

Abstract
Preeclampsia (PE) is characterized by new-onset hypertension during pregnancy and is associated with immune activation and placental oxidative stress. Mitochondrial dysfunction is a major source of oxidative stress and may play a role in the pathology of PE. We (Vaka VR, et al. Hypertension 72: 703–711, 2018. doi:10.1161/HYPERTENSIONAHA.118.11290.) have previously shown that placental ischemia is associated with mitochondrial oxidative stress in the reduced uterine perfusion pressure (RUPP) model of PE. Furthermore, we have also shown that placental ischemia induces natural killer (NK) cell activation in RUPP. Thus, we hypothesize that NK cell depletion could improve mitochondrial function associated with hypertension in the RUPP rat model of PE. Pregnant Sprague-Dawley rats were divided into three groups: normal pregnant (NP), RUPP, and RUPP+NK cell depletion rats (RUPP+NKD). On gestational day (GD)14, RUPP surgery was performed, and NK cells were depleted by administering anti-asialo GM1 antibodies (3.5 µg/100 µl ip) on GD15 and GD17. On GD19, mean arterial pressure (MAP) was measured, and placental mitochondria were isolated and used for mitochondrial assays. MAP was elevated in RUPP versus NP rats (119 ± 1 vs.104 ± 2 mmHg, P = 0.0004) and was normalized in RUPP+NKD rats (107 ± 2 mmHg, P = 0.002). Reduced complex IV activity and state 3 respiration rate were improved in RUPP+NKD rats. Human umbilical vein endothelial cells treated with RUPP+NKD serum restored respiration with reduced mitochondrial reactive oxygen species (ROS). The restored placental or endothelial mitochondrial function along with attenuated endothelial cell mitochondrial ROS with NK cell depletion indicate an important role of NK cells in mediating mitochondrial oxidative stress in the pathology of PE.
Keywords: mitochondria, natural killer cells, placental ischemia, reactive oxygen species
INTRODUCTION
Preeclampsia (PE) affects 5–7% of all pregnancies in the United States (1, 30, 31). Despite being the leading cause of maternal death and maternal and perinatal morbidity, there is no cure or standard treatment for this disease. Furthermore, the mechanisms responsible for the pathogenesis of PE are unclear. The syndrome is characterized by hypertension after the 20th week of pregnancy and is associated with endothelial dysfunction and systemic vasoconstriction (1, 26, 30, 31). Hypertension associated with PE often remits after delivery, implicating the placenta as a central culprit in the disease. An important initiating event is thought to be placental ischemia caused by reduced uteroplacental perfusion pressure (RUPP) that leads to the release of placental factors causing widespread maternal endothelial dysfunction, hypertension, and intrauterine growth restriction (IUGR) (17). Numerous factors have been implicated in the pathogenesis of PE, including immune activation and oxidative stress (12, 20). One potential mechanism is natural killer (NK) cell-mediated oxidative stress and tissue necrosis. NK cells play a critical role in maintenance of normal pregnancy (32). Uterine NK cells interact closely with trophoblast cells and secrete cytokines that promote trophoblast growth and mediate trophoblast differentiation, invasion, and spiral artery remodeling (15, 24, 27, 32). NK cells are activated by inflammatory cytokines and binding of their Fc receptors to IgG-bound cells, causing stimulation of perforin-granzyme-mediated cytotoxicity and oxidative stress. Recent studies show the importance of cytolytic NK cells in PE (10, 11). In fact, we (9) recently showed the importance of NK cells in mediating hypertension and fetal growth restriction in the RUPP rat model of PE. By depleting NK cells in RUPP rats with anti-asialo ganglio-N-tetraosylceramide (anti-asialo GM1) antibody, we were able to attenuate hypertension and reduce inflammatory cytokines tumor necrosis factor-α (TNF-α) and IL-17, in association with improved fetal outcomes in RUPP rats (9).
We (33) have recently shown that placental ischemia-induced hypertension is also associated with mitochondrial oxidative stress. Furthermore, mitochondrial antioxidants MitoQ/MitoTEMPO significantly reduced blood pressure and improved fetal weights in RUPP rats, which highlights the importance of mitochondrial oxidative stress in response to placental ischemia (33). The role of NK cells in contributing to mitochondrial oxidative stress during PE or in RUPP rats is unknown. However, since cytolytic NK cells are significantly elevated in PE and associated with the progression of the disease, we believe one mechanism of impaired mitochondrial function during PE is through NK cell cytotoxicity. Moreover, NK cells are elevated in RUPP rats, and to date there are no studies examining the role of NK cells in causing mitochondrial mediated oxidative stress in the RUPP rat. Therefore, we hypothesized that NK cell activation could mediate placental mitochondrial dysfunction, which contributes to the development of hypertension during PE. The objective of this study was to deplete NK cells by use of anti-asialo GM1 antibodies, as previously published, in RUPP rats and determine the effect on blood pressure and mitochondrial function. Moreover to examine a role for factors secreted in response to NK cell activation to cause peripheral mitochondrial reactive oxygen species (mtROS), we will examine the role of circulating factors from NK cell depleted RUPPs on mitochondrial function and oxidative stress in endothelial cells in vitro.
METHODS
Animals
Female pregnant Sprague-Dawley rats purchased from Envigo (Indianapolis, IN) were used in the present study. Rats were housed in a temperature-controlled room (23°C) with a 12:12-h light-dark cycle with free access to standard rat chow and water. This study complied with guidelines and principles published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the University of Mississippi Medical Center’s Institutional Care of Animals and the Institutional Animal Care and Use Committee (IACUC). All first-time pregnant rats used were age (12–13 wk) and weight (250 g) matched. On gestation day (GD)14, the average weight of the rats (before RUPP procedure or administration of anti-asialo GM1 antibodies) was 270 g. On GD19 (before euthanasia and harvest of tissues) the weights of the normal pregnant (NP) rats were 334 g, of the RUPP rats 281 g, and of the RUPP rats depleted of NK cells was 284 g.
Materials and Reagents
Glutamate (49621)/malate (M1000), oligomycin (O4876), FCCP (C2920), rotenone (R8875), antimycin A (A8674), sucrose (84097), HEPES (H3375), EGTA (E3889), BSA (A9647), MOPS (M1254), KPi (795488), TMPD (T3134), NADH (10107735001), KCN (60178), decyl-ubiquinone (D7911), DCPIP (D1878), DNTB (D218200), and M199 (M4530) were purchased from Sigma-Aldrich. Asialo-GM1 polyclonal antibody (16-6507-39), DMEM (10566-016), FBS (16000044), and antimycotic/antibiotic (15240062) were purchased from Thermo Fisher Scientific.
Effect of NK Cell Depletion on Placental Ischemia-Induced Mitochondrial-Mediated Oxidative Stress in Placenta
Reduced uterine perfusion pressure model.
Rats were divided into three groups: NPs, RUPPs, and RUPPs depleted of NK cells (RUPP+NKD). Briefly, in RUPPs, reduction in blood flow to the uteroplacental unit was achieved by placing silver clips (0.203 mm ID) on the abdominal aorta (1 clip) above the iliac bifurcation and around the ovarian arteries (2 clips) on both sides under isoflurane anesthesia on Gday 14.
NK cell depletion.
NK cells were depleted from RUPP rats using anti-asialo GM1 (3.5 µg/100 µl ip) antibodies on GD15 and GD 17, as previously published (9). On GD 18, catheters were inserted into carotid arteries, and mean arterial blood pressure was measured on GD19. Immediately after blood pressure measurements, placentas were harvested and processed for further experiments.
Isolation of intact mitochondria.
Intact placental mitochondria were isolated by a differential centrifugation method (33). Briefly, placentas (2 placentas per rat) were rinsed in ice-cold Mito I buffer (250 mM sucrose, 10 mM HEPES, 1 mM EGTA 0.1% BSA, pH 7.2), minced with fine scissors, and homogenized using a glass-glass dounce homogenizer (4 ml buffer, 5 manual stokes). The homogenate was subjected to centrifugation at 4,000 rpm for 3 min at 4°C. The supernatant containing mitochondria was collected and centrifuged at 10,000 rpm for 10 min at 4°C. Then, the pellet was washed with 1 ml of Mito I buffer followed by Mito II (250 mM sucrose, 10 mM HEPES, 0.1% BSA, pH 7.2) at the same settings as before. The final mitochondrial pellet was resuspended in 200 µl of Mito II buffer and used immediately for respiration measurements. A small fraction of mitochondrial sample was flash-frozen and stored at −80°C for electron transport chain (ETC) complex activity assays.
Mitochondrial respiration.
Oroboros Oxygraph-2K was used to perform respiration measurements in the isolated placental mitochondria. Basal oxygen consumption was collected from the isolated mitochondria suspended in respiration buffer (100 mM KCl, 5 mM KPi, 1 mM EGTA, BSA, 1 mg/ml, 50 mM MOPS, pH 7.4) with no added exogenous substrates. Then, glutamate (10 mM)/malate (2 mM, Complex I substrates) mixture was added to the chamber to collect state 2 respiration rate, and state 3 (coupled respiratory state) was measured by adding ADP (5 mM). This was followed by leak/state 4 and uncoupled states, obtained by adding oligomycin (2.5 µM) and FCCP (0.5 µM) respectively. At the end of the experiment, nonmitochondrial oxygen consumption was measured by the addition of rotenone (0.5 µM) and antimycin A (2.5 µM), which was subtracted from all the measured states to correct for nonmitochondrial respiration. The measurements were taken using a sample volume of 30 μl in 2 ml of respiration buffer. The collected data were normalized for the amount of protein and expressed as picomoles of oxygen consumed per second per milligram of mitochondrial protein.
Measurement of ETC activity.
Complex I and IV activities were measured using a spectrophotometer and Oxygraph-2K (OROBOROS-High Resolution respirometer) (33). Complex I (NADH dehydrogenase) activity was determined by measuring the oxidation of NADH at 340 nm (ε = 6.22 mM−1·cm−1) using a spectrophotometer (2800 UV/Vis Spectrophotometer no, 83057-30, Cole-Palmer). Briefly, a quartz cuvette containing placental mitochondrial membranes (10 µl), decyl ubiquinone (DUQ, 100 µM), KCN (2.5 mM), and antimycin A (5 mM) was incubated for 10 min in a at 25°C. After incubation, NADH (100 µM) was added to start the reaction. The reaction was monitored for 3 min at 340 nm to measure decrease in absorbance, an indicator of NADH oxidation. Additionally, rotenone sensitivity was measured in a separate assay by including rotenone (5 µM) in the above-mentioned reaction mixture. Absorbance in the presence of rotenone was subtracted from the absorbance from the reactions without rotenone. The activity of Complex I was calculated using a molar extinction coefficient (expressed as nmol of e−·mg−1·min−1). Complex IV (cytochrome c oxidase) activity was measured using the Oxygraph-2K. The reaction mixture containing 2 ml of Tris buffer (50 mM, pH 7.4) along with ascorbate (3 mM), N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD, 0.3 mM), and horse heart cytochrome c (40 μM) were added to the oxygraph chamber. The reaction was started by adding mitochondrial membranes (30 µl). Oxygen consumption before adding mitochondria was subtracted to correct for nonmitochondrial oxygen consumption. Complex IV activity was normalized per milligram (expressed as nmol of oxygen consumed, mg−1·min−1).
Effect of NK Cell Depletion on Placental Ischemia-Induced Soluble Factors-Mediated Vascular Endothelial Cell Mitochondrial Energetics
Cell culture.
Human umbilical vein endothelial cells (HUVECs) were purchased from ATCC. Cells were cultured in DMEM-M199 (50:50) with 10% FBS and 1% antimycotic/antibiotic in a humidified atmosphere with 5% CO2, 20% O2, and 75% N2 at 37°C. The cells at passage 4 were cultured in the appropriate T25 flasks or six-well plates for the following experiments.
Mitochondrial respiration in HUVECs.
Respiration was measured in HUVECs following incubation with 10% NP, RUPP, or RUPP+NKD serum overnight. In brief, HUEVCs were grown to 70% confluence in T25 culture flasks. Cells were serum starved for 4 h before incubation with 10% experimental NP or RUPP or RUPP+NKD serum overnight. Then, experimental serum was washed off twice with DPBS, and serum-free medium was added for an additional 4 h. Cells were harvested and counted after serum starvation. Immediately thereafter, cells were used to measure respiration using the Oxygraph-2K. Basal (cells), leak (Oligo), and maximal/uncoupled (FCCP) rates of respiration were measured. The rate of oxygen consumption was expressed as picomoles of oxygen consumed per second per million cells.
Mitochondrial mediated ROS production in HUVECs.
mtROS were measured using MitoSOX red, a novel fluorogenic dye specifically targeted to mitochondria in live cells. In brief, HUEVCs were grown to 70% confluence in six-well culture plates. Cells were serum starved for 4 h before incubation with 10% experimental NP or RUPP or RUPP+NKD serum overnight. Then, experimental serum was washed off and cells were incubated with MitoSOX red (5 µM) for 30 min at 37°C. Antimycin A (100 µM) was included in the experiment as a positive control. Serum-free medium was added after the cells were washed twice with DPBS, and the cells were incubated for an additional 4 h. Cells were collected and transferred to FACS tubes to run samples on the flow cytometer with 488-nm excitation to measure oxidized MitoSOX red in the FL2 and FL3 channels of a Gallios flow cytometer (Beckman Coulter, Brea, CA). At least 5,000 events for each sample were collected and analyzed.
Statistical Analysis
All data are expressed as means ± SE. Comparisons among the three groups were assessed by one-way ANOVA with Tukey’s post hoc multiple comparisons analysis. A value of P < 0.05 was considered statistically significant. When significance was not found among the groups, Student’s t-test was used to compare two groups: NP vs. RUPP, RUPP vs. RUPP+NKD, or NP vs. RUPP+NKD.
RESULTS
Effect of NK cell Depletion on Hypertension and Placental Mitochondrial Dysfunction in RUPP Rats
RUPP rats had higher blood pressures compared with NP rats on Gday 19 (Fig. 1; 119 ± 1 vs.104 ± 2 mmHg, P = 0.0004, n = 10 vs. 11). Depleting NK cells by the administration of anti-asialo GM1 antibodies in RUPPs normalized blood pressure to the level of NP rats (Fig. 1; 107 ± 2 mmHg, P = 0.0024, n = 13).
Fig. 1.
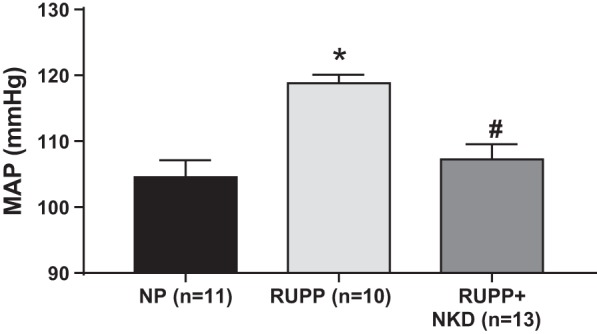
Natural killer (NK) cell depletion normalizes blood pressure in reduced uterine perfusion pressure (RUPP) rats. RUPP surgery was performed on gestation day (GDay) 14, anti-asialo ganglio-N-tetraosylceramide (anti-asialo GM1) antibodies (3.5 µg/100 µl ip) on Gday 15 and Gday 17. Conscious blood pressure was measured on GDay 19. Mean arterial pressure (MAP) is significantly elevated in RUPPs (n = 10) vs. normal pregnant (NP; n = 11, P = 0.0004) rats. Moreover, NK cell depletion attenuated this rise in blood pressure in RUPPs (n = 13, P = 0.002). Data are presented as means ± SE. One-way ANOVA with Tukey’s post hoc analysis was used to perform to statistical analysis. *Significance compare with NP; #significant compared with RUPP.
ETC activity is improved with NK cell depletion.
Complex I and IV activities were measured in the mitochondrial membranes to examine ETC activity. Complex I (Fig. 2A; 16 ± 0.2 vs. 29 ± 2 nmol e-·min−1·mg−1, P = 0.0046, Student’s t-test, n = 3 vs. 4) and complex IV (Fig. 2B; 323 ± 61 vs. 581 ± 86 nmol e-·min−1·mg−1, P = 0.0495, Student’s t-test, n = 4 vs. 4) activity was drastically reduced in RUPP placental mitochondria compared with NP placental mitochondria. NK cell depletion showed a trend in restoring complex IV activity (Fig. 2B; 554 ± 72 nmol e-·min−1·mg−1, P = 0.0579, Student’s t-test, n = 3) vs. RUPP controls. However, we did not see any improvement in Complex I activity with NK cell depletion (Fig. 2A; 25 ± 6.2 nmol e-·min−1·mg−1, P = 0.216, Student’s t-test, n = 3).
Fig. 2.

Electron transport chain (ETC) activity is improved in placenta of reduced uterine perfusion pressure (RUPP) rats depleted of natural killer (NK) cells. Complex I and Complex IV activities were measured in isolated mitochondrial membranes using spectrophotometer and Oxygraph-2K, respectively. Complex I and Complex IV activities were significantly reduced in RUPP (n = 4) placental mitochondria vs. normal pregnant rats (NP; n = 4, P = 0.0046 for Complex I and P = 0.0495 for Complex IV). Furthermore, NK cell depletion (NKD) shows a trend in improvement complex I (A; P = 0.216) and complex IV activity (B; P = 0.0579) in RUPP rats (n = 3). Data are presented as means ± SE. ANOVA with Tukeys post hoc analysis was used for comparisons of the 3 groups. Significant differences between NP and RUPP were found using Student’s t-test. *Significance compare with NP.
Mitochondrial respiration is improved in NK cell-depleted RUPPs.
Complex I-mediated state 3 respiration rate showed a trend in reduction in placental mitochondria of RUPP vs. NP (Fig. 3; 252 ± 49 vs. 352 ± 30 pmol·s−1·mg−1, P = 0.10, Student’s t-test, n = 4 vs. 6), and NK cell depletion improved state 3 respiration in RUPP placental mitochondria vs. RUPPs (Fig. 3; 374 ± 19, P = 0.03, Student’s t-test, n = 5). No changes in uncoupled respiration rates were observed between NP and RUPP mitochondria (Fig. 3; 190 ± 32 vs. 250 ± 21 pmol·s−1·mg−1, P = 0.14, Student’s t-test, n = 4 vs. 6). However, interestingly, RUPP+NKD mitochondria showed significantly higher uncoupled respiration rate vs. RUPP (283 ± 23 pmol·s−1·mg−1, P = 0.04, Student’s t-test, n = 5).
Fig. 3.

Natural killer (NK) cell depletion (NKD) improves mitochondrial respiration in reduced uterine perfusion pressure (RUPP) placental mitochondria. Highly coupled mitochondria were isolated from placenta, and oxygen consumption was measured using Oxygraph-2K. State 3 respiration rate show a trend in reduction in RUPP (n = 4, P = 0.094) placental mitochondria vs. normal pregnant rats (NP; n = 6). Placental mitochondria from RUPP+NKD rats (n = 5, P = 0.0207) show significant improvement in state 3 respiration (Student’s t-test). There were no differences observed in other states of respiration. Data are presented as means ± SE. Student’s t-test was used to perform statistical analysis between 2 respective groups. #Significant compared with RUPP.
Effect of NK Cell Depletion in RUPP Rats on HUVEC Mitochondrial Bioenergetics:
Sera from NK cell-depleted RUPPs improved HUVEC respiration and reduced mtROS in vitro compared with RUPP sera. Overnight incubation of HUVECs with 10% sera from RUPP rats reduced basal and uncoupled respiration rates compared with HUVECs treated with NP sera (Fig. 4; 7.2 ± 1.2 vs. 17.5 ± 3.8, P = 0.0114 and 24 ± 4.5 vs. 50 ± 1, pmol O2/million cells, P = 0.0264, Student’s t-test, n = 8 and 5, respectively). Interestingly, HUVECs treated with sera from NK cell-depleted RUPPs showed improved basal respiration rate compared with HUVECs treated with RUPP sera (Fig. 4; 19 ± 5 pmol O2/million cells, P = 0.0301, Student’s t-test, n = 6). However, uncoupled respiration (45 ± 14 pmol O2/million cells, P = 0.124, Student’s t-test, n = 6) was not significantly different. As we have previously reported (33), HUVECs treated with 10% RUPP sera show elevated mtROS (Fig. 5; 5.7 ± 0.6 vs. 2.3 ± 0.5 %gated, P = 0.0093, Student’s t-test, n = 3 vs. 4). Sera from NK cell-depleted RUPPs normalized mtROS in HUVECs compared with the RUPPs (Fig. 5; 2.7 ± 1 %gated, P = 0.0225, Student’s t-test, n = 7).
Fig. 4.
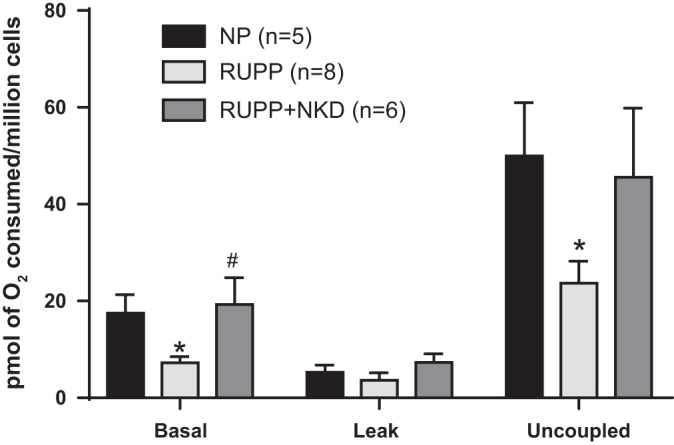
Natural killer (NK) cell depleted (NKD) reduced uterine perfusion pressure (RUPP) serum improves mitochondrial respiration in endothelial cells. Human umbilical vein endothelial cells (HUVECs) treated with 10% RUPP serum (n = 8) show a significant reduction in basal (P = 0.0114) and uncoupled respiration rates (P = 0.0264) vs. cells that were treated with NP serum (n = 5). Furthermore, RUPP+NKD serum (n = 6) restored basal respiration (P = 0.0301) with no improvement in uncoupled respiration (P = 0.124). Data are presented as means ± SE. Student’s t-test was used to perform to statistical analysis between 2 respective groups. *Significance compare with NP; #significant compared with RUPP.
Fig. 5.
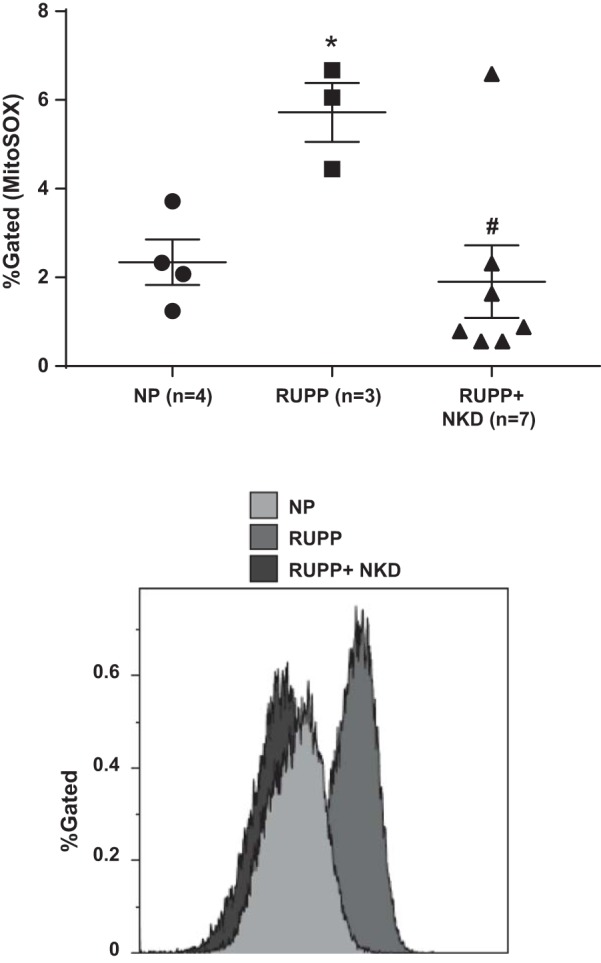
Natural killer (NK) cell depleted (NKD) reduced uterine perfusion pressure (RUPP) serum attenuates mitochondrial reactive oxygen species (mtROS) production in endothelial cells. Human umbilical vein endothelial cells (HUVECs) treated with 10% RUPP serum (n = 3) show a significant increase in mtROS vs. cells treated with normal pregnant (NP) serum (n = 4, P = 0.0093). Furthermore, RUPP+NKD serum (n = 7) attenuated mtROS in HUVECs vs. RUPP serum (P = 0.0225). Data are presented as means ± SE. Student’s t-test was used to perform to statistical analysis between 2 respective groups. *Significance compare with NP; #significant compared with RUPP.
DISCUSSION
Our study was performed to examine the role of NK cells in causing mitochondrial dysfunction, oxidative stress, and hypertension in response to placental ischemia. In our previously published study, we (33) reported that placental ischemia causes placental and renal mitochondrial dysfunction with increases in mtROS, which play a role in the development of hypertension in a rat model of PE. However, we had not explored the mechanisms that lead to mitochondrial dysfunction and elevated ROS production. Thus, the current study was performed to examine the role for NK cell activation in causing mitochondrial oxidative stress in the RUPP rat model of PE. Our novel findings in this study indicate that NK cell activation in response to placental ischemia causes hypertension, which is associated with placental and endothelial cell mitochondrial dysfunction and oxidative stress.
NK cells play a critical role in the innate immune response to target infected cells or tumor cells for destruction. Perforins released by activated NK cells cause pores on membrane of the target cells, spilling their intracellular contents while granzymes such as proteases and DNA-fragmenting enzymes enter the target cells causing apoptosis and release of oxidative stress molecules into the extracellular milieu (8). Furthermore, uterine NK cells play a critical role in maintenance of normal pregnancy by interacting closely with trophoblast cells and secreting cytokines that promote trophoblast growth, differentiation, invasion, and spiral artery remodeling (15, 27, 32). PE is associated with a shift in the NK cell population from uterine NK cells to cytolytic or activated NK cells (Type 1 shift) (24). Although increased activation of cytotoxic NK cells is associated with PE, the stimulus and signaling pathway for their activation has yet to be examined. We (14) have previously shown that the RUPP model of PE demonstrates increased total and cytolytic placental NK cells and that depleting NK cells reduced blood pressure along with inflammatory cytokines (interferon-γ and TNF-α) and improved fetal weight, suggesting an important role of NK cell activation in PE pathology.
NADPH oxidase has been studied extensively in the pathology of PE as a source of oxidative stress (6, 7, 18, 21). Similarly, another set of studies reported that mitochondrial oxidative stress is elevated in preeclamptic placentas (3, 14, 22, 29, 34). We (33) have recently shown that the RUPP rat model of PE is associated with placental and renal mitochondrial dysfunction and oxidative stress, which are associated with the development of hypertension and IUGR. However, the mechanisms that cause mitochondrial oxidative stress in PE are unknown. It is known that granzymes released by the activated NK cells can cause mitochondrial oxidative stress and apoptosis. The role for granzyme A and B in causing mitochondrial damage and apoptosis has been previously reported (13, 19). Moreover other immune factors such as TNF-α and IL-17 are also associated with mitochondrial dysfunction and PE. Hence, the goal of our current study was to determine whether depleting NK cells, which we have shown to reduce TNF-α and IL-17, will improve placental and peripheral endothelial cell mitochondrial function with reduced oxidative stress.
Consistent with the previously published data by our laboratory (9), MAP was normalized to the level of NP rats with NK cell depletion in RUPP rats. We (33) previously reported that the electron transport chain Complex I and Complex IV activities were significantly reduced in RUPP placental mitochondria. In the current study, we were able to reproduce these findings in RUPP placental mitochondria. Furthermore, depletion of NK cells in RUPP rats resulted in a trend toward improvement in Complex IV activity (P = 0.0579); however, we did not see changes in Complex I activity. We next sought to examine the functional activity of mitochondria by measuring respiration. As we previously showed (33), state 3 respiration was significantly reduced in RUPP placental mitochondria compared with the NP. Interestingly, state 3 respiration was restored in the mitochondria from RUPP+NKD rats compared with the RUPP placental mitochondria. However, we did not note any differences in basal, state 2, leak, and uncoupled respiration rates across the groups, suggesting that NK cell activation-mediated Complex IV damage might cause reduced state 3 respiration. Next, to examine the role of circulating factors downstream of NK cell activation in causing mitochondrial dysfunction in endothelial cells, we performed respiration and mtROS measurements in HUVECs that were treated with sera from NK cell-depleted RUPP or NP or control RUPP rats. RUPP sera reduced basal and uncoupled respiration rates in HUVECs compared with HUVECs treated with NP sera. Furthermore, RUPP+NKD sera restored basal respiration to the level of HUVECs treated with NP sera. However, the uncoupled respiration rate was not restored to the NP level. These findings suggested that NK cell activation causes endothelial cells to respire at significantly low rate. Furthermore, we wanted to determine whether the low respiration rate was associated with increased mtROS in these cells. As we previously showed in our published study (33), RUPP sera treatment causes mtROS generation in HUVECs. Interestingly, RUPP+NKD sera attenuated mtROS in HUVECs. Collectively, the restored basal respiration rate and reduced mtROS production in endothelial cells when treated with RUPP+NKD sera suggest an important role for NK cell activation in causing peripheral mitochondrial oxidative stress.
As PE pathology is complex, we suspect that mitochondrial dysfunction in response to activated cytolytic NK cells is complex and not solely attributed to one factor that is implicated in PE pathology. TNF-α has been shown to cause mitochondrial dysfunction in endothelial cells (5, 36). Furthermore, another inflammatory cytokine, IL-17, was shown to cause mitochondrial dysfunction in fibroblast-like synoviocytes (16). Interestingly, we previously showed that depletion of NK cells was associated with reduced circulating TNF-α and IL-17 levels in RUPP rats (9). Thus, we believe that mitochondrial oxidative stress in endothelial cells is possibly mediated by combined actions of granzymes and cytokines such as TNF-α and IL-17. However the direct role of these factors in causing mitochondrial dysfunction or mtROS was not examined in the current study, but it will be the subject of future investigations from our laboratory. Overall, our study reports that NK cell depletion is associated with attenuated hypertension and improved placental and endothelial cell mitochondrial function with reduced endothelial cell mtROS, thereby suggesting an important role for NK cell-mediated mitochondrial dysfunction in the pathology of PE.
Perspectives and Significance
Preeclamptic pathology is associated with oxidative stress. We and others (22, 33-35) have reported the evidence for mitochondrial dysfunction as one possible mechanism of hypertension in response to placental ischemia. Although clinical trials targeting oxidative stress (vitamin E and vitamin C) have not been successful, a mitochondrial targeted approach to combat oxidative stress (i.e., mitochondria-specific antioxidants) may be a promising therapeutic strategy. In the present study, as an attempt to examine the factors that drive mitochondrial oxidative stress, we studied the role of NK cell activation in mediating mitochondrial dysfunction. Our results demonstrate that activated NK cells indeed play a role in placental and endothelial cell mitochondrial dysfunction and mtROS. We have not examined the molecular mechanisms of NK cell-mediated mitochondrial dysfunction; however, we believe that cytokines such as TNF-α and IL-17 could be contributing factors. Our future studies will examine such mediators as a cause for mitochondrial oxidative stress and dysfunction in response to placental ischemia.
GRANTS
This research was supported by National Institutes of Health Grants RO1 HD-067541 and P20 GM-121334 (BL)/Office of Research, UMMC to B. LaMarca, R00 HL-130456 to D. C. Cornelius, and 17PRE33660592/AHA predoctoral fellowship to V. Vaka.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.R.V. and B.D.L. conceived and designed research; V.R.V., K.M.M., T.I., A.J., N.U., M.W.C., and L.M.A. performed experiments; V.R.V., K.M.M., D.C.C., T.I., A.J., M.W.C., L.M.A., and B.D.L. analyzed data; V.R.V., K.M.M., and B.D.L. interpreted results of experiments; V.R.V. prepared figures; V.R.V. and B.D.L. drafted manuscript; V.R.V., K.M.M., D.C.C., T.I., A.J., N.U., M.W.C., L.M.A., and B.D.L. edited and revised manuscript; V.R.V., K.M.M., D.C.C., T.I., A.J., N.U., M.W.C., L.M.A., and B.D.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Kramer for helping with the optimization of mitochondrial assays, and Dr. Hosler and K. Shirey for assistance with mitochondrial assays and analysis.
REFERENCES
- 1.Amaral LM, Cunningham MW Jr, Cornelius DC, LaMarca B. Preeclampsia: long-term consequences for vascular health. Vasc Health Risk Manag 11: 403–415, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J 5: 9–19, 2012. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brownfoot FC, Hastie R, Hannan NJ, Cannon P, Tuohey L, Parry LJ, Senadheera S, Illanes SE, Kaitu’u-Lino TJ, Tong S. Metformin as a prevention and treatment for preeclampsia: effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am J Obstet Gynecol 214: 356.e1–356.e15, 2016. doi: 10.1016/j.ajog.2015.12.019. [DOI] [PubMed] [Google Scholar]
- 4.Casasco A, Calligaro A, Casasco M, Tateo S, Icaro Cornaglia A, Reguzzoni M, Farina A.. Immunohistochemical localization of lipoperoxidation products in normal human placenta. Placenta 18: 249–253, 1997. [DOI] [PubMed] [Google Scholar]
- 5.Corda S, Laplace C, Vicaut E, Duranteau J. Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am J Respir Cell Mol Biol 24: 762–768, 2001. doi: 10.1165/ajrcmb.24.6.4228. [DOI] [PubMed] [Google Scholar]
- 6.Cui XL, Brockman D, Campos B, Myatt L. Expression of NADPH oxidase isoform 1 (Nox1) in human placenta: involvement in preeclampsia. Placenta 27: 422–431, 2006. doi: 10.1016/j.placenta.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dechend R, Viedt C, Müller DN, Ugele B, Brandes RP, Wallukat G, Park JK, Janke J, Barta P, Theuer J, Fiebeler A, Homuth V, Dietz R, Haller H, Kreuzer J, Luft FC. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation 107: 1632–1639, 2003. doi: 10.1161/01.CIR.0000058200.90059.B1. [DOI] [PubMed] [Google Scholar]
- 8.Eischen CM, Leibson PJ. Role for NK-cell-associated Fas ligand in cell-mediated cytotoxicity and apoptosis. Res Immunol 148: 164–169, 1997. doi: 10.1016/S0923-2494(97)84219-8. [DOI] [PubMed] [Google Scholar]
- 9.Elfarra J, Amaral LM, McCalmon M, Scott JD, Cunningham MW Jr, Gnam A, Ibrahim T, LaMarca B, Cornelius DC. Natural killer cells mediate pathophysiology in response to reduced uterine perfusion pressure. Clin Sci (Lond) 131: 2753–2762, 2017. doi: 10.1042/CS20171118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukui A, Funamizu A, Yokota M, Yamada K, Nakamua R, Fukuhara R, Kimura H, Mizunuma H. Uterine and circulating natural killer cells and their roles in women with recurrent pregnancy loss, implantation failure and preeclampsia. J Reprod Immunol 90: 105–110, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Fukui A, Yokota M, Funamizu A, Nakamua R, Fukuhara R, Yamada K, Kimura H, Fukuyama A, Kamoi M, Tanaka K, Mizunuma H. Changes of NK cells in preeclampsia. Am J Reprod Immunol 67: 278–286, 2012. 10.1111/j.1600-0897.2012.01120.x. [DOI] [PubMed] [Google Scholar]
- 12.Granger JP. Inflammatory cytokines, vascular function, and hypertension. Am J Physiol Regul Integr Comp Physiol 286: R989–R990, 2004. doi: 10.1152/ajpregu.00157.2004. [DOI] [PubMed] [Google Scholar]
- 13.Jacquemin G, Margiotta D, Kasahara A, Bassoy EY, Walch M, Thiery J, Lieberman J, Martinvalet D. Granzyme B-induced mitochondrial ROS are required for apoptosis. Cell Death Differ 22: 862–874, 2015. doi: 10.1038/cdd.2014.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin X, Xu Z, Cao J, Shao P, Zhou M, Qin Z, Liu Y, Yu F, Zhou X, Ji W, Cai W, Ma Y, Wang C, Shan N, Yang N, Chen X, Li Y. Proteomics analysis of human placenta reveals glutathione metabolism dysfunction as the underlying pathogenesis for preeclampsia. Biochim Biophys Acta Proteins Proteomics 1865: 1207–1214, 2017. doi: 10.1016/j.bbapap.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Karimi K, Arck PC. Natural killer cells: keepers of pregnancy in the turnstile of the environment. Brain Behav Immun 24: 339–347, 2010. 10.1016/j.bbi.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 16.Kim EK, Kwon JE, Lee SY, Lee EJ, Kim DS, Moon SJ, Lee J, Kwok SK, Park SH, Cho ML. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis 8: e2565, 2017. doi: 10.1038/cddis.2016.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamarca B. Endothelial dysfunction. An important mediator in the pathophysiology of hypertension during pre-eclampsia. Minerva Ginecol 64: 309–320, 2012. [PMC free article] [PubMed] [Google Scholar]
- 18.Lee VM, Quinn PA, Jennings SC, Ng LL. Neutrophil activation and production of reactive oxygen species in pre-eclampsia. J Hypertens 21: 395–402, 2003. doi: 10.1097/00004872-200302000-00032. [DOI] [PubMed] [Google Scholar]
- 19.Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22: 355–370, 2005. doi: 10.1016/j.immuni.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 20.Matsubara K, Higaki T, Matsubara Y, Nawa A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int J Mol Sci 16: 4600–4614, 2015. doi: 10.3390/ijms16034600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsubara S, Sato I. Enzyme histochemically detectable NAD(P)H oxidase in human placental trophoblasts: normal, preeclamptic, and fetal growth restriction-complicated pregnancy. Histochem Cell Biol 116: 1–7, 2001. [DOI] [PubMed] [Google Scholar]
- 22.McCarthy C, Kenny LC. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci Rep 6: 32683, 2016. doi: 10.1038/srep32683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris JM, Gopaul NK, Endresen MJ, Knight M, Linton EA, Dhir S, Anggard EE, Redman CW. Circulating markers of oxidative stress are raised in normal pregnancy and pre-eclampsia. Br J Obstet Gynaecol 105: 1195–1199, 1998. [DOI] [PubMed] [Google Scholar]
- 24.Peritt D, Robertson S, Gri G, Showe L, Aste-Amezaga M, Trinchieri G. Differentiation of human NK cells into NK1 and NK2 subsets. J Immunol 161: 5821–5824, 1998. [PubMed] [Google Scholar]
- 25.Raijmakers MT, Peters WH, Steegers EA, Poston L. NAD(P)H oxidase associated superoxide production in human placenta from normotensive and pre-eclamptic women. Placenta 25: S85–S89, 2004. 10.1016/j.placenta.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Roberts JM, Pearson G, Cutler J, Lindheimer M; NHLBI Working Group on Research on Hypertension During Pregnancy . Summary of the NHLBI Working Group on Research on Hypertension During Pregnancy. Hypertension 41: 437–445, 2003. doi: 10.1161/01.HYP.0000054981.03589.E9. [DOI] [PubMed] [Google Scholar]
- 27.Saito S, Shiozaki A, Sasaki Y, Nakashima A, Shima T, Ito M.. Regulatory T cells and regulatory natural killer (NK) cells play important roles in feto-maternal tolerance. Semin Immunopathol 29: 115–122, 2007. 10.1007/s00281-007-0067-2. [DOI] [PubMed] [Google Scholar]
- 28.Shibata E, Ejima K, Nanri H, Toki N, Koyama C, Ikeda M, Kashimura M. Enhanced protein levels of protein thiol/disulphide oxidoreductases in placentae from pre-eclamptic subjects. Placenta 22: 566–572, 2001. 10.1053/plac.2001.0693. [DOI] [PubMed] [Google Scholar]
- 29.Shibata E, Nanri H, Ejima K, Araki M, Fukuda J, Yoshimura K, Toki N, Ikeda M, Kashimura M. Enhancement of mitochondrial oxidative stress and upregulation of antioxidant protein peroxiredoxin III/SP-22 in the mitochondria of human pre-eclamptic placentae. Placenta 24: 698–705, 2003. doi: 10.1016/S0143-4004(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 30.Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet 365: 785–799, 2005. doi: 10.1016/S0140-6736(05)71003-5. [DOI] [PubMed] [Google Scholar]
- 31.Sibai BM, Caritis S, Hauth J; National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network . What we have learned about preeclampsia. Semin Perinatol 27: 239–246, 2003. doi: 10.1016/S0146-0005(03)00022-3. [DOI] [PubMed] [Google Scholar]
- 32.Vacca P, Moretta L, Moretta A, Mingari MC. Origin, phenotype and function of human natural killer cells in pregnancy. Trends Immunol 32: 517–523, 2011. doi: 10.1016/j.it.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 33.Vaka VR, McMaster KM, Cunningham MW, Ibrahim T, Hazlewood R, Usry N, Cornelius D, Amaral LM, LaMarca B. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertension 72: 703–711, 2018. doi: 10.1161/HYPERTENSIONAHA.118.11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vishnyakova PA, Volodina MA, Tarasova NV, Marey MV, Kan NE, Khodzhaeva ZS, Vysokikh MY, Sukhikh GT. Alterations in antioxidant system, mitochondrial biogenesis and autophagy in preeclamptic myometrium. BBA Clin 8: 35–42, 2017. doi: 10.1016/j.bbacli.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Walsh SW. Increased superoxide generation is associated with decreased superoxide dismutase activity and mRNA expression in placental trophoblast cells in pre-eclampsia. Placenta 22: 206–212, 2001. 10.1053/plac.2000.0608. [DOI] [PubMed] [Google Scholar]
- 36.Zhou P, Lu S, Luo Y, Wang S, Yang K, Zhai Y, Sun G, Sun X. Attenuation of TNF-α induced inflammatory injury in endothelial cells by Ginsenoside Rb1 via inhibiting NFκB, JNK and p38 signaling pathways. Front Pharmacol 8: 464, 2017. 10.3389/fphar.2017.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zusterzeel PL, Rütten H, Roelofs HM, Peters WH, Steegers EA. Protein carbonyls in decidua and placenta of pre-eclamptic women as markers for oxidative stress. Placenta 22: 213–219, 2001. doi: 10.1053/plac.2000.0606. [DOI] [PubMed] [Google Scholar]