

Abstract
Cell differentiation is driven by changes in gene expression that manifest as changes in cellular phenotype or function. Altered cellular phenotypes, stemming from genetic mutations or other perturbations, are widely assumed to directly correspond to changes in the transcriptome and vice versa. Here, we exploited the cytologically well-defined Prdm9 mutant mouse as a model of developmental arrest to test whether parallel programs of cellular differentiation and gene expression are tightly coordinated, or can be disassociated. By comparing cytological phenotype markers and transcriptomes in wild-type and mutant spermatocytes, we identified multiple instances of cellular and molecular uncoupling in Prdm9–/– mutants. Most notably, although Prdm9–/– germ cells undergo cytological arrest in a late-leptotene/zygotene stage, they nevertheless develop gene expression signatures characteristic of later developmental substages. These findings suggest that transcriptomic changes may not reliably map to cellular phenotypes in developmentally perturbed systems.
INTRODUCTION
Cellular differentiation unfolds via a combination of genetic, epigenetic, transcriptional, translational, and cytological subprograms that establish specific cellular identities. Transcriptional regulation is the best studied of these concurrent programs across diverse cell types (Chen and Dent, 2014), but the degree to which these subprograms are coordinated is not well understood. Often, progression through differentiation is signified by the hallmark expression of one or more well-established marker genes. Protein quantification of the expression of marker gene products is the most reliable method to stage cellular development; however, transcript abundance determined by deep sequencing allows for unbiased genome-wide analyses of gene expression. Although transcript abundance is frequently used as a surrogate for protein abundance, numerous examples have highlighted that transcript abundance does not necessarily correspond with protein abundance (Gan et al., 2013; Wu et al., 2014; Battle et al., 2015; Chick et al., 2016; Goncalves et al., 2017). Further, the reliability of transcript abundance of marker genes to reflect cytological cell state in abnormal cells is rarely assessed.
Mammalian gametogenesis is an instructive model system for cell differentiation in that it is characterized by marked transcriptomic changes paralleling morphologically dramatic stages of cytodifferentiation—particularly in the male germline. Spermatogenesis in mammals entails differentiation of committed germ cells from stem progenitor cells, followed by the germ cell-specific process of meiosis and then cytological transformation into the highly specialized sperm cells (Eddy, 1998; Hammoud et al., 2014). The cytological and developmental stages of spermatogenesis have been well characterized in the mouse and, in particular, meiotic prophase substages have been defined by precise cytological and molecular criteria (Handel and Schimenti, 2010; Bolcun-Filas and Handel, 2018), which can be related to transcriptomic states (Ball et al., 2016). During meiotic prophase, spermatocyte nuclei progressively pass through well-defined and unique structural configurations as chromosomes undergo synapsis and recombination, followed by desynapsis; together, these define the well-characterized prophase substages of leptonema, zygonema, pachynema, and diplonema (Handel and Schimenti, 2010; Baudat et al., 2013). Concurrent with the unfolding of these cytological events, a complex gene expression program that is highly correlated with cytologically defined meiotic prophase substages unfolds in a precise temporal pattern (Schultz et al., 2003; Ramskold et al., 2009; Fallahi et al., 2010; Soumillon et al., 2013; Ball et al., 2016; Green et al., 2018). Interestingly, an exceptionally large number and diverse array of transcripts are expressed in spermatocytes during meiotic substages, that is, more than 20,000 transcripts, including ∼5000 noncoding RNAs (Xie et al., 2014; Ball et al., 2016). This complex transcriptome supports spermatogenesis in at least two major ways: it provides templates for proteins that are required for meiotic progression, and it produces transcripts that are stored in an inactive state, but later support the postmeiotic differentiation known as spermiogenesis (Fallahi et al., 2010; Gan et al., 2013; Ball et al., 2016; da Cruz et al., 2016). Although expression of spermiogenic transcripts does not lead to immediate translation, their production is specific to cytologically defined meiotic prophase substages (Ball et al., 2016; da Cruz et al., 2016). Experimental analysis of the parallel programs of cytological differentiation and transcription is complicated by a practical problem: the extreme heterogeneity of both somatic and germ cell types in the mammalian testis makes isolation of purified cell populations and stage-specific transcript profiling difficult. Recently, this problem was addressed by a novel computational strategy, Permutation-based maximum covariance analysis (PMCA, which identifies transcripts that covary with proportions of meiotic substages in complex cell populations (Ball et al., 2016). Although PMCA analysis associates gene expression states and cytological states, it alone cannot determine whether the cytological or transcriptomic differentiation programs are sequential, or, more importantly, whether one drives the other. To infer these relationships, controlled perturbation of the system is helpful.
Genetic mutations that disrupt meiotic prophase differentiation provide experimental models to analyze the coupling between concurrent differentiation and transcriptomic programs. Here we investigate the relationship between transcript abundance and cellular programs in spermatocytes from mice bearing a null mutation in the Prdm9 gene, a key gene for meiotic prophase progression (Hayashi et al., 2005). This gene encodes PRDM9 (PR/SET Domain 9), a zinc-finger DNA-binding protein with histone methyltransferase activity that is expressed in early meiotic prophase, during leptonema and zygonema (Sun et al., 2015), and is required for activation of recombination (Parvanov et al., 2010). Recent work has demonstrated that, despite its deposition of classically gene-activating marks, PRDM9 does not directly regulate gene expression (Thibault-Sennett et al., 2018). Mice bearing inactivating mutations of Prdm9 (herein designated as Prdm9–/–) are infertile and meiosis is arrested at early to midprophase, with the failure of reciprocal recombination and absence of cytologically normal germ cells past the zygotene substage. In addition to effects on cytodifferentiation during meiosis, the molecular consequences of Prdm9 mutation could also directly alter the transcriptional program of meiotic prophase. The PRDM9 protein selects and binds to specific genomic sites, known as hotspots, which are subsequently recognized by the SPO11 protein for formation of the DNA double-strand breaks (DSBs) that initiate the molecular events of meiotic recombination. In the absence of functional PRDM9, DSBs are misplaced to other genomic sites, primarily, but not exclusively, gene promoters (Brick et al., 2012). The ectopic DSBs fail to be repaired, which is likely a part of the cause for meiotic arrest in Prdm9 mutant spermatocytes. It has also been shown that Prdm9–/– mutant spermatocytes fail to inactivate the sex chromosomes (Hayashi and Matsui, 2006). With its robust characterization, the Prdm9 mutant is an informative model in which to study the coupling of cellular differentiation and transcriptomic programs in the context of an arrested developmental program. Here, our goal was to determine how the transcriptomic phenotype in Prdm9–/– germ cells relates to well-characterized meiotic substages and meiotic arrest of Prdm9–/– spermatocytes. We studied the initial wave of spermatogenesis in newborn mutant and wild-type (WT) mice in order to compare similar populations of cells in both. We documented both cellular and transcriptomic states in the same cell populations. This strategy revealed that the cytological and transcriptomic programs become uncoupled during abnormal meiotic progression, with progression of the transcriptomic program in spite of disruption and arrest of the cytological program of differentiation. This observation suggests a complex degree of independent regulation of cooccurring programs of differentiation, a conclusion that underscores the importance of anchoring transcript abundance profiles to their cellular context in order to understand both processes.
RESULTS
Cytological staging sets parameters of the Prdm9 mutant phenotype and provides context for concurrent transcriptomic analyses
We characterized the cellular and molecular effects of loss of PRDM9 function in male germ cells undergoing meiosis. Germ cells were enriched from testes collected from Prdm9 WT, heterozygous (Prdm9+/–), and mutant (Prdm9–/–) male mice at 8, 12, and 16 d postpartum (dpp) (Materials and Methods) (Figure 1A; Supplemental Table S1), a time period when the first wave of differentiating germ cells progresses through meiotic prophase I substages. A small portion of each sample of enriched germ cells was used for cytological staging, with the remainder used for RNA-Seq (Figure 1A).
FIGURE 1:

Cytological phenotypes of Prdm9–/– spermatocytes reflect meiotic arrest. (A) Cytological analyses and RNA sequencing were performed on germ cells enriched from testes at 8, 12, and 16 dpp. (B) Representative images of spermatocytes in each meiotic substage across Prdm9+/+ and Prdm9–/– samples. Germ cells were immunostained for combinatorial arrays of marker proteins that are well established for cytological characterization of meiotic prophase substages in spermatocytes. (C) Quantification of average frequencies of spermatogenic and meiotic prophase substages represented at each time point in the samples of germ cells retrieved from Prdm9+/+ and Prdm9–/– testes.
Cytological substage-specific protein markers were used to characterize cellular morphology and determine the relative proportions of meiotic substages in each sample of Prdm9+/+ and Prdm9–/– germ cells (Figure 1, B and C). Spermatogonia, and multiple substages of spermatocytes—preleptotene (PL), early leptotene (EL), late leptotene/zygotene (LL/Z), early pachytene (EP), and late pachytene/diplotene (LP/D)—were scored by combinatorial application of antibodies recognizing previously established marker proteins (Materials and Methods) (Ball et al., 2016). As the juvenile mice age toward puberty, the time period when the first wave of germ cells develop, subsequent waves of spermatogenesis are also continuously initiated, resulting in the presence of each of substages preceding the most developed substage at each time point (Figure 1C). While all stages listed above were represented in the WT B6 samples, no cells in the Prdm9–/– samples met the cytological criteria for mid- to late-pachytene spermatocytes (e.g., full synapsis detected by labeling with SYCP3 antibody, γH2AX restricted to the XY chromosome pair, and the presence of histone H1t). Those cells exhibiting only some of these features, for example, only partial synapsis, with a diffuse labeling pattern of γH2AX labeling that is indicative of accumulated DSBs, were defined as “pachytene-like” (P-like) cells (Figure 1B). These observations are consistent with previous reports of the Prdm9–/– phenotype (Hayashi et al., 2005; Hayashi and Matsui, 2006). Each time point scored captured a key aspect of the mutant phenotype in Prdm9–/– mice.
The substage proportion differences between the WT and mutant germ cell populations were not severe at 8 dpp but became much more apparent by 12 dpp, with the greatest differences observed at 16 dpp. Thus, at 8 dpp, the germ cells present in both WT and mutant samples are in early meiotic prophase, and do not exhibit any apparent morphological phenotype in Prdm9–/– testes (Figure 1C, left). However, by 12 dpp, there is apparent delay in differentiation of Prdm9–/– germ cells, reflected in relatively lower proportions of later prophase spermatocytes compared with WT at the same age (Figure 1C, middle). Finally, at 16 dpp, when WT testes have many mid- to late-prophase spermatocytes (EP and LP/D), the germ cells in Prdm9–/– testes appear arrested, with no progress beyond a P-like stage (described above), reflected by both appearance of the spermatocytes and diminished proportion of stages represented in the samples collected at this time (Figure 1C, right). These cytological characterizations and determination of proportions of substages, which reflect prior knowledge of the phenotype, inform the interpretation of germ cell transcriptome data (see below).
Specific gene signatures reflect known mutant phenotypes
RNA-Seq was used to obtain genome-scale transcriptome states that correspond with the cellular states obtained at each time point (above). We sought to use an unbiased method to account for the multidimensional variation in our RNA-Seq data in order to reveal transcriptomic differences among Prdm9+/+, Prdm9+/–, and Prdm9–/– samples. We first identified and removed variability in the transcriptome data stemming from batch and litter differences (Materials and Methods and Supplemental Figures S1 and S2) (Johnson et al., 2007) and then performed principal component analysis (PCA) on the residual variation (Figure 2A). The first principal component (PC) separated samples by age, and the second PC separated samples by genotype (at 16 dpp), both parameters being expected determinants of transcriptome states. Further, PC3 segregated the 12 dpp samples by their Prdm9 genotype (Supplemental Figure S2A). Despite Prdm9+/– mice being fertile, they generally displayed intermediate cytological (Supplemental Figure S2B) and transcriptomic phenotypes (Figure 2A and Supplemental Figure S2, A and C–E). Differential expression analysis (Materials and Methods) between WT and Prdm9–/– transcriptomes at each time point revealed a number of differentially expressed genes (DEGs), with some down-regulated (Figure 2B), and some up-regulated (Figure 2C). The number of DEGs detected increased with age in both WT and mutant samples (Figure 2, B and C), reflecting the increase in complexity of cellular composition with increasing age.
FIGURE 2:
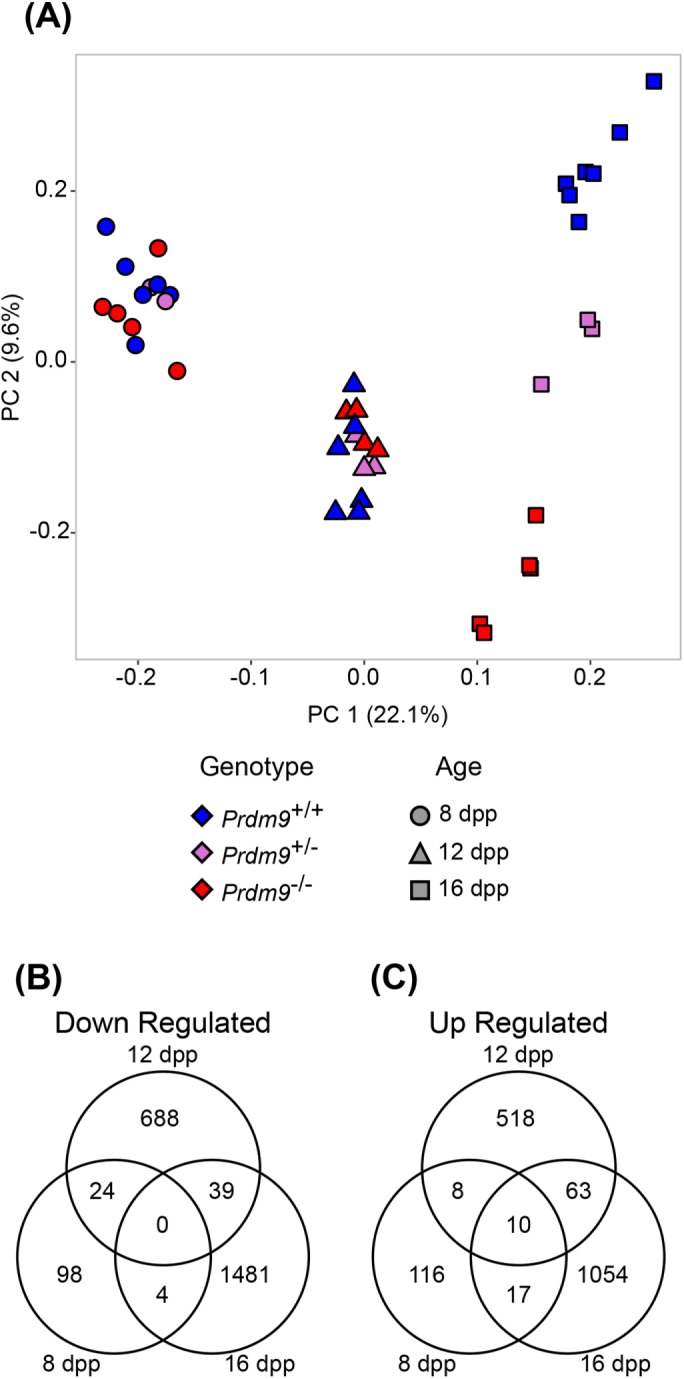
ComBat-adjusted data of gene expression across genotype and age conditions shows differential expression between Prdm9+/+ and Prdm9–/– samples. (A) Principal component 1 (PC1) vs. principal component 2 (PC2) from PCA of all ComBat-adjusted samples. Colors denote genotype, and shapes denote sample age, as indicated. (B, C) Shared and unique differentially expressed transcripts with decreased or increased abundance in Prdm9–/– samples compared with Prdm9+/+. FDR < 0.01 and LFC > 0.5 for B and C.
As expected, expression of the Prdm9 gene in Prdm9–/– samples was significantly less than in WT samples (Supplemental Figure S3A). Although low levels of Prdm9 transcripts were detected in the Prdm9–/– samples, no reads mapped to the exons deleted in the mutant (Supplemental Figure S3B), and PRDM9 has been shown to be absent from these mice (Sun et al., 2015). Other genes previously reported (Hayashi et al., 2005) to be differentially expressed in Prdm9–/– germ cells or key genes involved in processes expected to be disrupted in Prdm9–/– testes were validated in our data (Morc2b, Hspa2, and Piwil1; Figure 3A), as well as early meiotic regulatory genes not expected to change (Dmc1, Spo11, and Stra8; Figure 3B). As might be anticipated, given the decrease in Piwil1 expression between Prdm9+/+ and Prdm9–/– samples, Piwi-interacting RNA (piRNA) precursors were not expressed at WT levels in Prdm9–/– germ cells at 16 dpp (Figure 3C). In concordance with molecular patterns found in previous analyses of Prdm9 mutants, we found that XY-linked genes were enriched in overrepresented DEGs (p < 2.2 × 10–16) from Prdm9–/– germ cells at 16 dpp, corresponding with the failure of cells to progress to a stage with a fully formed and transcriptionally inactivated sex-body (Supplemental Figure S4A) (Hayashi et al., 2005; Hayashi and Matsui, 2006; Namekawa et al., 2006). Autosomal transcripts were not similarly overrepresented (Supplemental Figure S4B). Taken together, these transcriptomic data reflect both previous reports on the Prdm9 mutants (Hayashi et al., 2005) and known temporal patterns of gene expression in spermatocytes (Deng and Lin, 2002).
FIGURE 3:

Changes in expression of specific meiotic gene reflect abnormalities and meiotic arrest in Prdm9–/– germ cells. Colors denote genotypes, as indicated. (A) Log2(TPM+1) expression of Morc2b, Hspa2, and Piwil1 at 8, 12, and 16 dpp. (B) Log2(TPM+1) expression of Dmc1, Spo11, and Stra8 at 8, 12, and 16 dpp. (C) Relative expression of piRNA precursors at 8, 12, and 16 dpp. *** represents FDR < 0.0001.
FIGURE 4:

Substage specificity of transcripts determined from PMCA is different in Prdm9–/– germ cells than in WT germ cells. (A) Relative expression of transcripts assigned to LP/D substage based on their WT expression patterns. (B) Relative number of genes shared among substages assigned in WT and Prdm9–/– samples. The size of the circle represents the relative number of genes shared between two substage assignments. (C) Relative expression of transcripts assigned to LP/D in WT samples but to LL/Z in Prdm9–/– samples.
Transcriptomic changes precede cytological phenotypes in Prdm9–/– testes
We annotated the DEGs at each time point to compare how well mutant transcriptomic phenotypes coincide with cytological phenotypes. To determine whether there are early transcriptomic signatures of the Prdm9–/– cytological phenotype, we conducted Gene Ontology (GO) enrichment analyses and Ingenuity Pathway Analysis (IPA) on the DEGs at each time point (Materials and Methods). At 16 dpp, when the mutant testes exhibit the most drastic phenotype, genes annotated to spermatogenesis-related GO terms, specifically genes related to late spermatogenesis, were enriched in the down-regulated DEG lists, and terms related to defense and immune responses were highly enriched in the up-regulated DEG lists (all false discovery rate [FDR] < 0.05; Supplemental Table S3). (Although this analysis could be biased by increased representation of sex-chromosome transcripts, limiting GO enrichment analysis to autosomal DEGs did not substantially change the repertoire of enriched terms.) IPA identified “Cyclins and Cell Cycle Regulation” (p = 8.52 × 10–4) as an enriched pathway in the DEGs at 16 dpp, following “Gluconeogenesis I” (p = 8.19 × 10–5) and “LPS/IL-1 Mediated Inhibition of RXR Function” (p = 4.84 × 10–4). “Sperm Disorder” was an enriched function in this gene set as well (p = 8.10 × 10–5). These results build upon the GO term enrichment analysis to suggest that much of the signal seen at 16 dpp is due to the loss of late-prophase subtypes, as well as cell cycle arrest.
To identify transcriptomic changes that precede either the appearance of EP cells or the mutant phenotype of meiotic prophase arrest in the mutant, we analyzed DEGs at 8 and 12 dpp. No significantly enriched GO terms were found in either the up-regulated or the down-regulated gene sets at either 8 or 12 dpp (both time points are before detection of any cytological anomalies). We did find an enrichment of the pathways “Cell Cycle: G2/M DNA Damage Checkpoint Regulation” (p = 5.16 × 10–3) and “EIF2 Signaling” (p = 1.04 × 10–2) among the top 10 most significant pathways. The G2/M DNA Damage Checkpoint would logically be activated in these cells with unrepaired DSBs, and EIF2 Signaling, a translational regulation program, could indicate posttranscriptional regulation as a component of the molecular response to the absence of PRDM9. The highest-scoring network in this DEG list is “Cell Cycle, Cell Death and Survival, Endocrine System Disorders.” These findings demonstrate a transcriptomic signal for cell death even before the appearance of the EP cells where cell death may be manifest.
Differential expression of genes, especially down-regulated transcripts, could be due to a general response to genome-wide DNA damage in spermatocytes, or altered expression specifically of genes that undergo the ectopic DSBs that occur in Prdm9–/– germ cells (Brick et al., 2012). To discriminate between these alternatives, we compared the location of promoters of genes expressed in Prdm9–/– germ cells to the genomic locations of DSBs using previously published data (Brick et al., 2012) on localization of DMC1, which is a widely accepted surrogate for sites of DSBs. Most genes with DMC1 peaks within their promoters in Prdm9–/– testes (Brick et al., 2012) were expressed in our data set (74%); however, these genes were not biased toward being differentially expressed at any time point (Fisher’s exact test, p = 1). Within those genes exhibiting a promoter DMC1 peak, the magnitude of the peak (frequency within the sample) was not correlated with the coefficient of differential expression for the gene (Spearman’s ρ = 0.001, p = 0.98). This result suggests that promoter-localized DSBs do not contribute significantly to the observed gene expression changes in Prdm9–/– germ cells. Instead, DEGs could be a part of a general response to DNA damage and/or due to changes in frequencies of specific meiotic substages in the cell populations (Figure 1), given meiotic arrest. Because these data revealed that transcriptomic changes in germ cells precede the onset of the characteristic cellular phenotype, we next sought to determine whether cellular arrest was coupled with arrest of the spermatogenic transcriptomic program.
Transcriptomic progression is uncoupled from cellular progression in Prdm9–/– germ cells
Cytological arrest in a perturbed (mutant) system may a priori be expected to occur coincidentally with, or as an immediate consequence of, transcriptional arrest. To test this expectation, we compared the expression of substage-specific genes in WT versus Prdm9–/– spermatocytes. Transcripts detected in the RNA-Seq analysis derive from mixed pools of germ cells at different substages of meiotic progression. Therefore, any differences in transcript abundance due to Prdm9 genotype could stem either from changes in the relative proportion of cells in different meiotic substages (Figure 1C) or from intracellular changes in expression within individual cells at the same substage. PMCA (Materials and Methods) (Ball et al., 2016) utilizes two measurements made on the same sample to identify covarying modules. Here, spermatogenic substage frequencies and RNA abundance were processed by PMCA to identify substage-specific lists of transcripts. PMCA-based identification and analysis of substage-specific transcripts in both WT and mutant samples allowed us to detect changes in substage-specific transcriptomic progression that are independent of changes in meiotic substage proportions in each germ cell preparation.
We used previously defined lists of WT substage-specific genes (Ball et al., 2016) derived by PMCA (Materials and Methods) to compare the expression patterns of substage-specific genes between WT and Prdm9–/– germ cells (Supplemental Figure S5A and Figure 4, A and B), thereby determining the impact of the Prdm9–/– phenotype on developmentally unfolding transcriptomic program. Most differences in expression patterns of WT substage-specific genes in mutant germ cells reflected the differences in the relative substage frequencies in the mutant cell populations (Figure 1C) with only a few substage-specific transcripts being differentially expressed (Supplemental Table S4). However, a striking exception to this generalization is with respect to genes assigned by PMCA to WT EP and LP/D substages. Even though there was notable loss of these meiotic prophase substages among Prdm9–/– germ cells, there was no equivalent loss of expression of genes normally specific to these substages (Figure 4A and Supplemental Figure S5A). Indeed, despite the notable decrease in abundance of EP cells at 12 and 16 dpp Prdm9–/– germ cells (Figure 1C), only ∼17% of EP transcripts were differentially expressed between Prdm9+/+ and Prdm9–/– samples (Supplemental Figure S5A and Supplemental Table S4). And although there was a decrease in relative average expression of LP/D transcripts in 16 dpp Prdm9–/– samples (Figure 4A), only 33% of transcripts were differentially expressed (Supplemental Table S4). This was surprising because there are no cells cytologically characterized as LP/D cells in these samples (Figure 1C) and suggests that a spermatogenic transcriptomic program is being executed independently of the normally cooccurring cytological differentiation program. In general, the LP/D-specific transcripts detected in mutant spermatocytes exhibited either of two divergent expression patterns. Some 16 dpp LP/D transcripts in Prdm9–/– germ cells were at the same level as at 12 dpp. As mentioned above, only 33% of LP/D transcripts were differentially expressed between Prdm9+/+ and Prdm9–/– germ cells at this time point. These transcripts could simply reflect the loss of pachytene spermatocytes by 16 dpp. However, some LP/D-specific transcripts in Prdm9–/– samples exhibited an increase in expression level from 12 to 16 dpp, with ∼20% (n = 771) of LP/D transcripts differentially expressed between Prdm9–/– germ cells 12 and 16 dpp (FDR < 0.01), trending similarly to their pattern in the WT transcriptomic progression (Figure 4A). This aberrant expression of pachytene- and diplotene-specific transcripts in Prdm9–/– cell population, despite a dramatic decrease in the abundance of these cell types, suggests uncoupling of transcriptomic progression and cytological progression in Prdm9–/– germ cells.
To further investigate this apparent discrepancy between cytological stage and gene expression, we applied PMCA to assign transcripts to the specific substages detected by cytological markers in the Prdm9–/– germ cell samples (FDR < 0.01, Supplemental Figure S5B). We compared these Prdm9–/– substage-specific gene lists to those derived from PMCA analysis of WT data. First, relatively few transcripts from the mutants were assigned to early prophase substages due to low variation in the cytological frequency of those substages across biological samples (Figure 1C); this had been reported previously for WT substage-specific transcription (Ball et al., 2016). This resulted in few genes overlapping between PL and EL. Many transcripts in Prdm9–/– cell populations were identified with the same substage as they were in WT, despite the genetic perturbation (Figure 4B). However, a substantial number of genes that had been annotated to EP (n = 68) and LP/D (n = 85) in the original WT analysis were assigned to an earlier substage, LL/Z, in the PMCA analysis of the mutant transcriptomes (Figure 4C). This unanticipated observation is evidence for transcriptomic progression of the Prdm9–/– germ cells despite their apparent arrest or delay at the LL/Z substage. These transcripts contribute to the subset of LP/D genes that trend toward WT expression levels at 16 dpp in Prdm9–/– germ cells (Figure 4A). We compared these late meiotic prophase-specific genes expressed in mutants to genes expressed in more extensively purified WT pachytene spermatocytes (Ball et al., 2016). Forty-three of 68 EP transcripts that exhibit LL/Z specificity in the Prdm9–/– germ cells, and all 85 LP/D transcripts that instead exhibit LL/Z specificity in the mutant, were represented in the purified WT pachytene spermatocyte transcriptome. These analyses reveal that while cytology identifies a stage-specific meiotic arrest by EP in Prdm9–/– germ cells, some aspects of the spermatogenic transcriptomic program move forward unabated.
We used a bioinformatic approach to determine whether the expression of LP/D genes in LL/Z Prdm9–/– germ cells might be driven by the same regulators that normally control the repertoire of LP/D genes, or whether the evidence suggested a more stochastic response to the mutant phenotype. To identify candidate factors that might regulate the uncoupled molecular program in Prdm9–/– germ cells, we used iRegulon (Janky et al., 2014) to identify shared transcription factor (TF) binding sites among substage-specific genes. Analyses revealed enriched motifs for E2F1, REL, and YY1 at genes expressed in Prdm9–/– EL, while ZFP143, ETV6, and ETV5 motifs were identified by genes specifically expressed in LL/Z and EP (Figure 5). In the mutant data, NR3C1 and QSOX1 potential binding sites were enriched near genes from the P-like list (Figure 5), so these could potentially be driving some of the transcriptional and meiotic arrest. Analysis of the mutant data led to identification of TF motifs that had previously been annotated in WT data as gene-expression regulators for specific substages, particularly the early prophase substages (Ball et al., 2016), as well as potential novel regulators of the P-like stage. To identify prospective mechanisms underlying the uncoupling of transcriptomic programs from a cytological differentiation program, we conducted a TF motif analysis on the subset of genes that are specific to LP/D cells in WT testes, but aberrantly expressed in LL/Z cells in Prdm9–/– testes. Among the 56 TF motifs enriched at these genes was that of ETV4, which has been previously annotated as a regulator of the expression of LP/D genes (Ball et al., 2016). Interestingly, unlike Rfx4, encoding another regulator of LP/D, Etv4 is not differentially expressed in Prdm9–/– germ cells compared with WT (Figure 6). This suggests that while some TFs that regulate LP/D transcription, such as RFX4, are not activating their targets, another subset of TFs, including ETV4, continue to regulate transcription even though the Prdm9–/– germ cells do not reach the LP/D substage. This mixed and seemingly uncoordinated response to meiotic arrest suggests that the uncoupling of molecular and cellular pathways in the Prdm9–/– mutant is not tightly regulated (Figure 5).
FIGURE 5:
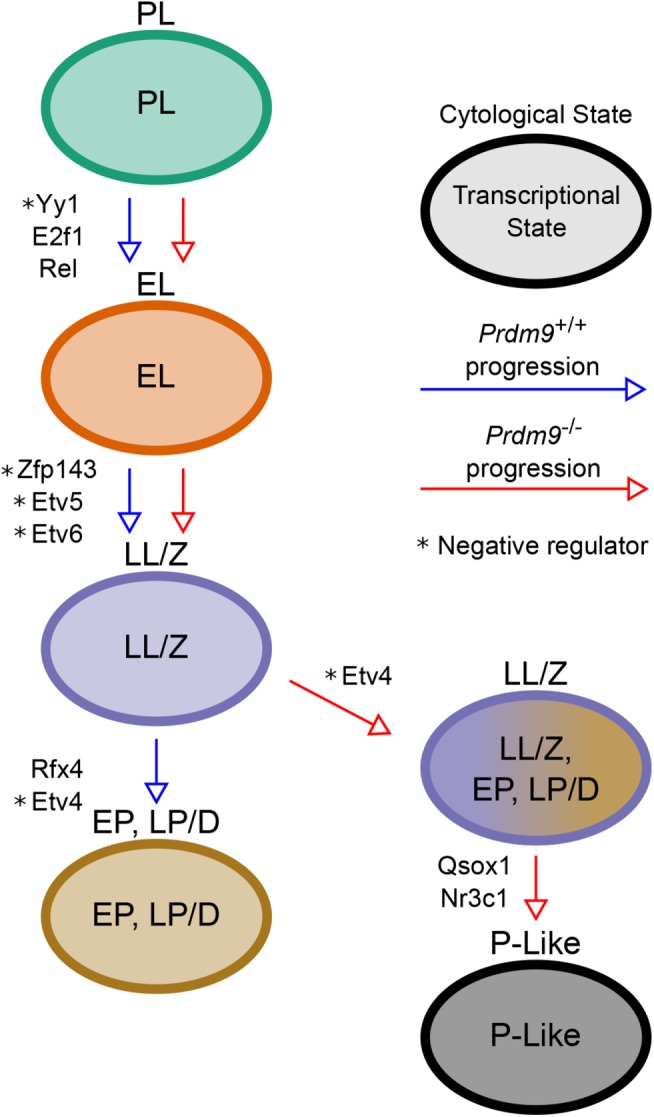
Summary model of cellular and molecular progression in Prdm9+/+ and Prdm9–/– germ cells. Arrows represent meiotic progression, colored by genotype as indicated. Text labels adjacent to the arrows indicate the transcriptional regulators of the genes expressed in the cell substage following the arrow.
FIGURE 6:

Upstream regulators of LP/D-specific genes show divergent expression changes in Prdm9–/– germ cells. (A) Log2(TPM+1) expression of activating transcriptional regulator Rfx4 at 8, 12, and 16 dpp. (B, C) Relative expression of RFX4 targets in Prdm9+/+ and Prdm9–/– samples, respectively. (D) Log2(TPM+1) expression of repressive transcriptional regulator Etv4 at 8, 12, and 16 dpp. (E, F) Relative expression of ETV4 targets in Prdm9+/+ and Prdm9–/– samples, respectively. *** represents FDR < 0.0001.
DISCUSSION
In this study, we used a mutant spermatogenesis phenotype as a model to determine the correspondence between molecular and cellular differentiation programs when the developmental process is perturbed and/or abrogated. The Prdm9 mutation is a robust model for this analysis because of the well-characterized morphological and cytological phenotypes caused by the absence of PRDM9, a key meiotic regulator protein. Normally, the spermatogenesis transcriptomic program and the meiotic gene expression programs are closely associated, running in parallel. However, by identifying transcriptomic signatures that match each of the known cytological substages and mutant phenotypes of Prdm9–/– germ cells, we found that the transcriptomic and cytodifferentiation programs of meiotic progression are partially uncoupled in mutant germ cells, most notably in two fundamental aspects. First, transcripts reflective of meiotic arrest and germ cell death phenotypes were detectable before the relevant characteristic cytological phenotypes appear. Second, mutant germ cells expressed transcripts typical of late-prophase substages in spite of being arrested before the onset of these substages; thus, a “spermatogenic transcriptome program” moves forward in spite of abrogation of the unfolding “meiotic cytodifferentiation program.” Overall, these two features yield stage-specific transcriptomes that are disassociated from their cell differentiation contexts. These findings not only further characterize the Prdm9–/– model of infertility but also are generally applicable to studies of developmental processes of differentiation and demonstrate the value of a combination of computational and cytological tools to address the challenges of assigning transcripts to cell types of origin, particularly in cases where single-cell RNA-Seq is not feasible.
A problem that frequently impedes biological interpretation of mutant versus WT transcriptomic analyses is the inability to differentiate the causal differences that propel the mutant phenotype by either primary or secondary effects of the mutation. This problem is exacerbated in heterogeneous cell populations, such as in testes, where it is difficult to purify cell stages in a developmental lineage. We used computational analysis to relate transcriptomic data to cellular differentiation in a heterogeneous cell population where the proportions of component cell stages are known. Specifically, we applied PMCA (Ball et al., 2016) to assign transcripts to their cells of origin, both in WT and mutant cell populations. By thus analyzing transcriptomic differences at the substage-specific level, we determined expression differences that were molecular phenotypes of specific cell types, rather than attributable to sample-level variation in the proportions of specific substages. Together these methods allowed interpretation of transcriptomic changes as preceding or following the observable phenotypes, thereby contributing to building a hierarchy of molecular and cytological phenotypes.
We showed that the transcriptomic and cytodifferentiation programs of meiotic progression are disassociated in the Prdm9–/– germ cells. We identified two distinct points where this asynchrony, or uncoupling, of the molecular and cytological programs is apparent. First, at 12 dpp in our samples, transcriptomic signatures of DNA damage checkpoint, activation, and repair appear in Prdm9–/– germ cells, preceding the specific cytological phenotypes that indicate unrepaired DNA damage (e.g., prevalence of the pH2AFX proteins indicative of DSBs). This seems biologically relevant, because we might expect that a future phenotype would be prefigured at the level of gene expression. The second example of asynchrony of transcriptomic and cytological phenotypes is not an anticipatory event prefiguring a phenotype, but instead is the unfolding of a transcriptome characteristic of later stages that never appear in the mutant. This is reflected in the shift in transcriptome profiles in Prdm9–/– germ cells as they exit the zygotene substage. Normally, this is followed cytologically by full chromosome synapsis, defining the pachytene substage. To the contrary, very few Prdm9–/– germ cells reach the EP substage and none reach the midpachytene substage. Instead, cells delay at LL/Z substages, and subsequently enter an abnormal P-like state, with subsequent cell death. As expected, the transcriptomic signature for cell death coincides with the appearance of the P-like germ cells. Additionally, however, the mutant germ cells delayed at the LL/Z substages express transcripts typical of the late spermatogenesis genes expressed in WT LP/D cells (Ball et al., 2016), representing disassociation of the ongoing transcriptome program from the delayed cytodifferentiation program. Indeed, the late-spermatogenesis transcriptomic signature of arrested spermatocytes suggests that regulatory programs promoting this transcription are at least partially functional in these germ cells, regardless of delay and arrest in cytological differentiation. The fact that the spermatogenic transcriptomic program is uncoupled from the differentiation program, and seemingly running on an independent clock, would not have been apparent without this twofold transcriptomic and cellular analysis of the meiotic arrest mutant phenotypes.
Transcriptional-factor analyses reveal that the late-spermatogenesis transcripts expressed in Prdm9–/– germ cells appear to share common TF-binding sites. This suggests that these transcripts are coregulated and that their WT-like expression in Prdm9–/– germ cells may be caused by the programmed processes, rather than random chance. One TF with an enriched binding motif among these genes is ETV4, which is negatively correlated with the expression of its targets. Therefore, the developmentally premature decline in Etv4 expression could lead to the observed up-regulation of spermiogenic transcripts. In contrast, the late-spermatogenesis transcripts that fail to be expressed (or are down-regulated) in the mutant spermatocytes are enriched for the binding motif of RFX4, which is positively correlated with the expression of its proposed targets. In this case, the root regulatory event may be failure to express the TFs in the mutant germ cells, but here, that failure also leads to the down-regulation of its targets. Together, these observations suggest that TF down-regulation can incongruently lead to both up-regulation and down-regulation of targets, explaining the apparent asynchrony between cellular and transcriptomic stages in mutant germ cells undergoing meiotic arrest. Thus, these findings put forth a model of meiotic arrest in which there is asynchrony of transcriptomic and cytological differentiation programs, each revealing independent autonomy.
In addition to the biological implications, divergent or asynchronous programs in phenotypes of developmental arrest present challenges for biological data interpretation. The premature expression of late-spermatogenesis transcripts in inappropriately early meiotic substages in Prdm9–/– germ cells is obviously not sufficient to avoid meiotic arrest or propel spermatocytes to the cellular stage appropriate for the transcript expression. Although it is not known whether the prematurely expressed transcripts are translated in Prdm9–/– spermatocytes, it is not likely that their translational state could rescue meiotic arrest, especially since some of the transcripts are destined for postmeiotic translation. Nonetheless, an unsolved problem that transcriptomic analyses cannot address is the degree of developmental autonomy and synchrony between cytological differentiation and the programs of protein translation and activation. Moreover, finding transcriptomic signatures at variance with the cytological stage has implications for the interpretation of bulk RNA-Seq from other mutants, especially those with less well-characterized cytological phenotypes. As this study reveals, where transcriptomic data may not reflect corresponding cellular changes, it is essential to have a complement of classical cytological measures and a method for integration of the two types of data. Much of the benefit of single-cell RNA-Seq (scRNA-Seq) relies on the ability to infer the cellular context of a cell based on its transcriptomic signature, but our study suggests that uncoupled molecular and cellular processes would complicate such inferences, absent a cytological characterization to accommodate the results. Determination of cellular context and autonomous transcriptomic programs in a developmental mutant might be facilitated with scRNA-Seq data; however, the possibility of cellular and molecular uncoupling should be considered even when analyzing single-cell data. In particular, cell-type-specific molecular markers may become unreliable identifiers of cellular states when standard transcriptional programs fragment. This phenomenon is unlikely to be unique to spermatogenesis and warrants investigation in other developmental and differentiation contexts.
MATERIALS AND METHODS
Experimental design
Sample acquisition.
All genotypes of mice collected for this study were bred from Prdm9tm1Ymat heterozygous mice, which are nearly congenic C57BL/6J (B6) mice, with <10% 129P2/OlaHsd remaining, and obtained from The Jackson Laboratory (JAX, Bar Harbor, ME). Samples were collected at 8, 12, and 16 dpp; 10 samples were collected at each time point, comprising various proportions of Prdm9+/+, Prdm9+/–, and Prdm9–/– mice. At each time point, five mice were Prdm9–/–. The testes of each mouse were pooled prior to germ cell enrichment. An aliquot of enriched germ cells was analyzed for the substage proportion in each sample through cytological methods, while the rest of the germ cells were used for our transcriptome analysis using RNA sequencing. All mice were maintained following protocols approved by the JAX Institutional Animal Care and Use Committee (IACUC).
Germ cell enrichment.
Interstitial cells were removed with collagenase from seminiferous tubules, which were subsequently enzymatically digested to yield dispersed cells. Germ cells were isolated from this population by size filtration. Details of the procedure are outlined in La Salle et al. (2009) and Ball et al. (2016). The resulting cell populations were relatively pure, with no more than 10% of each sample comprised of somatic cells (recognized as not expressing SYCP3; phosphorylated histone H2AX, STRA8, or H1T; and showing a distinct DAPI staining pattern) (Figure 1B).
Cytological methods: chromatin spread preparation and immunostaining of spread chromatin
Briefly, as previously described (Ball et al., 2016), spread chromatin of germ cells were fixed in 1% paraformaldehyde (PFA) in H2O containing 0.015% SDS and 0.02% Photo-Flo (Kodak, Rochester, NY) for 1 h at room temperature, and cells were further fixed in 2% PFA in H2O with or without 0.03% SDS for 3 min each. For immunostaining, prepared chromatin preparation was incubated with 10% antibody dilution buffer (ADB) blocking solution (phosphate-buffered saline containing 2% bovine serum albumin and 0.05% Triton X-100) and immunostained with rat anti-SYCP3 antibody (1:1000 dilution; Handel Laboratory), mouse anti-phosphorylated histone H2AX antibody (1:200 dilution, 05-636; Millipore, Billerica, MA), rabbit anti-STRA8 antibody (1:1000 dilution, ab49405; Abcam, Cambridge, England), guinea pig anti-H1t antibody (1:500 dilution; Handel lab), and Alexa Fluor (488, 594, or 647)-conjugated secondary antibodies (1:500 dilution; Molecular probes, Thermo Fisher Scientific, Waltham, MA); nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were observed using a Zeiss AxioImager.Z2 epifluorescence microscope equipped with a Zeiss AxioCam MRm CCD camera (Carl Zeiss, Jena, Germany). Approximately 450 germ cells were staged per sample. Owing to their similar frequency patterns, as well as to match the previously published data set (Ball et al., 2016), LL and Z were combined, as well as LP and D, for all analyses.
RNA methods: isolation of RNA and sequencing library preparation and RNA sequencing
As outlined previously (Ball et al., 2016), cells were resuspended and homogenized before RNA was purified from each sample. The quality of the isolated RNA was assessed and then the mRNA-sequencing libraries were prepared and subsequently tested for quality; 100-base paired-end reads were sequenced and filtered by quality. Technical replicates were run in different lanes and merged for the final samples used for later analyses.
Computational methods
Data and ode availability.
All codes used to produce major findings for this article can be found at https://github.com/AFine1/Uncoupling-in-Prdm9KO. All transcriptomic data are available through Gene Expression Omnibus, accession GSE110703. An R Shiny web app is available at https://shinyapps.jax.org/d86f1ec60d6c596dfc1eab16e5d68aca for the visualization of gene expression levels from our data set.
Alignment and expression.
The transcripts from each RNA-Seq sample were aligned and quantified on a custom-built pseudotranscriptome, comprising the mm10 reference transcriptome of Ensembl Genome Reference Consortium, build 38, release 75 (Flicek et al., 2014); NONCODE v4 lncRNA (Xie et al., 2014); and piRNA precursor transcripts (Li et al., 2013). The sequences and genomic positions of the piRNA precursors were acquired from Ball et al. (2016). NONCODE and piRNA transcripts were defined on mm9 and converted to mm10 coordinates using liftOver (Fujita et al., 2011). Alignment of our RNA-Seq samples was performed using Bowtie 1.0.0 (Langmead et al., 2009), and the estimation of expression was calculated using RSEM (Li and Dewey, 2011). Transcript expression was quantified as log, base 2, of transcripts per million (TPM) from RSEM, log2(TPM+1). Transcripts were excluded from further analysis if the expression was <1 for all samples, for example, we required ComBat-adjusted (Johnson et al., 2007) log2(TPM+1) ≥1 in at least one sample. From the expression of Prdm9, it appeared that the genotype of two samples at 12 dpp had been mislabeled, so neither sample was included in the analyses.
RNA-Seq sample integration.
To increase the number of samples at each time point, we also utilized published data from previously collected Prdm9+/+ samples (Supplemental Table S1) that had been collected with the same protocols as above (Ball et al., 2016) (GSE72833). These mice were all C57BL/6J mice obtained from JAX. Substage proportion and transcriptome profiles had been collected. Three samples were utilized from 8 dpp and five samples were utilized from 12 and 16 dpp, respectively. To distinguish the origin of each sample, samples that were collected for the purpose of this study are referred to as being in the Prdm9 Dataset, while samples collected previously (Ball et al., 2016) are part of the Background Dataset.
PC analysis.
To visualize the transcriptomic variation among samples, a PCA was run by performing singular value decomposition (SVD) on transcript expression data, where each transcript’s expression was centered and scaled. The PCA was performed using the function svd(x) using the R statistical framework (R Core Team, 2015).
ComBat adjustment.
We used ComBat (Johnson et al., 2007) to adjust for known systematic variation in our data set. The systematic variation among our samples, as visualized by PCA, showed that samples segregate by data set (Baseline vs. Prdm9, Supplemental Figure S1A), as well as by litter (Supplemental Figure S1, B–D). Since this variation can be directly attributed to these known cofactors, we used ComBat in R from the package sva (Leek et al., 2017) to adjust for these batch and litter effects. First, we ran the expression data through ComBat to adjust for data set variation (Prdm9 vs. Baseline), keeping variation that could be attributed to genotype and age. Then, for each time point, we corrected for variation by litter. Genotype, data set, and litter information can be found in Supplemental Table S1. The litter information for samples from the Baseline data set was unknown, so these were considered to be part of a single litter at each time point.
Differential expression analysis.
We used a regression model to identify genes that were differentially expressed in Prdm9–/– germ cells, compared with Prdm9+/+ germ cells. We ran the function lm(x) from the stats package in R (R Core Team, 2015). This linear model fits variation in gene expression across samples to given variables of interest. For this analysis, the variables that we were interested in were genotype, age, and genotype-by-age. We then used the function contrast(x) (Kuhn et al., 2016) to identify the significance of changes in gene expression among conditions at each time point. The p values were corrected for multiple testing using p.adjust(x) (R Core Team, 2015). Transcripts with an FDR < 0.01 and a log fold change >0.5 were considered to be significantly differentially expressed. Differential expression coefficients, unadjusted p values, as well as FDR values are in Supplemental Table S2.
Epigenetic integration.
Published ChIP-Seq data (Brick et al., 2012) were downloaded and integrated into our analysis. We downloaded DMC1 peaks from the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO; GSE35498). Using DMC1 as a marker for DSBs in germ cells, we identified which DSBs in Prdm9–/– germ cells were found at promoters. DSBs were defined as the central 1 kb region of DMC1 peaks and promoters were defined as 1 kb upstream or downstream of transcription start sites. We used Fisher’s exact test to test the enrichment of DSBs at the promoters of DEGs (R Core Team, 2015)
PMCA.
We assigned transcripts to substages of prophase in the Prdm9–/– samples with PMCA (Ball et al., 2016). This robust statistical method identifies common patterns across measurements of coupled samples and provides a FDR for each association. In this study, PMCA identified transcripts whose abundances followed the same pattern as the proportions of a substage’s cells across time points, thus identifying substage-specific gene sets. We applied PMCA to the Prdm9 data set and identified substage-specific gene sets for the Prdm9 mutant, with FDR ≤ 0.01 for all substages. Substage assignment for transcripts is indicated in Supplemental Table S2. Next, we compared these gene sets to previously identified WT substage-specific gene sets, similarly identified with PMCA in a previously published study (Ball et al., 2016).
Bioinformatic methods
GO analysis.
GO term enrichment analysis was implemented on sets of DEGs at each time point using the GO enRIchment anaLysis and visuaLizAtion (GOrilla) tool (Harris et al., 2004; Eden et al., 2009). Because the GO analysis we used does not take into account direction or magnitude of differential expression, DEGs at each time point were analyzed separately based on the directionality of their differential expression and provided to GOrilla as an unranked list of genes. We used the list of all genes expressed in our data set as the background set, to avoid overrepresentation of germ cell- and meiosis-related terms.
Pathway analysis.
To further assess the functionality of DEGs, we used IPA (QIAGEN; www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) (Krämer et al., 2014). This program uses curated gene annotations, associations, and functions to provide pathways that are enriched in a given gene list. We used IPA to analyze our DEG lists from 12 and 16 dpp respectively. We initially used FDR < 0.05 for our significance threshold for DEGs for these analyses; however, due to the number of DEGs at 16 dpp, a large number of pathways were enriched that made the interpretation of these results challenging. Therefore, we reran this analysis after further limiting our DEGs at 16 dpp to FDR < 0.01 in order to identify only the most relevant pathways enriched.
TF analysis.
TFs were identified for gene lists using iRegulon, version 1.3 (Janky et al., 2014) in Cytoscape, version 3.4.0 (Shannon et al., 2003). For all analyses, identified TFs were limited to those that were expressed in our data set and had a normalized enrichment score ≥4, corresponding to an FDR < 0.01. TFs identified in this study were compared with previously published TFs (Ball et al., 2016).
Supplementary Material
Acknowledgments
We thank the Handel and Carter laboratories for discussions and gratefully acknowledge Sabrina Petri for animal care. We also thank J. Trowbridge, A. Yee, C. Cowan, and S. Munger for feedback on the project and comments on the manuscript. This work was supported by National Institutes of Health/National Institute of General Medical Sciences (NIGMS) Grant no. P01 GM099640 (G.W.C. and M.A.H.) and by a fellowship from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, Grant no. T32 HD007065 (A.D.F.).
Abbreviations used:
- D
diplotene
- DEG
differentially expressed gene
- dpp
days postpartum
- DSB
double-strand break
- EL
early leptotene
- EP
early pachytene
- FDR
false discovery rate
- GO
Gene Ontology
- LL
late leptotene
- LP
late pachytene
- PCA
principal component analysis
- piRNA
Piwi-interacting RNA
- PL
preleptotene
- P-Like
pachytene-like
- PMCA
permutation-based maximum covariance analysis
- Sp
spermatogonia
- TF
transcription factor
- TPM
transcripts per million
- WT
wild type
- Z
zygotene
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-10-0681) on January 16, 2019.
REFERENCES
- Ball RL, Fujiwara Y, Sun F, Hu J, Hibbs MA, Handel MA, Carter GW. (2016). Regulatory complexity revealed by integrated cytological and RNA-seq analyses of meiotic substages in mouse spermatocytes. BMC Genomics , 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A, Khan Z, Wang SH, Mitrano A, Ford MJ, Pritchard JK, Gilad Y. (2015). Genomic variation. Impact of regulatory variation from RNA to protein. Science , 664–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Imai Y, de Massy B. (2013). Meiotic recombination in mammals: localization and regulation. Nat Rev Genet , 794–806. [DOI] [PubMed] [Google Scholar]
- Bolcun-Filas E, Handel MA. (2018). Meiosis: the chromosomal foundation of reproduction. Biol Reprod , 112–126. [DOI] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV. (2012). Genetic recombination is directed away from functional genomic elements in mice. Nature , 642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Dent SY. (2014). Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet , 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick JM, Munger SC, Simecek P, Huttlin EL, Choi K, Gatti DM, Raghupathy N, Svenson KL, Churchill GA, Gygi SP. (2016). Defining the consequences of genetic variation on a proteome-wide scale. Nature , 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz I, Rodriguez-Casuriaga R, Santinaque FF, Farias J, Curti G, Capoano CA, Folle GA, Benavente R, Sotelo-Silveira JR, Geisinger A. (2016). Transcriptome analysis of highly purified mouse spermatogenic cell populations: gene expression signatures switch from meiotic- to postmeiotic-related processes at pachytene stage. BMC Genomics , 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Lin H. (2002). miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev Cell , 819–830. [DOI] [PubMed] [Google Scholar]
- Eddy EM. (1998). Regulation of gene expression during spermatogenesis. Semin Cell Dev Biol , 451–457. [DOI] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics , 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallahi M, Getun IV, Wu ZK, Bois PR. (2010). A global expression switch marks pachytene initiation during mouse male meiosis. Genes (Basel) , 469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al (2014). Ensembl 2014. Nucleic Acids Res , D749–D755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al (2011). The UCSC Genome Browser database: update 2011. Nucleic Acids Res , D876–D882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H, Cai T, Lin X, Wu Y, Wang X, Yang F, Han C. (2013). Integrative proteomic and transcriptomic analyses reveal multiple post-transcriptional regulatory mechanisms of mouse spermatogenesis. Mol Cell Proteomics , 1144–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves E, Fragoulis A, Garcia-Alonso L, Cramer T, Saez-Rodriguez J, Beltrao P. (2017). Widespread post-transcriptional attenuation of genomic copy-number variation in cancer. Cell Syst , 386–398 e384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CD, Ma Q, Manske GL, Shami AN, Zheng X, Marini S, Moritz L, Sultan C, Gurczynski SJ, Moore BB, et al (2018). A comprehensive roadmap of murine spermatogenesis defined by single-cell RNA-Seq. Dev Cell , 651–667.e610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud SS, Low DH, Yi C, Carrell DT, Guccione E, Cairns BR. (2014). Chromatin and transcription transitions of mammalian adult germline stem cells and spermatogenesis. Cell Stem Cell , 239–253. [DOI] [PubMed] [Google Scholar]
- Handel MA, Schimenti JC. (2010). Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat Rev Genet , 124–136. [DOI] [PubMed] [Google Scholar]
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, et al (2004). The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res , D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Matsui Y. (2006). Meisetz, a novel histone tri-methyltransferase, regulates meiosis-specific epigenesis. Cell Cycle , 615–620. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Yoshida K, Matsui Y. (2005). A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature , 374–378. [DOI] [PubMed] [Google Scholar]
- Janky R, Verfaillie A, Imrichova H, Van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D, et al (2014). iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol , e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics , 118–127. [DOI] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J, Jr, Tugendreich S. (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics , 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M, Weston CFS, Wing J, Forester J, Thaler T. (2016). Contrast: A Collection of Contrast Methods. R package version 0.21.
- Langmead B, Trapnell C, Pop M, Salzberg SL. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol , R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Salle S, Sun F, Handel MA. (2009). Isolation and short-term culture of mouse spermatocytes for analysis of meiosis. Methods Mol Biol , 279–297. [DOI] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Fertig EJ, Jaffe AE, Storey JD. (2017). sva: Surrogate Variable Analysis. R package version 3.26.0.
- Li B, Dewey CN. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics , 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XZ, Roy CK, Dong X, Bolcun-Filas E, Wang J, Han BW, Xu J, Moore MJ, Schimenti JC, Weng Z, Zamore PD. (2013). An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes. Mol Cell , 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa SH, Park PJ, Zhang LF, Shima JE, McCarrey JR, Griswold MD, Lee JT. (2006). Postmeiotic sex chromatin in the male germline of mice. Curr Biol , 660–667. [DOI] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K. (2010). Prdm9 controls activation of mammalian recombination hotspots. Science , 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramskold D, Wang ET, Burge CB, Sandberg R. (2009). An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol , e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2015). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing.
- Schultz N, Hamra FK, Garbers DL. (2003). A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc Natl Acad Sci USA , 12201–12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res , 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthes P, Kokkinaki M, Nef S, Gnirke A, et al (2013). Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep , 2179–2190. [DOI] [PubMed] [Google Scholar]
- Sun F, Fujiwara Y, Reinholdt LG, Hu J, Saxl RL, Baker CL, Petkov PM, Paigen K, Handel MA. (2015). Nuclear localization of PRDM9 and its role in meiotic chromatin modifications and homologous synapsis. Chromosoma , 397–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault-Sennett S, Yu Q, Smagulova F, Cloutier J, Brick K, Camerini-Otero RD, Petukhova GV. (2018). Interrogating the functions of PRDM9 domains in meiosis. Genetics , 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Williams EG, Dubuis S, Mottis A, Jovaisaite V, Houten SM, Argmann CA, Faridi P, Wolski W, Kutalik Z, et al (2014). Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell , 1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Yuan J, Li H, Li M, Zhao G, Bu D, Zhu W, Wu W, Chen R, Zhao Y. (2014). NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Res , D98–D103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.