

Abstract
Whereas many studies have examined the properties of the compromised neocortex in the first several days following ischemia, there is less information regarding the initial 12 h poststroke. In this study we examined live mouse neocortical slices harvested immediately and 12 h after a 30-min middle cerebral artery occlusion (MCAo). We compared nonischemic and ischemic hemispheres with regard to the propensity for tissue swelling and for generating spreading depolarization (SD), as well as evoked synaptic responses and single pyramidal neuron electrophysiological properties. We observed spontaneous SD in 7% of slices on the nonstroked side and 25% in the stroked side following the 30-min MCAo. Spontaneous SD was rare in 12-h recovery slices. The region of the ischemic core and surround in slices was not susceptible to SD induced by oxygen and glucose deprivation. At the neuronal level, neocortical gray matter is surprisingly unaltered in brain slices harvested immediately poststroke. However, by 12 h, the fields of pyramidal and striatal neurons that comprise the infarcted core are electrophysiologically silent because the majority are morphologically devastated. Yet, there remains a subset of diffusely distributed “healthy” pyramidal neurons in the core at 12 h post-MCAo that persist for days poststroke. Their intact electrophysiology and dendritic morphology indicate a surprisingly selective resilience to stroke at the neuronal level.
NEW & NOTEWORTHY It is generally accepted that the injured core region of the brain resulting from a focal stroke contains no functioning neurons. Our study shows that some neurons, although surrounded by devastated neighbors, can maintain their structure and electrical activity. This surprising finding raises the possibility of discovering how these neurons are protected to pinpoint new strategies for reducing stroke injury.
Keywords: ATPase, hypothalamus, Na+-K+ pump, neuroprotection, pyramidal, spreading depolarization, spreading depression, stroke, traumatic brain injury, whole cell recording
INTRODUCTION
During focal ischemia, cerebral blood flow (CBF) declines precipitously in brain areas supplied by an occluded artery. Within 1–2 min, Na+-K+ pump failure leads to gray matter depolarization that propagates across the neocortex (Busch et al. 1996; Dijkhuizen et al. 1999; Hossmann 1994; Takano et al. 1996) and subcortically (Brisson et al., 2014). This ischemic “spreading depolarization” (SD) manifests as neurons (and glia) suddenly lose their membrane potential, driving a large negative extracellular voltage that migrates at 2–5 mm/min across gray matter (Dreier 2011; Somjen 2001). Within 1–2 min, ischemic neurons begin losing their spines as the dendrites swell and form beaded processes, as observed in vivo following photothrombosis (Murphy et al., 2008) and in acute brain slices after oxygen and glucose deprivation (OGD) (Andrew et al. 2007; Jarvis et al. 2001; Obeidatet al. 2000).
Infarct expansion is correlated with increased SD incidence (Back et al. 1996; Mies et al. 1994). Furthermore, KCl-triggered SD () waves traverse into partially ischemic gray matter and increase infarct volume (Busch et al. 1996; Dijkhuizen et al. 1999; Takano et al. 1996). SD incidence is common up to 2 h after middle cerebral artery occlusion (MCAo)-induced ischemia and is then followed by a 6-h period of few SD events (Back et al. 1996; Dijkhuizen et al. 1999; Koroleva and Bures 1996). A later secondary phase of spontaneous SD follows 10–16 h after 30 min of MCAo (Hartings et al. 2003).
There have been many investigations of postischemic SD, but few with observations during the first 12 h after a focal stroke. Because of the historical focus on the peri-infarct region, the cellular fate of the ischemic core has been poorly defined (Jiang et al. 2017). In this study, we harvested brain slices immediately following a 30-min MCAo or 12 h later. We compared the effects of elevated extracellular potassium concentration ([K+]ext) and elevated extracellular glutamate concentration ([glu]ext), as well as 10 min of OGD in these postischemic brain slices. This enabled an examination of gray matter responsivity in the absence of inherent CBF fluctuations that follow stroke in vivo (Shin et al. 2006; von Bornstädt et al. 2015) and that can confound SD properties (Sukhotinsky et al., 2010).
Superfusion of neocortical slices with elevated [K+]ext, elevated [glu]ext, or OGD simulates aspects of the extracellular milieu in the postischemic brain. A [K+]ext of ~10 mM arises just before SD during ischemia (Nedergaard and Hansen 1993; Tanaka et al. 1997) and helps promote SD in hippocampal slices. As a result of MCAo, [glu]ext becomes elevated (Morimoto et al. 1996; Sciotti et al. 1992) and glutamate release evoked by SD contributes to the propagation of (Zhang et al. 2015; Zhou et al. 2013). OGD initiates SD in cortical slices by mimicking the decrease in glucose and oxygen coincident with CBF decline. By applying elevated [K+]ext., elevated [glu]ext, or OGD in slices harvested after MCAo, we can observe their individual effects on postischemic gray matter in the absence of changes in CBF.
While examining the progression of neuronal injury during the first 12 h of MCAo using Golgi-Cox staining, we observed a subset of apparently healthy pyramidal neurons within the core, a recent observation made independently using histochemical and immunohistochemical techniques (Jiang et al. 2017). We demonstrate that these cells display a surprisingly intact morphology with robust and near-normal electrophysiological properties but dampened synaptic input.
MATERIALS AND METHODS
MCAo and Brain Slice Preparation
The protocol was approved by the University Animal Care Committee of Queen’s University. Male C57/BL6 mice (20–25g) were anesthetized using isoflurane (3% initial, 1– 1.5% maintenance) in 80% air and 20% O2. The animal remained under general anesthesia for the duration of the surgical procedure. The time under anesthesia was 80–90 min. Although isoflurane can inhibit SD initiation in the intact animal, SD onset in our harvested brain slices on the nonstroked side was similar to that in nonanesthetized mice in previous studies (Joshi and Andrew 2001). Focal cerebral ischemia was then induced by intraluminal occlusion of the left middle cerebral artery (MCA) for 30 min (Barber et al. 2004). The silicon-coated nylon suture (diameter 180–220 mm) was introduced into the external carotid artery (ECA) and pushed up the internal carotid artery (ICA) until resistance was felt. The filament was inserted 9–10 mm from the carotid bifurcation, blocking the MCA for 30 min, after which it was removed and the ECA was permanently tied. Brains were harvested either immediately or following 12 h of in vivo reperfusion. The anesthetized mouse was decapitated and its brain quickly removed and immersed in ice-cold and oxygenated (95% O2-5% CO2) artificial cerebral spinal fluid (aCSF) composed of (in mM) 240 sucrose, 3.3 KCl, 26 NaHCO3, 1.3 MgSO4·7H2O, 1.23 NaH2PO4, 11 d-glucose, and 1.8 CaCl2. With the use of a Leica 1200-T vibratome, 350-μm slices were cut in the sucrose aCSF through the coronal plane and then incubated in regular aCSF (equimolar NaCl replacing sucrose above) at 35°C for at least 1 h before slice experiments. The aCSF osmolality of ~290 mosmol/kgH2O was increased to 310 mosmol/kgH2O using mannitol, and pH was 7.4. Brain slices were recorded during the following 4 h. Mannitol increased the rat aCSF from 290 to 310 mosmol/kgH2O to reflect in vivo plasma levels in the mouse.
Imaging Changes in Light Transmittance
Brain slices were transferred to a recording/imaging chamber mounted on an inverted microscope (Axoscope 2FS; Zeiss) with a ×10 objective lens and zoom lens. Slices were submerged in flowing aCSF (3.5 ml/min) at 34 ± 0.5°C. Video images were captured with a cooled charge-coupled device (model C4742; Hamamatsu) using Imaging Workbench 6 software (Indec Biosystems). Each image of a video series consisted of 16 averaged frames acquired at 10 Hz. The first image of the series was the control transmittance (Tcont), which was subtracted from each of the subsequent images (Texp) in the series. The difference signal was normalized by dividing by Tcont, which varies across the slice depending on the zone sampled. For example, Tcont was lower in white matter than in gray matter. This value was then presented as a percentage of the digital intensity of the control image of that series. That is, the change in light transmittance (ΔLT) = [(Texp − Tcont)/Tcont] × 100 = (ΔT/T)%. The ΔLT was displayed using a pseudocolor intensity scale. The slice image in bright field was displayed using a gray intensity scale. ΔLT was recorded in the hemisphere ipsi- and contralateral to the lesion during bath application of OGD, 500 µM glutamate, or 9.7 mM [K+]ext aCSF solutions. The OGD aCSF was of similar composition to control aCSF, except for substitution of carbon dioxide and oxygen (95% O2-5% CO2) bubbling of aCSF with nitrogen and oxygen (95% N2-5% CO2). In addition, 11 mM glucose was reduced to 1 mM glucose with osmotic adjustment using NaCl.
For imaging the effects of bath-applied KCl or glutamate, an LT of ~5% arises during superfusion with hyposmotic (−30 mosmol/kgH2O) aCSF (Andrew and MacVicar 1994). A 5% threshold value was selected to record a non-SD tissue swelling event that exceeded baseline fluctuations. Data were obtained from one region of interest in the medial and lateral parietal cortex in each hemisphere, the predicted region of the MCAo on the ischemic side.
Electrophysiology
Whole cell patch clamp.
Visually guided whole cell patch-clamp recordings were obtained using micropipettes pulled from borosilicate glass (outside diameter 1.2 mm, inside diameter 0.68 mm; World Precision Instruments) to a resistance of 4–6 MΩ. Pyramidal neurons were identified in neocortical gray using Dodt optics based on their size and shape. The internal pipette solution contained (in mM) 125 K-gluconate, 10 KCl, 2 MgCl2, 5.5 EGTA, 10 HEPES, 2 Na-ATP, and 0.1 CaCl2 (pH adjusted to 7.3 with KOH). The junction potential was measured as 14 mV by measuring the potential of the electrode within the bath. This value was subtracted from the negative resting potential measurements during data analysis. All recordings were acquired in current-clamp mode of an Axoclamp 2A 23 amplifier and a Digidata 1322 analog-to-digital converter. Clampex 10 software was used for data acquisition, with subsequent analysis using Clampfit 10 software. Sampling frequency was 5 kHz, and low-pass filtering was done with an external Bessel filter (LPF 202a) at 10 kHz. Action potential width, amplitude, and threshold were measured from the first evoked action potential by a depolarizing current pulse. Fast afterhyperpolarizations (fAHP) were measured as the voltage deviation from the beginning of the upstroke to the trough following an action potential to within 5 ms. The time constants of ischemic and nonischemic neocortical pyramidal neurons were derived from a “curve of best fit” of membrane potential exponential decay following the second hyperpolarizing pulse of stepped current pulses (200 ms, −0.1 nA). Whole cell input resistance was derived from the linear portion of the current-voltage plot 170 ms from the beginning of constant-current pulses whose strength was increased stepwise from −0.5 to 0 nA.
Evoked synaptic responses.
To record evoked field potentials, a micropipette (2–4 MΩ) was pulled from thin-walled capillary glass, filled with 200 mM NaCl, and mounted on a three-dimensional (3-D) micromanipulator. It was connected by a chloride-coated silver wire to an amplifier probe, and output was monitored on an online oscilloscope. The tip was placed in layers II/III of the neocortex, and a concentric bipolar electrode (Rhodes Electronics) was placed in layer VI to stimulate the immediately overlying layers. A single-current pulse (0.1-ms duration; 0.1 Hz) was applied at increasingly stronger voltages, peaking at 50 mV, to maximize an evoked response if detected. The amplified signals were digitized, displayed, and plotted using pCLAMP 10 software (Axon Instruments).
Golgi-Cox Staining
Following 30 min of MCAo, the mouse was decapitated either immediately or after 12 h of reperfusion. The brain was extracted, immersed in freshly prepared impregnation solution, and processed according to the protocol provided by FD NeuroTechnologies (2014) using the FD Rapid GolgiStain kit. Coronal brain sections were then cut at 50 μm and further processed as per the kit instructions. Ischemic and nonischemic hemispheres were imaged at low magnification to observe gross tissue morphology and at higher magnification to observe individual neuronal morphology.
Statistical Analysis
Field excitatory postsynaptic potentials (EPSPs), baseline electrophysiological properties, latency to SD onset, speed of SD propagation, and LT changes during and after SD were compared in the ischemic vs. nonischemic hemispheres of poststroke brain slices using a paired Student’s t-test. SD incidence, evoked synaptic responses, and increased cell swelling represented by elevated LT values (Jarvis et al. 2001) were compared in the ischemic vs. nonischemic hemisphere using Mann-Whitney U nonparametric analyses. Electrophysiological parameters were described using means ± SE.
RESULTS
Light Transmittance Imaging
During ischemia, oxygen and glucose levels decrease significantly in brain areas, whereas extracellular K+ and glutamate become elevated as a result of SD generation. Independently of blood flow and its fluctuations, we bath-applied aCSF containing elevated [K+]ext, elevated [glu]ext, or OGD aCSF to postischemic brain slices harvested immediately or 12 h after a 30-min MCAo. We compared the number of slices that displayed SD. Both ischemic and nonischemic sides were imaged simultaneously. Immediately post-MCAo, spontaneous SD events at a baseline of 3.2 mM [K+]ext over a period of ~15 min were observed in 7% (n = 1 of 14 slices) in the nonischemic hemisphere and 25% (n = 5 of 20 slices) in the ischemic hemisphere. At 9.6 mM [K+]ext, the SD incidence in the nonischemic hemisphere was 80% of slices (n = 11 of 14) vs. 35% (n = 7 of 20) in the ischemic hemisphere. When 9.6 mM [K+]ext was bath-applied to brain slices harvested 12 h after MCAo, the SD incidence in the nonischemic hemisphere was 89% (n = 8 of 9 slices) vs. 33% (n = 3 of 9 slices) in the ischemic hemisphere. So, surprisingly, a significantly decreased incidence of in the ischemic hemisphere vs. the nonischemic hemisphere was observed 12 h after MCAo (Mann-Whitney U-test, P < 0.05) as shown in Fig. 1. initiated and propagated both within and outside neocortex supplied by the MCA in brain slices harvested immediately after MCAo. However, only three events occurred within MCA territory of the ischemic hemisphere in slices 12 h post-MCAo.
Fig. 1.
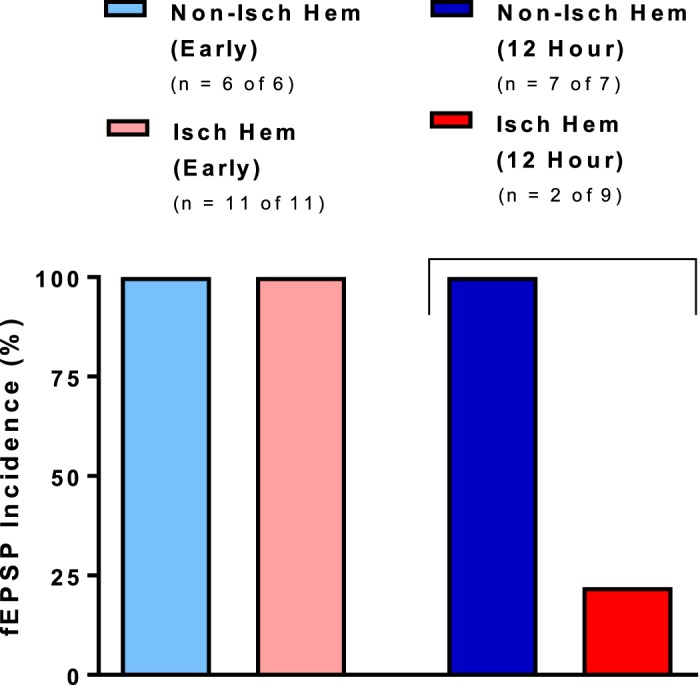
Incidence of external K+ ([K+]ext)-induced spreading depolarization (SD) in brain slices harvested immediately and 12 h after middle cerebral artery occlusion (MCAo). In either case, slices rarely display SD spontaneously within the ischemic hemisphere when superfused with 3.2 mM K+-saline. The ischemic hemisphere displays a significantly decreased incidence of SD 12 h after MCAo (Mann-Whitney U-test, P < 0.05). Values are means; n = no. of slices. fEPSP, fast excitatory postsynaptic potential; Isch hem, ischemic hemisphere; Non-isch hem, nonischemic hemisphere.
In vivo, extracellular glutamate levels become significantly elevated immediately after ischemia. In the nonischemic hemisphere, bath application of glutamate (500 µM) onto brain slices harvested immediately after MCAo elevated LT by >5% in 64% of slices (n = 7 of 11). After 12 h of reperfusion, 65% of slices (n = 11 of 19) displayed such LT increases on the nonischemic side. As shown in Fig. 2, LT increases of >5% were never observed in the lateral parietal cortex in the ischemic hemisphere (i.e., neocortex affected by the MCAo). LT also did not increase at either time point near the central sulcus outside of MCA territory, which is less affected by MCAo blood flow (i.e., medial motor cortex and cingulate cortex). A Mann-Whitney U statistical analysis showed the incidence of swelling evoked by 500 µM glutamate was significantly lower in the ischemic than in the nonischemic hemisphere both immediately (P < 0.01, n = 11 slices) and 12 h after MCAo (P < 0.01, n = 19 slices) as shown in Fig. 3. No SD was evoked in any slice region, as expected (see discussion).
Fig. 2.
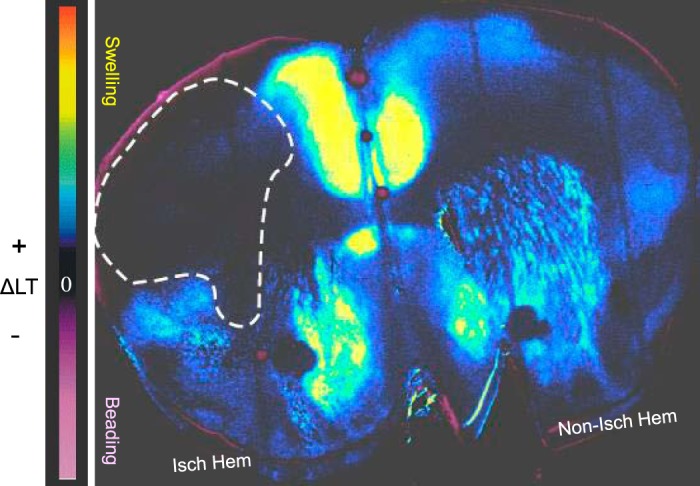
KCl-triggered spreading depolarization () was initiated outside middle cerebral artery (MCA) territory (dashed line) when brain slices were harvested after 12 h of reperfusion following MCA occlusion (MCAo). Change in light transmittance (ΔLT) was imaged during superfusion of brain slices with 9.6 mM external K+. In this pseudocolored image, blue/yellow depicts increased ΔLT and cell swelling. SD initiates and propagates throughout the entire right (nonischemic hemisphere) but excludes the estimated MCAo region of the ischemic hemisphere (dashed line). Isch hem, ischemic hemisphere; Non-isch hem, nonischemic hemisphere.
Fig. 3.
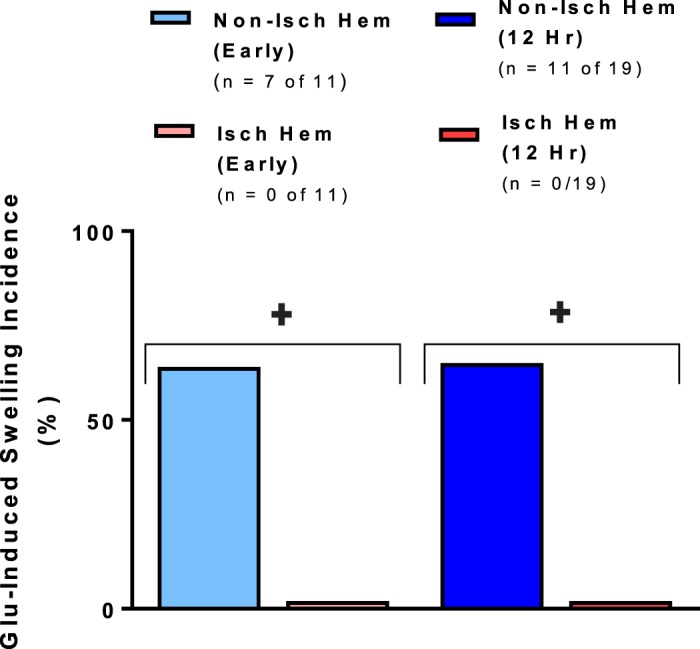
Reduced swelling as measured by increased change in light transmittance (ΔLT) induced by bath application of 500 µM glutamate in the predicted ischemic core region of brain slices harvested both immediately and 12 h after middle cerebral artery occlusion. The ischemic hemisphere displays a significantly lower incidence of glutamate-induced tissue swelling that exceeds a 5% threshold Mann-Whitney U-test, (+P < 0.001) than the homotypic contralateral region. Values are means; n = no. of slices. Isch hem, ischemic hemisphere; Non-isch hem, nonischemic hemisphere.
When OGD aCSF was bath-applied to slices immediately after MCAo, SD was evoked in every slice in both the ischemic (31 of 31 slices) and the nonischemic hemispheres (31 of 31 slices) (Fig. 4). SD initiated within neocortex supplied by the MCA (i.e., lateral parietal neocortex) and anterior cerebral artery (i.e., medial motor and cingulate neocortex). In contrast, slices harvested 12 h post-MCAo generated significantly fewer SD events in the ischemic hemisphere (42%, 5 of 12 slices) compared with the nonischemic hemisphere (100%, 12 of 12 slices) (Mann Whitney U-test, P < 0.01). SD usually initiated outside of MCA territory (i.e., medial motor cortex and cingulate cortex).
Fig. 4.
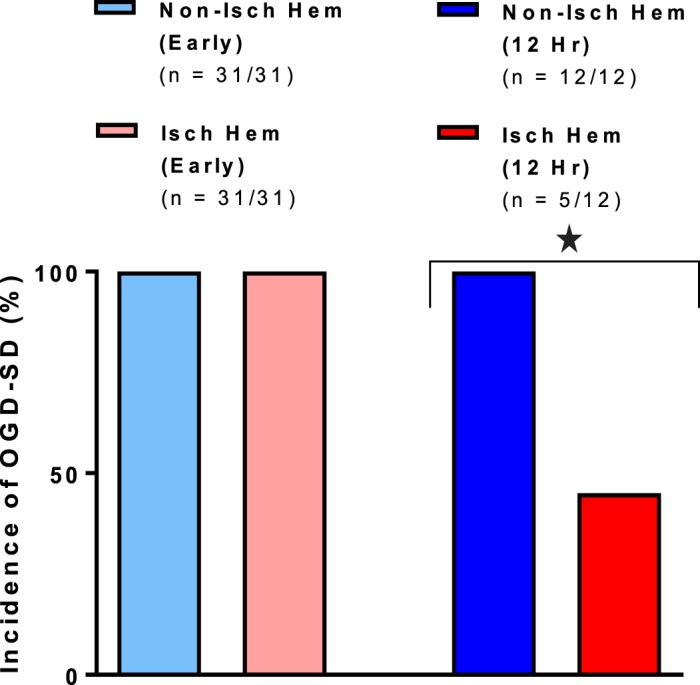
Spreading depolarization (SD) incidence in the ischemic hemisphere is reduced by 42% at 12 h, but not immediately, post-middle cerebral artery occlusion (*P < 0.01, Mann-Whitney U-test). Values are means; n = no. of slices. Isch hem, ischemic hemisphere; Non-isch hem, nonischemic hemisphere.
Electrophysiological Recording
Whole cell recording.
We patch-clamped pyramidal neurons of neocortical layer II/III in the ischemic or nonischemic hemispheres. As viewed under a ×40 magnification objective, there were abundant pyramidal neurons with a normal “healthy”-looking morphology under contrast optics when viewed immediately poststroke in the approximate region of future infarct. Within this region, action potential threshold (Fig. 5A), as well as amplitude and width (Fig. 5B), were compared between neocortical pyramidal neurons in ischemic and nonischemic hemispheres. In addition, the fAHP that followed a depolarizing pulse evoking a spike train (Fig. 5A) was measured. None of these properties were significantly different between the two hemispheres. In addition, resting membrane potential, whole cell input resistance, and the time constant of membrane potential decay following an injected hyperpolarizing pulse were not significantly different between neurons in the ischemic hemisphere (n = 21) and neocortical neurons in the nonischemic hemisphere (n = 16) (Table 1).
Fig. 5.

Electrophysiological properties of whole cell patched pyramidal cells were similar in the ischemic and nonischemic hemispheres. A: the input resistance, fast afterhyperpolarization, and time constant obtained from a series of hyperpolarizing and depolarizing current steps were similar in the ischemic and nonischemic hemispheres. B: the action potential threshold, amplitude, and width were similar in the ischemic and nonischemic hemispheres. C: the evoked synaptic responses were also similar in the ischemic and nonischemic hemispheres. See Table 1. MCAo, middle cerebral artery occlusion.
Table 1.
Baseline electrophysiological properties of whole cell patched pyramidal cells in the ischemic and nonischemic hemispheres of brain slices harvested immediately after MCAo
| n | RMP, mV | AP Threshold, mV | AP Amplitude, mV | AP Width, ms | fAHP, mV | Rin, MΩ | Tau, ms | |
|---|---|---|---|---|---|---|---|---|
| Nonischemic hemisphere | 16 | −72 ± 5 | −37 ± 3 | 83 ± 6 | 0.7 ± 0.1 | 9 ± 3 | 122 ± 38 | 16 ± 4 |
| Ischemic hemisphere | 21 | −71 ± 7 | −34 ± 6 | 79 ± 14 | 0.7 ± 0.2 | 10 ± 5 | 113 ± 31 | 16 ± 6 |
| P value | 0.60 | 0.11 | 0.16 | 0.64 | 0.54 | 0.43 | 0.73 |
Values are means ± SE (n = no. of slices) for resting membrane potential (RMP), action potential (AP) threshold, amplitude, and width, post-train fast afterhyperpolarization (fAHP), whole cell input resistance (Rin), and whole cell time constant (tau). None of the parameters differed significantly between nonischemic and ischemic hemispheres. Values were compared using a paired Student’s t-test.
We next searched through the ischemic core of live brain slices harvested 12 h after MCAo to locate any viable neurons for electrophysiological recordings in whole cell configuration under current clamp. Compared with nonischemic neocortex in either hemisphere, only rare and scattered, normal-appearing pyramidal neurons (triangle-shaped, obvious apical dendrite) could be visualized with contrast optics within what was obviously infarcted neocortex. It was impossible to maintain patch recordings from neighboring neurons that were not triangular with a distinct apical dendrite. In five mice, it was possible (with some difficulty) to patch onto five pyramidal neurons of normal appearance but with necrotic-appearing neighbors. Generally, the single-cell properties of these neurons all appeared surprisingly normal (Fig. 6A). The average resting membrane potential of the five neurons in the ischemic hemisphere was slightly but significantly depolarized in comparison to the six cells in the nonischemic hemisphere (−65 ± 8 vs. −72 ± 3 mV; P > 0.05). Otherwise, the mean values for the post-train fAHP, whole cell input resistance, and whole cell time constant as well as action potential threshold, amplitude, and width (Fig. 6B) were not significantly different between ischemic and nonischemic hemispheres (Table 2). However, notably, the pyramidal neurons in either side of the 12-h post-MCAo mice did not support repetitive discharge driven by steady depolarizing current injection (Fig. 6A), unlike immediately post-MCAo (Fig. 5A).
Fig. 6.

Recordings from scattered pyramidal surviving neurons in the ischemic core 12 h post-middle cerebral artery occlusion (MCAo). Neurons displayed intact electrophysiological properties but weak or absent synaptic connectivity. Whole cell patched pyramidal cells were similar in the ischemic and nonischemic hemispheres. A: the input resistance, fast afterhyperpolarization, and time constant obtained from a hyperpolarizing and depolarizing series of current steps were similar in the ischemic and nonischemic hemispheres. B: the action potential threshold, amplitude, and width were similar in the ischemic and nonischemic hemisphere. C: the evoked synaptic response was attenuated in the nonischemic hemisphere and absent in infarcted regions within the ischemic hemisphere. See Table 2.
Table 2.
Baseline electrophysiological properties of pyramidal neurons 12 h post-MCAo in the nonischemic hemisphere and of 5 surviving neurons within the core of the ischemic hemisphere
| n | RMP mV | AP Threshold, mV | AP Amplitude, mV | AP Width, ms | fAHP, mV | Rin, MΩ | Tau, ms | |
|---|---|---|---|---|---|---|---|---|
| Nonischemic hemisphere | 6 | −72 ± 3 | −34 ± 5 | 77 ± 4 | 0.7 ± 0.2 | 14 ± 5 | 108 ± 13 | 16 ± 3 |
| Ischemic hemisphere | 5 | −65 ± 8 | −34 ± 6 | 69 ± 11 | 0.6 ± 0.2 | 11 ± 5 | 109 ± 41 | 27 ± 15 |
| P value | 0.05 | 0.44 | 0.16 | 0.62 | 0.11 | 0.95 | 0.18 |
Values are means ± SE (n = no. of slices) for resting membrane potential (RMP), action potential (AP) threshold, amplitude, and width, post-train fast afterhyperpolarization (fAHP), whole cell input resistance (Rin), and whole cell time constant (tau). None of the parameters differed significantly between nonischemic and ischemic hemispheres. Values compared using a paired Student’s t-test.
Synaptic function.
We tested whether the reduction in SD events recorded in the ischemic hemisphere at the early and late time points could involve a reduction in synaptic function. Evoked field EPSPs were measured at multiple recording sites in cortical layers II/III nearby and within neocortical regions supplied by the MCA. The viability of synaptic communication in the nonischemic vs. ischemic hemispheres of brain slices harvested after MCAo was assessed. Stimulation of neocortical layer VI resulted in field EPSPs evoked in cortical layers II/III in the ischemic (0.4 ± 0.1 mV, n = 11) and nonischemic hemispheres (0.7 ± 0.1 mV, n = 6) that were not significantly different (P = 0.07), as shown in Fig. 5C. This provided support for reasonably intact synaptic communication in the ischemic hemisphere of brain slices harvested immediately after MCAo. Slices harvested immediately post-MCAo displayed intact evoked synaptic responses at every stimulation site.
In contrast, the ischemic hemisphere 12 h post-MCAo had a significantly decreased number of responses (33%, 3 of 9 recordings) compared with the nonischemic hemisphere (100%, 7 of 7 recordings), as shown in Figs. 6C and 7 (Mann-Whitney U-test, P < 0.05). Moreover, synaptic responses on the nonischemic side were diminished in amplitude compared with slices from immediate post-MCAo mice (Fig. 5C).
Fig. 7.
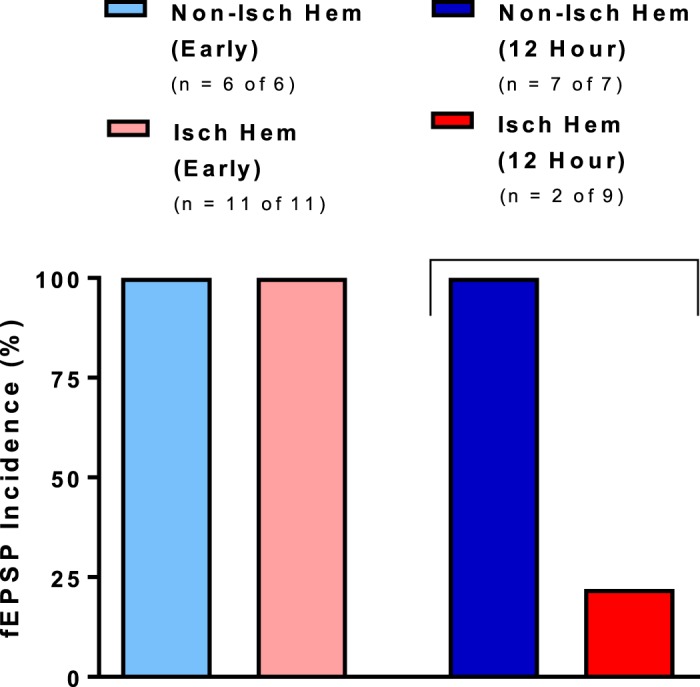
Evoked synaptic response incidence was significantly lower in the ischemic hemisphere of brain slices harvested 12 h, but not immediately (early), after 30-min middle cerebral artery occlusion (MCAo). Ischemic hemisphere of brain slices harvested 12 h, but not immediately, after 30-min MCAo had a significantly decreased incidence of evoked synaptic responses (23%) than the nonischemic hemisphere (100%) (Mann-Whitney U-test, P < 0.05). Values are means; n = no. of slices. Isch hem, ischemic hemisphere; Non-isch hem, nonischemic hemisphere.
Ischemic Core Morphology Fixed Immediately Post-MCAo
The Golgi-Cox technique randomly stains perhaps 1% of the neurons present, but all parts of each single neuron are revealed. Three mice underwent left MCAo for 30 min and then were immediately fixed and processed. To search for morphological evidence of neuronal deterioration immediately following MCAo, three brains were sectioned following Golgi-Cox treatment. The region of the future infarct was indistinguishable from surrounding gray matter (Fig. 8A). Medium spiny neurons in the striatal region of the predicted infarct appeared intact, possessing multiple dendrites (Fig. 8B) with abundant spines. They represent 95% of the neuronal population of mouse striatum. In the overlying neocortex, pyramidal neurons were likewise not morphologically different in the ischemic vs. nonischemic hemispheres of three mouse brains harvested immediately post-MCAo. A qualitative comparison of both neocortical and striatal regions between hemispheres showed no obvious swelling, dendritic beading, or necrosis of stained neurons throughout the ischemic hemisphere (Figs. 8 and 9).
Fig. 8.

A: Golgi-Cox-stained coronal section of a brain that underwent a 30-min left middle cerebral artery occlusion (MCAo) immediately followed by euthanization and fixation. Staining reveals no apparent irregularities to neuronal structure as examined in the predicted MCAo territory (dotted line). The future lesion involves the left striatum (Str) and variable parts of the overlying neocortex (NC), but not hippocampus (Hipp), thalamus (Thal), or hypothalamus (Hyp). B: higher magnification of medium spiny neurons in the ischemic (left) and nonischemic (right) striatum. The neurons on each side are qualitatively similar with no structural irregularities.
Fig. 9.
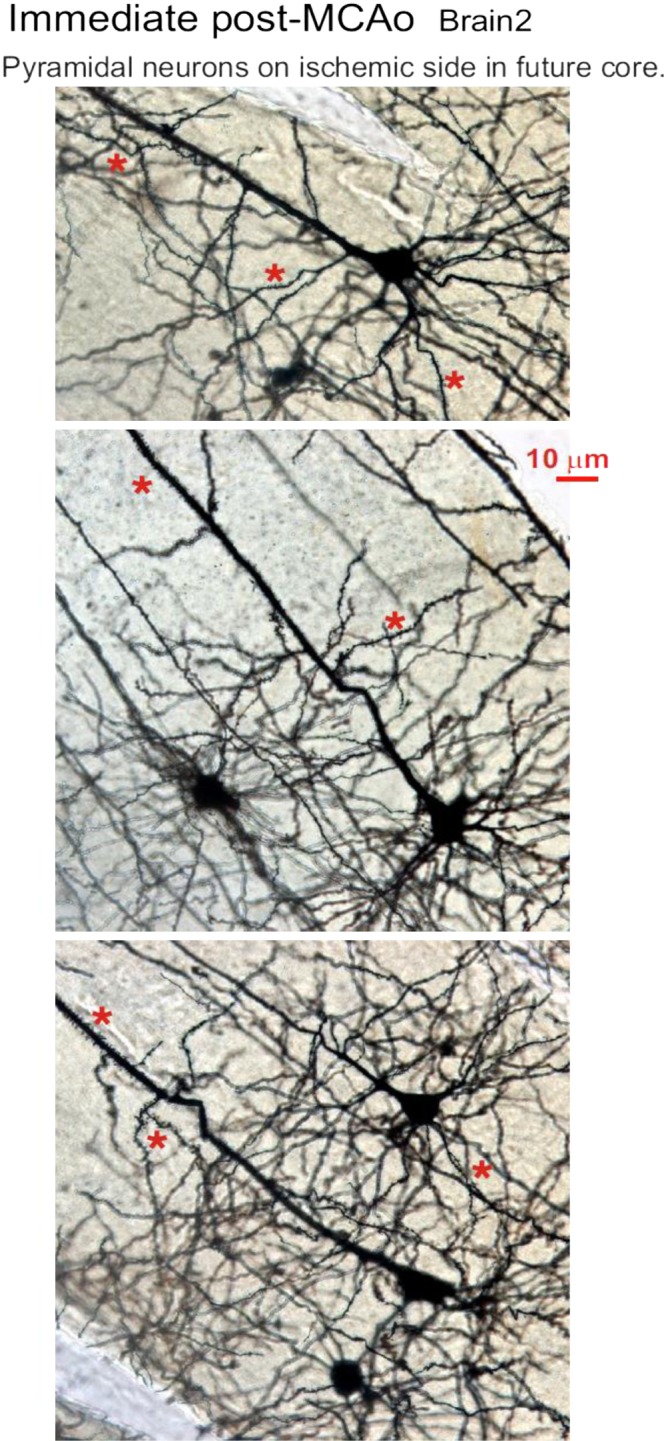
Neuronal morphology immediately poststroke. The neocortex of the ischemic hemisphere within the prospective middle cerebral artery occlusion (MCAo) core (see Fig. 8) contains normal-appearing pyramidal neurons and interneurons that are qualitatively indistinguishable from cells in the neocortex of the nonischemic side (not shown). None of the neuronal somata are swollen or dysmorphic, nor are their dendrites beaded, which would indicate injury. Instead, the pyramidal neurons have an extensive array of dendritic spines (*).
Ischemic Core Morphology Fixed 12 Hours Post-MCAo
Three additional mice were allowed to recover for 12 h before fixation and Golgi-Cox processing. Coronal sections of each brain in Fig. 10 are shown with outlining of the ischemic core based on the presence of dysmorphic neurons within the boundaries as observed at higher magnification. In the nonischemic hemisphere, stained pyramidal neurons in neocortex displayed extended apical dendrites with secondary and tertiary branches possessing dendritic spines (Fig. 11C, left), similar to those in Fig. 9. Various nonpyramidal neurons also stained, in particular a spherical cell type with naturally varicose dendrites (not shown). The underlying striatum also appeared normal, comprising the multipolar medium spiny neurons (Fig. 11C, right).
Fig. 10.
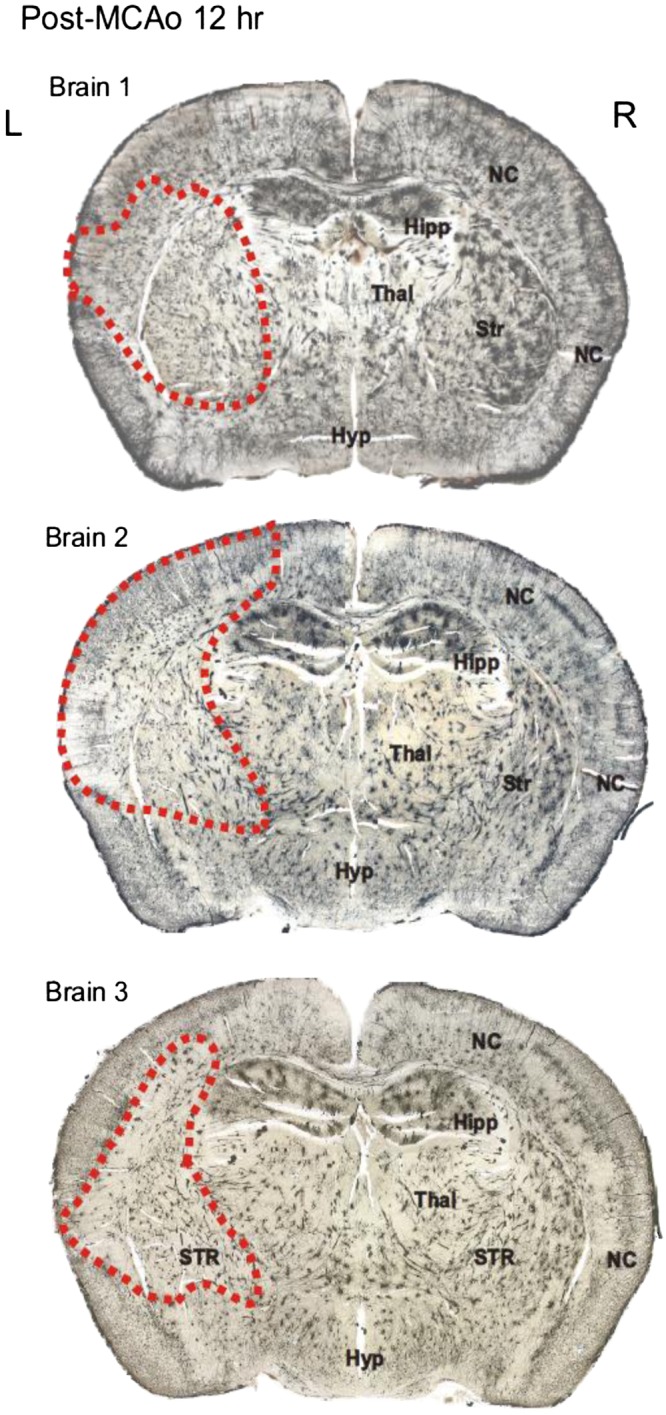
Coronal sections through the brains of 3 mice that each underwent a left hemisphere middle cerebral artery occlusion (MCAo) lasting 30 min, followed by a 12-h recovery period. Golgi-Cox staining revealed an obvious reduction in staining within the MCAo core (dashed lines). The lesion involved the left striatum (Str) and variable parts of the overlying neocortex (NC), but not hippocampus (Hipp), thalamus (Thal), or hypothalamus (Hyp). L, left; R, right.
Fig. 11.

A: the nonischemic hemisphere contains neurons with extensive dendritic arbors, in both neocortex (NC) and striatum (Str). B: in contrast, the ischemic hemisphere shows punctate cell bodies and reduced dendritic branching in the middle cerebral artery occlusion (MCAo) core in both NC and Str. C: at higher magnification, pyramidal cells in the NC are abundant. The spider-like medium spiny neurons in Str also appear healthy, with short but numerous dendrites decorated with spines (white arrows; inset right). D: in contrast, the ischemic hemisphere is populated with dysmorphic, necrotic cell bodies with stunted dendrites in both NC and Str. Some striatal neurons have lost most of their dendrites, whereas the remainder have stunted, beaded dendrites devoid of spines (inset right). MCAo, middle cerebral artery occlusion.
In contrast, 12 h post-MCAo, most neurons in the core region (Fig. 10, dashed lines) had lost their highly branched neurons. Every one of the striatum’s principle cells were shrunken with severely stunted dendrites completely devoid of spines (Fig. 11D, left). Neocortical neuron damage was more variable. Some core regions contained uniformly swollen cell bodies (Figs. 12A and 13A) that generally retained their pyramidal cell shape. The dendrites had lost their spines and taken on a beaded appearance, typical of ischemic neurons in the early phase of deterioration. More commonly, the remaining neocortical neurons appeared shrunken/necrotic with retracted and limited dendrites that lacked spines (Figs. 11D and 13B). This contrasted with gray matter immediately outside the MCAo infarct region that appeared normal. It was difficult to distinguish an intermediate area of injury that could represent the ischemic penumbra at 12 h following stroke. The dendritic beading connected to either swollen or dysmorphic neurons was dramatic and extensive in all three animals.
Fig. 12.

Isolated but intact pyramidal neurons surrounded by dysmorphic neurons within the infarct core 12 h post-middle cerebral artery occlusion (MCAo). In this region most cells are swollen pyramidal neurons (red arrows). Interspersed among these damaged cells are normal-appearing pyramidal cell bodies (yellow arrows) with extensive dendritic arbors displaying numerous dendritic spines.
Fig. 13.
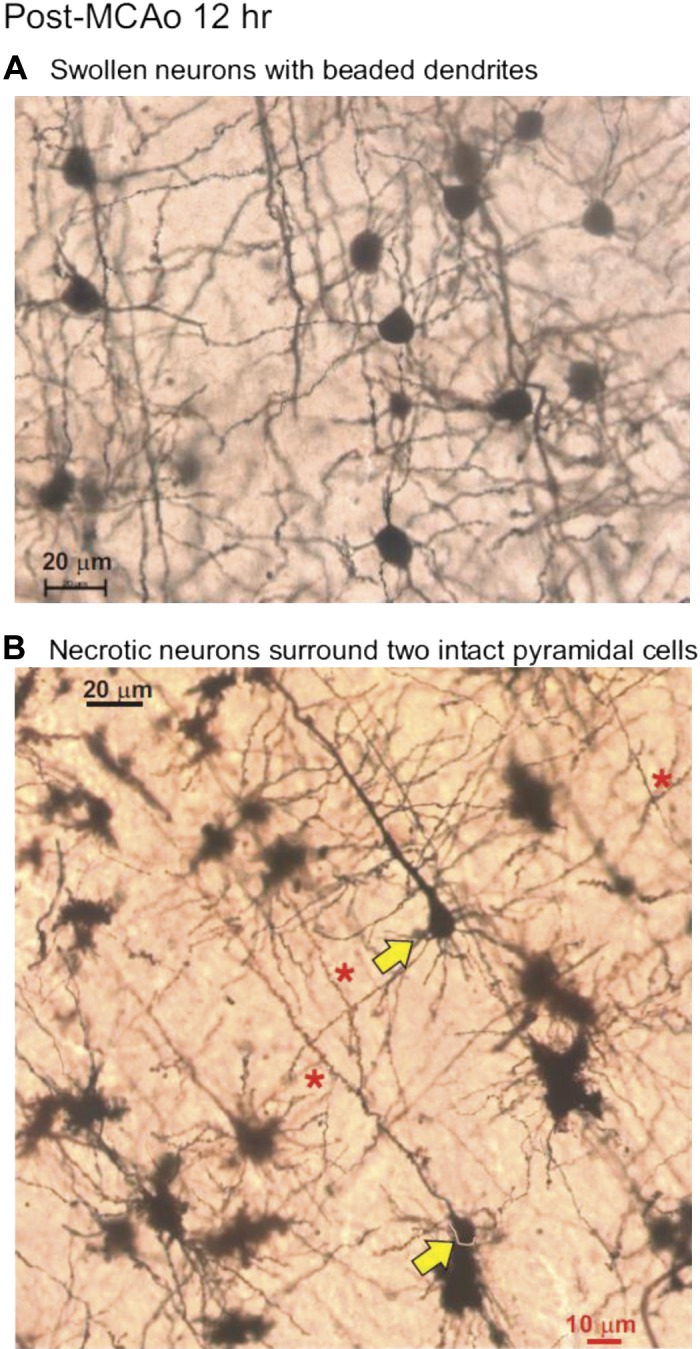
In the neocortical territory of the ischemic core 12 h post-middle cerebral artery occlusion (MCAo), neurons may be swollen with dendrites that are beaded (A) or necrotic with stunted and beaded dendrites (B). Also observed are rare intact pyramidal neurons (yellow arrows) displaying numerous dendritic spines (*), unlike their necrotic neighbors, which are constricted with beaded and retracted dendrites.
As predicted from the “normal” electrophysiological properties recorded in five pyramidal cells (Table 2) and their representative electrophysiological traces (Fig. 6, A and B), it was possible to identify some neurons that had survived 12 h post-MCAo with intact axon, dendrites, and pyramidal cell body shape. They were observed in the MCAo core containing swollen pyramidal neurons (Fig. 12A) or necrotic pyramidal neurons (Fig. 13B). These sparsely distributed neurons displayed an extensive dendritic tree with branches where spines, rather than beads, predominated. Some minor dendritic beading with loss of spines was often apparent (not shown). These cells were clearly within the core, because they were surrounded by morphologically devastated neighbors. Pyramidal neurons in this same region, but in the contralateral nonischemic hemisphere, displayed normal dendritic structure (not shown).
DISCUSSION
Postischemic SD and Leão’s Initial Observations of SD
Leão initially observed a slowly propagating wave of depressed EEG activity following electrical stimulation of rabbit and cat neocortex (Leão 1944). This so-called cortical spreading depression (CSD) was later determined to be reversible and repeatable, and could be evoked by focally elevating K+ levels or with brief hypoxia (Somjen 2001). Leão then interrupted the cerebral circulation of rabbits and noted the similarity of electrically induced CSD to a “change of the same nature as one resulting from prolonged interruption of the circulation occurs in the cerebral cortex” (Leão 1947). In live brain slices, the depolarizing wave was subsequently determined to result from inhibition of the Na+-K+ pump, either by OGD or by ouabain binding (Somjen 2001). These events are now collectively referred to as spreading depolarization (SD) (Dreier 2011; Hartings et al. 2017).
In intact rats, ATP, lactate, and creatine phosphate levels vary along with CBF after ischemia (Lauritzen et al. 1990; Obrenovitch et al. 1988). Postischemic SD events also induce a stepwise decrease in CBF (Shin et al. 2006; von Bornstädt et al. 2015). CBF fluctuations are also influenced by [K+]ext., nitric oxide (Dreier et al. 1998; Windmüller et al. 2005), hypoxia, and hypotension (Sukhotinsky et al. 2008) and can alter the properties of SD (Sukhotinsky et al. 2010). In our slice experiments, LT changes at SD initiation and propagation are easily imaged as changes in LT evoked by OGD (Andrew et al. 2007; Jarvis et al. 2001) or elevated K+ (Anderson and Andrew 2002). We could observe SD in post-MCAo slices in the absence of postischemic CBF fluctuations (Shin et al. 2006; von Bornstädt et al. 2015). We found that SD was induced by elevated [K+]ext, by OGD, or by ouabain. However, elevation of [glu]ext in the slice bath only evoked cell swelling in brain slices but not SD, confirming previous observations (Obeidat and Andrew 1998; Polischuk and Andrew 1996).
Effects of Elevated [K+]ext, Elevated [glu]ext, or OGD on Postischemic Slices
During ischemia, [K+]ext is measured at ~10–12 mM leading up to SD initiation (Nedergaard and Hansen 1993) and similarly in brain slices (Tanaka et al. 1997). However, [K+]ext itself does not evoke CSD at <15 mM in slices (Tang et al. 2014). Thus 9.6 mM [K+]ext should promote SD. When we applied aCSF with 9.6 mM [K+]ext to brain slices harvested immediately after MCAo, SD was more difficult to induce in the ischemic vs. nonischemic hemisphere. This contrasts with KCl application to ischemic cortex in vivo, which increases ATP depletion and diffusion-imaged lesion volume (Busch et al. 1996; Dijkhuizen et al. 1999) as well as the SD incidence (Busch et al. 1996; Dijkhuizen et al. 1999; Takano et al. 1996). However, the 25% incidence of K+-evoked SD events in our slices harvested immediately after MCAo was similar to observations during in vivo ischemia (Takano et al. 1996). Although SD incidence is high initially after ischemia in vivo, spontaneous and KCl-induced SD incidence peaks in the initial 20 min after MCAo and then declines sharply (Back et al. 1996; Dijkhuizen et al. 1999; Koroleva and Bures 1996). A quiescent phase of minimal spontaneous SD ensues 4–6 h following reperfusion after ischemia (Hartings et al. 2003). Therefore, this decreased postischemic SD incidence is congruent with our findings of reduced SD incidence induced by elevated [K+]ext in the ischemic hemisphere of brain slices immediately following 30-min MCAo.
Slices of the ischemic hemisphere 12 h post-MCAo also displayed decreased tissue swelling during 9.6 mM [K+]ext superfusion. This is not surprising, given the obvious damage in the ischemic hemisphere in Golgi-stained brain sections at 12 h post-MCAo. Widespread neuronal injury and/or astrocytes already swollen from ischemia could account for this reduction in the swelling signal. SD was evident in brain regions outside the infarcted neocortex supplied by the anterior cerebral artery. The SD events originated within the contralateral nonischemic hemisphere and seemed to propagate across the longitudinal fissure and into the ischemic hemisphere. This does not occur in the intact brain during ischemia in vivo (Nedergaard and Hansen 1993) and is likely an artifact of the slice preparation where released K+ can promote SD initiation in nearby gray matter. Nevertheless, it was apparent that an ischemic “core” that displayed decreased propensity to cell swelling and to SD was juxtaposed with a medial penumbra-like region that could sustain following 12 h of reperfusion post-MCAo.
In addition to [K+]ext release during ischemia (Branston et al. 1977; Nedergaard and Hansen 1993; Strong et al. 1983), [glu]ext also increases following stroke for 1–2 h (Benveniste et al. 1984; Hillered et al. 1989; Phillis et al. 1991; Sciotti et al. 1992) from a baseline concentration of ~20 to ~200 µM during anoxia-induced SD and to ~80 µM during ischemia-induced SD (Iijima et al. 1998; Satoh et al. 1999). Elevated [glu]ext also promotes the propagating wave front of SD (Zhang et al. 2015; Zhou et al. 2013). However, directly applying glutamate to normal brain slices produces only a graded, general swelling of gray matter (Obeidat and Andrew 1998; Polischuk and Andrew 1996). This swelling does not resemble the characteristic propagating SD front of cell swelling that traverses gray matter on exposure to ischemia, OGD, elevated [K+]ext, or ouabain (Anderson and Andrew 2002; Anderson et al. 2005; Dietz et al. 2008; Dohmen et al. 2008; Tanaka et al. 1997). When we bath-applied 500 µM [glu]ext to brain slices harvested immediately and 12 h after MCAo, we observed an LT increase >5% only in the nonischemic hemisphere. This 5% ΔLT response is comparable to superfusion with hyposmotic (−30 mosmol/kgH2O) aCSF (Andrew and MacVicar 1994). It takes a 5 mM concentration of glutamate to elicit a similar regional increased ΔLT response and tissue swelling to only 100 µM of N-methyl-d-aspartate (Andrew et al. 1996; Obeidat and Andrew 1998), a specific agonist at ionotropic glutamate receptors. This is because uptake mechanisms constantly remove the extracellular glutamate (Obeidat and Andrew, 1998). This glutamate uptake may be enhanced in brains harvested immediately after a 30-min MCAo, similar to in vivo observations of increased glutamate transporter expression and decreased neuronal death after ischemic postconditioning (Zhang et al. 2010). In contrast, brain slices harvested after 12 h post-MCAo display minimal glutamate-evoked swelling because neuronal death and preexisting swollen neurons (both evident in Golgi-stained tissue) preclude further depolarization and swelling.
Unlike elevated [glu]ext and [K+]ext, which cause reversible swelling of gray matter, OGD causes irreversible damage in live slices of gray matter from the higher brain (Anderson and Andrew 2002; Anderson et al. 2005; Brisson et al. 2014; Obeidat and Andrew 1998). The decreased likelihood of OGD-induced SD in the ischemic hemisphere of brain slices harvested 12 h after MCAo likely results from the sparse number of remaining intact neurons (evident with Golgi-Cox staining) that are unable to drive SD.
Postischemic Microscopy and Electrophysiology
In brain sections harvested immediately after MCAo, there was no morphological evidence of infarction in Golgi-stained tissue. These findings support our electrophysiological recordings indicating that single neocortical pyramidal neurons were not significantly altered in their intrinsic or synaptic properties immediately post-MCAo and were not different between the two hemispheres. Supporting these results, the electrophysiological properties and synaptic communication in pyramidal CA1 neurons were not significantly different from those in preischemic controls up to 5 h after reperfusion following 19 min of global ischemia (Gao et al. 1998, 1999). The threshold, amplitude, and width of action potentials as well as the decay of the time constant following an action potential train in postischemic brain slices were also similar to those of regular-spiking neocortical pyramidal neurons in control animals from other studies (McCormick et al. 1985; Silva et al. 1991). The properties of 30-min recovery pyramidal neurons (which went through the identical stroke as the 12-h recovery neurons) were essentially identical to nonstroked slices whose properties we and others have previously described (Brisson and Andrew 2012; White et al. 2012). This argues that the inability of the 12-h neurons to fire a train of impulses is not related to surgical procedure or anesthesia.
In our brain slices harvested 12 h after MCAo, evoked synaptic responses were lost within and near the ischemic core. The ischemic hemisphere in those brain slices displayed a decreased incidence of OGD-induced SD, K+-induced SD, and glutamate-evoked swelling compared with the nonischemic hemisphere. There was also dramatic neuronal swelling or necrosis in Golgi-stained pyramidal neurons in the neocortical portion of the MCAo core.
The few remaining healthy-appearing pyramidal neurons found in the core could not be expected to override the greatly diminished synaptic responses caused by the obvious and extensive neuronal injury, yet these surviving neurons are intriguing because they are neuroprotected while their immediate neighbors are destroyed. During our study, we wondered how long these cells might survive. A recent study (Jiang et al. 2017) showed that neurons are present 7–14 days post-MCAo. Our work identifies the survivors as pyramidal cells, the projection neurons of the neocortex. Moreover, we show that these cells display extensive arborization with high numbers of dendritic spines. However, an intact spine does not necessarily mean a functioning presynaptic axon. Given the many sources of synaptic input to a single neocortical pyramidal neuron, it is unlikely that the entire arbor is still receiving synaptic input by 12 h post-MCAo. Perhaps some spines are “waiting” for alternate or revived input. It is important to note that the reduced or lost field EPSP on the stroked side only demonstrates that evoked postsynaptic activity is greatly reduced. This is not surprising given that the great majority of core neurons are morphologically devastated, but we cannot rule out the possibility that damaged presynaptic axons within the core also contribute to signal loss.
We cannot yet explain why spike train firing is impaired on the contralateral side at 12-h recovery post-MCAo. The two sides are synaptically connected. Silasi and Murphy (2014) showed that homotypic cortex is connected through corpus callosum as well as via the lateral striatum. Projecting axons through white matter are particularly sensitive to ischemia. We intend to examine this issue further in our ongoing follow-up studies of 24- and 72-h recovery times. We expect that the contralateral neurons will mostly recover their train-firing ability.
Conclusions
Although many studies have examined the properties of the neocortex several days following stroke, there is little information regarding the state of the neurons in the ischemic core at 12 h poststroke. In this study we examined mouse neocortex immediately and 12 h after a 30-min MCAo, comparing nonischemic and ischemic hemispheres with regard to propensity for cell swelling and SD, as well as evoked synaptic responses and single pyramidal neuron properties. We found that neocortical gray matter is surprisingly unaltered in brain slices harvested immediately poststroke even though the propensity to induce cell swelling or SD is dramatically reduced. By 12 h, an evoked synaptic response in the core is absent and most of pyramidal and striatal neurons are morphologically devastated. Yet, surprisingly, scattered pyramidal neurons in the neocortical core retain their normal electrophysiological and neuroanatomical properties 12 h after MCAo. Apparently, these neurons continue to survive for 72 h or more, based on histological staining techniques (Jiang et al. 2017). That study noted that it was not possible to determine whether the surviving neurons were functional and able to contribute to tissue repair. In the present study, we show that by 12 h, when the majority of neurons in the core are essentially devastated, the intrinsic electrophysiological properties of the remaining pyramidal neurons are relatively intact even as synaptic connectivity fails. We are interested in discovering the fate of the pool of survivors that Jiang et al. (2017) have shown still survive 72 h poststroke. Our data provide important evidence showing that these cells appear uninjured and discharge normally (except for what is probably a temporary inability to fire spike trains). Researching the mechanism supporting their neuroprotection will inform on new strategies or pharmacological targets for neuroprotection.
GRANTS
This work was supported by Heart and Stroke Foundation of Canada Grant G-14-0006260 (to R. David Andrew), Natural Sciences and Engineering Research Council of Canada Grant RGPIN-2017-04624 (R. David Andrew), King Abdulaziz University of Saudi Arabia (to R. H. Mehder), and Canadian Institutes of Health Grant PJT 153013 (to B. M. Bennett).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.Y.J. and R.D.A. conceived and designed research; D.P., P.J.G., R.H.M., and R.D.A. performed experiments; D.P., A.Y.J., and R.D.A. analyzed data; D.P., P.J.G., B.M.B., and R.D.A. interpreted results of experiments; D.P., R.H.M., and R.D.A. prepared figures; D.P. and R.D.A. drafted manuscript; B.M.B., A.Y.J., and R.D.A. edited and revised manuscript; D.P., P.J.G., R.H.M., B.M.B., A.Y.J., and R.D.A. approved final version of manuscript.
REFERENCES
- Anderson TR, Andrew RD. Spreading depression: imaging and blockade in the rat neocortical brain slice. J Neurophysiol 88: 2713–2725, 2002. doi: 10.1152/jn.00321.2002. [DOI] [PubMed] [Google Scholar]
- Anderson TR, Jarvis CR, Biedermann AJ, Molnar C, Andrew RD. Blocking the anoxic depolarization protects without functional compromise following simulated stroke in cortical brain slices. J Neurophysiol 93: 963–979, 2005. doi: 10.1152/jn.00654.2004. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Adams JR, Polischuk TM. Imaging NMDA- and kainate-induced intrinsic optical signals from the hippocampal slice. J Neurophysiol 76: 2707–2717, 1996. doi: 10.1152/jn.1996.76.4.2707. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA. Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex 17: 787–802, 2007. doi: 10.1093/cercor/bhk032. [DOI] [PubMed] [Google Scholar]
- Andrew RD, MacVicar BA. Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience 62: 371–383, 1994. doi: 10.1016/0306-4522(94)90372-7. [DOI] [PubMed] [Google Scholar]
- Back T, Ginsberg MD, Dietrich WD, Watson BD. Induction of spreading depression in the ischemic hemisphere following experimental middle cerebral artery occlusion: effect on infarct morphology. J Cereb Blood Flow Metab 16: 202–213, 1996. doi: 10.1097/00004647-199603000-00004. [DOI] [PubMed] [Google Scholar]
- Barber PA, Hoyte L, Colbourne F, Buchan AM. Temperature-regulated model of focal ischemia in the mouse: a study with histopathological and behavioral outcomes. Stroke 35: 1720–1725, 2004. doi: 10.1161/01.STR.0000129653.22241.d7. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem 43: 1369–1374, 1984. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- Branston NM, Strong AJ, Symon L. Extracellular potassium activity, evoked potential and tissue blood flow. Relationships during progressive ischaemia in baboon cerebral cortex. J Neurol Sci 32: 305–321, 1977. doi: 10.1016/0022-510X(77)90014-4. [DOI] [PubMed] [Google Scholar]
- Brisson CD, Andrew RD. A neuronal population in hypothalamus that dramatically resists acute ischemic injury compared to neocortex. J Neurophysiol 108: 419–430, 2012. doi: 10.1152/jn.00090.2012. [DOI] [PubMed] [Google Scholar]
- Brisson CD, Hsieh YT, Kim D, Jin AY, Andrew RD. Brainstem neurons survive the identical ischemic stress that kills higher neurons: insight to the persistent vegetative state. PLoS One 9: e96585, 2014. doi: 10.1371/journal.pone.0096585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch E, Gyngell ML, Eis M, Hoehn-Berlage M, Hossmann KA. Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J Cereb Blood Flow Metab 16: 1090–1099, 1996. doi: 10.1097/00004647-199611000-00002. [DOI] [PubMed] [Google Scholar]
- Dietz RM, Weiss JH, Shuttleworth CW. Zn2+ influx is critical for some forms of spreading depression in brain slices. J Neurosci 28: 8014–8024, 2008. doi: 10.1523/JNEUROSCI.0765-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen RM, Beekwilder JP, van der Worp HB, Berkelbach van der Sprenkel JW, Tulleken KA, Nicolay K. Correlation between tissue depolarizations and damage in focal ischemic rat brain. Brain Res 840: 194–205, 1999. doi: 10.1016/S0006-8993(99)01769-2. [DOI] [PubMed] [Google Scholar]
- Dohmen C, Sakowitz OW, Fabricius M, Bosche B, Reithmeier T, Ernestus RI, Brinker G, Dreier JP, Woitzik J, Strong AJ, Graf R; Co-Operative Study of Brain Injury Depolarisations (COSBID) . Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol 63: 720–728, 2008. doi: 10.1002/ana.21390. [DOI] [PubMed] [Google Scholar]
- Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17: 439–447, 2011. doi: 10.1038/nm.2333. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Körner K, Ebert N, Görner A, Rubin I, Back T, Lindauer U, Wolf T, Villringer A, Einhäupl KM, Lauritzen M, Dirnagl U. Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab 18: 978–990, 1998. doi: 10.1097/00004647-199809000-00007. [DOI] [PubMed] [Google Scholar]
- Gao TM, Pulsinelli WA, Xu ZC. Prolonged enhancement and depression of synaptic transmission in CA1 pyramidal neurons induced by transient forebrain ischemia in vivo. Neuroscience 87: 371–383, 1998. doi: 10.1016/S0306-4522(98)00150-X. [DOI] [PubMed] [Google Scholar]
- Gao TM, Pulsinelli WA, Xu ZC. Changes in membrane properties of CA1 pyramidal neurons after transient forebrain ischemia in vivo. Neuroscience 90: 771–780, 1999. doi: 10.1016/S0306-4522(98)00493-X. [DOI] [PubMed] [Google Scholar]
- Hartings JA, Rolli ML, Lu XC, Tortella FC. Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: relation to infarct growth and neuroprotection. J Neurosci 23: 11602–11610, 2003. doi: 10.1523/JNEUROSCI.23-37-11602.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA, Shuttleworth CW, Kirov SA, Ayata C, Hinzman JM, Foreman B, Andrew RD, Boutelle MG, Brennan KC, Carlson AP, Dahlem MA, Drenckhahn C, Dohmen C, Fabricius M, Farkas E, Feuerstein D, Graf R, Helbok R, Lauritzen M, Major S, Oliveira-Ferreira AI, Richter F, Rosenthal ES, Sakowitz OW, Sánchez-Porras R, Santos E, Schöll M, Strong AJ, Urbach A, Westover MB, Winkler MK, Witte OW, Woitzik J, Dreier JP. The continuum of spreading depolarizations in acute cortical lesion development: examining Leão’s legacy. J Cereb Blood Flow Metab 37: 1571–1594, 2017. doi: 10.1177/0271678X16654495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillered L, Hallström A, Segersvärd S, Persson L, Ungerstedt U. Dynamics of extracellular metabolites in the striatum after middle cerebral artery occlusion in the rat monitored by intracerebral microdialysis. J Cereb Blood Flow Metab 9: 607–616, 1989. doi: 10.1038/jcbfm.1989.87. [DOI] [PubMed] [Google Scholar]
- Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol 36: 557–565, 1994. doi: 10.1002/ana.410360404. [DOI] [PubMed] [Google Scholar]
- Iijima T, Shimase C, Iwao Y, Sankawa H. Relationships between glutamate release, blood flow and spreading depression: real-time monitoring using an electroenzymatic dialysis electrode. Neurosci Res 32: 201–207, 1998. doi: 10.1016/S0168-0102(98)00090-X. [DOI] [PubMed] [Google Scholar]
- Jarvis CR, Anderson TR, Andrew RD. Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cereb Cortex 11: 249–259, 2001. doi: 10.1093/cercor/11.3.249. [DOI] [PubMed] [Google Scholar]
- Jiang MQ, Zhao YY, Cao W, Wei ZZ, Gu X, Wei L, Yu SP. Long-term survival and regeneration of neuronal and vasculature cells inside the core region after ischemic stroke in adult mice. Brain Pathol 27: 480–498, 2017. doi: 10.1111/bpa.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi I, Andrew RD. Imaging anoxic depolarization during ischemia-like conditions in the mouse hemi-brain slice. J Neurophysiol 85: 414–424, 2001. doi: 10.1152/jn.2001.85.1.414. [DOI] [PubMed] [Google Scholar]
- Koroleva VI, Bures J. The use of spreading depression waves for acute and long-term monitoring of the penumbra zone of focal ischemic damage in rats. Proc Natl Acad Sci USA 93: 3710–3714, 1996. doi: 10.1073/pnas.93.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ, Kronborg D, Wieloch T. Cortical spreading depression is associated with arachidonic acid accumulation and preservation of energy charge. J Cereb Blood Flow Metab 10: 115–122, 1990. doi: 10.1038/jcbfm.1990.14. [DOI] [PubMed] [Google Scholar]
- Leão AA. Spreading depression of activity in the cerebral cortex. J Neurophysiol 7: 359–390, 1944. doi: 10.1152/jn.1944.7.6.359. [DOI] [PubMed] [Google Scholar]
- Leão AA. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol 10: 409–414, 1947. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Connors BW, Lighthall JW, Prince DA. Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J Neurophysiol 54: 782–806, 1985. doi: 10.1152/jn.1985.54.4.782. [DOI] [PubMed] [Google Scholar]
- Mies G, Kohno K, Hossmann KA. Prevention of periinfarct direct current shifts with glutamate antagonist NBQX following occlusion of the middle cerebral artery in the rat. J Cereb Blood Flow Metab 14: 802–807, 1994. doi: 10.1038/jcbfm.1994.100. [DOI] [PubMed] [Google Scholar]
- Morimoto T, Globus MY, Busto R, Martinez E, Ginsberg MD. Simultaneous measurement of salicylate hydroxylation and glutamate release in the penumbral cortex following transient middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab 16: 92–99, 1996. doi: 10.1097/00004647-199601000-00011. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Li P, Betts K, Liu R. Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci 28: 1756–1772, 2008. doi: 10.1523/JNEUROSCI.5128-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Hansen AJ. Characterization of cortical depolarizations evoked in focal cerebral ischemia. J Cereb Blood Flow Metab 13: 568–574, 1993. doi: 10.1038/jcbfm.1993.74. [DOI] [PubMed] [Google Scholar]
- Obeidat AS, Andrew RD. Spreading depression determines acute cellular damage in the hippocampal slice during oxygen/glucose deprivation. Eur J Neurosci 10: 3451–3461, 1998. doi: 10.1046/j.1460-9568.1998.00358.x. [DOI] [PubMed] [Google Scholar]
- Obeidat AS, Jarvis CR, Andrew RD. Glutamate does not mediate acute neuronal damage after spreading depression induced by O2/glucose deprivation in the hippocampal slice. J Cereb Blood Flow Metab 20: 412–422, 2000. doi: 10.1097/00004647-200002000-00024. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP, Garofalo O, Harris RJ, Bordi L, Ono M, Momma F, Bachelard HS, Symon L. Brain tissue concentrations of ATP, phosphocreatine, lactate, and tissue pH in relation to reduced cerebral blood flow following experimental acute middle cerebral artery occlusion. J Cereb Blood Flow Metab 8: 866–874, 1988. doi: 10.1038/jcbfm.1988.144. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Walter GA, Simpson RE. Brain adenosine and transmitter amino acid release from the ischemic rat cerebral cortex: effects of the adenosine deaminase inhibitor deoxycoformycin. J Neurochem 56: 644–650, 1991. doi: 10.1111/j.1471-4159.1991.tb08198.x. [DOI] [PubMed] [Google Scholar]
- Polischuk TM, Andrew RD. Real-time imaging of intrinsic optical signals during early excitotoxicity evoked by domoic acid in the rat hippocampal slice. Can J Physiol Pharmacol 74: 712–722, 1996. doi: 10.1139/y96-066. [DOI] [PubMed] [Google Scholar]
- FD NeuroTechnologies FD Rapid GolgiStain Kit: User Manual. Columbia, MD: FD NeuroTechnologies Consulting & Services, 2014, p. 1–32. [Google Scholar]
- Satoh M, Asai S, Katayama Y, Kohno T, Ishikawa K. Real-time monitoring of glutamate transmitter release with anoxic depolarization during anoxic insult in rat striatum. Brain Res 822: 142–148, 1999. doi: 10.1016/S0006-8993(99)01141-5. [DOI] [PubMed] [Google Scholar]
- Sciotti VM, Roche FM, Grabb MC, Van Wylen DG. Adenosine receptor blockade augments interstitial fluid levels of excitatory amino acids during cerebral ischemia. J Cereb Blood Flow Metab 12: 646–655, 1992. doi: 10.1038/jcbfm.1992.89. [DOI] [PubMed] [Google Scholar]
- Shin HK, Dunn AK, Jones PB, Boas DA, Moskowitz MA, Ayata C. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab 26: 1018–1030, 2006. doi: 10.1038/sj.jcbfm.9600252. [DOI] [PubMed] [Google Scholar]
- Silasi G, Murphy TH. Stroke and the connectome: how connectivity guides therapeutic intervention. Neuron 83: 1354–1368, 2014. doi: 10.1016/j.neuron.2014.08.052. [DOI] [PubMed] [Google Scholar]
- Silva LR, Gutnick MJ, Connors BW. Laminar distribution of neuronal membrane properties in neocortex of normal and reeler mouse. J Neurophysiol 66: 2034–2040, 1991. doi: 10.1152/jn.1991.66.6.2034. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81: 1065–1096, 2001. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Venables GS, Gibson G. The cortical ischaemic penumbra associated with occlusion of the middle cerebral artery in the cat: 1. Topography of changes in blood flow, potassium ion activity, and EEG. J Cereb Blood Flow Metab 3: 86–96, 1983. doi: 10.1038/jcbfm.1983.11. [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Dilekoz E, Moskowitz MA, Ayata C. Hypoxia and hypotension transform the blood flow response to cortical spreading depression from hyperemia into hypoperfusion in the rat. J Cereb Blood Flow Metab 28: 1369–1376, 2008. doi: 10.1038/jcbfm.2008.35. [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Yaseen MA, Sakadzić S, Ruvinskaya S, Sims JR, Boas DA, Moskowitz MA, Ayata C. Perfusion pressure-dependent recovery of cortical spreading depression is independent of tissue oxygenation over a wide physiologic range. J Cereb Blood Flow Metab 30: 1168–1177, 2010. doi: 10.1038/jcbfm.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano K, Latour LL, Formato JE, Carano RA, Helmer KG, Hasegawa Y, Sotak CH, Fisher M. The role of spreading depression in focal ischemia evaluated by diffusion mapping. Ann Neurol 39: 308–318, 1996. doi: 10.1002/ana.410390307. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H. Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol 78: 891–902, 1997. doi: 10.1152/jn.1997.78.2.891. [DOI] [PubMed] [Google Scholar]
- Tang YT, Mendez JM, Theriot JJ, Sawant PM, López-Valdés HE, Ju YS, Brennan KC. Minimum conditions for the induction of cortical spreading depression in brain slices. J Neurophysiol 112: 2572–2579, 2014. doi: 10.1152/jn.00205.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bornstädt D, Houben T, Seidel JL, Zheng Y, Dilekoz E, Qin T, Sandow N, Kura S, Eikermann-Haerter K, Endres M, Boas DA, Moskowitz MA, Lo EH, Dreier JP, Woitzik J, Sakadžić S, Ayata C. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron 85: 1117–1131, 2015. doi: 10.1016/j.neuron.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SH, Brisson CD, Andrew RD. Examining protection from anoxic depolarization by the drugs dibucaine and carbetapentane using whole cell recording from CA1 neurons. J Neurophysiol 107: 2083–2095, 2012. doi: 10.1152/jn.00701.2011. [DOI] [PubMed] [Google Scholar]
- Windmüller O, Lindauer U, Foddis M, Einhäupl KM, Dirnagl U, Heinemann U, Dreier JP. Ion changes in spreading ischaemia induce rat middle cerebral artery constriction in the absence of NO. Brain 128: 2042–2051, 2005. doi: 10.1093/brain/awh545. [DOI] [PubMed] [Google Scholar]
- Zhang W, Miao Y, Zhou S, Wang B, Luo Q, Qiu Y. Involvement of glutamate transporter-1 in neuroprotection against global brain ischemia-reperfusion injury induced by postconditioning in rats. Int J Mol Sci 11: 4407–4416, 2010. doi: 10.3390/ijms11114407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XL, Shuttleworth CW, Moskal JR, Stanton PK. Suppression of spreading depolarization and stabilization of dendritic spines by GLYX-13, an NMDA receptor glycine-site functional partial agonist. Exp Neurol 273: 312–321, 2015. doi: 10.1016/j.expneurol.2015.07.025. [DOI] [PubMed] [Google Scholar]
- Zhou N, Rungta RL, Malik A, Han H, Wu DC, MacVicar BA. Regenerative glutamate release by presynaptic NMDA receptors contributes to spreading depression. J Cereb Blood Flow Metab 33: 1582–1594, 2013. doi: 10.1038/jcbfm.2013.113. [DOI] [PMC free article] [PubMed] [Google Scholar]