

Abstract
Background:
Genetic variants currently known to affect coronary artery disease (CAD) risk explain less than a quarter of disease heritability. The heritability contribution of regulatory gene networks (RGNs) in CAD, which are modulated by both genetic and environmental factors, is unknown.
Objective:
To determine the heritability contributions of single nucleotide polymorphisms affecting gene expression (eSNPs) in RGNs causally linked to CAD.
Methods:
Seven vascular and metabolic tissues collected in two independent genetics-of-gene expression studies of patients with CAD were used to identify eSNPs and to infer co-expression networks. To construct RGNs with causal relations to CAD, the prior information of eSNPs in the co-expression networks was used in a Bayesian algorithm. Narrow-sense CAD heritability conferred by the RGNs was calculated from individual-level genotype data from nine European genome-wide association studies (GWAS, 13,612 cases, 13,758 controls).
Results:
We identified and replicated 28 independent RGNs active in CAD. The genetic variation in these networks contributed to 10.0% of CAD heritability beyond the 22% attributable to risk loci identified by GWAS. RGNs in the atherosclerotic arterial wall (n=7) and subcutaneous or visceral abdominal fat (n=9) were most strongly implicated, jointly explaining 8.2 % of CAD heritability. In all, these 28 RGNs (each contributing to >0.2% of CAD heritability) comprised 24 to 841 genes, whereof 1 to 28 genes had strong regulatory effects (key disease drivers) and harbored many relevant functions previously associated with CAD. The gene activity in these 28 RGNs also displayed strong associations with genetic and phenotypic cardiometabolic disease variations both in humans and mice, indicative of their pivotal roles as mediators of gene–environmental interactions in CAD.
Conclusions:
RGNs capture a major portion of genetic variance and contribute to heritability beyond that of genetic loci currently known to affect CAD risk. These networks provide a framework to identify novel risk genes/pathways and study molecular interactions within and across disease-relevant tissues leading to CAD.
Keywords: Heritability, coronary artery disease, systems genetics, regulatory-gene networks
Graphical Abstract
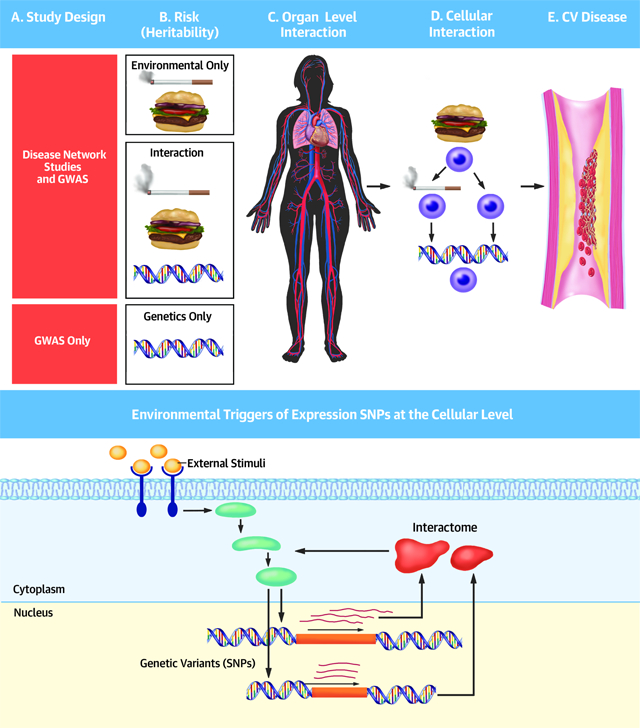
Central Illustration. Environmentally triggered eSNPs of cellular gene networks contribute to CAD heritability. (A) Schematics of the macro- and micro levels of gene-environmental interactions. Macro-environmental factors (e.g., lifestyle, food intake and smoking) interact with genetic variants in organs to change the micro-environment in cellular networks leading to CAD. (B)Schematics of environmental and genetic risk for complex diseases over a lifetime. One fraction (10–25%) of inherited risk for complex diseases (H2) with the strongest penetrance is characterized by genetic variants promoting disease development independent of parallel environmental risk factors and thus, likely influence disease development already from early stages in life (“G”, upper life span axis). Similarly, there may also be a smaller fraction (10–25%) of penetrant environmental risk factors driving disease independently of the genetic makeup of an individual (“E”, lower life span axis). However, research in cell cultures has shown that a large fraction of SNPs affecting gene expression (eSNPs) in genetically identical cells is altered depending on parallel environmental perturbations.(47) Accordingly, it is feasible that the largest fraction (50–80%) of inherited risk (H2) for complex diseases constitutes SNPs which disease-driving effects only transpire at stages in a life when triggering environmental factors are present (“GxE”, middle life span axis). An example of a triggering environmental factor can be that of fatty diet or smoking, but also a certain age. (C) Fractions of genetic factors contributing to the heritability (H2) of CAD.
Condensed Abstract:
Coronary artery disease (CAD), the cause of myocardial infarction, is partly inherited. Yet less than 25% of CAD heritability has thus far been accounted for by genome-wide association studies (GWAS). Using two recent genetics of gene expression studies, we sought additional heritability contributions from regulatory gene networks (RGNs) active in CAD. Genetic variants of the RGNs contribute to additional 10 % of CAD heritability beyond the 25% previously identified by GWAS. This novel fraction of genetic variants should help to improve clinical risk predictions of CAD and myocardial infarction, and the RGNs provide new mechanistic insights into the etiology of CAD.
Introduction
The risk of developing coronary artery disease (CAD) is partly inherited. According to family-based studies carried out in Western populations (1,2), the overall contribution of inherited/genetic factors (i.e., the broad sense heritability (H2)) to total CAD varianceis estimated to be in the range of 40–60% (3). Over recent years, single nucleotide polymorphisms (SNPs) underlying H2 by additive genetic effects (i.e., by narrow sense heritability (h2)) have been identified by genome-wide association studies (GWAS).(4, 5) However, the 302 most significant SNPs in genetic loci identified by GWAS currently account for less than a quarter of CAD heritability (H2). Thus, the majority of genetic variants underlying CAD heritability can be considered “missing”.
The lack of success in identifying a greater portion of genetic variance despite the enormous size and statistical power of recent meta-analyses of GWAS suggests that some of the missing heritability may not be detectable using a GWAS data analysis design solely focusing on SNPs with genome-wide significance.(3) For instance, DNA variants that increase risk of CAD only in the presence of environmental risk factors (e.g., environmental-triggered risk SNPs (Central Illustration; Online Figure 1) may result in sub-significant, or weak associations in GWAS data(6). Similarly, rare familial variants(3) are also not detectable. Another major limitation of GWAS is that they provide no mechanistic insights into how identified risk variants ultimately affect a late-onset disease such as CAD (4, 5).
An extension to GWAS are genetics-of-gene-expression-studies, which introduce RNA expression in an intermediary layer capturing both the effects of genetic variability and environmental perturbations driving disease phenotypes. This complementary, data-driven approach can identify not only disease-causal genes but also CAD-relevant pathways in the form of regulatory gene networks (RGNs).(6–12) In these RGNs, the directionality of gene interactions is disclosed by applying probabilistic Bayesian network algorithms(13) using genetic modifiers (i.e., expression quantitative trait loci (eQTLs)) as priors. In addition, certain “hub” genes are identifiable as being highly connected and located in the top of the RGN hierarchy (i.e., regulating many down-stream network genes). Accordingly, perturbation of these key network drivers by either increasing or inhibiting their level of expression in relevant in vitro model systems has demonstrated their hierarchical ability to modulate and impact the activity of the entire network, as well as any downstream network-associated phenotype such as CAD (14,15). This latter characteristic has also prompted the term “key disease drivers” (14,16). It has been suggested that the causal nature of RGNs(17), including their key drivers, may provide a valuable mechanistic framework to decipher complex disease biology.
Here, we determined whether SNPs associated with expression levels of genes (eSNPs/eQTLs) in RGNs inferred from two genetics-of-gene-expression studies of CAD(8, 11) contribute to heritability not accounted for by known genetic risk loci identified by GWAS. Moreover, both at the tissue and molecular level, we functionally characterize these networks and determine their individual contributions to CAD heritability.
Methods
Study Populations
The Stockholm Atherosclerosis Gene Network study (STAGE)(8, 11) is a genetics-of-gene-expression study of atherosclerotic aortic wall, non-atherosclerotic internal mammary artery, liver, skeletal muscle, visceral abdominal fat, subcutaneous fat and whole blood. Tissues were obtained from CAD patients during coronary artery bypass surgery at the Karolinska Hospital in Stockholm, Sweden (Ethical Approval Dnr 2002–04). RNA samples were used for gene expression profiling with a custom Affymetrix array (HuRSTA-2a520709).(18) In the Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task study (STARNET)(12) (Ethical Approval Dnr 2007/1521–32), a continuation of STAGE, the same seven tissues were collected from 600 bypass patients and analyzed by RNA sequencing (RNAseq), mainly using the ribo-zero protocol (50–100 bp read-length, pair ends, 20–50 M read depth). Standard platforms for genotyping DNA in STAGE and STARNET are shown in Online Table 1.
Individual-level GWAS data were obtained from nine case-control studies of CAD, representing 13,612 CAD cases and 13,758 controls. Most of the subjects were from Germany (the German Myocardial Infarction Family Studies [GerMIFS] I,(19) II,(20) III (KORA),(21) IV,(22) and V(23)) and England (Wellcome Trust Case Control Consortium [WTCCC]).(24) Others included subjects from France (Cardiogenics)(25) and Italy and the US (Myocardial Infarction Genetics Consortium [MIGen]).(26) All subjects were of Western European descent and gave broad written informed consent before participating in these studies to understand the genetic underpinnings of cardiovascular disease. All GWAS were approved by their local Ethical Committees. In the GerMIF studies, Cardiogenics, WTCCC, and MIGen, information on CAD manifestations was validated by medical records. MIGen data were from the database of Genotypes and Phenotypes(27) (project ID 49717–3). Genotyping was done with commercially available arrays (Online Table 1).
Mouse Data
The Hybrid Mouse Diversity Panel (HMDP) has been described previously as a set of over 100 different inbred mouse strains, which were studied under chow diet, high-fat diet and on the background of transgenic implementation of human APOE-Leiden and cholesteryl ester transfer protein to generate strain specific degrees of atherosclerosis (28).
Inference and Validation of Regulatory Gene Networks
Using data from the STAGE(11) and STARNET(12) studies, genetic variants affecting gene expression in the same locus (cis eSNP/eQTLs) and 171 co-expression networks inferred simultaneously to capture gene–gene interactions both within and across tissues(29) were identified. By applying a linear Gaussian Bayesian algorithm, RGNs were inferred separately for each co-expression network using eQTLs and transcription factors as priors. Applying a Bayesian information criterion,(30) a multiple-restart greedy hill-climbing algorithm, with edge additions, deletions, and reversals, was used to search optimal RGN models for each co-expression module. Performed as described(11) were network; key driver identification(31), Gene Ontology (GO)(32), eigengene/phenotype association, enrichment in genetic association with CAD risk factors (e.g., blood lipids and glucose levels) according to GWAS, and cross-species eigengene/phenotype associations in the HMDP data.(28).
Replication of Stage Co-Expression Networks in Starnet
To assess reproducibility of STAGE co-expression networks, gene symbols for microarray probes in each tissue were mapped to normalized STARNET RNAseq data. For genes matching the STAGE networks, pairwise Pearson’s correlations in STARNET data were computed. The average absolute correlation was used as a measure of module connectivity. For each candidate module, connectivity was compared to an empirical null distribution from 1,000 random permutations with identical numbers of genes per tissue. To account for differences in RNA biases between microarray and RNA-seq technologies, permutations were also matched to yield identical distributions of RNA categories (e.g., protein coding, pseudogenes, and lincRNA) in each tissue. A Benjamini-Hochberg test(33) was used to adjust for multiple testing.
Assessment of CAD Heritability
To calculate heritability contributions from eSNPs in the 98 CAD networks inferred from STAGE(8, 11) and STARNET,(12) we used individual-level genotype data from a pool of GWAS datasets (Online Table 1). For SNPs not originally available in these GWAS datasets, ±500-kb flanking regions were assessed for the best proxy with a linkage disequilibrium (LD) (r2>0.8). Genome relationship matrixes for each eSNP list were calculated with LDAK(34) and adjusted for LD and minor-allele frequency of 5%. To eliminate potential population stratification or study batch biases, we made further adjustments based on the top-20 multiple dimensions derived from individual-level genotype data. With a CAD population prevalence set at 5% and the portion of CAD heritability at 40% (i.e. H2) CAD variance explained in a liability model was calculated with the restricted maximum likelihood method (REML) with GCTA.(35) Unlike traditional multifactorial liability threshold models typically used for heritability assessments of independent lead SNPs in GWAS,(5) REML enables assessing heritability from groups of multiple SNPs (35). Importantly, REML produced similar assessments of CAD heritability from lead SNPs as previously reported using the multifactorial liability threshold model in GWAS (4, 5) (Online Figure 2). In most recent GWAS(4, 5, 36), 302 so-called “lead SNPs” were identified associated with CAD at the genome-wide significant (P<5×10−8) or a false discovery rate (FDR) <5% levels. These 302 SNPs jointly contribute to ~22% of H2. Thus, the origin of ~78% of H2 in CAD remains unaccounted for.
Results
We used 171 RGNs previously derived using co-expression and Bayesian algorithms applied to genotype and gene expression data from seven metabolic and vascular tissues in the STAGE study.(8, 11) By assessing the average connectivity (absolute Pearson’s correlation) compared to the null distribution for network genes matched by tissue, we first replicated 98 of these STAGE networks in STARNET RNA sequencing data(12) (FDR<0.2, Online Figure 3). We then used eSNPs also derived from STARNET(12) to assess heritability contributions to CAD from the replicated RGNs by applying REML(35) to 13,612 CAD cases and 13,758 controls. Prior to this analysis, to ensure that any contribution to CAD heritability from the RGNs is independent of that from the lead SNPs found in GWAS, we excluded from the analysis all SNPs with known genome-wide significant and FDR<5%(4, 5) associations with CAD as well as all SNPs in their LD (r2>0.2).
eSNPs in the 98 RGNs replicated in STARNET were found contribute to an additional 11.9% of CAD heritability (i.e., H2) in addition to the ~22% from known GWAS loci for CAD (Figure 1). The majority of this substantial addition to CAD heritability was identifiable in the top-28 networks (10.0% of H2), each with a relative contribution to CAD heritability >0.2% (Figure 1, Online Table 2). Compared to all independent imputed SNPs (n=1 million) in GWAS(37) with a background CAD H2/SNP contribution of 0.0001% (and a total CAD H2 contribution of 78%, Online Table 2), the average H2 contribution of eQTLs of the top-28 RGNs (with a total CAD H2 contribution of 10.0%) is 0.0072% (Online Table 2). Compared to randomized groups (n=100) of SNPs regulating expression of genes matched by tissue in GTEx (38), the H2 contributions of eQTLs in networks isolated from patients with CAD in STAGE/STARNET are on average 2–5 fold higher (P<0.001, Online Table 2).
Figure 1. Contribution to CAD heritability from expression (e)SNPs of regulatory gene networks identified in STAGE/STARNET.

Bar plots of CAD heritability (H2) contributions from 98 regulatory-gene networks identified in the STAGE study(11) and replicated in STARNET(12) (blue). The yellow bar indicates contribution to H2 from the top-28 networks, each contributing >0.2% CAD heritability. The following bars show H2 contributions from subsets of these 28 regulatory gene networks according to their tissue of origin (n = number of networks). Non-fat refers to networks found in tissues other than fat; H2 error bars are standard error of mean.
Analysis of the 28 RGNs by tissue of origin showed that 9 in fat (subcutaneous or visceral abdominal) and 7 in atherosclerotic arterial wall respectively contribute to 5.0% and 3.1% of CAD heritability (H2) (Figure 1). The contributions to CAD heritability from each RGN in relation to size are shown in Figure 2. As might be expected, larger RGNs with more genes tended to contribute to larger fractions of CAD heritability than smaller RGNs. However, this tendency was not significant (Spearman rank correlation, r=0.36, P=0.06), a fact that may relate to differences in the number of eSNPs per network and the individual contributions of these eSNPs to CAD heritability. The individual contributions of eQTLs in the top 28 networks to CAD heritability are shown in Online Table 3.
Figure 2. Individual contributions to heritability from top-28 regulatory-gene networks associated with CAD.

Bar plots of CAD heritability (H2) contributions (blue, left y-axis) and network-gene number (red, right y-axis) of the top-28 regulatory-gene networks (>0.2% h2/network, Online Figures 4 and 5).
We next assessed the 28 RGNs by their gene members for functional relevance according to GO. Fourteen of the 28 RGNs were enriched for biological processes previously implicated in CAD/atherosclerosis, such as cell adhesion, immune and defense responses, scavenger receptor activity, apoptosis and blood coagulation. Four networks had no GO annotation (Figures 3 and 4, Online Figures 4 and 5 and Online Tables 3–6).
Figure 3. Examples of two regulatory gene networks in the arterial wall and whole blood with substantial contributions to CAD heritability.


Shown are two tissue-specific regulatory-gene networks contributing to 0.64% and 0.41% to CAD heritability, respectively. These networks were identified in gene expression data from the internal mammary artery (IMA) (A) and whole blood (B) isolated from CAD patients in the STAGE study(11) and replicated in RNAseq data from corresponding tissues in STARNET.(12) Circles in the network illustrations indicate individual genes. Squares indicate key disease drivers.
Figure 4. A regulatory-gene network identified in abdominal fat with major contribution to CAD heritability.

This regulatory-gene network example contributing to 0.57% of CAD heritability was identified in gene expression data from visceral abdominal fat (VAF) from CAD patients in the STAGE study (11) and replicated in RNAseq data from the same tissue in STARNET.(12) Circles in the network illustration indicate individual genes. Squares indicate key disease drivers.
In order to further analyze the functional implications of these RGNs, we assessed their associations with CAD-relevant phenotypes. In this respect, the principal components of the expression levels of all network genes (i.e., their eigengene values) were associated with continuous phenotypes including the extent of coronary atherosclerosis as assessed in preoperative angiographs and established CAD risk factors such as levels of plasma lipids and glucose. Twenty-six out of the 28 RGNs were significantly associated with CAD, or at least one CAD-relevant phenotype (Figures 3 and 4, Online Figures 4 and 5 and Online Tables 3–6).
In principle, an association between a RGN eigengene and a given phenotype cannot unequivocally distinguish a causal relationship from a reactive one, where the phenotype is affecting the activity of the network (e.g., levels of plasma lipid impacting the gene activity of an arterial wall network).(39) In contrast, RGNs controlled by eSNPs that contribute to heritability (i.e., those associated with disease in GWAS data) imply a causal relationship, since the affected phenotype always is downstream of the regulatory eSNPs.(13) eSNP in sixteen of the top-28 networks were, in addition to CAD, also associated with at least one CAD-relevant phenotype such as plasma lipids and glucose levels according to corresponding GWAS data.(11) In addition, fourteen of the top-28 networks were enriched in genes previously implicated in research of CAD or atherosclerosis according to text mining(11) (Figures 3 and 4, Online Figures 4 and 5 and Online Tables 3–6).
Lastly, using the Hybrid Mouse Diversity Panel (HMDP) data(28), we examined if the described network eigengene-phenotype associations in humans are conserved across species. We assessed this by investigating if the network eigengene values of corresponding gene orthologs are conserved in equivalent tissues isolated from 105 strains of mice bread onto an atherosclerosis susceptible background (i.e., ApoE4 Leiden back crosses).(28) We found that, in mice, seven of the top-28 networks were associated with at least on similar CAD-relevant phenotype also found in CAD patients (Figures 3 and 4, Online Figures 4 and 5 and Online Tables 3–6).
The relatively high number of genes in most RGNs makes experimental validation challenging. We therefore also inferred the key disease drivers in each network.(31) These genes have proven to be good targets for experimental validation(14) and, as suggested,(40) for pharmaceutical interventions. Within the top-28 RGNs, we identified a total of 188 key drivers, averaging 6–7 per network, and ranging from 1–28 key drivers per network (Figures 3 and 4, Online Figures 4 and 5 and Online Tables 3–6).
Two examples from the top-28 RGNs (Online Figures 4 and 5)—one active in the arterial wall and one in whole blood—are shown in Figure 3. One of these networks identified in the non-atherosclerotic internal mammary artery comprised 122 genes and was also found to be associated with the extent of coronary atherosclerosis (P<0.05) in STAGE and contributes to as much as 0.64% of CAD heritability (H2). It also contains five key disease drivers (CCDC55 (NSRP1), TRIP11 ZNF37A, ZNF83, ZNF138) suggesting a role in transcriptional regulation, which was confirmed by GO analysis (Figure 3A). Transcriptional regulation has been sparsely studied in relation to CAD/atherosclerosis, which was confirmed by the observation that only 12.5% of the genes in this network have been implicated in previous studies of CAD or atherosclerosis.(11) Another network found in whole blood with 100 genes and 8 key disease drivers (GUCY1A1, GUCY1B1, ABLIM3, GFI1B, LY6G6F, MFAP3I, PTCRA, TAL1) contributed to 0.41% of CAD heritability. According to GO enrichment, genes in this network are involved in blood coagulation (P=1.27e−19). In its center is soluble guanylate cyclase, a heterodimeric protein formed by GUCY1A1 and GUCY1B1 and activated by nitric oxide, that catalyzes the conversion of GTP to 3’,5’-cyclic GMP and pyrophosphate, thereby causing vasodilation and inhibiting platelet function, i.e. central disease processes affected in atherosclerosis. Prior studies have implicated as many as 52 of the 100 genes in this network in CAD or atherosclerosis (P=4.66e−8).
Nine of the top-28 RGNs contributing to 5.0% of CAD heritability were identified in fat (Figure 1). One interesting example among these adipose networks is shown in Figure 4. This visceral abdominal fat RGN was found to contribute to as much as 0.57% of CAD heritability and to comprise 139 genes, including 10 key disease drivers (C2orf63, BCLAF1, MLL5, SCLT1, SP100, THOC1, XAF1, ZNF33A, ZNF92, ZNF136) (Figure 4). GO analysis indicated strong involvement of this network in RNA processing (P=1.17e−7), similarly as noted above for the arterial wall network (Figure 3A), a biological process that has scarcely been studied in relation to CAD or atherosclerosis (Figure 4). Nonetheless, eSNPs in this network were strongly enriched in associations with plasma levels of HDL (2.8-fold, P<8.93e−33), LDL (4.5-fold, P=1.99e−117), and pro-insulin (4.02-fold, P=3.75e−84) according to integrative analysis with corresponding GWAS,(41, 42) suggesting an indirect role in CAD by modifying levels of plasma lipids and pro-insulin. This notion is further supported in the HMDP, where orthologs of the 139 human genes in mouse adipose were found to be associated with levels of plasma LDL (P<0.003).
The individual contributions of the top-28 RGNs and their eQTLs to CAD heritability, along with key disease drivers, main gene ontologies, phenotype associations, possible enrichment in genetic association according to genotype data from GWAS and cross-species conservation assessments using the HMDP are shown in detail in Online Figures 4 (tissue-specific networks) and 5 (cross-tissue networks) and Online Tables 3–6.
Discussion
In this study, three main observations that broaden the view of CAD pathophysiology were made. First, we identified a number of regulatory gene networks contributing to a substantial proportion of CAD heritability of CAD. Second, of all tissues studied fat and arterial wall harbored the regulatory gene networks that exerted the strongest influence on the risk of CAD. Third, these regulatory gene networks (and their eSNPs) define, in addition to known CAD risk factors, a number of biological functions involving DNA binding, RNA metabolism, and blood coagulation with causal roles in CAD pathogenesis. From a broader perspective, the CAD heritability contribution of these regulatory gene networks establishes systems genetics as a “top-down” interpretation of complex disease biology – complementary to established approaches mostly with a gene/pathway “bottom-up” perspective – with the common goal of unbiased characterization of molecular interactions within and across specific disease-relevant tissues leading to CAD.
Risk factors for common complex disorders are traditionally divided into two categories: genetic (inherited) and environmental (e.g., smoking, sedentary life style, food intake) (Central Illustration) each contributing about 40–60% of disease risk.(3) However, in reality, it is unlikely that genetic and environmental risk portions operate separately (other than perhaps a small fraction). Instead, it is inevitable that they mostly interact(10, 43) in that a substantial number of risk alleles exert their disease-causal effects only after interactions with certain environmental factors (“environmentally-triggered risk alleles”) (Central Illustration; Online Figure 1). Such interactions may become evident at the level of transcriptional regulation and ultimately are likely to affect the regulatory gene networks studied here. Importantly, since the genetic variants affecting these regulatory gene networks account for a large proportion of disease heritability, it is reasonable to conclude that these networks reflect causal mechanisms driven by both genetic and environmental factors. Thus, further analysis of the most significant networks may help explaining CAD etiology and give novel insights on how to prevent its development.
The current study adds a substantial number of genetic variants (Online Table 3) contributing to CAD heritability beyond the 160 chromosomal loci with genome-wide significant association signals identified by GWAS of CAD to date.(2) The most immanent challenge following identification of these associations is to unravel their downstream functional consequences. The joint analysis of genetic variants affecting expression levels (eSNPs) and interacting transcriptional networks, as carried out in this analysis, offers a first step in this direction. In fact, regulatory gene networks harboring genetic variance of CAD constitute the beginning of a novel framework of gene-gene interactions across metabolic and vascular tissues that will be essential to enable linking DNA risk variants at the top of information flow to the end stage phenotypic changes in the forms of CAD and myocardial infarction.
In CAD GWAS data, environmentally triggered risk SNPs will not display genome-wide significant association. Rather, they will more likely present themselves with weak statistical significance due to the fact that the presence of macro- or microenvironmental triggering factors (unlike clinically significant CAD) are not uniquely distributed in the case or control groups of GWAS. In this study, we discovered that cis eQTLs of genes in networks built from both genetic and environmental variation in CAD patients are useful to filter sub-significant SNPs in GWAS data that truly contribute to CAD variation from SNPs with marginal or no contribution to H2.
The understanding of biological networks in complex disease biology is still in its infancy.(10) Indeed, RGNs identified herein (Online Figures 4 and 5) both strongly align with our current understanding of CAD biology but in many ways also challenges the same: The RGN affecting coagulation in whole blood (Figure 3B) makes strong sense in relation to our current understanding of CAD and risk for myocardial infarction,(44) In contrast, the arterial wall and fat networks (Figure 3A and 5) with major contributions to CAD heritability respectively involve transcriptional regulation and RNA processing (the latter suggesting non-coding RNA involvement), which have not been widely studied in relation to CAD. The conceptual novelty of the identified networks is particularly evident looking at those consisting of gene/nodes from several tissues (Online Figure 5). As a group these cross-tissue networks had the third largest contribution to CAD heritability after those in fat and the arterial wall (Figure 1). The nature of cross-tissue networks may at first seem obscure. However, cross-tissue gene interactions may appear less surprising when one considers that key risk factors for CAD such as plasma lipid and glucose levels are not uniquely regulated based on gene activity in single tissues, but depend on synthesis, secretion and uptake taking place in several tissues. The underlying biological explanation(s) for gene interactions across tissues remain to be determined but a recent study suggests that a panoply of largely uncharacterized secretory proteins may play central endocrine roles for cross-tissue communication of genes (45).
Despite remaining challenges such as lack of detailed pathway information, regulatory gene networks described in the literature have proven to be evolutionary well-conserved(46) and contain key disease driver genes,(16) some of which have been experimentally validated.(14, 15) In this study, we extensively evaluated the CAD networks besides enrichments in biological processes according to gene ontology, first by replication using the independent STARNET RNA sequence data collection, then in relation to relevant CAD phenotypes using both network eigengene associations and integrative analyses of network (e)SNPs in GWAS data for risk factors of CAD. Last, we found evidence for cross-species conservation of identified networks using data from evolutionary diversity in mice (28).
Study Limitations
Additional molecular, anatomical, and functional data with temporal resolution at the tissue, cell-type and single-cell levels in a range of CAD-model systems and from spectrums of human ethnicities are needed to achieve a complete understanding of the framework of RGNs active within and across tissues leading to CAD. We believe that with increasing accuracy and resolution of regulatory-gene network models for complex diseases, the more they will be found to contribute to heritability and thus, help us to improve our understanding of complex disease biology.(7–9, 11) Importantly, network eQTLs may also capture non-additive inheritance, including gene-environment, gene-gene and gene-age interactions.
Conclusions
We found that genetic variants in disease-causal gene networks contribute to a major portion of previously unidentified CAD heritability.(11, 12) The regulatory gene networks with the strongest influence on CAD risk were found in fat and the arterial wall, which by inference signifies these tissues as being particularly important in understanding the pathobiology of CAD. A major step in the future battle of CAD, we believe, will be to assess how our knowledge and understanding of these network models can achieve earlier prevention, diagnosis and more effective network-focused therapies.
Supplementary Material
Clinical Perspectives.
Competency in Medical Knowledge:
Single nucleotide polymorphisms (SNPs) in regulatory networks increase the heritability relevant of coronary artery disease (CAD) contribute to beyond the amount explained by genome-wide association studies (GWAS).
Translational Outlook:
Identified network expression SNPs should be included in polygenetic risk scores to improve the precision of CAD risk prediction, and key driver genes in identified CAD networks explored as novel therapeutic targets.
Funding:
Johan LM Björkegren acknowledges research support from NIH R01HL125863, American American Heart Association (A14SFRN20840000), Swedish Research Council (2018–02529) and Heart Lung Foundation (20170265), Foundation Leducq(PlaqueOmics: Novel Roles of Smooth Muscle and Other Matrix Producing Cells in Atherosclerotic Plaque Stability and Rupture, 18CVD02; and CADgenomics: Understanding CAD Genes, 12CVD02]) and Astra-Zeneca. Heribert Schunkert’s and Lingyao Zeng’s work is funded by the Deutsche Forschungsgemeinschaft (DFG) as part of the Sonderforschungsbereich CRC 1123 (B2). Further grants were received from the Fondation Leducq [CADgenomics: Understanding CAD Genes, 12CVD02], the German Federal Ministry of Education and Research (BMBF) within the framework of ERA-NET on Cardiovascular Disease [Joint Transnational Call 2017, 01KL1802], within the framework of target validation [BlockCAD: 16GW0198K], within the framework of the e:Med research and funding concept [AbCD-Net: grant 01ZX1706C], and DigiMed Bayern. Jason C. Kovacic acknowledges research support from the National Institutes of Health (R01HL130423) and Foundation Leducq.
Abbreviations
- CAD
Coronary artery disease
- RGN
Regulatory gene network
- GWAS
Genome-wide association study
- SNP
Single nucleotide polymorphism
- eSNP
Expression SNP
- eQTL
Expression quantitative expression trait
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: Li-Ming Gan - Employee of AstraZeneca R&D; Johan Bjorkegren - Founding CEO of Sema4, On board of directors of CGN; Tom Michoel - Clinical Gene Networks AB, shareholder & consultant. The remaining authors have nothing to disclose.
References
- 1.Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994. April 14;330(15):1041–6. [DOI] [PubMed] [Google Scholar]
- 2.Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018. July 15;114(9):1241–1257. doi: 10.1093/cvr/cvy084. [DOI] [PubMed] [Google Scholar]
- 3.McPherson R, Tybjaerg-Hansen A. Genetics of Coronary Artery Disease. Circ Res. 2016. February 19;118(4):564–78. doi: 10.1161/CIRCRESAHA.115.306566. [DOI] [PubMed] [Google Scholar]
- 4.Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015. October;47(10):1121–1130. doi: 10.1038/ng.3396.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson CP, Goel A, Butterworth AS et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017. September;49(9):1385–1391. doi: 10.1038/ng.3913. [DOI] [PubMed] [Google Scholar]
- 6.Schadt EE, Bjorkegren JL. NEW: network-enabled wisdom in biology, medicine, and health care. Sci Transl Med. 2012. January 4;4(115):115rv1. doi: 10.1126/scitranslmed.3002132. [DOI] [PubMed] [Google Scholar]
- 7.Skogsberg J, Lundström J, Kovacs A,et al. Transcriptional profiling uncovers a network of cholesterol-responsive atherosclerosis target genes. PLoS Genet. 2008. March 14;4(3):e1000036. doi: 10.1371/journal.pgen.1000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hägg S, Skogsberg J, Lundström J,et al. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and LIM domain binding 2: the Stockholm Atherosclerosis Gene Expression (STAGE) study. PLoS Genet. 2009. December;5(12):e1000754. doi: 10.1371/journal.pgen.1000754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Björkegren JL, Hägg S, Talukdar HA et al. Plasma cholesterol-induced lesion networks activated before regression of early, mature, and advanced atherosclerosis. PLoS Genet. 2014. February 27;10(2):e1004201. doi: 10.1371/journal.pgen.1004201. eCollection 2014 Feb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Björkegren JLM, Kovacic JC, Dudley JT, Schadt EE. Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J Am Coll Cardiol. 2015. March 3;65(8):830–845. doi: 10.1016/j.jacc.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talukdar HA, Foroughi Asl H, Jain RK et al. Cross-Tissue Regulatory Gene Networks in Coronary Artery Disease. Cell Syst. 2016. March 23;2(3):196–208. doi: 10.1016/j.cels.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franzén O, Ermel R, Cohain A et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science. 2016. August 19;353(6301):827–30. doi: 10.1126/science.aad6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schadt EE, Lamb J, Yang X et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005. July;37(7):710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shang MM, Talukdar HA, Hofmann JJ et al. Lim domain binding 2: a key driver of transendothelial migration of leukocytes and atherosclerosis. Arterioscler Thromb Vasc Biol. 2014. September;34(9):2068–77. doi: 10.1161/ATVBAHA.113.302709. [DOI] [PubMed] [Google Scholar]
- 15.Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013. April 25;153(3):707–20. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang IM et al. , Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol 8, 594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schadt EE, Molecular networks as sensors and drivers of common human diseases. Nature 461, 218–223 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Foroughi Asl H, Talukdar HA, Kindt AS et al. Expression quantitative trait Loci acting across multiple tissues are enriched in inherited risk for coronary artery disease. Circ Cardiovasc Genet. 2015. April;8(2):305–15. doi: 10.1161/CIRCGENETICS.114.000640. [DOI] [PubMed] [Google Scholar]
- 19.Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007. August 2;357(5):443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erdmann J, Grosshennig A, Braund PS et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009. March;41(3):280–2. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erdmann J, Willenborg C, Nahrstaedt J, et al. Genome-wide association study identifies a new locus for coronary artery disease on chromosome 10p11.23. Eur Heart J. 2011. January;32(2):158–68. doi: 10.1093/eurheartj/ehq405. [DOI] [PubMed] [Google Scholar]
- 22.Nikpay M, Goel A, Won HH, et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015. October;47(10):1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myocardial Infarction Genetics Consortium Investigators, Stitziel NO, Won HH, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014. November 27;371(22):2072–82. doi: 10.1056/NEJMoa1405386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.W. T. C. C. Consortium., Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.C. A. D. Consortium et al. , Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet 45, 25–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kathiresan S et al. , Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet 41, 334–341 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tryka KA et al. , NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 42, D975–979 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett BJ et al. , A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 20, 281–290 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang B, Horvath S, A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol 4, Article17 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Schmidt M, Niculescu-Mizil A, Murphy K, Learning graphical model structure using L1-regularization paths. AAAI’07 Proceedings of the 22nd National Conference on Artificial Intelligence 2, 1278–1283 (2007). [Google Scholar]
- 31.Zhang B, Zhu J, Identification of key causal regulators in gene networks. Proceedings of the World Congress on Engineering 2013 II, 1309–1312 (2013). [Google Scholar]
- 32.Ashburner M et al. , Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glueck DH, Mandel J, Karimpour-Fard A, Hunter L, Muller KE, Exact calculations of average power for the Benjamini-Hochberg procedure. Int J Biostat 4, Article 11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Speed D, Hemani G, Johnson MR, Balding DJ, Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet 91, 1011–1021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J et al. , Common SNPs explain a large proportion of the heritability for human height. Nat. Genet 42, 565–569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howson JMM et al. , Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat. Genet 49, 1113–1119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.International HapMap C et al. , A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.The GTEx Consortium, The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vilne B et al. , Network analysis reveals a causal role of mitochondrial gene activity in atherosclerotic lesion formation. Atherosclerosis 267, 39–48 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Braenne I et al. , Prediction of Causal Candidate Genes in Coronary Artery Disease Loci. Arterioscler Thromb Vasc Biol 35, 2207–2217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teslovich TM et al. , Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dupuis J et al. , New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet 42, 105–116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khera AV et al. , Genetic Risk, Adherence to a Healthy Lifestyle, and Coronary Disease. N Engl J Med 375, 2349–2358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wobst J, Schunkert H, Kessler T, Genetic alterations in the NO-cGMP pathway and cardiovascular risk. Nitric Oxide 76, 105–112 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Seldin MM, Koplev S, Rajbhandari P et al. A Strategy for Discovery of Endocrine Interactions with Application to Whole-Body Metabolism. Cell Metab. 2018. May 1;27(5):1138–1155.e6. doi: 10.1016/j.cmet.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hinman VF, Nguyen AT, Cameron RA, Davidson EH, Developmental gene regulatory network architecture across 500 million years of echinoderm evolution. Proc. Natl. Acad. Sci. U S A 100, 13356–13361 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orozco LD, Bennett BJ, Farber CR, et al. Unraveling inflammatory responses using systems genetics and gene-environment interactions in macrophages. Cell. 2012. October 26;151(3):658–70. doi: 10.1016/j.cell.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.