

Abstract
The role of hyperphosphatemia in the pathogenesis of secondary hyperparathyroidism, cardiovascular disease, and progression of renal failure is widely known. Here we studied effects of dietary phosphate restriction on mortality and vascular calcification in uremic rats. Control and uremic rats were fed a high-phosphate diet and at 3 months a portion of rats of each group were killed. Serum phosphate and the calcium phosphate product increased in uremic rats, as did aortic calcium. Of the rats, 56% had positive aortic staining for calcium (von Kossa), RUNX2, and osteopontin. The remaining uremic rats were continued on diets containing high phosphate without and with sevelamer, or low phosphate, and after 3 more months they were killed. Serum phosphate was highest in uremic rats on high phosphate. Serum PTH and FGF-23 were markedly lower in rats on low phosphate. Mortality on high phosphate was 71.4%, with sevelamer reducing this to 37.5% and phosphate restriction to 5.9%. Positive aortic staining for von Kossa, RUNX2, and osteopontin was increased, but phosphate restriction inhibited this. Kidneys from low-phosphate and sevelamer-treated uremic rats had less interstitial fibrosis, glomerulosclerosis, and inflammation than those of uremic rats on high phosphate. Importantly, kidneys from rats on low phosphate showed improvement over kidneys from high-phosphate rats at 3 months. Left ventricles from rats on low phosphate had less perivascular fibrosis and smaller cardiomyocyte size compared to rats on high phosphate. Thus, intensive phosphate restriction significantly reduces mortality in uremic rats with severe vascular calcification.
Keywords: FGF-23, mortality, phosphate, PTH, renal failure, vascular calcification
Cardiovascular disease is a frequent complication of renal disease and is the leading cause of death in patients with chronic kidney disease (CKD).1 These patients have increased morbidity and mortality as a result of cardiovascular disease, which may be partly due to excess vascular calcification.2 Vascular calcification occurs much earlier in life in CKD patients.3 Hyperphosphatemia has been shown to be a predictor of cardiovascular death in these patients, as is secondary hyperparathyroidism and an elevated calcium × phosphorus (Ca × P) product.4–7 Vascular calcification has been shown to be associated with both elevated serum P and Ca × P product, as well as ingested Ca.8,9 Vascular calcification is now known to be an active process whereby osteogenic changes occur in vascular cells. In vascular calcification, there is a downregulation of vascular smooth muscle cell markers concomitant with an upregulation of bone-related markers such as osteocalcin (OC), osteopontin (OPN), cbfa1/Runx2, matrix γ-carboxyglutamic acid protein, and osteoprotegerin.10–12 Osteocalcin, found at sites of vascular calcification, is secreted solely by osteoblasts, binds to hydroxyapatite, and is the most abundant noncollagenous protein in the bone.13 Cbfa1/Runx2 is a transcription factor that is essential for osteoblastic differentiation.14,15 Elevated extracellular P levels accelerate vascular calcification and are associated with the induction of cbfa1/Runx2, as well as increases in osteocalcin, OPN, and alkaline phosphatase.16–18
Control of serum phosphate (P) has long been a goal of CKD treatment. Sevelamer carbonate, a non-Ca-based phosphate binder, is currently in use in CKD patients. Sevelamer has been shown to reduce mortality in CKD patients,19 possibly by reducing low-density lipoprotein cholesterol levels and decreasing the frequency of vascular calcifications.20–26 Sevelamer improves insulin resistance, increases levels of fetuin A, is an inhibitor of vascular calcification, and has anti-inflammatory properties, such as the lowering of serum C-reactive protein levels.24–27
Here, our primary aim was to evaluate the effect of lowering serum P (either by dietary P restriction or by the use of the phosphate binder, sevelamer carbonate) on mortality using the 5/6 nephrectomized rat model. We first created a uremic rat model with increased vascular calcification and renal damage by feeding uremic rats a high-P diet (1.4% P). After 3 months, a portion of the rats were killed. The remaining uremic rats were split into three groups that were fed the following diets: (1) high-P (HP) diet, (2) high-P diet to which 4% sevelamer was added, or (3) a low-P (LP) diet (0.1%). The study was continued for an additional 3 months. Mortality was documented, and serum chemistries, vascular calcification, left ventricular perivascular fibrosis, and renal damage were evaluated.
RESULTS
Serum chemistries, PTH, and FGF-23
The protocol for the study is shown in Figure 1, and serum chemistries, parathyroid hormone (PTH), and fibroblast growth factor-23 (FGF-23) are shown in Table 1. Uremic rats that were fed a 1.4% P diet for 3 months (UHP-3M) and 6 months (UHP-6M) had significantly lower body weights than their respective normal control rats (NC-3M and NC-6M), which were also fed the 1.4% P diet (UHP-3M: 234.6 ± 7.6 vs. NC-3M: 277.0 ± 7.3 g, P < 0.01; and UHP-6M: 259.5 ± 8.7 vs. NC-6M 307.5±8.6 g, P < 0.01). In contrast, the weights of rats that were switched to LP diet (283.4 ± 7.4 g) or to sevelamer (Sev; 289.0 ± 13.5 g) were not different from normal. Serum creatinine was higher after 3 months of uremia compared with normal (NC-3M: 0.45 ± 0.02 vs. UHP-3M: 1.16 ± 0.06 mg/dl, P < 0.001). Serum ionized and total Ca were not different between these two groups, but serum P was higher with uremia (NC-3M: 5.90 ± 0.34 vs. UHP-3M: 10.27 ± 0.76 mg/ml, P < 0.001), resulting in a much higher Ca × P product (56.7 ± 3.4 vs. 94.3 ± 7.1 mg2/ml2, P < 0.01). Similarly, PTH increased from 173 ± 63 to 5850 ± 727 pg/ml (P < 0.001) and FGF-23 increased from 720 ± 31 to 9044 ± 2233 pg/ml (P < 0.05). The remaining rats were continued on the 1.4% P diet (UHP-6M) or switched to either a low-P (0.1%) diet (uremic rats that were fed a 0.1% P diet (ULP)) or to the 1.4% P diet containing 4% sevelamer carbonate (UHP + Sev) for an additional 3 months. The study lasted for a total of 6 months. After 6 months, serum creatinine was significantly higher in all groups of uremic rats compared with normal animals (NC-6M: 0.43 ± 0.02 vs. UHP-6M: 1.48 ± 0.23, P < 0.01; UHP + Sev: 1.16 ± 0.07, P < 0.001; and ULP: 1.08 ± 0.13 mg/dl, P < 0.01). Serum ionized and total Ca were higher in rats switched to a low-P diet compared with their uremic control counterparts (P < 0.001). Serum P was markedly higher in UHP-6M rats compared with normal (11.99 ± 1.35 vs. 3.90 ± 0.30 mg/dl, P < 0.001). Switching rats to a 0.1% P diet or treatment with sevelamer decreased serum P compared with UHP-6M rats (ULP: 3.69 ± 0.23 mg/dl, P < 0.001 and UHP-Sev: 6.96 ± 0.82 mg/dl, P < 0.01). Concomitant with the higher serum P, the Ca × P in UHP-6M rats was also sharply elevated (UHP-6M: 112.2 ± 12.9 vs. NC-6M: 38.6 ± 3.1 mg2/ml2, P < 0.001). Despite the higher calcium seen in ULP rats, the Ca × P was virtually identical to normal rats (ULP: 40.4 ± 3.3 mg2/ml2, P < 0.001 vs. UHP-6M). Treatment with sevelamer also blunted the increase in Ca × P (69.0 ± 7.8 mg2/ml2, P < 0.05). As expected, after 6 months of uremia, PTH was markedly elevated compared with normal (UHP-6M: 6574 ± 1641 vs. NC-6M: 67 ± 8 pg/ml, P < 0.01). Switching rats to a low-P diet markedly reduced serum PTH (289 ± 70 pg/ml, P < 0.001). Although PTH levels were lower in the group receiving sevelamer compared with their uremic counterparts, this decrease did not reach significance (4333 ± 1816 pg/ml). After 3 months, serum FGF-23 levels also increased from 720 ± 31 in NC-3M rats to 9044 ± 2233 pg/ml in UHP-3M animals (P < 0.05). FGF-23 was even higher after 6 months of uremia (UHP-6M: 22,244 ± 4049 vs. NC-6M: 392 ± 64 pg/ml, P < 0.001). Switching rats to a low-P diet markedly reduced FGF-23 levels (1079 ± 162 pg/ml, P < 0.001). Sevelamer treatment induced a slight decrease (18,429 ± 3765 pg/ml) compared with UHP-6M (not significant (NS)).
Figure 1. Treatment protocol for the study.
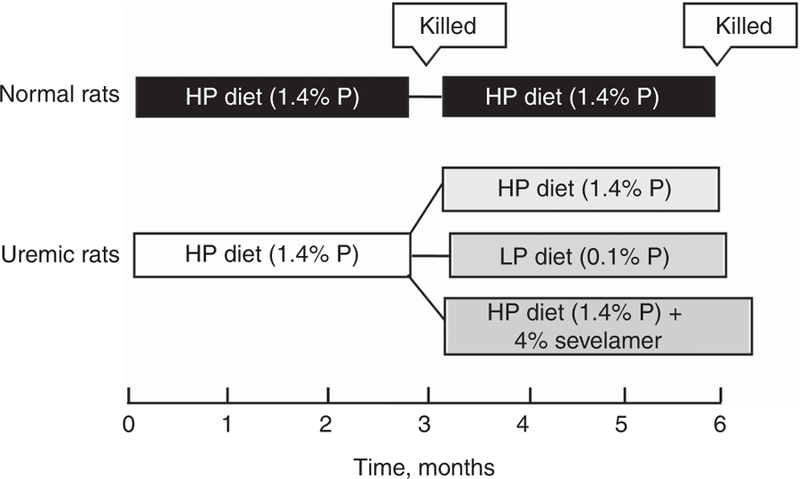
After induction of uremia, rats were fed a high phosphate (HP, 1.4%) diet for 3 months. A group of rats was killed at this point, and the remaining were split into three groups that were fed the HP diet, a low P (LP, 0.1%) diet, or the HP diet to which 4% sevelamer carbonate was added. The study continued for an additional 3 months. Normal rats fed the 1.4% P diet served as controls.
Table 1 |.
Serum chemistries
| Group | Body wt (g) | Cr (mg/dl) | Ica++ (mg/dl) | Total Ca (mg/dl) | P (mg/dl) | Ca_P (mg2/dl2) | PTH (pg/ml) | FGF-23 (pg/ml) |
|---|---|---|---|---|---|---|---|---|
| NC-3M (n=6) | 277.0±7.3 | 0.45±0.02 | 4.72±0.04 | 9.61±0.15 | 5.90±0.34 | 56.7±3.4 | 173±63 | 720±31 |
| NC-6M (n=6) | 307.5±8.6 | 0.43±0.02 | 4.92±0.02 | 9.90±0.11 | 3.90±0.30 | 38.6±3.1 | 67±8 | 392±64 |
| UHP-3M (n=9) | 234.6±6.7# | 1.16±0.06* | 4.36±0.14 | 9.18±0.14 | 10.27±0.76* | 94.3±7.1# | 5850±727* | 9044±2233† |
| UHP-6M (n=10) | 259.5±8.7# | 1.48±0.23# | 4.41±0.18† | 9.40±0.26 | 11.99±1.35* | 112.2±12.9* | 6574±1641# | 22,244 ±4049* |
| ULP (n=16) | 283.4±7.4 | 1.08±0.13# | 5.20±0.03*‡ | 10.78±0.22†‡ | 3.69±0.23‡ | 40.4±3.3‡ | 289±70‡ | 1079±162†‡ |
| UHP+Sev (n=5) | 289.0±13.5 | 1.16±0.07* | 4.65±0.14 | 9.96±0.26 | 6.96±0.82#§ | 69.0±7.8#¶ | 4333±1816† | 18,429±3765* |
Abbreviations: Ca, calcium; Ca×P, calcium×phosphorus; Cr, creatinine; FGF-23, fibroblast growth factor-23; HP, high phosphate (1.4%); ICa++, ionized calcium; LP, low phosphate (0.1%); 3M, 3 months; 6M, 6 months; NC-3M, normal control rats that were fed a 1.4% P diet for 3 months; NC-6M, normal control rats that were fed a 1.4% P diet for 6 months; P, phosphate; PTH, parathyroid hormone; Sev, sevelamer; UHP-3M, uremic rats that were fed a 1.4% P (HP) diet for 3 months; UHP-6M, uremic rats that were fed a 1.4% P (HP) diet for 6 months; ULP, uremic rats that were fed a 0.1% P (LP) diet; wt, weight.
Serum chemistries in normal and uremic rats. Normal rats were fed a 1.4% P diet and killed at 3 or 6 months (NC-3M and NC-6M, respectively). Uremic rats were fed a 1.4% P diet for 3 months. One group was killed at this point (UHP-3M). The remaining rats were continued for an additional 3 months and either maintained on the 1.4% P diet (UHP-6M), switched to a 0.1% P diet (ULP), or switched to the 1.4% P diet containing 4% sevelamer (UHP + Sev).
P < 0.001 versus NC
P < 0.01 versus NC
P < 0.05 versus NC
P < 0.001 versus UHP
P < 0.01 versus UHP, and
P < 0.05 versus UHP. NC-3M is compared with UHP-3M and NC-6M is compared with all other uremic groups.
Results are expressed as mean±s.e.m.
Aortic calcium deposition
Aortic Ca content and the percentage of rats showing positive aortic von Kossa staining are shown in Table 2. Aortic Ca content was higher after 3 months of uremia (NC-3M: 0.50 ± 0.06 vs. UHP-3M: 46.2 ± 14.2 μg per/mg dry wt, P < 0.05). After 6 months of uremia, aortic Ca content was even more elevated (NC-6M: 0.76 ± 0.06 vs. UHP-6M: 51.7 ± 16.3 μg per mg dry wt, P < 0.05). Sevelamer treatment had no effect (54.7 ± 24.3 μg per mg dry weight), but switching rats to a low-P diet decreased aortic Ca content (28.0 ± 9.7 μg per mg dry weight). This value was even lower than that of the 3-month UHP-3M rats, although not significant. Figure 2 shows representative von Kossa staining in normal and uremic rats. The percentage of rats showing positive aortic von Kossa staining in uremic rats increased from 56.0% at 3 months to 70.0% by 6 months. Sevelamer treatment showed a slight decrease in positive aortic von Kossa staining (60%), whereas only 31.3% of the rats that were switched to the low-P diet showed staining. Aortas were retrieved from several UHP-6M rats that died in the last few weeks of the study. All aortas from these rats showed increased Ca content (data not shown).
Table 2 |.
Aortic calcium
| Group | NC-3M | NC-6M | UHP-3M | UHP-6M | ULP | UHP + Sev |
|---|---|---|---|---|---|---|
| Aortic Ca content (mg per g dry wt) | 0.50±0.06 | 0.76±0.06 | 46.2±14.2* | 51.7±16.3* | 28.0±9.7 | 54.7±24.3* |
| Von Kossa (%) | 0.0 | 0.0 | 56.0 | 70.0 | 31.3 | 60.0 |
Abbreviations: Ca, calcium; HP, high phosphate (1.4%); LP, low phosphate (0.1%); 3M, 3 months; 6M, 6 months; NC-3M, normal control rats that were fed a 1.4% P diet for 3 months; NC-6M, normal control rats that were fed a 1.4% P diet for 6 months; P, phosphate; Sev, sevelamer; UHP-3M, uremic rats that were fed a 1.4% P (HP) diet for 3 months; UHP-6M, uremic rats that were fed a 1.4% P (HP) diet for 6 months; ULP, uremic rats that were fed a 0.1% P (LP) diet; wt, weight.
Aortic calcium (Ca) content and aortic von Kossa staining in normal and uremic rats. Normal rats were fed a 1.4% P diet and killed at 3 or 6 months (NC-3M and NC-6M, respectively). Uremic rats were fed a 1.4% P diet for 3 months. One group was killed at this point (UHP-3M). The remaining rats were continued for an additional 3 months and were either maintained on the 1.4% P diet (UHP-6M), switched to a 0.1% P diet (ULP), or switched to the 1.4% P diet containing 4% sevelamer (UHP + Sev).
P < 0.05 versus NC. NC-3M is compared with UHP-3M and NC-6M is compared with all other uremic groups.
Results are expressed as mean±s.e.m.
Figure 2. The effect of a high-phosphate (P) diet on indicators of aortic calcification.
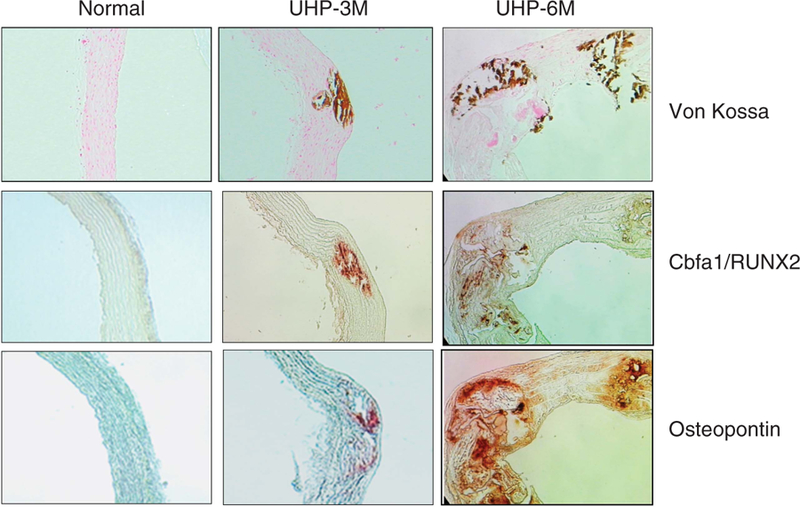
Representative microphotograph of aortic tissue from normal, UHP-3M, and UHP-6M rats stained for von Kossa, cbfa/RUNX2, and osteopontin (OPN). Normal control rats the were fed 1.4% P diet for 6 months; UHP-3M, uremic rats that were fed a 1.4% P (HP) diet for 3 months; UHP-6M, uremic rats that were fed a 1.4% P (HP) diet for 6 months.
Runx2 and OPN expression in aorta
Figure 2 also shows representative immunohistochemical staining for cbfa/Runx2 and OPN in the aorta. Both cbfa/Runx2 and OPN colocalized with areas of the aorta that were also positive for von Kossa staining in uremic rats. In addition, cbfa/Runx2 and OPN expression was also seen in areas that were not positive for von Kossa.
Mortality
Kaplan–Meier analysis (Figure 3) shows the mortality and survival rates for the last four groups of rats (NC-6M, UHP-6M, ULP, and UHP + Sev) during the final 3-month period. The mortality rate for uremic rats fed a 1.4% P diet (UHP-6M) was very high (71.4%). Of the 35 rats that were initially placed in this group, only 10 survived. Conversely, rats that were switched to the 0.1% P diet, ULP, experienced a mortality rate of only 5.9% (P < 0.0001 vs. UHP). In the ULP-6M group, 16 of the 17 rats that started in this group survived. Rats fed sevelamer, UHP + Sev, also had a lower mortality rate compared with the UC-6M group (37.5%), with five of the original eight rats surviving (NS vs. UHP-6M; P < 0.05 vs. ULP-6M).
Figure 3. Kaplan-Meier analysis of mortality.
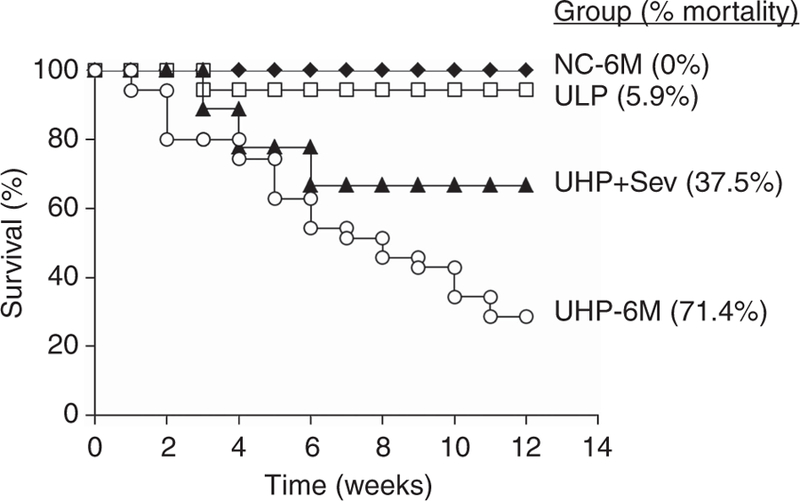
Uremic rats were fed a high phosphate (P) diet (1.4%) for 3 months. They were then divided into three groups that were fed 1.4% P diet (UHP-6M), a low P diet (ULP), or the 1.4% P diet to which 4% sevelamer carbonate was added (UHP + Sev). Tracking of mortality was begun at this point and continued for an additional 3 months. Normal rats fed the 1.4% P diet served as controls. HP, high phosphate (1.4%); LP, low phosphate (0.1%); NC-6M, normal control rats that were fed a 1.4% P diet for 6 months; P, phosphate; Sev, sevelamer; UHP-6M, uremic rats that were fed a 1.4% P (HP) diet for 6 months; ULP, uremic rats that were fed a 0.1% P (LP) diet.
Histological assessment of kidneys
Figure 4 shows representative trichrome-stained sections of kidneys from the NC-6M (Figure 4a), UHP-3M (Figure 4b), UHP-6M (Figure 4c), UHP + Sev (Figure 4d), and ULP (Figure 4e) groups. Normal kidneys (Figure 4a) had no interstitial fibrosis. They show intact glomeruli and tubulointerstitium. However, after 3 months of uremia and a 1.4% P diet, UHP-3M (Figure 4b) kidneys show considerable interstitial fibrosis, increased tubular atrophy, some interstitial inflammation, and minimal glomerulosclerosis. After an additional 3 months on a 1.4% P diet, kidneys from the UHP-6M group (Figure 4c) display severe interstitial fibrosis and glomerulosclerosis (thick arrow). They also have severe interstitial inflammation and calcifications (thin arrow). In comparison, sevelamer-treated animals (UHP + Sev; Figure 4d) displayed only mild to moderate interstitial fibrosis, chronic inflammation, and tubular dilatation. The glomeruli in these kidneys are intact. In addition, dietary phosphate restriction, ULP (Figure 4e), showed marked improvement compared with UHP-6M rats. Kidneys from ULP rats showed minimal interstitial fibrosis, no interstitial inflammation, and no glomerulosclerosis. More importantly, the kidneys from these rats also show a substantial improvement compared with those from UHP-3M rats, displaying much less interstitial fibrosis and tubular atrophy.
Figure 4. Kidney histology.
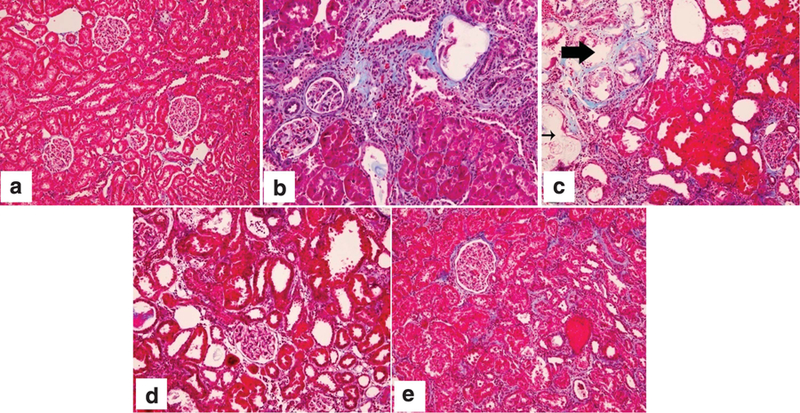
Representative kidney sections stained with Masson’s trichrome from (a) NC-6M, normal control kidney, which displayed intact glomeruli and no interstitial fibrosis; (b) UHP-3M, which showed considerable interstitial fibrosis, increased tubular atrophy, some interstitial inflammation, and minimal glomerulosclerosis; (c) UHP-6M, which displayed severe interstitial fibrosis and glomerulosclerosis (thick arrow) and interstitial inflammation and calcifications (thin arrow); (d) UHP + Sev, which showed mild to moderate interstitial fibrosis, chronic inflammation, and tubular dilatation; and (e) ULP, which had minimal interstitial fibrosis, no interstitial inflammation, and no glomerulosclerosis. Original magnification ×200. Please see text for further description of findings. HP, high phosphate (1.4%); LP, low phosphate (0.1%); NC-6M, normal control rats that were fed a 1.4% P diet for 6 months; P, phosphate; Sev, sevelamer; UHP-3M, uremic rats that were fed a 1.4% P (HP) diet for 3 months; UHP-6M, uremic rats that were fed a 1.4% P (HP) diet for 6 months; ULP, uremic rats that were fed a 0.1% P (LP) diet.
Effect on inflammatory cell infiltration as measured by expression of ED-1 in the kidney
Renal inflammation was assessed by ED-1-positive macrophages (Figure 5). ED-1-positive macrophages were markedly higher in UHP-3M (6.24 ± 1.36; P < 0.05), UHP-6M (6.12 ± 2.08), and the UHP + Sev (6.16 ± 1.81) groups compared with NC (0.85 ± 0.151). However, 3 months of P restriction partially reversed ED-1 expression in the kidney (ULP: 2.24 ± 1.26 positive cells per field).
Figure 5. Macrophage infiltration in the kidney.
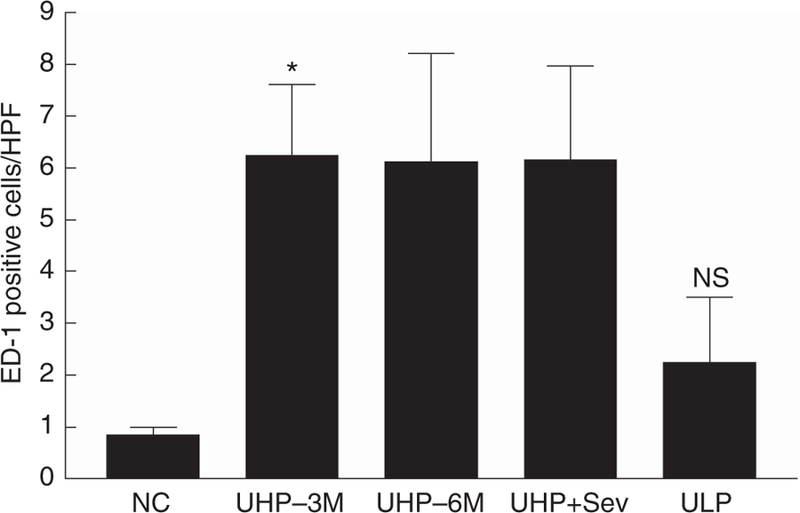
ED-1 expression (positive cells/high-powered field (HPF)) is shown in normal rats fed a 1.4% P diet for 6 months (NC-6M; n = 6), uremic rats fed a HP diet for 3 months (UHP-3M; n = 9) or 6 months (UHP-6M; n = 5), uremic rats fed a HP diet for 3 months and then treated with HP diet containing sevelamer (UHP + Sev; n = 5), or switched to an LP diet (ULP; n = 7) for an additional 3 months. Data expressed as mean ± s.e.m. P < 0.05 by ANOVA; *P < 0.05 NC versus UHP-3M by post hoc Dunnett’s multiple comparison test. ANOVA, analysis of variance; HP, high phosphate (1.4%); LP, low phosphate (0.1%); NC, normal controls; NC-6M, normal control rats that were fed a 1.4% P diet for 6 months; NS, not significantly different from normal control; P, phosphate; Sev, sevelamer; UHP-3M, uremic rats that were fed a 1.4% P (HP) diet for 3 months; UHP-6M, uremic rats that were fed a 1.4% P (HP) diet for 6 months; ULP, uremic rats that were fed a 0.1% P (LP) diet.
Myocardial histology
Perivascular fibrosis in the left ventricle was reported as the percentage of collagen surrounding the vessel to the total perivascular area. Representative images of collagen deposition (blue color) are shown in Figure 6a, and quantification is shown in Figure 6b. Perivascular fibrosis was significantly higher in uremic high-P rats (34.92% ± 5.02; P < 0.01) and in uremic rats treated with sevelamer (30.22% ± 4.91; P < 0.01) compared with normal control rats (7.56% ± 2.06). Dietary P restriction resulted in a marked improvement in perivascular fibrosis (22.03% ± 5.9; NS vs. control).
Figure 6. Left ventricular perivascular fibrosis was visualized by Masson’s trichrome staining.
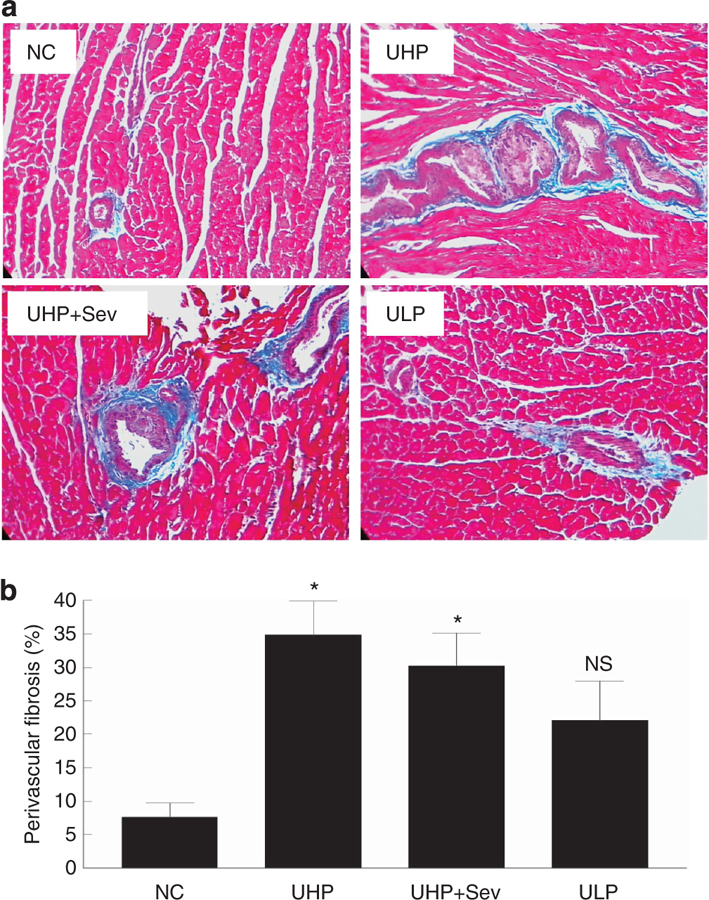
(a) Representative images (original magnification ×200) showing collagen deposition (blue) surrounding the vasculature of normal rats (NCs), uremic rats fed a HP diet for 6 months (UHP), uremic rats fed a HP diet for 3 months and then treated with HP diet containing sevelamer (UHP + Sev), or switched to a low-phosphate diet (ULP) for 3 months. (b) Quantification of analysis; P < 0.01 by ANOVA, *P < 0.01 versus NC by post hoc Dunnett’s multiple comparison test; n = 5 rats per group. ANOVA, analysis of variance; HP, high phosphate (1.4%); LP, low phosphate (0.1%); NC, normal controls; NS, not significantly different from normal control; P, phosphate; Sev, sevelamer; UHP, uremic rats that were fed a 1.4% P (HP) diet; ULP, uremic rats that were fed a 0.1% P (LP) diet.
Representative images of the effect of treatment on cardiomyocyte size is shown in Figure 7a, and quantification is shown in Figure 7b. Myocardial size (unit area in reference to pixel number) was significantly increased in UHP (1391.2 ± 84.7 pixels; P < 0.05) and UHP + Sev (1505.6 ± 135.9 pixels; P < 0.01) compared with NC (1025.7 ± 42.7 pixels). Myocardial size was decreased to near-normal levels with dietary P restriction (1117.7 ± 74.1; NS vs. control).
Figure 7. Effect of treatment on left ventricular cardiomyocyte size.
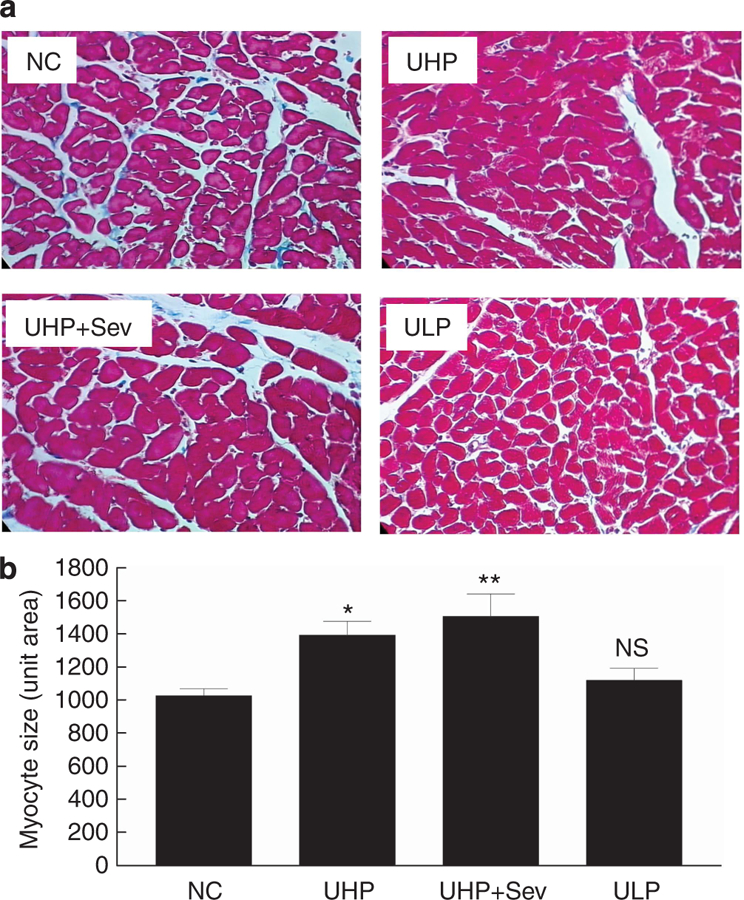
(a) Representative images (original magnification ×400) of cardiomyocytes of normal rats (NC; n = 4), uremic rats fed a HP diet for 6 months (UHP; n = 5), uremic rats fed a HP diet for 3 months and then treated with HP diet containing sevelamer for 3 months (UHP + Sev; n = 5), or switched to a low-phosphate diet for 3 months (ULP; n = 5). (b) Quantification of analysis; P < 0.01 by ANOVA, *P < 0.05 and **P < 0.01 versus NC by post hoc Dunnett’s multiple comparison test. ANOVA, analysis of variance; HP, high phosphate (1.4%); LP, low phosphate (0.1%); NC, normal controls; NS, not significantly different from normal control; P, phosphate; Sev, sevelamer; UHP, uremic rats that were fed a 1.4% P (HP) diet; ULP, uremic rats that were fed a 0.1% P (LP) diet.
DISCUSSION
In this study, we have demonstrated that P restriction reduces mortality in uremic rats. It also decreases vascular calcification, normalizes serum P and Ca × P, and lowers serum PTH and FGF-23. In addition, we show that P restriction not only prevents further renal damage but may even reverse the renal damage seen after 3 months of uremia and a high-P diet. In the rats switched to the low-P diet (ULP), improvement in renal damage and vascular calcification most certainly contributed to the marked reduction in mortality seen in this group (only 5.9% compared with 71.4% in the high-P group (UHP-6M)). The kidneys from ULP rats showed minimal interstitial fibrosis, no interstitial inflammation, and no glomerulosclerosis when compared with the UHP-6M group. However, the most important point of this study is that even after vascular calcification and renal damage has occurred, P restriction reverses both of these abnormalities. Lopez et al.28 have shown that just a 1-month reduction in P intake can partially resolve established extraosseous calcification, including vascular calcification. Not only was there less vascular calcification in ULP rats, but the kidneys from the ULP group also showed a marked improvement over those from the 3-month baseline (UC-3M). There was a substantial decrease in renal inflammation after P restriction, as assessed by decreased infiltration of ED-1-positive macrophages into kidney tissue, as well as much less interstitial fibrosis and tubular atrophy. Other studies have shown that, under circumstances other than dietary P restriction, renal damage can be reversed. Adamczac et al.29 showed a partial reversal of glomerulosclerosis and interstitial lesions after high-dose enalapril treatment in uremic rats. Ortmann et al.30 demonstrated that endothelin receptor antagonists can also reverse proteinuria and glomerulosclerosis in a rat model of aging. To our knowledge, however, the reversal of kidney damage has not previously been reported with dietary P restriction. In addition, kidneys from sevelamer-treated rats displayed less severe interstitial fibrosis and no glomerulosclerosis compared with UHP-6M rats. This may also have had a role in the decreased mortality seen in this group (only 37.5%).
It is well known that P promotes the progression of renal failure and that P restriction inhibits that progression.31–33 In this study, vascular calcification increased with the length of time that rats were fed the high-P diet. Although this evaluation was done at the end point of the study in only rats that survived, the aortas of several of the rats from the UHP-6M group that died before the end of the study were analyzed for Ca content. All had a marked elevation in aortic Ca content. The addition of 4% sevelamer carbonate to the high-P diet (UHP + Sev) did lower serum P, but not as efficiently as P restriction. Although mortality in the sevelamer-treated group was higher than that with dietary phosphate restriction, it was still lower than that in rats receiving no intervention (UHP-6M)—only 37.5%. The sevelamer-treated rats had less renal damage than UHP-6M rats, but not less vascular calcification. We have previously shown that sevelamer can attenuate kidney and cardiovascular calcification in uremic rats.34 Other researchers have also reported that sevelamer protects against deterioration of renal function and decreases vascular calcification in CKD patients.35–38 One reason for not seeing a decrease in vascular calcification in sevelamer-treated rats is that the diet used was very high in P (1.4%), which may have been too high for even 4% sevelamer to overcome. Although our previous study34 in uremic rats was also 6 months in length, the diet used was significantly lower in P (only 0.9%). In addition, in light of the high aortic Ca content seen in rats from the UHP-6M group that died before the end of the study, we presume that the differences seen between the UHP-6M and UHP + Sev groups would have been even greater if the rats that died before the end of the study could have been evaluated, especially as the mortality rate in UHP + Sev rats was much lower. A number of studies have shown that sevelamer reduces total and low-density lipoprotein cholesterol levels.20,24–26,38 Wilkes et al.20 reported a 35.9% decrease in serum low-density lipoprotein cholesterol in hemodialysis patients treated with sevelamer. Sevelamer has also been shown to have anti-inflammatory properties. C-reactive protein is a well-known marker of inflammation, is produced by the liver in response to cytokines, and is elevated in the serum of CDK patients. Studies by Ferramosca et al.,24 as well as by several other researchers, reported a significant lowering of serum C-reactive protein in hemodialysis patients with sevelamer treatment.24–26 In a more recent study in diabetic CKD patients, Vlassara et al.39 showed that not only did sevelamer carbonate improve the lipid profile of these patients, but it also lowered serum hemoglobin A1c and FGF-23 compared with calcium carbonate. One important mechanism for decreased mortality and apparent prevention of further renal damage seen in sevelamer-treated rats may simply be the reduction in serum P seen in this group. Although this decrease was not as great as that seen with phosphorus restriction, it may have been sufficient to still explain the sevelamer results.
Serum FGF-23 may also have an important role in cardiovascular disease. It has been shown that serum FGF-23 positively correlates with cardiovascular disease in patients with CKD.40–42 In addition, a role for FGF-23 in endothelial function was shown by Yilmaz et al.43 In sevelamer-treated CKD patients, they found a decrease in serum FGF-23 that was associated with an increase in flow-mediated vasodilation. In our study, serum FGF-23 was markedly lower in P-restricted rats, and may have improved cardiovascular function in these rats.
We showed that in aortas of rats displaying vascular calcification (positive von Kossa staining), cbfa1/Runx2 and OPN expression colocalized with the vascular calcification. In addition, these two proteins are also expressed at sites where there was no von Kossa staining. OPN is an inhibitor of calcification. It is abundant at sites of medial calcification44 and has been shown to inhibit hydroxyapatite formation45, as well as vascular smooth muscle cell calcification46 in vitro. Its presence at sites of calcification, as well as at sites that do not overtly show calcification, suggests a compensatory mechanism to combat osteogenic changes in vascular smooth muscle cells.
Cardiac hypertrophy and perivascular fibrosis are typical pathological findings in uremic cardiomyopathy. In this study, we show for the first time that dietary P restriction improves left ventricular perivascular fibrosis and decreases cardiomyocyte size in rats with established uremia. A study by Aman et al.47 showed that uremic rats fed a high-P diet developed an increase in cardiac fibrosis, whereas uremic rats fed a low-P diet did not. In addition, serum phosphate has been independently associated with left ventricular mass in patients with CKD,48 and reductions in left ventricular mass are associated with reduced mortality in patients at high cardiovascular risk.49
The low-P diet was clearly effective in markedly reducing serum phosphorus, PTH, and FGF-23, but it is beyond the scope of this study to determine with certainty what the individual role of each of these factors was in affecting the beneficial changes seen in vasculature, kidney, and myocardium.
In conclusion, this study clearly demonstrates the role of hyperphosphatemia in promoting vascular calcification and mortality. Moreover, we have shown that the kidneys of uremic rats fed a high-P diet (1.4%) for 3 months develop considerable interstitial fibrosis and increased tubular atrophy and interstitial inflammation. Importantly, this kidney damage was reversed by dietary P restriction, and further damage was inhibited by sevelamer. Indicators of uremic cardiomyopathy were significantly reduced with dietary P restriction. In addition, we were able to show that mortality was markedly reduced by the addition of sevelamer to the diet, and further reduced when serum P returned to normal by dietary P restriction. This study clearly indicates the importance of the control of phosphorus in CKD patients beginning in the earliest stages.
MATERIALS AND METHODS
Experimental protocol
All experimental protocols were approved by the Animal Studies Committee at Washington University School of Medicine in accordance with federal regulations. The protocol for the study is shown in Figure 1. Uremia was induced in a group of female Sprague–Dawley rats (225–250 g) by 5/6 nephrectomy. Uremic rats were placed on a 1.4% P and a 0.8% Ca diet (Dyets, Bethlehem, PA) and given enalapril in their drinking water (25 mg/l). Normal rats served as control and were fed the same diet, but were not given enalapril. After 3 months, a blood sample was taken from each uremic rat, and they were divided into four experimental groups, each with similar serum creatinine, Ca, and P. Group 1 rats, UHP-3M, were killed at this point along with half of the normal rats, NC-3M. The rats were killed by exsanguination via the dorsal aorta, and blood was taken for determination of serum chemistries, PTH, and FGF-23. The other three uremic groups were as follows: group 2, uremic + 1.4% P diet (UHP-6M); group 3, uremic + 1.4% P diet + 4% sevelamer carbonate (UHP + Sev; Genzyme Corpora-tion/Sanofi, Cambridge, MA); and group 4, uremic + 0.1% P, 0.2% Ca diet (ULP), also from Dyets. The study was continued for an additional 3 months, during which time mortality was documented. The remaining rats were then killed.
The section of aorta from the thoracic branch to the renal branch was dissected out. A portion was weighed and used for measurement of Ca content; another portion was fixed in 10% buffered formalin for histological examination. Portions of the remnant kidney and left ventricle were fixed in 10% buffered formalin for histological examination.
Analytical determinations
Serum P, creatinine, and total and ionized calcium were measured as previously reported.50 Intact PTH and FGF-23 levels were measured by two-site enzyme-linked immunosorbent assays (Immutopics, San Clemente, CA) according to the manufacturer’s specifications.
Chemical and morphologic assessment of calcification
Aortic Ca content was measured after acid digestion as described previously.50 Results were corrected by dry tissue weight (wt) and expressed as μg calcium per mg dry wt of tissue. Formalin-fixed paraffin sections of aorta were examined for Ca deposition using a Von Kossa kit (Polysciences, Warrington, PA).
Immunohistochemical evaluation for RUNX2 and OPN in aorta
Formalin-fixed paraffin sections of aorta were examined for RUNX2 and OPN expression. Deparaffinized, rehydrated sections were blocked with 2.5% horse serum and incubated with rabbit anti-RUNX2 (Santa Cruz Biotechnology, Santa Cruz, CA) or mouse anti-OPN antibody (American Research Products, Waltham, MA) for 1 h at room temperature. Sections were incubated for 30 min with the appropriate Impress secondary antibody (Vector Laboratories, Burlingame, CA), and visualized with AEC substrate.
Histochemistry and assessment of glomerulosclerosis and interstitial infiltration
Formalin-fixed paraffin sections of kidney tissue were stained with hematoxylin and eosin and Masson’s trichrome reagent. Kidney sections were assessed for glomerulosclerosis, interstitial inflammation, interstitial fibrosis, tubular atrophy, tubular dilatation, and calcifications. Each was scored 1–4 as follows: 1, 10% of surface area involved; 2, 10–24%; 3, 25–50%; and 4, ⩾50%.
Immunohistochemical evaluation for ED-1 expression in kidney
Immunohistochemical staining for ED-1 was performed as previously described,51 with the exception that 2.5% horse serum was used for blocking, and mouse Impress as the secondary antibody. The ED-1-positive cells were counted manually in 10–20 renal cortical areas including glomeruli (×400). Positive cells were expressed as the number per high-powered field, and results were reported as mean ± s.e.m.
The number of ED-1-positive cells was counted manually in 10 renal cortical areas including the glomeruli (×400).
Myocardial histology
Cross-sectional cuts of paraffin-embedded left ventricle were stained with Masson’s trichrome reagent and analyzed using the Image-Pro Plus software, Version 7.0 (Media Cybernetics, Silver Spring, MD). Perivascular fibrosis was assessed by calculating the percentage of trichrome-stained collagen deposit surrounding the vessel to the total perivascular area, using the color cube function of the software. Approximately 10 vessels (×200) were assayed for each rat (n = 5 rats/group). The area (in pixels) of individually circumscribed cardiomyocytes was determined using the ImagePro software. Approximately 100 cells from each rat sample were used to obtain the average cardiomyocyte size, expressed as arbitrary units (unit area).
Statistical analysis
All data are expressed as mean ± s.e.m. One-way analysis of variance with Tukey’s post test, unless stated otherwise, was used for comparison between uremic groups. Student’s nonpaired t-test was used to compare the normal control groups with individual uremic groups or when only two groups were compared. The Kaplan–Meier method was used to construct survival curves, and a log-rank (Mantel–Cox) test was used to determine whether differences between the groups were significant (GraphPad Prism 6.0; GraphPad Software, La jolla, CA). P < 0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Kevin Muñoz for his technical support. This research was supported in part by grants from Research in Renal Diseases, Washington University, from Genzyme Corporation, and from the WUCKDR O’Brien Center (P30DK079333).
Footnotes
DISCLOSURE
ES is a consultant for Abbott Pharmaceuticals and was a consultant for Genzyme Corporation.
All the other authors declared no competing interests.
REFERENCES
- 1.Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis 1988; 32: S112–S119. [DOI] [PubMed] [Google Scholar]
- 2.London GM, Guerin AP, Marchais SJ et al. Arterial media calcification in end stage renal disease: impact on all cause and cardiovascular mortality. Nephrol Dial Transplant 2003; 18: 1731–1740. [DOI] [PubMed] [Google Scholar]
- 3.Goodman WG, London G, Amman K et al. Vascular calcification in kidney disease. Am J Kidney Dis 2004; 43: 572–579. [DOI] [PubMed] [Google Scholar]
- 4.Block GA, Hulbert-Shearon TE, Levin NW et al. Association of serum phosphorus and calcium phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31: 601–617. [DOI] [PubMed] [Google Scholar]
- 5.Block GA, Port FK. Re-evaluation of risks associated with hyperphosphatemia and hyperparathyroidism in dialysis patients: recommendations for a change in management. Am J Kidney Dis 2000; 35: 1226–1237. [DOI] [PubMed] [Google Scholar]
- 6.Marco MP, Craver L, Betriu A et al. Higher impact of mineral metabolism on cardiovascular mortality in a European hemodialysis population. Kidney Int Suppl 2003; 85: S111–S114. [DOI] [PubMed] [Google Scholar]
- 7.Noordzij M, Korevaar JC, Boeschoten EW et al. The Kidney Disease Outcomes Quality Initiative (K/DOQI) guideline for bone metabolism and disease in CKD: association with mortality in dialysis patients. Am J Kidney Dis 2005; 46: 925–932. [DOI] [PubMed] [Google Scholar]
- 8.Goldsmith DJ, Covic A, Sambrook PA et al. Vascular calcification in longterm haemodialysis patients in a single unit: a retrospective analysis. Nephron 1997; 77: 7–43. [DOI] [PubMed] [Google Scholar]
- 9.Guerin AP, London GM, Marchais SJ et al. Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol Dial Transplant 2000; 15: 1014–1021. [DOI] [PubMed] [Google Scholar]
- 10.Shanahan CM, Cary N, Salisbury JR et al. Medial localization of mineralization-regulating proteins in association with Mönckeberg’s sclerosis: evidence for smooth muscle cell–mediated vascular calcification. Circulation 1999; 100: 2168–2176. [DOI] [PubMed] [Google Scholar]
- 11.Shanahan CM, Cary NR, Metcalfe JC et al. High expression of genes for calcification-regulating proteins in human atherosclerotic plaques. J Clin Invest 1994; 93: 2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schoppet M, Al-Fakhri N, Franke FE et al. Localization of osteoprotegerin, tumor necrosis factor-related apoptosis-inducing ligand, and receptor activator of nuclear factor-kappaB ligand in Monckeberg’s sclerosis and atherosclerosis. J Clin Endocrinol Metab 2004; 89: 4104–4112. [DOI] [PubMed] [Google Scholar]
- 13.Levy RJ, Gundberg C, Scheinman R. The identification of the vitamin K-dependent bone protein osteocalcin as one of the gammacarboxyglutamic acid containing proteins present in calcified atherosclerotic plaque and mineralized heart valves. Atherosclerosis 1983; 46: 49–56. [DOI] [PubMed] [Google Scholar]
- 14.Otto F, Thornell AP, Crompton T et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997; 89: 765–771. [DOI] [PubMed] [Google Scholar]
- 15.Franceschi RT, Xiao G. Regulation of the osteoblast-specific transcription factor, Runx2: responsiveness to multiple signal transduction pathways. J Cell Biochem 2003; 88: 446–454. [DOI] [PubMed] [Google Scholar]
- 16.Giachelli CM, Speer MY, Li X et al. Regulation of vascular calcification: roles of phosphate and osteopontin. Circ Res 2005; 96: 717–722. [DOI] [PubMed] [Google Scholar]
- 17.Jono S, Peinado C, Giachelli CM. Phosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification. J Biol Chem 2000; 275: 20197–20203. [DOI] [PubMed] [Google Scholar]
- 18.Steitz SA, Speer MY, Curinga G et al. Smooth muscle cell phenotypic transition associated with calcification: up regulation of Cbfa 1 and down regulation of smooth muscle lineage markers. Circ Res 2001; 89: 1147–1154. [DOI] [PubMed] [Google Scholar]
- 19.Panichi V, Bigazzi R, Paoletti S et al. Impact of calcium, phosphate, PTH abnormalities and management on mortality in hemodialysis: results from the RISCAVID study. J Nephrol 2010; 23: 556–562. [PubMed] [Google Scholar]
- 20.Wilkes BM, Reiner D, Kern M et al. Simultaneous lowering of serum phosphate and LDL-cholesterol by sevelamer hydrochloride (Rena Gel) in dialysis patients. Clin Nephrol 1998; 50: 381–386. [PubMed] [Google Scholar]
- 21.Chertow GM, Burke SK, Raggi P. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62: 245–252. [DOI] [PubMed] [Google Scholar]
- 22.Block GA, Spiegel DM, Erlich J et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2002; 68: 1815–1824. [DOI] [PubMed] [Google Scholar]
- 23.Raggi P, Bommer J, Chertow GM. Valvular calcification in hemodialysis patients randomized to calcium-based phosphorus binders or sevelamer. J Heart Valve Dis 2004; 13: 134–141. [PubMed] [Google Scholar]
- 24.Ferramosca E, Burke S, Chasan-Taber S et al. Potential antiatherogenic and anti-inflammatory properties of sevelamer in maintenance hemodialysis patients. Am Heart J 2005; 149: 820–825. [DOI] [PubMed] [Google Scholar]
- 25.Yamada K, Fujimoto S, Tokura T et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27: 361–365. [PubMed] [Google Scholar]
- 26.Shantouf R, Budoff MJ, Ahmadi N et al. Effects of sevelamer and calcium-based phosphate binders on lipid and inflammatory markers in hemodialysis patients. Am J Nephrol 2008; 28: 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caglar K, Yilmaz MI, Saglam M et al. Short-term treatment with sevelamer increases serum fetuin-a concentration and improves endothelial dysfunction in chronic kidney disease stage 4 patients. Clin J Am Soc Nephrol 2008; 3: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez I, Mendoza F, Guerrero F et al. The calcimimetic AMG 641 accelerates regression of extraosseous calcification in uremic rats. Am J Physiol Renal Physiol 2009; 296: F1376–F1385. [DOI] [PubMed] [Google Scholar]
- 29.Adamczac M, Gross ML, Krtil J et al. Reversal of glomerulosclerosis after high-dose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol 2003; 14: 2833–2842. [DOI] [PubMed] [Google Scholar]
- 30.Ortmann J, Amann K, Brandes RP et al. Role of podocytes for reversal of glomerulosclerosis and proteinuria in the aging kidney after endothelin inhibition. Hypertension 2004; 44: 974–981. [DOI] [PubMed] [Google Scholar]
- 31.Ibels LS, Alfrey AC, Haut L et al. Preservation of function in experimental renal disease by dietary restriction of phosphate. N Engl J Med 1978; 298: 122–126. [DOI] [PubMed] [Google Scholar]
- 32.Lau K Phosphate excess and progressive renal failure: the precipitation-calcification hypothesis. Kidney Int 1989; 36: 918–937. [DOI] [PubMed] [Google Scholar]
- 33.Loghmen-Adham M Role of phosphate retention in the progression of renal failure. J Lab Clin Med 1993; 122: 15–25. [PubMed] [Google Scholar]
- 34.Cozzolino M, Staniforth ME, Liapis H et al. Sevelamer hydrochloride attenuates kidney and cardiovascular calcifications in long-term experimental uremia. Kidney Int 2003; 64: 1653–1661. [DOI] [PubMed] [Google Scholar]
- 35.Nagano N, Miyata S, Obana S et al. Sevelamer hydrochloride, a phosphate binder, protects against deterioration of renal function in rats with progressive chronic renal insufficiency. Nephrol Dial Transplant 2003; 18: 2014–2023. [DOI] [PubMed] [Google Scholar]
- 36.Kakuta T, Tanaka R, Hyodo T et al. Effect of sevelamer and calcium-based phosphate binders on coronary artery calcification and accumulation of circulating advanced glycation end products in hemodialysis patients. Am J Kidney Dis 2010; 57: 422–431. [DOI] [PubMed] [Google Scholar]
- 37.Russo D, Miranda I, Ruocco C et al. The progression of coronary artery calcification in predialysis patients on calcium carbonate or sevelamer. Kidney Int 2007; 72: 1255–1261. [DOI] [PubMed] [Google Scholar]
- 38.Chertow GM, Burke SK, Dillon MA et al. Long-term effects of sevelamer hydrochloride on the calcium phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14: 2907–2914. [DOI] [PubMed] [Google Scholar]
- 39.Vlassara H, Uribarri J, Weijing C et al. Effects of sevelamer on Hba1c, inflammation, and advanced glycation end products in diabetic kidney disease. Clin J Am Soc Nephrol 2012; 7: 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zisman AL, Wolf M. Recent advances in the rapidly evolving field of fibroblast growth factor 23 in chronic kidney disease. Curr Opin Nephrol Hypertens 2010; 19: 335–342. [DOI] [PubMed] [Google Scholar]
- 41.Desjardins L, Liabeuf S, Renard C et al. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos Int 2012; 23: 2017–2025. [DOI] [PubMed] [Google Scholar]
- 42.Wolf M Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int 2012; 82: 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yilmaz M, Sonmez A, Saglam M et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59: 177–185. [DOI] [PubMed] [Google Scholar]
- 44.Ahmed S, O’Neill KD, Hood AF et al. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Kidney Dis 2001; 37: 1267–1276. [DOI] [PubMed] [Google Scholar]
- 45.Denhardt DT, Guo X. Osteopontin: a protein with diverse functions. FASEB J 1993; 7: 1475–1482. [PubMed] [Google Scholar]
- 46.Wada T, McKee MD, Steitz S et al. Calcification of vascular smooth muscle cell cultures. Inhibition by osteopontin. Circ Res 1999; 84: 166–178. [DOI] [PubMed] [Google Scholar]
- 47.Aman K, Törnig J, Kugel B et al. Hyperphosphatemia aggravates cardiac fibrosis and microvascular disease in experimental uremia. Kidney Int 2003; 63: 1296–1301. [DOI] [PubMed] [Google Scholar]
- 48.Chue CD, Edwards NC, Moody WE et al. Serum phosphate is associated with left ventricular mass in patients with chronic kidney disease: a cardiac magnetic resonance study. Heart 2012; 98: 219–224. [DOI] [PubMed] [Google Scholar]
- 49.Devereux RB, Wachtell K, Gerdts E et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA 2004; 292: 2350–2356. [DOI] [PubMed] [Google Scholar]
- 50.Mizobuchi M, Finch JL, Slatopolsky E. Differential effects of vitamin D receptor activators on vascular calcification in uremic rats. Kidney Int 2007; 72: 709–715. [DOI] [PubMed] [Google Scholar]
- 51.Mizobuchi M, Morrissey J, Finch JL et al. Combination therapy with an angiotensin-converting enzyme inhibitor and a vitamin D analog suppresses the progression of renal insufficiency in uremic rats. J Am Soc Nephrol 2007; 18: 1796–1806. [DOI] [PubMed] [Google Scholar]