

Abstract
Acute myeloid leukemia (AML) is a lethal hematologic malignancy characterized by an immunosuppressive milieu in the tumor microenvironment (TME) that fosters disease growth and therapeutic resistance. Hypomethylating agents (HMAs) demonstrate clinical efficacy in AML patients and exert immunomodulatory activities. In the present study, we show that guadecitabine augments both antigen processing and presentation resulting in increased AML susceptibility to T cell-mediated killing. Exposure to HMA results in the activation of the endogenous retroviral pathway with concomitant downstream amplification of critical mediators of inflammation. In an immunocompetent murine leukemia model, guadecitabine negatively regulates inhibitory accessory cells in the TME by decreasing PD-1 expressing T cells and reducing AML-mediated expansion of myeloid derived suppressor cells. Therapy with guadecitabine results in enhanced leukemia-specific immunity as manifested by increased CD4 and CD8 cells targeting syngeneic leukemia cells. We have previously reported that vaccination with AML/dendritic cell fusions elicits the expansion of leukemia-specific T cells and protects against disease relapse. In the present study, we demonstrate that vaccination in conjunction with HMA therapy results in enhanced anti-leukemia immunity and survival. The combination of a novel personalized DC/AML fusion vaccine and an HMA has therapeutic potential, and a clinical trial investigating this combination is planned.
Keywords: hypomethylating agent, dendritic cell vaccine, acute myeloid leukemia, immunotherapy, endogenous retroviral pathway
Introduction
Acute myeloid leukemia (AML) is a lethal hematologic malignancy (Döhner et al, 2010; Derolf et al, 2009). Standard therapy is often inadequate in sustaining durable remissions due to the emergence of chemotherapy-resistant clones (Ding et al, 2012). AML is characterized by an immunosuppressive tumor microenvironment (TME) that promotes immune tolerance and disease escape (Isidori et al, 2014; Coles et al, 2015; Austin et al, 2016). A major area of investigation is the development of immunotherapeutic strategies that enhance the targeting of AML by immune effector cells and the development of memory responses that protect from subsequent relapse (Beyar-Katz & Gill, 2018). The unique potency of cellular immunity in targeting AML is highlighted by the observation that allogeneic transplantation (alloSCT) is curative for a subset of patients due to the targeting of leukemic cells by alloreactive lymphocytes. However, due to the lack of donor lymphocyte specificity for malignant cells, morbid sequelae of alloSCT includes graft-versus-host-disease and opportunistic infections (Copelan, 2006). Developing effective leukemia-specific immunotherapies is further impeded by the immunosuppressive milieu including impaired antigen presentation, increased regulatory T-cells, dysregulation of checkpoint pathways, and an increased burden of myeloid-derived suppressor cells (MDSCs) (Andersen, 2014; Schlößer et al, 2014; Sharma & Allison, 2015; Isidori et al, 2014). In contrast, therapeutic agents with immunomodulatory properties can potentially overcome immune tolerance, enhance tumor immunogenicity, and induce the expansion of functionally competent leukemia-specific T cells (Lichtenegger et al, 2015).
Hypomethylating agents (HMAs) demonstrate potent anti-leukemia effects with the capacity to induce disease remission in a subset of AML patients (Kantarjian et al, 2012; Ritchie et al, 2013; Lübbert et al, 2011). While the mechanism of action of HMAs in AML is diverse, there is increasing appreciation of immunomodulatory effects in mediating response. In solid tumor models, HMAs exert regulatory effects on the antigen presenting properties of malignant cells (Karpf, 2006; Ørskov et al, 2015a; Almstedt et al, 2010; Akers et al, 2010; Woloszynska-Read et al, 2008; Goodyear et al, 2010; Srivastava et al, 2014). Pre-treatment of tumor cells with HMAs primes tumors to induce potent immune responses. Azacitidine exposure yielded an enhanced response to checkpoint antibody therapy in a lung cancer model (Juergens et al, 2011). Moreover, HMA treatment accentuated responses to anti-CTLA-4 immunotherapy in a melanoma model through activation of the endogenous retroviral pathway (ERV) (Chiappinelli et al, 2015). The ERV pathway is an evolutionary conserved pathway that when stimulated, culminates in T cell activation through IFN production (Stoye, 2012). In addition, HMAs potentially impact functional properties of accessory and immune effector cells that populate the TME and mediate tumor-associated immune suppression.
In the present study, we characterize the immunoregulatory effects of the second generation HMA, guadecitabine (SGI-110), on antigen presentation by leukemia cells, recruitment of MDSCs, and expansion of functionally-competent leukemia-specific T cells. We demonstrate that SGI-110 enhances presentation of the leukemia antigen, PR-1, associated with increased expression of the antigen processing enzyme, TAP2. Treatment with SGI-110 in an immunocompetent murine leukemia model decreases infiltrating PD-1 expressing T cells and MDSCs at the tumor site resulting in increased levels of functionally-active leukemia-specific T cells. Immunoregulatory effects of SGI-110 are associated with the signaling via the ERV pathway providing a pro-inflammatory environment promoting T cell activation.
We have developed a personalized tumor vaccine in which patient-derived tumor cells are fused with autologous dendritic cells (DCs) such that a broad array of tumor antigens are presented in the context of DC-mediated co-stimulation (Rosenblatt et al, 2005; Rosenblatt & Avigan, 2006). In a phase I/II clinical trial of 17 AML patients achieving a chemotherapy-induced remission, vaccination resulted in the durable expansion of leukemia-specific T cells. Despite a median age of 63 years, 71% of patients remained alive without disease recurrence at a 57-month median follow-up (Rosenblatt et al, 2016a). Despite these encouraging results, vaccine durability remains uncertain as responses may diminish over time due to immune tolerance. We hypothesized that HMA therapy would enhance response to vaccination by increasing the immunogenicity of the leukemia cell target and altering the immunosuppressive milieu of the TME. In a murine leukemia model, we observed that vaccination in conjunction with SGI-110 results in increased expansion of leukemia-specific T cells and improved survival in mice treated with SGI-110 plus the fusion vaccine as compared to mice treated with either agent alone.
Methods
Cell culture
The AML cells lines THP-1, MV4–11, MOLM-14 (human) and TIB-49 (murine) were purchased from ATCC, cultured at 37 °C in a humidified 5% CO2 incubator and maintained in RPMI 1640 media (Cellgro, Manassas, VA) supplemented with heat-inactivated 10% fetal calf serum albumin (Sigma, St. Louis, MO) and 100 IU/ml penicillin, 100 μg/ml streptomycin (Cellgro). Primary human AML cells were cultured similarly in a concentration of 1 million cells per mL.
Flow cytometry
Cells were analyzed for GFP, PD-1, CD11b, and Gr1 expression by multichannel flow cytometry. Cells were incubated with FcR blocking reagent (Miltenyi, Bergisch Gladbach, Germany) for 10 min at room temperature. Subsequently, cells were incubated with the anti-PD-1-APC (eBioscience, San Diego, CA), anti-CD11b-APC-Cy7 (BioLegend, San Diego, CA) and anti-Gr1-PE (BioLegend, San Diego, CA) or isotype control. The cells were analyzed using FACS Aria (BD Biosciences, San Jose, CA). Kaluza software (Beckman Coulter, Brea, CA) was used to analyze the obtained data.
The expression of IFN-γ was analyzed by intracellular flow cytometry. Cells were pulsed with GolgiStop (1 μg/ml; BD Pharmingen) for 4–6 h at 37 °C before analysis. Cells were next harvested and labeled with CD4-PB and CD8-FITC. Cells were then permeabilized by incubation in Cyto-fix/Cytoperm plus (BD Pharmingen) containing formaldehyde and saponin for 30 min at 4 °C, washed twice in Perm/Wash solution, and incubated with PE-conjugated IFN-γ (Invitrogen, Camarillo, CA), or a matched isotype control for 30 min. Cells were washed in 1 × Perm/Wash solution prior to analysis.
T cell receptor-like (TCRL) antibody assessment
The HLA-A2 positive cell line, THP-1, was treated with SGI-110 1 μM twice daily for two days. The cells were analyzed for PR-1 presentation using a TCRL Ab (8F4 mAb) as described [Ref 28]. HLA-A2 expression was assessed with anti–HLA-A2 mAb BB7.2.
Immunohistochemistry
AML cells were obtained from the BM of patients with disease. Cytospins were prepared after isolation using anti-CD34 magnetic beads. Cells were stained with anti-TAP2 (Novus Biologicals) or goat-anti-mouse IgG using the Vectastain ABC Kit (Vector Laboratories). Cells were fixed in 2% paraformaldehyde (Sigma-Aldrich) and visualized by phase contrast light microscopy (Olympus AX70 microscope) using an oil immersion objective lens (100X).
Western blot
MOLM-14 and MV4–11 were treated with low-dose decitabine (25nM and 50nM) daily X 3 days. Cells were assessed for ERV-3 (anti-ERV-3 Everest Biotech, Oxfordshire, UK, 30 kD) and IFNβ (anti-IFNβ Abcam, Cambridge, UK, 22 kD) expression.
Leukemia-specific immunity
Autologous DC/AML fusions were generated. Leukemia cells were obtained from BM aspirates from AML patients with active disease and cryopreserved as per an IRB-approved protocol. PB samples were subsequently obtained from patients achieving complete remission and PBMCs were isolated via ficoll density centrifugation. DCs were generated from adherent mononuclear cells cultured with GM-CSF, IL-4 and TNF alpha. DC and AML cells were fused by co-culture with polyethylene glycol (PEG). Fusions were cultured at a 1:10 ratio with autologous T cells for 6 days with 1μ;M SGI-110 or with diluent control. CD4+ and CD8+ populations expressing IFN-γ production following stimulation with the vaccine were quantified. To assess for cytolytic capacity, autologous patient-derived T cells were cultured with fusions for 5–7 days. Fusion-stimulated T cells were assessed for their capacity to lyse leukemia cells using a cytotoxic T lymphocyte (CTL) assay.
CTL assay
Fusion cells were generated as described above. Autologous T cells were stimulated with the fusion vaccine for 6 days. Vaccine-stimulated T cells were co-cultured with control autologous AML blasts or AML blasts exposed to twice daily treatment with 1 μM SGI-110 for 2 days and left in culture for an additional 2 days. The lysis of AML blasts, with and without SGI-110, by vaccine-stimulated T cells was detected using a CTL flourochrome assay (OncoImmunin Inc., Gaithersburg, MD, USA). Target AML cells were incubated in APC labeled phosphate-buffered saline (1 μl of reconstituted TFL4 in phosphate-buffered saline at 1:3000 ratio) at 1 × 106 cells/ml for 30 min at 37 °C. Labeled cells were washed twice in phosphate-buffered saline. Using SGI-110 treated or control human AML cells obtained through an IRB-approved protocol, autologous PBMCs were co-incubated with labeled target cells in the presence of a fluorogenic granzyme B substrate for 1–2 h at 37 °C. Cells were washed and analyzed by flow cytometry. Dead target cells were identified through dual staining for granzyme B and APC label. As a negative control, granzyme B positive tumor cells not co-cultured with T cells were quantified.
TME and leukemia-specific immunity in-vivo
C57BL/6J mice were inoculated with 1 X 105 GFP-transduced TIB-49 syngeneic AML cells via retro-orbital injection. Animals were treated with SGI-110 or diluent for 3 days, sacrificed on day 18 and assessed for tumor burden, MDSCs, and T cell PD-1 expression by flow cytometry. AML-specific T cells were quantified in BM and spleen by flow cytometry for intracellular IFN-γ expression following exposure to TIB-49 lysate.
Synergy between fusions and HMA therapy in-vivo
Murine syngeneic DC/AML fusion cells were generated. DCs were generated from the BM mononuclear cells harvested from C57BL/6J mice cultured with IL-4 and GM-CSF for 5–7 days and fused with GFP+ syngeneic TIB-49 cells in the presence of PEG. Fusion cells were isolated by flow cytometric sorting for cells co-expressing CD86/GFP. C57BL/6J mice were retro-orbitally inoculated with 50 × 103 GFP-transduced syngeneic TIB-49 cells. The animals were treated with SGI-110 alone, fusions alone or the combination. Mice were bled to assess disease burden via GFP+ in the PB. Mice were observed for survival. Spleen cells were harvested and assessed for tumor burden and T cell PD-1 expression by flow cytometry. Cells were also stimulated ex-vivo with TIB-49 lysate. Following 6 days of stimulation, CD4+ and CD8+ T cells underwent flow cytometric analysis for intracellular IFN-γ expression.
Results
HMA treatment results in an enhanced inflammatory anti-tumor immune response while downregulating immune inhibition.
HMA-mediated enhancement of antigen presentation has been documented in solid tumor models. Accordingly, we first examined whether exposure to HMA in-vitro could impact the immunogenicity of AML cells via its effect on antigen presentation. Co-culture of the HLA-A2.1 positive human AML cell line, THP-1, with SGI-110 resulted in a statistically significant increase in MHC HLA-A2.1 expression (Fig 1A). We subsequently examined the impact of HMA exposure on presentation of the leukemia-associated antigen using a T-cell receptor mimic antibody (8F4) that recognizes the PR-1-MHC-HLA-A2.1 complex on the surface of leukemic cells where PR-1 is a known leukemia-associated antigen (Sergeeva et al, 2011). Exposure to SGI-110 resulted in an increase in PR-1 presentation (Fig 1B) as measured by flow cytometric analysis in THP-1 cells (n=4; p< 0.05). Of note, treatment of patient-derived AML cells with SGI-110 led to increase in TAP2 expression (8% to 75%) (Fig 1C). In contrast, co-stimulatory molecules expression of HLA-DR, CD80 and CD86 on patient-derived AML cells following exposure to SGI-110 was not altered (Fig S1A). Furthermore, the cytokine profile of AML cells was not affected following SGI-110 treatment (Fig S1B).
Figure 1. SGI-110 increases antigen presentation and processing.
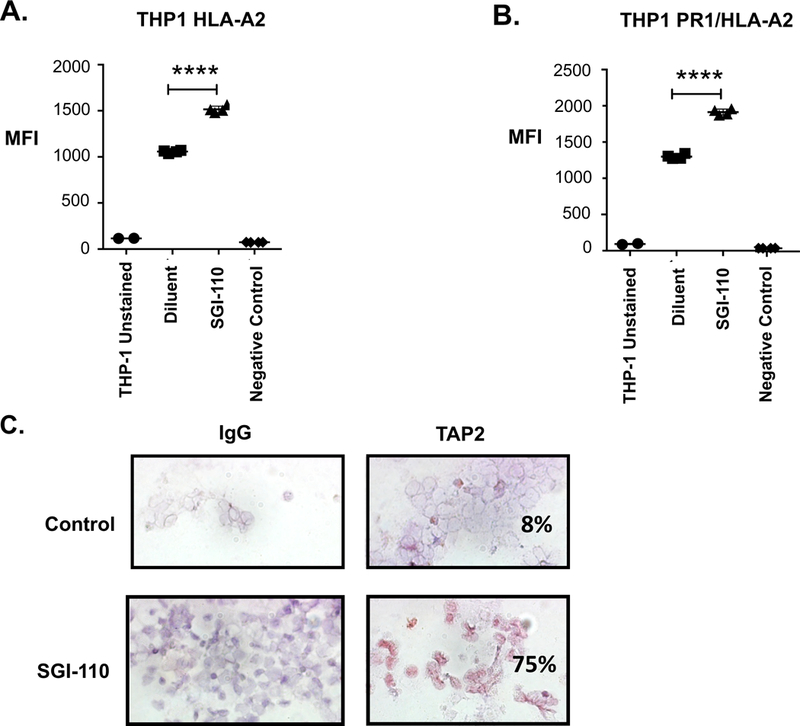
The HLA-A2 expressing AML cell line, THP-1, was treated for two days with four doses of 1μM SGI-110 added twice daily and then cultured for an additional two days. The cells were then analyzed for binding capacity of a unique T cell receptor-like (TCRL) antibody. The TCRL Ab is a TCR mimic that recognizes the PR1-HLA-A2-MHC complex on the surface of leukemic cells where PR-1 is a known leukemic antigen. Following exposure of SGI-110 to THP-1, enhanced tumor antigen presentation as evidenced by HLA-A2 upregulation (A) as well as increased binding of TCRL antibody observed via flow cytometric analysis (B). An HLA-A2 negative cell line U937 (histiocytic lymphoma) was used as a negative control. (C) AML patient-derived tumor cells were treated for two days with four doses of 1μM SGI-110 added twice daily and then cultured for additional two days. TAP2 expression was assessed using immunohistochemical staining with and without treatment with SGI-110 as shown in this representative illustration.
We subsequently examined the effect of SGI-110 on the TME in an immunocompetent murine model. C57BL/6J mice were retro-orbitally inoculated with 100,000 syngeneic TIB-49 cells stably transduced with GFP. Following establishment of disease as determined by the presence of AML cells in the peripheral blood (PB) on day 12, mice were treated with SGI-110 (1mg/kg) or vehicle daily for 3 days. Prior dose escalation in-vivo experiments were conducted to select appropriate SGI-110 dosing in our model. We aimed to select a dose that would preserve leukemic engraftment in order to best study the immune effects of SGI-110 on the AML cell. To this end, there was no significant difference in the engraftment of GFP+ TIB-49 cells in the spleens and bone marrow (BM) between the SGI-110 (1mg/kg) treated and control animals (data not shown). However, exposure to SGI-110 resulted in a significant decrease in T cells expressing PD-1 levels among CD4+ splenocytes as detected by flow cytometric analysis with mean values 6.3% and 3.1% of vehicle control and SGI-110-treated mice, respectively (n=4; p=0.01) (Fig 2A). Furthermore, a statistically significant decrease in the number of MDSCs as measured by quantification of CD11b+Gr1+ cells was observed in both the BM (n=5; p=0.01) and spleens (n=4; p=0.03) of SGI-110-treated mice, as compared to vehicle (Fig 2B). We subsequently examined whether the effect of SGI-110 on the immunogenicity of AML cells and the microenvironment results in enhanced recognition of leukemia cells by the native T cell repertoire in the immunocompetent murine leukemia model. Treatment with SGI-110 resulted in higher levels of CD4 and CD8 splenocytes expressing IFN-γ upon exposure to syngeneic TIB-49 lysate (Fig 2C).
Figure 2. SGI-110 alters the tumor microenvironment and enhances leukemia-specific immunity.
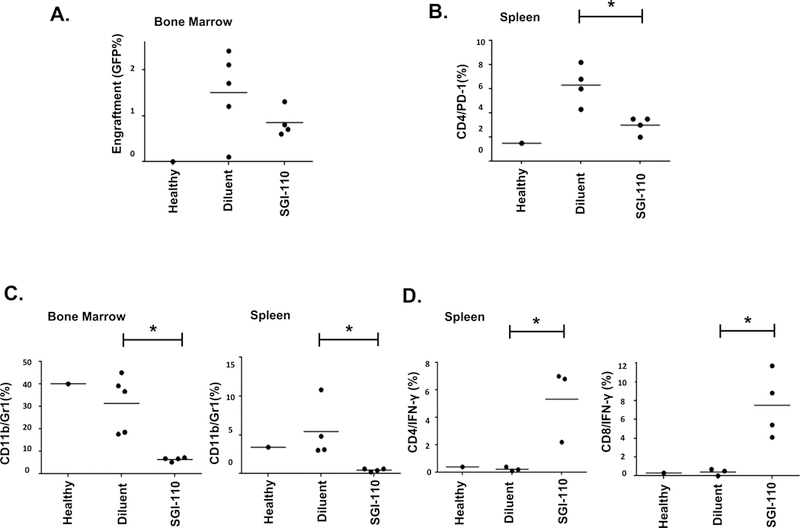
C57BL/6J mice were retro-orbitally inoculated with 100,000 syngeneic TIB-49 AML cells that were stably transduced with GFP. The mice were then treated with 1mg/kg SGI-110 or diluent control. At day 12 following inoculation, BM and spleen cells were isolated and evaluated using flow cytometric analysis for (A) BM engraftment, (B) PD-1 expression of CD4+ cells in the spleen, (C) Myeloid-derived suppressor cell (MDSC) burden in the bone marrow and spleen, and (D) intracellular IFN-γ expression after exposure to autologous tumor lysate in the bone marrow and spleen.
Exposure to HMAs activates the endogenous retroviral pathway
HMAs induce a pro-inflammatory state via the activation of native ERV pathways. Activation of the ERV pathway results in enhanced IFNß expression leading to increased MHC HLA Class I expression and subsequently, immune stimulation in a solid tumor model (Chiappinelli et al, 2015). In the present study, MOLM-14 and MV4–11 AML cell lines were co-cultured in-vitro with decitabine (DAC) at 25nM and 50nM for 3 days and analyzed for ERV-3 expression by western blotting. The results demonstrate significant up-regulation of ERV-3 expression after treatment with 50 nM DAC (Fig 3A,B). Due to activation of the ERV pathway, we examined the expression of IFNβ following HMA treatment. Indeed, IFNβ expression was shown to be upregulated following treatment (Fig 3A,B).
Figure 3. HMA upregulates the endogenous retroviral (ERV) pathway.
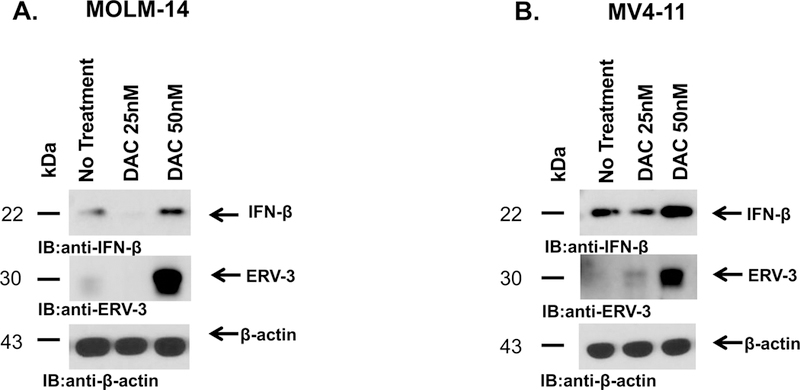
MOLM-14 (A) and MV4–11 (B) cells were exposed to decitabine (DAC) at 25nM or 50nM daily for 3 days and analyzed for ERV-3 expression using western blotting on day 4 of treatment (WB). The results showed significant upregulation of ERV-3 expression following treatment. Due to activation of the ERV pathway, increased IFNß expression was observed with WB analysis.
In-vitro stimulation of PBMCs with a fusion vaccine plus HMA increases tumor-specific T cells and tumor-specific cytolytic capacity in AML patients.
We have developed a personalized leukemia vaccine that demonstrates the potent capacity to expand leukemia-specific T cells and protection from relapse.(Rosenblatt et al, 2016b) Given its effect on antigen presentation and the leukemia-associated microenvironment, we postulated that HMA therapy would demonstrate synergy with the vaccine in generating protective leukemia-specific immunity. To interrogate this hypothesis, fusion cells were generated from patient-derived leukemic cells fused with autologous DCs and cultured at a 1:10 ratio with autologous T cells for 6 days in the presence and absence of 1μ;M SGI-110. Exposure to SGI-110 resulted in increased CD4+ population expressing IFN-γ following stimulation with the vaccine (n=4; p<0.05) (Fig 4A,B).
Figure 4. SGI-110 enhances T cell leukemia-specific response and cytolytic capacity in-vitro.
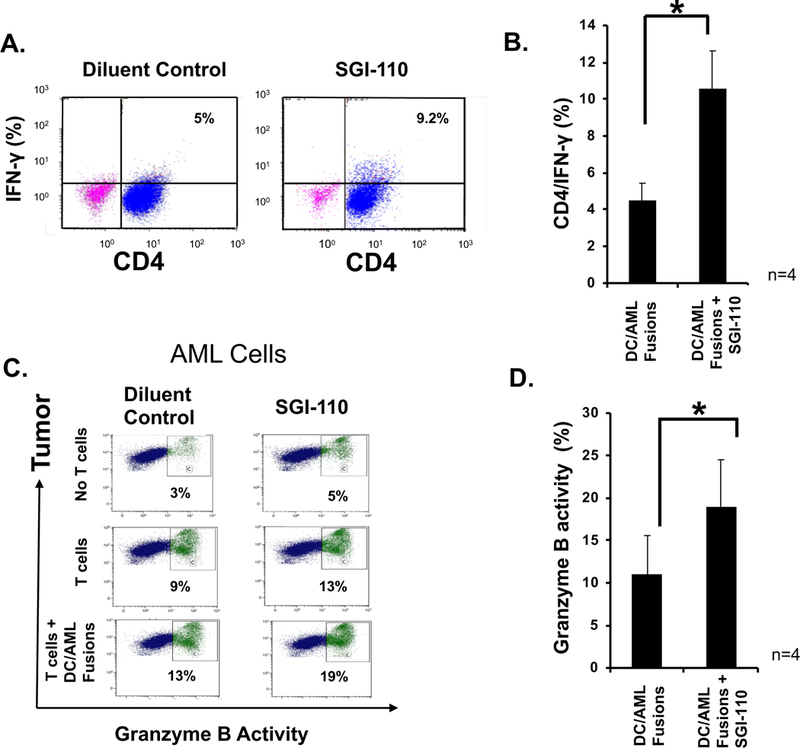
DC/AML fusion cells were generated as described. DC/AML fusions were co-cultured at a 1:10 ratio with autologous T cells for 5–6 days in the presence and absence of four doses of 1uM SGI-110 added every 12 hours. IFN-γ expression was then detected in autologous T cells using intracellular staining as shown in a (A) representative FACS plot and (B) a summary of 4 independent experiments. AML patient-derived tumor cells were treated for two days with four doses of 1uM SGI-110 added twice daily and then cultured for additional two days. Tumor cell lysis following addition of autologous T cells with and without exposure to SGI-110 was measured using cleavage of the granzyme B substrate that results in increased green fluorescence in the tumor that was detected using multi-channel flow cytometry. AML cells were identified using an APC fluorescence label. The percent of lysed cells is calculated out of total labeled target AML cells as shown in a representative FACS plot (C) and summary of 4 independent experiments (D).
Given our finding that exposure to HMAs resulted in enhanced presentation of leukemia-associated antigens, we hypothesized that in-vitro exposure to SGI-110 would augment the targeting of AML cells by vaccine-stimulated T cells. Autologous patient-derived T cells were cultured with fusions for 5–7 days. Fusion-stimulated T cells were assessed for their capacity to lyse leukemia cells in a standardized flourochrome cytotoxic T lymphocyte (CTL) assay. First, autologous AML cells were exposed to 4 doses of 1μM SGI-110 or diluent and then incubated for an additional 2 days. Results show that lysis of leukemia cells by resting T cells and vaccine-stimulated T cells is augmented by pre-incubation of the AML cells with SGI-110 (n=4; p<0.05) (Fig 4C,D).
Vaccination with DC/AML fusions induces tumor-specific immunity and prevents leukemia engraftment in an immunocompetent murine leukemia model.
We subsequently examined whether the combination of HMAs and the fusion vaccine would result in enhanced leukemia-specific immunity and effective targeting of AML cells in-vivo in an immunocompetent syngeneic murine model. Fusion cells were generated as discussed in the methods section. Fusion cells were identified through sorting by co-expression of CD86 and GFP. Black mice were inoculated retro-orbitally with 150×103 GFP+ TIB-49 cells and 24 hours later, mice were injected subcutaneously with 100×103 sorted fusion cells. Two weeks following inoculation with TIB-49 cells, control mice exhibited significant tumor growth at the injection site as opposed to vaccine-treated mice (Fig 5A). The BM of the mice was harvested and examined for tumor infiltration using GFP expression. Whereas all mice in the non-vaccinated group showed BM AML involvement, the vaccinated mice did not demonstrate any BM-infiltrating blasts (Fig 5B; p<0.05). Moreover, stimulation of these BM-derived cells with autologous tumor lysate leads to an expansion in tumor-specific CD4+ T cells as detected by intracellular IFN-γ expression (Fig 5C; p<0.05).
Figure 5. DC/AML fusion vaccine prevents engraftment and increases leukemia-specific immunity in murine syngeneic AML model.
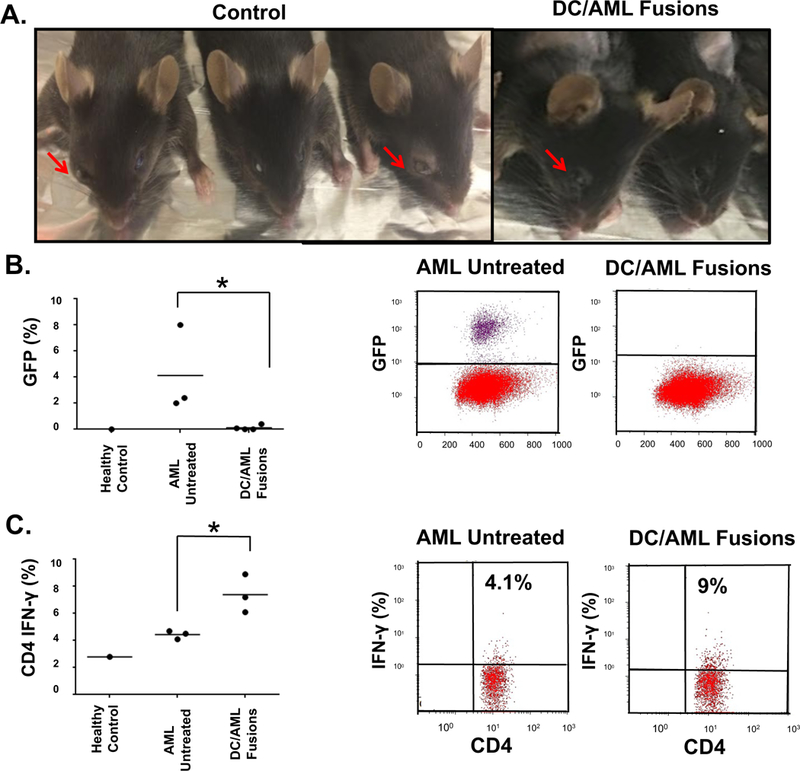
DCs were generated from the BM cells harvested from femurs of C57BL/6J black mice. BM cells were cultured in the presence of IL-4 and GM-CSF for 5–7 days to induce DC differentiation and maturation. To generate the fusion vaccine, the DCs were then collected and fused with GFP+ syngeneic TIB-49 AML cells in the presence of PEG. Fusion cells were identified through sorting by co-expression of CD86 and GFP. Black mice were inoculated retro-orbitally with GFP+ TIB-49 cells and 24 hours later, mice were injected subcutaneously with the sorted DC/AML fusion vaccine. Two weeks following inoculation with TIB-49 cells, control mice exhibited significant tumor growth at the injection site as opposed to mice treated with the DC/AML fusion vaccine (A). Two weeks following inoculation with TIB-49 cells, BM of the animals was harvested and examined for GFP expression. The percent of BM-infiltrating blasts (B) is depicted. Stimulation of those BM-derived cells with autologous tumor lysate as detected by intracellular IFN-γ expression is shown in (C).
Vaccination with DC/AML fusions in combination with HMA increases survival and induction of tumor-specific immunity in an immunocompetent murine leukemia model.
We next studied whether treatment with HMA was associated with enhanced response to DC/AML fusion vaccination in the murine leukemia model. C57BL/6J mice were inoculated retro-orbitally with GFP+ TIB-49 cells on day 1. Mice were divided into 4 separate cohorts of 5 mice each. The 4 cohorts were as follows: 1) vehicle-treated mice, 2) SGI-110 (1mg/kg) on days 6–10, 3) vaccine alone on day 12, 4) SGI-110 (1mg/kg) on days 6–10 and vaccine on day 12. Untreated mice were used as an additional control. Vaccine was administered after the establishment of AML. The experimental schema is summarized in Fig 6A. To assess the induction of tumor-specific immune response, splenocytes were harvested from animals at the time of euthanasia. IFN-γ expression by CD4 and CD8 cells was quantified following exposure to syngeneic tumor lysate. Splenocytes from healthy mice were used as baseline for IFN-γ expression. Therapy with SGI-110 plus the vaccine shows a significant increase in tumor-specific T cells as detected by IFN-γ production in CD4 and CD8 splenocytes as compared to treatment with the fusion vaccine (n=5; p=0.02) or SGI-110 monotherapy (n=5; p=0.02) (Fig 6B). Furthermore, CD4 and CD8 splenocytes were evaluated for PD-1 expression. The data demonstrates a significant decrease in PD-1 levels following treatment with SGI-110. However, addition of the vaccine did not further alter PD-1 expression (Fig 6C). Survival studies demonstrate that animals treated with SGI-110 or the vaccine had improved survival as compared to control animals (p= 0.001). Consistent with the above findings, combination therapy with the vaccine and SGI-110 extends survival as compared to either therapy alone (Fig 6D).
Figure 6. HMA with fusion vaccination enhances leukemia-specific immunity and survival in murine syngeneic AML model.
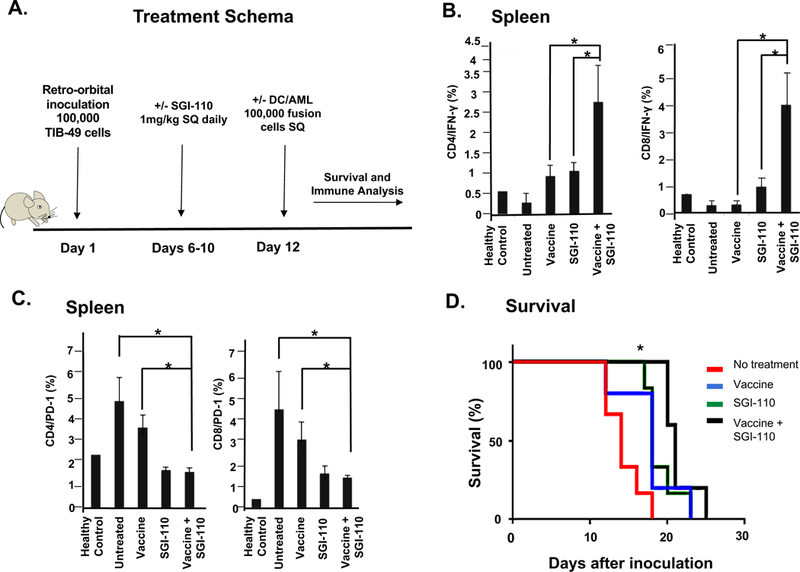
C57BL/6J mice were retro-orbitally inoculated with 100,000 syngeneic TIB-49 AML cells that were stably transduced with GFP. DC/AML fusion cells were generated. The mice were then treated with either vehicle control, SGI-110 (1mg/kg) X 5 days, vaccine alone, or combination of SGI-110 and the fusion vaccine as shown in the treatment schema (A). The mice were followed for survival and splenocytes were harvested. The cells were then exposed ex-vivo to autologous tumor lysate for 5 days. The cells underwent intracellular flow cytometric analysis for IFN-γ expression in both CD4+ and CD8+ T cells (B) and PD-1 expression (C). Mice treated with the combination of SGI-110 plus vaccine had prolonged survival compared to either agent alone (D).
Discussion
Immune evasion is a critical aspect of AML disease progression characterized by the loss of immunogenicity of the AML cell and the creation of an immunosuppressive TME that fosters tolerance and disease growth. The potency of cellular immunotherapy as a uniquely effective therapeutic strategy for AML is highlighted by the observation that alloSCT is curative for a subset of patients due to the graft-versus-disease effect mediated by alloreactive lymphocytes (Copelan, 2006). However, the lack of specificity of this response is associated with significant morbidity due to graft-versus-host disease and relapse (Soiffer, 2008). A major area of investigation is the development of strategies to induce leukemia-specific T cell immunity that selectively targets leukemia cells.
We have developed a promising personalized cancer vaccine in which patient-derived AML cells are fused with autologous DCs (Rosenblatt et al, 2016a). Vaccination is associated with a persistent expansion of leukemia-specific T cells with enhanced expression of IFN-γ and protection from relapse. In a phase II clinical trial, vaccination of AML patients following chemotherapy-induced remission resulted in 71% of patients demonstrating remission with nearly 5 years of follow-up despite a median age of 63. Despite these promising findings, there is ongoing concern that potency leukemia-specific effector cells to mediate durable clinical responses is subject to the immunosuppressive regulation of the TME which promotes inactivation of tumor-reactive lymphocytes fostering immune tolerance (Armand, 2015; Andersen, 2014; Schlößer et al, 2014; Isidori et al, 2014; Sharma & Allison, 2015). Of note, targeting the microenvironment alone with checkpoint inhibitors alone has been ineffective in AML likely due to the lack of a significant presence of leukemia-specific effector cells to activate. As such, the rational pairing of strategies to expand leukemia-specific T cells with efforts to effectively target critical factors of the immunosuppressive microenvironment is vital.
HMAs demonstrate therapeutic activity against AML and are increasingly recognized as immunomodulators (Srivastava et al, 2014). Through demethylation, HMAs increase tumor antigen expression by allowing for the expression of genes otherwise transcriptionally repressed due to promoter methylation (Srivastava et al, 2014; Almstedt et al, 2010). Pre-treatment of cancer cells with low-dose HMAs enhances responses to subsequent immune checkpoint antibody therapy, and HMA exposure has been demonstrated to increase the expression of immune-related genes that regulate antigen processing and presentation, interferon signaling, and cytokine production in various tumor models (Juergens et al, 2011; Li et al, 2014).
In the present study, HMA exposure resulted in enhanced leukemia-specific immunity in an in-vitro model and an immunocompetent syngeneic AML murine model. The data herein suggests that the increase in leukemia-specific immunity following HMA therapy is due in part to HMA-mediated reversal of critical immunosuppressive components of the TME and the tumor itself. We demonstrated that HMA exposure results in enhanced antigen presentation evidenced by increased ligation of MHC1-PR-1 complex to a TCRL antibody mimicking signal one necessary for T cell activation as well as increased MHCI expression. Increased MHCI expression following HMA therapy has been observed in breast cancer (Luo et al, 2018). Moreover, culture of patient-derived AML cells in the presence of SGI-110 increased TAP2 expression where TAP2 is imperative for antigen presentation and functions to translocate the peptide MHC complex to the cell surface (Leone et al, 2013).
While other groups have reported that HMAs upregulate T cell PD-1 expression, we found that the SGI-110 decreases circulating T cell PD-1 expression (Wrangle et al, 2013; Yang et al, 2014; Ørskov et al, 2015a). It has been described that PD-1 gene transcription is in part regulated through promoter methylation where methylation results in transcriptional silencing of PD-1 (Youngblood et al, 2011). In hematologic malignancies, 44% of patients (n=27) treated with a median of 4 HMA cycles showed a loss of DNA methylation in the PD-1 promoter, and methylation of the promoter was inversely correlated with PD-1 gene expression in T cells. However, flow cytometry quantified increased PD-1 CD8 T cells expression in only 4 patients (Ørskov et al, 2015b). Treatment of the KG-1 AML cell line with decitabine resulted in demethylation of PD-1 in a dose-dependent manner as observed at concentrations of greater than 1uM (Yang et al, 2014). Given these findings, upregulation of PD-1 following repeated HMA exposure has been explored as a mechanism of possible HMA resistance in hematologic malignancies, and clinical trials assessing the combination of HMA with checkpoint blockade are underway. It is likely that PD-1 T cell promoter hypermethylation is a dynamic process and demethylation of the PD-1 promoter requires repeated adequate concentrations of drug exposure which may be achieved with repeated cycles in patients. It is possible that with one cycle of therapy, as observed in our murine model, the threshold for promoter demethylation was not fully met, and PD-1 expression was therefore not increased. Our finding of PD-1 T cell surface upregulation peaked seven days following one cycle of HMA therapy.
We also noted decreased MDSC burden following HMA treatment in the murine immunocompetent model, which has not been previously reported in AML. MDSCs are immature myeloid precursors with immunosuppressive capabilities (Gabrilovich & Nagaraj, 2009). In a breast cancer model, decitabine depletes MDSCs through the induction of apoptosis whereas another model demonstrates the differentiation of MDSCs into mature tumor-derived APCs following HMA therapy (Zhou et al, 2017; Daurkin et al, 2010).
Recently, an additional pathway by which HMAs elicit immune activation in cancer cells through viral mimicry has been described (Chiappinelli et al, 2015; Roulois et al, 2015). HMAs activate the ERV defense pathway in tumor cells where ERV genes are remnants of inactive retrovirus DNA that account for about 8% of the human genome. Genes in the ERV pathway are normally silenced by promoter hypermethylation. Activation of this pathway through demethylation culminates in T cell activation with inflammatory cytokine production, enhanced T cell activation and subsequently, cancer cell death (Strick et al, 2016; Chiappinelli et al, 2015; Roulois et al, 2015). Specifically, activation of this pathway leads to transcription of IFN-stimulated genes that causes IFN-γ receptor expression and increased MHCI expression, both leading to T cell activation/IFN-γ release and subsequent tumor death. Activation of the ERV pathway by HMAs also results in dsRNA-elicited IFNβ production. In a colon cancer model, HMAs induced the transcription of endogenous dsRNAs activating this IFN response pathway causing decreased proliferation of colorectal cancer initiating cells (Roulois et al, 2015). In the present study, we observed a significant upregulation in ERV-3 and IFNβ by WB following HMA exposure in leukemia cells. Consistent with activation of this pathway, we report increased MHC1 expression on AML blasts following HMA exposure.
We subsequently examined whether the combination of SGI-110 and vaccination could synergistically result in leukemia cell death through the reversal of tolerance in the TME. Here we demonstrate that fusion vaccination with SGI-110 enhances leukemia-specific immunity both in-vitro and an in our murine model. We did not observe a synergistic decrease in MDSC burden with the combination of vaccine and HMA, suggesting this effect is solely HMA-mediated. Similarly, there was no additive effect on T cell checkpoint inhibitory expression.
For decades, many groups have utilized dsRNA mimetics such as poly I:C to enhance vaccine immunogenicity. Interestingly, it has been shown that dsRNA-elicited IFNβ production via these mimetics improves DC functionality (Gatti et al, 2013). Hence, HMAs theoretically enhance DC functionality through the production of IFNβ via ERV pathway activation. This may additionally explain why we observed enhance leukemia-specific immunity in our combinatorial fusion plus HMA murine model, augmenting the immunological eradication of AML cells and resulting in a survival benefit.
In conclusion, we have demonstrated that the HMA SGI-110 modulates critical factors contributing to the immunosuppressive milieu in AML resulting in enhanced anti-leukemic immunity. HMAs markedly enhance antigen presentation and the immunogenicity of AML cells through increased antigen processing and presentation. This effect is mediated in part though the activation of endogenous retroviral sequences and the resultant expression of inflammatory markers such as IFNß. In addition, SGI-110 augments the immune response to DC/AML vaccination in a murine model resulting in a survival benefit. The combination of a novel personalized fusion vaccine and HMA holds great potential, and a clinical trial using these therapies is planned.
Supplementary Material
(A) AML patient-derived tumor cells were treated for two days with four doses of 1uM SGI-110 added twice daily and then cultured for additional two days. The expression of co-stimulatory molecules was detected using flow-cytometry, as demonstrated in a representative FACS plot (n=4). (B) AML patient-derived tumor cells were treated for two days with four doses of 1uM SGI-110 added twice daily and then cultured for additional two days. The production of cytokines was analyzed by cytokine array and expressed as an average of two independent experiments.
Acknowledgements
This study was supported by research funding from Astex Pharmaceuticals to D.A and by research funding from the WES (When Everyone Survives) Foundation to J.R.
D.A. has received research funding from Astex Pharmaceuticals.
Footnotes
Disclosure of Conflicts of interest
The remaining authors declare no competing financial interests.
References:
- Akers SN, Odunsi K & Karpf AR (2010) Regulation of cancer germline antigen gene expression: implications for cancer immunotherapy. Future oncology (London, England), 6, 717–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almstedt M, Blagitko-Dorfs N, Duque-Afonso J, Karbach J, Pfeifer D, Jäger E & Lübbert M (2010) The DNA demethylating agent 5-aza-2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leukemia Research, 34, 899–905 Available at: http://linkinghub.elsevier.com/retrieve/pii/S0145212610000822. [DOI] [PubMed] [Google Scholar]
- Andersen MH (2014) The targeting of immunosuppressive mechanisms in hematological malignancies. Leukemia, 28, 1784–1792 Available at: http://www.nature.com/doifinder/10.1038/leu.2014.108. [DOI] [PubMed] [Google Scholar]
- Armand P (2015) Immune checkpoint blockade in hematologic malignancies. Blood, 125, 3393–3400 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25833961. [DOI] [PubMed] [Google Scholar]
- Austin R, Smyth MJ & Lane SW (2016) Harnessing the immune system in acute myeloid leukaemia. Critical reviews in oncology/hematology, 103, 62–77 Available at: http://www.ncbi.nlm.nih.gov/pubmed/27247119. [DOI] [PubMed] [Google Scholar]
- Beyar-Katz O & Gill S (2018) Novel Approaches to Acute Myeloid Leukemia Immunotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research Available at: http://www.ncbi.nlm.nih.gov/pubmed/29903894. [DOI] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, Makarov V, Buhu S, Slamon DJ, Wolchok JD, Pardoll DM, Beckmann MW, Zahnow CA, Mergoub T, Chan TA, Baylin SB, et al. (2015) Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell, 162, 974–986 Available at: http://linkinghub.elsevier.com/retrieve/pii/S009286741500848X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles SJ, Gilmour MN, Reid R, Knapper S, Burnett AK, Man S, Tonks A & Darley RL (2015) The immunosuppressive ligands PD-L1 and CD200 are linked in AML T-cell immunosuppression: identification of a new immunotherapeutic synapse. Leukemia, 29, 1952–4 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25748687. [DOI] [PubMed] [Google Scholar]
- Copelan EA (2006) Hematopoietic Stem-Cell Transplantation. New England Journal of Medicine, 354, 1813–1826 Available at: http://www.nejm.org/doi/abs/10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- Daurkin I, Eruslanov E, Vieweg J & Kusmartsev S (2010) Generation of antigen-presenting cells from tumor-infiltrated CD11b myeloid cells with DNA demethylating agent 5-aza-2’-deoxycytidine. Cancer immunology, immunotherapy : CII, 59, 697–706 Available at: http://www.ncbi.nlm.nih.gov/pubmed/19882154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derolf AR, Kristinsson SY, Andersson TM-L, Landgren O, Dickman PW & Bjorkholm M (2009) Improved patient survival for acute myeloid leukemia: a population-based study of 9729 patients diagnosed in Sweden between 1973 and 2005. Blood, 113, 3666–3672 Available at: http://www.bloodjournal.org/cgi/doi/10.1182/blood-2008-09-179341. [DOI] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, McMichael JF, Wallis JW, Lu C, Shen D, Harris CC, Dooling DJ, Fulton RS, Fulton LL, Chen K, Schmidt H, et al. (2012) Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature, 481, 506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Löwenberg B & Bloomfield CD (2010) Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood, 115, 453–74. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI & Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nature reviews. Immunology, 9, 162–174 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2828349&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti G, Nuñez NG, Nocera DA, Dejager L, Libert C, Giraudo C & Maccioni M (2013) Direct effect of dsRNA mimetics on cancer cells induces endogenous IFN-β production capable of improving dendritic cell function. European journal of immunology, 43, 1849–61. [DOI] [PubMed] [Google Scholar]
- Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, Vyas P, Cavenagh J, Stankovic T, Moss P & Craddock C (2010) Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood, 116, 1908–18. [DOI] [PubMed] [Google Scholar]
- Isidori A, Salvestrini V, Ciciarello M, Loscocco F, Visani G, Parisi S, Lecciso M, Ocadlikova D, Rossi L, Gabucci E, Clissa C & Curti A (2014) The role of the immunosuppressive microenvironment in acute myeloid leukemia development and treatment. Expert review of hematology, 7, 807–18. [DOI] [PubMed] [Google Scholar]
- Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, Lee B, Tsai S, Delgado IE, Rudek MA, Belinsky SA, Herman JG, Baylin SB, Brock MV & Rudin CM (2011) Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer discovery, 1, 598–607 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3353724&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, Gau JP, Chou WC, Buckstein R, Cermak J, Kuo CY, Oriol A, Ravandi F, Faderl S, Delaunay J, Lysák D, Minden M & Arthur C (2012) Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. Journal of Clinical Oncology, 30, 2670–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpf AR (2006) Potential role for epigenetic modulatory drugs in the enhancement of cancer/germ-line antigen vaccine efficacy. Epigenetics, 1, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone P, Shin E-C, Perosa F, Vacca A, Dammacco F & Racanelli V (2013) MHC Class I Antigen Processing and Presenting Machinery: Organization, Function, and Defects in Tumor Cells. JNCI Journal of the National Cancer Institute, 105, 1172–1187 Available at: https://academic.oup.com/jnci/article-lookup/doi/10.1093/jnci/djt184. [DOI] [PubMed] [Google Scholar]
- Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen R-WC, Vatapalli R, Topper MJ, Luo J, Connolly RM, Azad NS, Stearns V, Pardoll DM, Davidson N, Jones PA, Slamon DJ, Baylin SB, Zahnow CA & Ahuja N (2014) Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget, 5, 587–598 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3996658&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenegger FS, Krupka C, Köhnke T & Subklewe M (2015) Immunotherapy for Acute Myeloid Leukemia. Seminars in hematology, 52, 207–14. [DOI] [PubMed] [Google Scholar]
- Lübbert M, Suciu S, Baila L, Rüter BH, Platzbecker U, Giagounidis A, Selleslag D, Labar B, Germing U, Salih HR, Beeldens F, Muus P, Pflüger K-H, Coens C, Hagemeijer A, Eckart Schaefer H, Ganser A, Aul C, de Witte T & Wijermans PW (2011) Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Rese. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 29, 1987–96. [DOI] [PubMed] [Google Scholar]
- Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, Zahnow CA, Nickels ML, Liu F, Tantawy MN, Sanders ME, Manning HC & Balko JM (2018) DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nature communications, 9, 248 Available at: http://www.ncbi.nlm.nih.gov/pubmed/29339738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørskov AD, Treppendahl MB, Skovbo A, Holm MS, Friis LS, Hokland M & Grønbæk K (2015a) Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget, 6, 9612–26 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25823822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørskov AD, Treppendahl MB, Skovbo A, Holm MS, Friis LS, Hokland M & Grønbæk K (2015b) Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget, 6, 9612–26 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25823822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie EK, Feldman EJ, Christos PJ, Rohan SD, Lagassa CB, Ippoliti C, Scandura JM, Carlson K & Roboz GJ (2013) Decitabine in patients with newly diagnosed and relapsed acute myeloid leukemia. Leukemia & Lymphoma, 54, 2003–2007 Available at: http://www.tandfonline.com/doi/full/10.3109/10428194.2012.762093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J & Avigan D (2006) Can leukemia-derived dendritic cells generate antileukemia immunity? Expert review of vaccines, 5, 467–72. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Kufe D & Avigan D (2005) Dendritic cell fusion vaccines for cancer immunotherapy. Expert opinion on biological therapy, 5, 703–715 Available at: http://www.ncbi.nlm.nih.gov/pubmed/15934845. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Stone RM, Uhl L, Neuberg D, Joyce R, Levine JD, Arnason J, McMasters M, Luptakova K, Jain S, Zwicker JI, Hamdan A, Boussiotis V, Steensma DP, DeAngelo DJ, Galinsky I, Dutt PS, Logan E, Bryant MP, Stroopinsky D, et al. (2016a) Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Science translational medicine, 8, 368ra171 Available at: http://www.ncbi.nlm.nih.gov/pubmed/27928025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J, Stone RM, Uhl L, Neuberg D, Joyce R, Levine JD, Arnason J, McMasters M, Luptakova K, Jain S, Zwicker JI, Hamdan A, Boussiotis V, Steensma DP, DeAngelo DJ, Galinsky I, Dutt PS, Logan E, Bryant MP, Stroopinsky D, et al. (2016b) Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Science translational medicine, 8, 368ra171 Available at: http://www.ncbi.nlm.nih.gov/pubmed/27928025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H., Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, O’Brien C & De Carvalho DD (2015) DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell, 162, 961–973 Available at: http://linkinghub.elsevier.com/retrieve/pii/S0092867415009721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlößer HA, Theurich S, Shimabukuro-Vornhagen A, Holtick U, Stippel DL & Bergwelt-Baildon M von (2014) Overcoming tumor-mediated immunosuppression. Immunotherapy, 6, 973–988 Available at: http://www.futuremedicine.com/doi/abs/10.2217/imt.14.58. [DOI] [PubMed] [Google Scholar]
- Sergeeva A, Alatrash G, He H, Ruisaard K, Lu S, Wygant J, McIntyre BW, Ma Q, Li D, St John L, Clise-Dwyer K & Molldrem JJ (2011) An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood, 117, 4262–72 Available at: http://www.ncbi.nlm.nih.gov/pubmed/21296998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P & Allison JP (2015) The future of immune checkpoint therapy. Science, 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Soiffer R (2008) Immune modulation and chronic graft-versus-host disease. Bone marrow transplantation, 42 Suppl 1, S66--S69 Available at: http://www.ncbi.nlm.nih.gov/pubmed/18724307. [DOI] [PubMed] [Google Scholar]
- Srivastava P, Paluch BE, Matsuzaki J, James SR, Collamat-Lai G, Karbach J, Nemeth MJ, Taverna P, Karpf AR & Griffiths EA (2014) Immunomodulatory action of SGI-110, a hypomethylating agent, in acute myeloid leukemia cells and xenografts. Leukemia Research, 38, 1332–1341 Available at: http://linkinghub.elsevier.com/retrieve/pii/S0145212614002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoye JP (2012) Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nature reviews. Microbiology, 10, 395–406 Available at: http://www.ncbi.nlm.nih.gov/pubmed/22565131. [DOI] [PubMed] [Google Scholar]
- Strick R, Strissel PL, Baylin SB & Chiappinelli KB (2016) Unraveling the molecular pathways of DNA-methylation inhibitors: human endogenous retroviruses induce the innate immune response in tumors. Oncoimmunology, 5, e1122160 Available at: http://www.ncbi.nlm.nih.gov/pubmed/27467919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woloszynska-Read A, Mhawech-Fauceglia P, Yu J, Odunsi K & Karpf AR (2008) Intertumor and intratumor NY-ESO-1 expression heterogeneity is associated with promoter-specific and global DNA methylation status in ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research, 14, 3283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, Vancriekinge W, Demeyer T, Du Z, Parsana P, Rodgers K, Yen R-W, Zahnow CA, Taube JM, Brahmer JR, Tykodi SS, Easton K, Carvajal RD, Jones PA, Laird PW, et al. (2013) Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget, 4, 2067–2079 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3875770&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng Q-R, Fang Z, Nguyen M, Pierce S, Wei Y, Parmar S, Cortes J, Kantarjian H & Garcia-Manero G (2014) Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia, 28, 1280–8 Available at: http://www.ncbi.nlm.nih.gov/pubmed/24270737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngblood B, Oestreich KJ, Ha S-J, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, Boss JM & Ahmed R (2011) Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity, 35, 400–12 Available at: http://www.ncbi.nlm.nih.gov/pubmed/21943489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Yao Y, Shen Q, Li G, Hu L & Zhang X (2017) Demethylating agent decitabine disrupts tumor-induced immune tolerance by depleting myeloid-derived suppressor cells. Journal of cancer research and clinical oncology, 143, 1371–1380 Available at: http://www.ncbi.nlm.nih.gov/pubmed/28321548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) AML patient-derived tumor cells were treated for two days with four doses of 1uM SGI-110 added twice daily and then cultured for additional two days. The expression of co-stimulatory molecules was detected using flow-cytometry, as demonstrated in a representative FACS plot (n=4). (B) AML patient-derived tumor cells were treated for two days with four doses of 1uM SGI-110 added twice daily and then cultured for additional two days. The production of cytokines was analyzed by cytokine array and expressed as an average of two independent experiments.