the HaloTag JF 656 ligand, Dustin Updike (MDI Biological Laboratory) for sharing the PGL-1::GFP strain, Geraldine Seydoux (Johns Hopkins) for sharing the MEG-3::meGFP strain, and Dan Dickinson (UT Austin) for sharing the C. elegans codon-optimized HaloTag plasmid (pDD378) and for advice on labeling with Halo ligands. We thank Younan Li and Ed Munro (University of Chicago) for help with single-molecule detection and tracking. The TIRF microscope used in this study was funded by National Institutes of Health (NIH) Grant S10OD018046. The FCS microscope was funded by National Science Foundation award DBI-1039423. Y.W. was supported by a Copenhaver Fellowship from Dartmouth. Research in the Griffin lab is supported by NIH Grant R01GM110194 to E.E.G.
Abbreviations used:
- A/P axis
anterior/posterior axis
- FCS
fluorescence correlation spectroscopy
- NEBD
nuclear envelope breakdown.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-06-0402) on December 12, 2018.
REFERENCES
Boldface names denote co–first authors.
- Alberti S, Carra S. (2018). Quality control of membraneless organelles. J Mol Biol , 4711–4729. [DOI] [PubMed] [Google Scholar]
- Andralojc KM, Campbell AC, Kelly AL, Terrey M, Tanner PC, Gans IM, Senter-Zapata MJ, Khokhar ES, Updike DL. (2017). ELLI-1, a novel germline protein, modulates RNAi activity and P-granule accumulation in Caenorhabditis elegans. PLoS Genet , e1006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, Fire AZ. (2014). Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans . Genetics , 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg HC, Purcell EM. (1977). Physics of chemoreception. Biophys J , 193–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi B, Lorenzo Gotor N, Dhar R, Cirillo D, Baldrighi M, Tartaglia GG, Lehner B. (2016). A concentration-dependent liquid phase separation can cause toxicity upon increased protein expression. Cell Rep , 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, Hyman AA. (2009). Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science , 1729–1732. [DOI] [PubMed] [Google Scholar]
- Brenner S. (1974). The genetics of Caenorhabditis elegans . Genetics , 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase D, Serafinas C, Ashcroft N, Kosinski M, Longo D, Ferris DK, Golden A. (2000). The polo-like kinase PLK-1 is required for nuclear envelope breakdown and the completion of meiosis in Caenorhabditis elegans . Genesis , 26–41. [DOI] [PubMed] [Google Scholar]
- Crocker JC, Grier DG. (1996). Methods of digital video microscopy for colloidal studies. J Colloid Interf Sci , 298–310. [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. (2013). Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods , 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S, Kim Y, Szczepaniak K, Chen CC, Eckmann CR, Myong S, Brangwynne CP. (2015). The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci USA , 7189–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo CM, Wang JT, Motegi F, Seydoux G. (2010). Cytoplasmic partitioning of P granule components is not required to specify the germline in C. elegans .. Science , 1685–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods , 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. (1991). Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem , 262–267. [DOI] [PubMed] [Google Scholar]
- Han B, Antkowiak KR, Fan X, Rutigliano M, Ryder SP, Griffin EE. (2018). Polo-like kinase couples cytoplasmic protein gradients in the C. elegans zygote. Curr Biol , 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. (2008). Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods , 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell , 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi Y, Rogers E, Robertson SM, Lin R. (2008). Polo kinases regulate C. elegans embryonic polarity via binding to DYRK2-primed MEX-5 and MEX-6. Development , 687–697. [DOI] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G. (2015). High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics , 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KM, Ishidate T, Nakamura K, Shirayama M, Trzepacz C, Schubert CM, Priess JR, Mello CC. (2004). The minibrain kinase homolog, mbk-2, is required for spindle positioning and asymmetric cell division in early C. elegans embryos. Dev Biol , 127–139. [DOI] [PubMed] [Google Scholar]
- Pellettieri J, Reinke V, Kim SK, Seydoux G. (2003). Coordinate activation of maternal protein degradation during the egg-to-embryo transition in C. elegans . Dev Cell , 451–462. [DOI] [PubMed] [Google Scholar]
- Purice MD, Taylor JP. (2018). Linking hnRNP function to ALS and FTD pathology. Front Neurosci , 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintin S, Mains PE, Zinke A, Hyman AA. (2003). The mbk-2 kinase is required for inactivation of MEI-1/katanin in the one-cell Caenorhabditis elegans embryo. EMBO Rep , 1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami M, Taylor JP, Parker R. (2013). Altered ribostasis: RNA-protein granules in degenerative disorders. Cell , 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin FB, McFadden WM, Yao B, Munro EM. (2014). Single-molecule analysis of cell surface dynamics in Caenorhabditis elegans embryos. Nat Methods , 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Weber CA, Nousch M, Adame-Arana O, Hoege C, Hein MY, Osborne-Nishimura E, Mahamid J, Jahnel M, Jawerth L, et al. (2016). Polar positioning of phase-separated liquid compartments in cells regulated by an mRNA competition mechanism. Cell , 1572–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailer A, Anneken A, Li Y, Lee S, Munro E. (2015). Dynamic opposition of clustered proteins stabilizes cortical polarity in the C. elegans zygote. Dev Cell , 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert CM, Lin R, de Vries CJ, Plasterk RH, Priess JR. (2000). MEX-5 and MEX-6 function to establish soma/germline asymmetry in early C. elegans embryos. Mol Cell , 671–682. [DOI] [PubMed] [Google Scholar]
- Sheth U, Pitt J, Dennis S, Priess JR. (2010). Perinuclear P granules are the principal sites of mRNA export in adult C. elegans germ cells. Development , 1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, Brangwynne CP. (2017). Liquid phase condensation in cell physiology and disease. Science , eaaf4382. [DOI] [PubMed] [Google Scholar]
- Smith J, Calidas D, Schmidt H, Lu T, Rasoloson D, Seydoux G. (2016). Spatial patterning of P granules by RNA-induced phase separation of the intrinsically-disordered protein MEG-3. Elife , e21337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strome S, Updike D. (2015). Specifying and protecting germ cell fate. Nat Rev Mol Cell Biol , 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Fire A. (1998). Specific interference by ingested dsRNA. Nature , 854. [DOI] [PubMed] [Google Scholar]
- Updike D, Strome S. (2010). P granule assembly and function in Caenorhabditis elegans germ cells. J Androl , 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina E. (2012). The diverse functions of germline P-granules in Caenorhabditis elegans. Mol Reprod Dev , 624–631. [DOI] [PubMed] [Google Scholar]
- Wang JT, Smith J, Chen BC, Schmidt H, Rasoloson D, Paix A, Lambrus BG, Calidas D, Betzig E, Seydoux G. (2014). Regulation of RNA granule dynamics by phosphorylation of serine-rich, intrinsically disordered proteins in C. elegans .. Elife , e04591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Han B, Li Y, Munro E, Odde DJ, Griffin EE. (2018). Rapid diffusion-state switching underlies stable cytoplasmic gradients in the Caenorhabditis elegans zygote. Proc Natl Acad Sci USA , E8440–E8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Elbaum-Garfinkle S, Langdon EM, Taylor N, Occhipinti P, Bridges AA, Brangwynne CP, Gladfelter AS. (2015). RNA Controls PolyQ Protein Phase Transitions. Mol Cell , 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.