

Abstract
Background
This is an updated version of the original Cochrane Review published in Issue 12, 2016. This review is one in a series of Cochrane Reviews investigating pair‐wise monotherapy comparisons.
Epilepsy is a common neurological condition in which abnormal electrical discharges from the brain cause recurrent unprovoked seizures. It is believed that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to become seizure‐free and go into long‐term remission shortly after starting drug therapy, the majority of which may be able to achieve remission with a single antiepileptic drug (AED).
The correct choice of first‐line AED for individuals with newly diagnosed seizures is of great importance and should be based on the highest‐quality evidence available regarding the potential benefits and harms of various treatments for an individual.
Topiramate and carbamazepine are commonly used AEDs. Performing a synthesis of the evidence from existing trials will increase the precision of results of outcomes relating to efficacy and tolerability, and may help inform a choice between the two drugs.
Objectives
To review the time to treatment failure, remission and first seizure with topiramate compared with carbamazepine when used as monotherapy in people with focal onset seizures (simple or complex focal and secondarily generalised), or generalised onset tonic‐clonic seizures (with or without other generalised seizure types).
Search methods
For the latest update we searched the Cochrane Register of Studies (CRS Web), which includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL); MEDLINE (Ovid); ClinicalTrials.gov; and the WHO International Clinical Trials Registry Platform (ICTRP) to 22 May 2018. We imposed no language restrictions. We also contacted pharmaceutical companies and trial investigators.
Selection criteria
Randomised controlled trials (RCTs) comparing monotherapy with either topiramate or carbamazepine in children or adults with focal onset seizures or generalised onset tonic‐clonic seizures (with or without other generalised seizure types).
Data collection and analysis
This was an individual participant data (IPD), review. Our primary outcome was time to treatment failure. Our secondary outcomes were time to first seizure post‐randomisation, time to six‐month remission, time to 12‐month remission, and incidence of adverse events. We used Cox proportional hazards regression models to obtain trial‐specific estimates of hazard ratios (HRs), with 95% confidence intervals (CIs), using the generic inverse variance method to obtain the overall pooled HR and 95% CI.
Main results
IPD were available for 1151 of 1239 eligible individuals from two of three eligible studies (93% of the potential data). A small proportion of individuals recruited into these trials had 'unclassified seizures;' for analysis purposes, these individuals are grouped with those with generalised onset seizures. For remission outcomes, a HR < 1 indicated an advantage for carbamazepine, and for first seizure and treatment failure outcomes, a HR < 1 indicated an advantage for topiramate.
The main overall results for the primary outcome, time to treatment failure, given as pooled HR adjusted for seizure type were: time to failure for any reason related to treatment 1.16 (95% CI 0.97 to 1.38); time to failure due to adverse events 1.02 (95% CI 0.82 to 1.27); and time to failure due to lack of efficacy 1.46 (95% CI 1.08 to 1.98). Overall results for secondary outcomes were time to first seizure 1.11 (95% CI 0.96 to 1.29); and time to six‐month remission 0.88 (0.76 to 1.01). There were no statistically significant differences between the drugs. A statistically significant advantage for carbamazepine was shown for time to 12‐month remission: 0.84 (95% CI 0.71 to 0.99).
The results of this review are applicable mainly to individuals with focal onset seizures; 81% of individuals included within the analysis experienced seizures of this type at baseline. For individuals with focal onset seizures, a statistically significant advantage for carbamazepine was shown for time to failure for any reason related to treatment (HR 1.21, 95% CI 1.01 to 1.46), time to treatment failure due to lack of efficacy (HR 1.47, 95% CI 1.07 to 2.02), and time to 12‐month remission (HR 0.82, 95% CI 0.69 to 0.99). There was no statistically significant difference between topiramate and carbamazepine for 'time to first seizure' and 'time to six‐month remission'.
Evidence for individuals with generalised tonic‐clonic seizures (9% of participants contributing to the analysis), and unclassified seizure types (10% of participants contributing to the analysis) was very limited; no statistically significant differences were found but CIs were wide; therefore we cannot exclude an advantage to either drug, or a difference between drugs.
The most commonly reported adverse events with both drugs were drowsiness or fatigue, "pins and needles" (tingling sensation), headache, gastrointestinal disturbance and anxiety or depression. The rate of adverse events was similar across the two drugs.
We judged the methodological quality of the included trials generally to be good; however, there was some evidence that the open‐label design of the larger of the two trials may have influenced the treatment failure rate within the trial. Hence, we judged the certainty of the evidence for treatment failure to be moderate for individuals with focal onset seizures and low for individuals with generalised onset seizures. For efficacy outcomes (first seizure, remission), we judged the certainty of evidence from this review to be high for individuals with focal onset seizures and moderate for individuals with generalised onset or unclassified seizures.
Authors' conclusions
For individuals with focal onset seizures, there is moderate‐certainty evidence that carbamazepine is less likely to be withdrawn and high‐certainty evidence that 12‐month remission will be achieved earlier than with topiramate. We did not find any differences between the drugs in terms of the other outcomes measured in the review and for individuals with generalised tonic‐clonic seizures or unclassified epilepsy; however, we encourage caution in the interpretation of results including small numbers of participants with these seizure types.
Future trials should be designed to the highest quality possible and take into consideration masking, choice of population, classification of seizure type, duration of follow‐up, choice of outcomes and analysis, and presentation of results.
Plain language summary
Topiramate versus carbamazepine as single drug treatment for epilepsy
This is an updated version of the Cochrane Review previously published in Issue 12, 2016 of the Cochrane Database of Systematic Reviews.
Background
Epilepsy is a common disorder of the nervous system in which abnormal electrical discharges from the brain cause recurrent seizures (physical convulsions or thought disturbances or a combination of these symptoms). We studied two types of epileptic seizures in this review: generalised onset seizures in which electrical discharges begin in one part of the brain and move throughout the brain, and focal onset seizures (also known as partial onset seizures) in which the seizure is generated in and affects the same part of the brain. Focal onset seizures may become generalised (secondary generalisation) and move from one part of the brain to throughout the brain. Up to 70% of individuals with active epilepsy have the potential to go into long‐term remission shortly after starting drug therapy and around 70% of these individuals can achieve seizure freedom using a single antiepileptic drug.
This review applies to people with focal onset seizures (with or without secondary generalisation) and people with tonic‐clonic seizures, a specific type of generalised onset seizure, as the recommended treatments for these seizure types are similar.
Objective
Topiramate and carbamazepine are commonly used treatments for individuals with epilepsy. The aim of this review was to compare how effective these drugs are at controlling recently diagnosed seizures, whether they are associated with side effects that may result in individuals stopping the drug and to inform a choice between these drugs.
Methods
We assessed the evidence from three clinical trials that compared topiramate with carbamazepine. We were able to combine data for 1151 people from two trials; we were not able to use the data from the remaining trial, which included 88 participants.
Results
Most (81%) of the people included in the two trials experienced focal seizures, so the results of this review apply mainly to people with this seizure type. Many of the remaining 19% of people experienced a seizure type which was difficult to classify as focal or generalised (unclassified seizures). Considering only people with focal seizures, the results showed that those taking carbamazepine were more likely to take their treatment for longer and to achieve a remission of 12 months duration earlier than those taking topiramate. No differences were found between the drugs in individuals with generalised onset or unclassified epilepsy.
The most common side effects reported by the participants during the trials were fatigue, 'pins and needles' (tingling sensation), headache, gastrointestinal problems and anxiety or depression. These side effects were reported a similar number of times by people taking topiramate or carbamazepine.
Certainty of the evidence
For people with focal onset seizures, we judged the certainty of the evidence to be moderate to high. The design of the trials (whether the people and treating clinicians knew which drug they were taking) may have influenced how long a participant stayed on their treatment. For the small number of people with generalised onset or unclassified seizures, we judged the certainty of the evidence to be low to moderate. The evidence is current to May 2018.
Conclusions
Carbamazepine is currently recommended by experts for the treatment of individuals who are newly diagnosed with focal onset seizures and the results of this review do not provide any evidence to contradict this. More information is needed for people with generalised onset or unclassified seizures. All future trials comparing these drugs, or any other antiepileptic drugs, should be designed using high‐quality methods, and the types of seizure of the people included in any trials should be classified very carefully to ensure that the results are of high quality.
Summary of findings
Background
This is an updated version of the Cochrane Review previously published in Issue 12, 2016 of the Cochrane Database of Systematic Reviews (Nolan 2016a).
Description of the condition
Epilepsy is a common neurological condition in which abnormal electrical discharges from the brain cause recurrent, unprovoked seizures. Epilepsy is a disorder comprising many heterogeneous seizure types, with an estimated incidence of 33 to 57 per 100,000 person‐years worldwide (Annegers 1999; Hirtz 2007; MacDonald 2000; Olafsson 2005; Sander 1996), accounting for between 1% and 5% of the global burden of disease (Murray 1994; Sander 1996). The lifetime risk of epilepsy onset is estimated to be 1300 to 4000 per 100,000 person‐years (Hauser 1993; Juul‐Jenson 1983). Recently, around 42 million individuals worldwide were reported to have active epilepsy worldwide (Global Burden of Disease Study 2013); however, country‐specific prevalence and incidence rates are thought to vary considerably, with higher rates in resource‐poor countries (Bell 2014). It is thought that the lifetime prevalence could be as much as 70 million people worldwide (Ngugi 2010). Experts believe that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to go into long‐term remission shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), and around 70% of these individuals can achieve seizure freedom using antiepileptic drug (AED) monotherapy (Cockerell 1995). Current UK National Institute for Health and Care Excellence (NICE) guidelines recommend that both adults and children with epilepsy be treated with monotherapy, wherever possible (NICE 2012). The remaining 30% of individuals who experience refractory or drug‐resistant seizures will often require treatment with combinations of AEDs or alternative therapies, such as epilepsy surgery (Kwan 2000).
We studied two seizure types in this review; generalised onset seizures in which electrical discharges begin in one part of the brain and move throughout the brain, and focal onset seizures in which the seizure is generated in and affects one part of the brain (the whole hemisphere of the brain or part of a lobe of the brain).
Description of the intervention
Carbamazepine was amongst the earliest of the 'traditional' drugs licensed for the treatment of epileptic seizures and has been commonly used as monotherapy for focal onset and generalised onset seizures for over 30 years (Shakir 1980). Topiramate is a second‐generation AED, licensed as monotherapy for epileptic seizures following demonstrations of efficacy in dose‐controlled studies compared with 'traditional' AEDs, such as carbamazepine and sodium valproate (Gilliam 2003; Privitera 2003; SANAD A 2007; SANAD B 2007). Comparative trials have also shown newer AEDs, such as topiramate, to be generally well‐tolerated as monotherapy in both adults and children and associated with fewer adverse events, fewer serious adverse events, and fewer drug interactions with concomitant AEDs and other concomitant medications than traditional first‐line AEDs, such as carbamazepine (French 2007).
Evidence regarding the teratogenic effects (disturbances to foetal development) of carbamazepine and topiramate is inconclusive. Experts believe that the risk of congenital malformation may be higher in women taking carbamazepine than in the general population (Meador 2008; Morrow 2006; Weston 2016), and studies have associated carbamazepine with neural tube defects (Matlow 2012). The risk of malformations is thought to be lower for women taking topiramate monotherapy than for those taking carbamazepine monotherapy (Hunt 2008; Meador 2008; Morrow 2006), but the risk of malformation may increase in women taking topiramate as a component of polytherapy (Hunt 2008). It is unclear whether taking topiramate or carbamazepine during pregnancy has any negative neurodevelopmental effects on the child (Bromley 2014).
Current UK guidelines for adults and children recommend carbamazepine or lamotrigine as a first‐line treatment for newly onset focal seizures, and sodium valproate for newly onset generalised tonic‐clonic seizures (with or without other generalised seizure types) (NICE 2012). Carbamazepine may be a suitable second‐line treatment for generalised onset tonic‐clonic seizures, but may exacerbate myoclonic or absence seizures (Liporace 1994; Shields 1983; Snead 1985). Topiramate is mainly recommended for adjunctive use, but may be considered as a second‐line treatment for both focal and generalised seizures if first‐line treatments have failed or are unsuitable.
How the intervention might work
AEDs suppress seizures by reducing neuronal excitability (disruption of the usual mechanisms of a neurone within the brain, which may lead to an epileptic seizure) (MacDonald 1995). Both topiramate and carbamazepine are considered broad‐spectrum treatments, suitable for many seizure types. Carbamazepine has an anticonvulsant mechanism that works by blocking ion channels, binding with neurotransmitter receptors, or inhibiting the metabolism or reuptake of neurotransmitters (Brodie 1996; Ragsdale 1991). The mechanisms of action of topiramate are not fully understood but may include the inhibition of voltage‐dependent sodium channels and the enhancement or modulation of gamma‐aminobutyric acid‐A by action at a unique modulatory site (Coulter 1993; White 1997).
Why it is important to do this review
With evidence that up to 70% of individuals with active epilepsy have the potential to go into long‐term remission of seizures shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), the correct choice of first‐line antiepileptic therapy for individuals with newly diagnosed seizures is of great importance. It is important that clinicians are able to choose the most appropriate AED for an individual using the highest‐quality evidence available regarding the potential benefits and harms of various treatments. It is also important to compare the efficacy and tolerability of AEDs appropriate to given seizure types. Performing a synthesis of the evidence from existing trials will increase the precision of the results of outcomes relating to efficacy and tolerability, and may help inform a choice between drugs.
There are difficulties in undertaking a systematic review of epilepsy monotherapy trials, as the important efficacy outcomes require analysis of time‐to‐event data (e.g. time to first seizure after randomisation). Although methods have been developed to synthesise time‐to‐event data using summary information (Parmar 1998; Williamson 2002), the appropriate statistics are not commonly reported in published epilepsy trials (Nolan 2013a). Furthermore, although most epilepsy monotherapy trials collect seizure data, the definitions and reporting of outcomes are inconsistent. For example, trials may report time to 12‐month remission but not time to first seizure or vice versa, or some trials may define time to first seizure from the date of randomisation whereas others use the date of achieving maintenance dose. Trial investigators have also adopted differing approaches to data analysis, particularly with respect to the censoring of time‐to event data. For these reasons, we performed this review using individual participant data (IPD), which helps to overcome these problems. This review is one in a series of Cochrane IPD reviews investigating pair‐wise monotherapy comparisons (Marson 2000; Nevitt 2017b; Nevitt 2018a; Nevitt 2018b; Nevitt 2018c; Nevitt 2018d; Nolan 2013b). These data have also been included in IPD network meta‐analyses of AED monotherapy (Nevitt 2017a; Tudur Smith 2007).
Objectives
To review the time to treatment failure, remission and first seizure with topiramate compared with carbamazepine when used as monotherapy in people with focal onset seizures (simple or complex focal and secondarily generalised), or generalised onset tonic‐clonic seizures (with or without other generalised seizure types).
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) using either an adequate method of allocation concealment (e.g. sealed opaque envelopes) or a quasi‐randomised method of allocation (e.g. allocation by date of birth)
Trials may have been double‐blind, single‐blind, or unblinded
Trials must be of parallel design; cross‐over studies are not an appropriate design for measuring the long‐term outcomes of interest in this review (see Types of outcome measures)
Trials must include a comparison of topiramate monotherapy with carbamazepine monotherapy in individuals with epilepsy; therefore, cluster‐randomised studies are not an eligible design
Types of participants
We included children or adults with focal onset seizures (simple focal, complex focal or secondarily generalised tonic‐clonic seizures), or generalised onset tonic‐clonic seizures (with or without other generalised seizure types, i.e. those who had only generalised tonic‐clonic seizures and those who had both generalised onset tonic‐clonic seizures and generalised seizures of other types, e.g. absence, myoclonic etc.)
We excluded individuals with other generalised seizure types alone without generalised tonic‐clonic seizures (e.g. those who had only absence seizures without any generalised tonic‐clonic seizures), due to differences in first‐line treatment guidelines for other generalised seizure types (NICE 2012).
We included individuals who had a new diagnosis of epilepsy or who had experienced a relapse following antiepileptic monotherapy withdrawal only, due to differences in first‐line treatment guidelines for individuals with refractory epilepsy (NICE 2012).
Types of interventions
Included studies had to have made a randomised comparison of topiramate and carbamazepine (of any dose) as monotherapy. If studies included additional arms of treatments, other than topiramate and carbamazepine, we did not include these treatment arms in our analysis of the review.
Types of outcome measures
Below is a list of outcomes we investigated in this review. Reporting of these outcomes in the original study report was not an eligibility requirement for this review.
Primary outcomes
Time to treatment failure (retention time). This was a combined outcome reflecting both efficacy and tolerability, as the following may have led to failure of treatment: continued seizures, side effects, non‐compliance or the initiation of additional add‐on treatment. This is an outcome to which the participant makes a contribution and is the primary outcome measure recommended by the Commission on Antiepileptic Drugs of the International League Against Epilepsy (ILAE 1998; ILAE 2006).
Time to treatment failure is considered according to the following three definitions.
Time to treatment failure for any treatment‐related reason (continued seizures, side effects, non‐compliance or the initiation of additional add‐on treatment)
Time to treatment failure due to adverse events (i.e. side effects)
Time to treatment failure due to lack of efficacy (i.e. continued seizures)
Secondary outcomes
Time to first seizure recurrence post‐randomisation
Time to achieve six‐month remission (seizure‐free period) post‐randomisation
Time to achieve 12‐month remission (seizure‐free period) post‐randomisation
Incidence of adverse events (all reported, whether related or unrelated to treatment) and adverse events leading to treatment withdrawal
Search methods for identification of studies
Electronic searches
We ran searches for the original review on 14 April 2016, and subsequent searches on 22 May 2018. For the latest update we searched the following databases.
Cochrane Register of Studies (CRS Web, 22 May 2018) using the search strategy shown in Appendix 1. This includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL).
MEDLINE (Ovid, 1946 to 22 May 2018) using the search strategy shown in Appendix 2.
ClinicalTrials.gov (22 May 2018) using the search strategy shown in Appendix 3.
WHO International Clinical Trials Registry Platform (ICTRP, 22 May 2018) using the search strategy shown in Appendix 4.
Searching other resources
We handsearched the reference lists of retrieved studies for additional reports of relevant studies. We contacted Novartis (formerly Ciba Geigy, manufacturers of carbamazepine), Janssen Pharmaceuticals (manufacturers of topiramate) and the original investigators of relevant trials to identify any additional published or unpublished data.
Data collection and analysis
Selection of studies
Two review authors (SJN, AGM) independently assessed studies for inclusion, resolving any disagreements by discussion.
Data extraction and management
We requested the following IPD for all studies meeting our inclusion criteria.
-
Trial design and methods
Method of generation of random list
Method of allocation concealment
Stratification factors
Blinding methods
-
Participant covariates
Sex
Age
Seizure types
Time between first seizure and randomisation
Number of seizures prior to randomisation (with dates)
Presence of neurological signs
Electroencephalographic (EEG) results
Computerised tomography/magnetic resonance imaging (CT/MRI) results
-
Follow‐up data
Treatment allocation
Date of randomisation
Dates of follow‐up
Dates of seizures post‐randomisation or seizure frequency data between follow‐up visits
Dates of treatment withdrawal or failure and reasons for treatment withdrawal or failure
Dose
Dates of dose changes
If IPD were not available for a trial, we intended to carry out an assessment to see whether the trial reported any relevant aggregate level data or whether we could indirectly estimate such data using the methods of Parmar 1998 and Williamson 2002. Where graphical time‐to‐event data (e.g. Kaplan‐Meier curves) were published, with or without corresponding effective numbers at risk, we intended to use a macro‐enabled Microsoft Excel spreadsheet to indirectly estimate hazard ratios (HRs) or make use of graphical digitising software, if appropriate, and the quality of the published graph(s) allowed (Excel 2010; Tierney 2007).
We accepted follow‐up and outcome data in any format provided. One trial provided dates of seizures after randomisation (Privitera 2003), and one study provided the number of seizures recorded at each follow‐up visit (SANAD A 2007). To enable the calculation of time‐to‐event outcomes for studies that provided seizure data only in terms of the number of seizures recorded between each follow‐up visit, rather than the specific dates of seizures, we applied linear interpolation to approximate dates of seizures between follow‐up visits. For example, if the trial recorded four seizures between two visits that occurred on 1 March 2010 and 1 May 2010 (interval of 61 days), then we estimated that the first seizure took place around 13 March 2010. This method allowed the computation of an estimate of the time to six‐ and 12‐month remission for studies of sufficient length.
We calculated time to first seizure from the date of randomisation to the date that we estimated the first seizure to have occurred. If seizure data were missing for a particular visit, we censored these outcomes at the previous visit. We also censored these outcomes if the individual died or if follow‐up ceased prior to the occurrence of the event of interest.
We calculated time to six‐ and 12‐month remission from the date of randomisation to the date (or estimated date) that the individual had first been free of seizures for six or 12 months, respectively (e.g. 365 days for those who achieve 12‐month remission immediately). If the person had one or more seizure(s) during the trial, a six‐ or 12‐month seizure‐free period could also occur between the estimated date of the last seizure during the trial and a period of six or 12 months of seizure freedom.
We calculated time to treatment failure as the date of randomisation to the date of withdrawal from the trial or treatment failure. For the time‐to‐event analysis, we defined an 'event' as the failure or withdrawal of the allocated treatment because of reasons related to the treatment (i.e. lack of efficacy, occurrence of adverse events, or both; non‐compliance with the treatment regimen; withdrawal of consent from the trial; etc). We censored the outcome if treatment was withdrawn for reasons not related to the trial treatment (i.e. loss to follow‐up, death that was not treatment‐ or epilepsy‐related, etc). We also censored individuals who were still on allocated treatment at the date of the end of follow‐up.
We considered documented reasons for withdrawal or treatment failure on a case‐by‐case basis for relation to treatment; two authors (SJN, MS) independently classified reasons for failures as events or censored, and resolved any disagreements by discussion. If included trials classified the reasons for withdrawal or failure as events or censored differently from our definitions, we conducted sensitivity analyses to account for differences in the definition of a treatment failure 'event'.
Assessment of risk of bias in included studies
Two review authors (SJN, MS) independently assessed all included trials for risk of bias according to the Cochrane 'Risk of bias' tool (Higgins 2017), resolving any disagreements by discussion. We rated each of the following six domains as low, unclear or high risk of bias: method of generating random sequence, allocation concealment, blinding methods, incomplete outcome data, selective outcome reporting, and other sources of bias. Any discrepancies in the two authors' 'Risk of bias' judgements were resolved by discussion.
In the event of the presence of a high risk of bias in included trials (due to inadequate allocation concealment or lack of blinding), we intended to conduct sensitivity analyses excluding these trials.
Measures of treatment effect
We measured all outcomes in this review as time‐to‐event outcomes using the HR as the measure of treatment effect. We calculated 95% confidence intervals (CIs) to provide a measure of precision of the treatment effect estimate. We calculated all outcomes from IPD provided, where possible, and if IPD were not available, we intended to use extracted or estimated aggregate data from published trials if possible.
We considered adverse events narratively rather than formally in analyses due to anticipated differences in the format of adverse event reporting in the included studies.
Unit of analysis issues
Cross‐over and cluster‐randomised studies were not an eligible design for this review (see Types of studies).
If eligible studies included multiple treatment arms of different topiramate or carbamazepine doses, we pooled study arms of the same treatment in primary analyses to allow a comparison of topiramate and carbamazepine. For one trial, which randomised participants to two doses of topiramate (100 mg/day or 200 mg/day), we performed a secondary analysis to analyse the different doses compared with carbamazepine (Privitera 2003).
It was not within the scope of this review to compare directly different doses of the same treatment (e.g. the two doses of topiramate).
Dealing with missing data
For each trial that supplied IPD, we performed the following consistency checks.
We cross‐checked study details against any published report of the study and contacted the data providers if we found missing data, errors or inconsistencies.
If the data providers could not resolve inconsistencies between IPD and published data, we intended to either perform sensitivity analyses or exclude the data from the meta‐analysis, depending on the extent of the inconsistencies.
If possible, we reviewed the chronological randomisation sequence and checked the balance of prognostic factors, taking account of any stratification factors in the randomisation procedure.
Assessment of heterogeneity
We assessed heterogeneity statistically using the Q test (P < 0.10 for significance) and the I2 statistic (values greater than 50% indicating considerable heterogeneity, Higgins 2003), with output produced using the generic inverse variance approach available in Review Manager (Review Manager 2014). We also assessed heterogeneity visually by inspecting forest plots.
Assessment of reporting biases
Two review authors (SJN, MS) undertook full quality and 'Risk of bias' assessments according to methods outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2017). In theory, a review using IPD should overcome issues of reporting biases, as unpublished data can be provided and unpublished outcomes calculated. We requested all study protocols with IPD. If we suspected any selective reporting bias, we intended to explore the extent of the bias using the Outcome Reporting Bias In Trials (ORBIT) classification system (Kirkham 2010).
Data synthesis
We carried out our analysis on an intention‐to‐treat basis (that is, we analysed participants in the group to which they were randomised, irrespective of which treatment they actually received). Therefore, for the time‐to‐event outcomes 'time to six‐month remission', 'time to 12‐month remission', and 'time to first seizure post‐randomisation', we did not censor participants if treatment was withdrawn or failed. Intention‐to‐treat analyses often tend to suggest equivalence between treatments (i.e. no statistically significant difference), so we intended to undertake a secondary per‐protocol analysis as a sensitivity analysis if the primary analyses suggest equivalence. In this case, participants would be censored at the time of treatment failure for seizure outcomes.
For all outcomes, we investigated the relationship between the time‐to‐event and treatment effect of the AEDs. We used Cox proportional hazards regression models to obtain trial‐specific estimates of log (HR), or treatment effect and associated standard errors in Stata Statistical Software, version 14 (Stata 2015). The model assumes that the ratio of hazards (risks), between the two treatment groups is constant over time (i.e. hazards are proportional). We tested this proportional hazards assumption of the Cox regression model for each outcome of each trial by testing the statistical significance of a time‐varying covariate in the model. We evaluated overall estimates of HRs (with 95% CIs), using the generic inverse variance method in MetaView. We expressed results as a HR and a 95% CI. We used a fixed‐effect model and, if considerable heterogeneity was present (I2 statistic > 50%), we intended to repeat the analysis using a random‐effects model.
By convention, a HR greater than 1 indicated that an event was more likely to occur earlier with topiramate than with carbamazepine. Hence, for time to treatment failure or time to first seizure, a HR greater than 1 indicates a clinical advantage for carbamazepine (e.g. a HR of 1.2 would suggest a 20% increase in risk of treatment failure from topiramate compared with carbamazepine), and for time to six‐month, 12‐month and 24‐month remission, a HR greater than 1 indicates a clinical advantage for topiramate (i.e. the seizure‐free period occurs earlier with topiramate than with carbamazepine).
Subgroup analysis and investigation of heterogeneity
Due to the strong clinical belief that some AEDs are more effective for some seizure types than for others (see Description of the intervention and How the intervention might work), we intended to stratify all analyses by epilepsy type (focal onset versus generalised onset), according to the classification of main seizure type at baseline. We classified focal seizures (simple or complex) and focal secondarily generalised seizures as focal epilepsy. We classified primarily generalised tonic‐clonic seizures (with or without other seizure types), as generalised epilepsy.
Seizure type was missing (unclassified) for 89 participants from SANAD A 2007 and 13 participants were classified as having generalised onset seizures, even though the trial was designed to include only participants with focal onset seizures. Also, only 88 participants from Privitera 2003 were classified as having generalised onset seizures (by design the majority of participants forming the comparison of carbamazepine and topiramate had focal onset seizures, see Characteristics of included studies for more details) and seizure type was missing (unclassified) for 22 participants from Privitera 2003.
Therefore, for the purposes of subgroup analysis, we felt it would be more appropriate to compare the subgroup of participants with focal onset epilepsy and the subgroup with 'generalised onset or unclassified epilepsy'. We conducted a Chi2 test of interaction between treatment and epilepsy type.
If further trials recruiting individuals with generalised seizure types are included in updates of this review, we hope to perform a subgroup analysis of focal onset versus generalised onset epilepsy.
If we deemed considerable statistical heterogeneity to be present (I2 statistic > 50%), we intended to perform meta‐analyses using a random‐effects model in addition to a fixed‐effect model and present the results of both models. Also, if possible, we considered investigating factors that could contribute to heterogeneity (e.g. participant covariates, trial design as described in Data extraction and management) via further subgroup analyses or via meta‐regression models.
Sensitivity analysis
We intended to perform sensitivity analyses if we considered studies to be at high risk of bias (see Assessment of risk of bias in included studies), if we found inconsistencies between published study reports and the IPD provided (see Dealing with missing data) or if trials included multiple treatment arms (see Unit of analysis issues). We also intended to perform several sensitivity analyses to test the robustness of our results in relation to the characteristics of the included trials.
Definition of time to treatment failure: we classified reasons for treatment failure or withdrawal that were related to the trial treatment as 'events' and reasons not related to treatment as 'censored' in analyses of 'time to treatment failure.' If included trials classified the reasons for withdrawal or failure as events or censored differently from us, we conducted sensitivity analyses to account for differences in the definition of a withdrawal or failure 'event' (SANAD A 2007).
Aggregate data: this is an IPD review; we will include IPD only in all primary analyses. We were unable to extract any aggregate data from the one trial included in this review for which no IPD were available (Resendiz‐Aparicio 2004), but if we are able to extract aggregate data from trials included in future updates of this review (see Data extraction and management), we intend to combine aggregate data with IPD in sensitivity analyses and examine the differences between the IPD and combined analyses.
Open‐label extension: one included trial comprised a six‐month, double‐blind phase followed by an open‐label extension phase (Privitera 2003). As both blinded and open‐label trials are eligible for inclusion in this review, by our intention‐to‐treat approach, we included the entire follow‐up period in analysis. We also performed a sensitivity analysis of outcomes of time to treatment failure, time to first seizure and time to six‐month remission, censoring these outcomes at the end of the double‐blind phase and comparing results to those from the primary analysis (we note that in this analysis, time to six‐month remission becomes time to immediate six‐month remission when considered over a six‐month period).
Misclassification of seizure type: this is a recognised problem in epilepsy, whereby some people with generalised seizures have been mistakenly classed as having focal onset seizures and vice versa. Such misclassification had an impact on the results of three reviews in a series of pair‐wise reviews of monotherapy in epilepsy comparing carbamazepine, phenobarbitone, phenytoin and sodium valproate, in which around 30% to 50% of participants analysed may have had their seizure type misclassified as generalised onset (Nevitt 2017b; Nevitt 2018b; Nevitt 2018d). Given the potential biases introduced into these three reviews, we examined the distribution of age at onset for individuals with generalised seizures in the trials included in this review, to assess the potential impact of misclassification of seizure type on the outcomes. (There is clinical evidence that individuals with generalised onset seizures are unlikely to have an 'age of onset' greater than 25 to 30 years (Malafosse 1994)). Given that most of the individuals recruited to the trials included in the present review experienced focal onset seizures, this sensitivity analysis was not appropriate for this review and instead we performed a subgroup analysis of focal onset versus generalised onset or unclassified epilepsy (see Subgroup analysis and investigation of heterogeneity).
For updates of the review, if future trials recruit more individuals with generalised onset seizures, we intend to perform a sensitivity analysis in two ways:
we will reclassify individuals with generalised seizure types and age at onset greater than 30 years as having focal onset seizures, and we will repeat subgroup analyses;
we will reclassify individuals with generalised seizure types and age at onset greater than 30 years into an 'uncertain seizure type' group, and we will repeat subgroup analyses with three groups.
'Summary of findings' table
We have presented two 'Summary of findings' tables (Table 1; Table 2). The first presents the summary of the main comparison reporting the primary outcome of 'time to treatment failure' in the subgroups of participants with focal onset epilepsy and generalised onset or unclassified epilepsy overall for all participants, adjusted by epilepsy type.
Summary of findings for the main comparison. Topiramate compared with carbamazepine for epilepsy (time to treatment failure).
| Topiramate compared with carbamazepine for epilepsy (time to treatment failure) | ||||||
|
Population: adults and children with newly onset focal or generalised epilepsy Settings: outpatients Intervention: topiramate Comparison: carbamazepine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Carbamazepine | Topiramate | |||||
|
Time to treatment failure (any reason related to treatment) All participants ‐ adjusted by seizure type Range of follow‐up: 0 to 2420 days |
The median time to treatment failure was 1144 days in the carbamazepine group | The median time to treatment failure was 614 days (530 days shorter) in the topiramate group | HR 1.16 (0.97 to 1.38)a |
1129 (2 studies) |
⊕⊕⊕⊝ Moderateb | HR < 1 indicates a clinical advantage for topiramate Treatment failure due to lack of efficacy (HR 1.46, 95% CI 1.08 to 1.98, P = 0.01), occurred significantly earlier on topiramate compared to carbamazepine and there was no difference between the drugs for treatment failure due to adverse events (HR 1.02, 95% CI 0.82 to 1.27, P = 0.84) |
|
Time to treatment failure (any reason related to treatment)Subgroup: focal onset seizures Range of follow‐up: 0 to 2420 days |
The median time to treatment failure was 1149 days in the carbamazepine group | The median time to treatment failure was 505 days (644 days shorter) in the topiramate group | HR 1.21 (1.01 to 1.46) |
937 (2 studies) |
⊕⊕⊕⊝ Moderateb | HR < 1 indicates a clinical advantage for topiramate Treatment failure due to lack of efficacy (HR 1.47, 95% CI 1.07 to 2.02, P = 0.02), occurred significantly earlier on topiramate compared to carbamazepine and there was no difference between the drugs for treatment failure due to adverse events (HR 1.08, 95% CI 0.85 to 1.36, P = 0.53) |
|
Time to treatment failure (any reason related to treatment) Subgroup: generalised onset tonic‐clonic seizures or unclassified epilepsy Range of follow‐up: 0 to 1446 days |
The median time to treatment failure was 1056 days in the carbamazepine group | The median time to treatment failure was 1448 days (392 days longer) in the topiramate group | HR 0.88 (0.56 to 1.39) |
192 (2 studies) |
⊕⊕⊝⊝ Lowc, d | HR < 1 indicates a clinical advantage for topiramate There was also no statistically significant difference between drugs in treatment failure due to adverse events (HR 0.72, 95% CI 0.39 to 1.31, P = 0.28) or treatment failure due to lack of efficacy (HR 1.41, 95% CI 0.54 to 3.67, P = 0.48) |
| *Illustrative risks in the topiramate and carbamazepine groups are calculated at the median time to treatment failure (i.e. the time to 50% of participants failing or withdrawing from allocated treatment) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'time to treatment failure' between the treatment groups. CI: confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low certainty: we are very uncertain about the estimate | ||||||
aPooled HR for all participants adjusted for seizure type. bDowngraded once for risk of bias; the larger of the two studies was open‐label (SANAD A 2007), and may have influenced the withdrawal rates of the trial. cDowngraded once for imprecision and applicability; limited information on generalised seizure types and most participants do not have a classified seizure type in this subgroup so the interpretation of this seizure type is unclear.
Summary of findings 2. Topiramate compared with carbamazepine for epilepsy (secondary outcomes).
| Topiramate compared with carbamazepine for epilepsy (secondary outcomes) | ||||||
|
Population: adults and children with newly onset focal or generalised epilepsy Settings: outpatients Intervention: topiramate Comparison: carbamazepine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Carbamazepine | Topiramate | |||||
|
Time to first seizure after randomisation All participants Range of follow‐up: 0 to 2420 days |
The median time to first seizure after randomisation was 154 days in the carbamazepine group | The median time to first seizure after randomisation was 124 days (30 days shorter) in the topiramate group | HR 1.11 (0.96 to 1.29)a |
1115 (2 studies) |
⊕⊕⊕⊕ High | HR < 1 indicates a clinical advantage for topiramate |
|
Time to first seizure after randomisation Subgroup: focal onset seizures Range of follow‐up: 0 to 2420 days |
The median time to first seizure after randomisation was 95 days in the carbamazepine group | The median time to first seizure after randomisation was 90 days (5 days shorter) in the topiramate group | HR 1.12 (0.95 to 1.31) |
925 (2 studies) |
⊕⊕⊕⊕ High | HR < 1 indicates a clinical advantage for topiramate |
|
Time to first seizure after randomisation Subgroup: generalised onset tonic‐clonic seizures or unclassified epilepsy Range of follow‐up: 0 to 853 days |
The median time to first seizure after randomisation was 495 days in the carbamazepine group | The median time to first seizure after randomisation was 393 days (102 days shorter) in the topiramate group | HR 1.08 (0.70 to 1.66) |
190 (2 studies) |
⊕⊕⊕⊝ Moderateb | HR < 1 indicates a clinical advantage for topiramate |
|
Time to 12‐month remission of seizures All participants Range of follow‐up: 0 to 2420 days |
The median time to achieve 12‐month remission was 484 days in the carbamazepine group | The median time to achieve 12‐month remission was 537 days (53 days longer) in the topiramate group | HR 0.84 (0.71 to 0.99)a |
1115 (2 studies) |
⊕⊕⊕⊕ High | HR < 1 indicates a clinical advantage for carbamazepine |
|
Time to 12‐month remission of seizures Subgroup: focal onset seizures Range of follow‐up: 0 to 2420 days |
The median time to achieve 12‐month remission was 533 days in the carbamazepine group | The median time to achieve 12‐month remission was 582 days (49 days longer) in the topiramate group | HR 0.82 (0.69 to 0.99) |
925 (2 studies) |
⊕⊕⊕⊕ High | HR < 1 indicates a clinical advantage for carbamazepine |
|
Time to 12‐month remission of seizures Subgroup: generalised onset tonic‐clonic seizures or unclassified epilepsy Range of follow‐up: 0 to 853 days |
The median time to achieve 12‐month remission was 365 days in the carbamazepine group | The median time to achieve 12‐month remission was 365 days (0 days longer) in the topiramate group | HR 0.92 (0.61 to 1.41) |
190 (2 studies) |
⊕⊕⊕⊝ Moderateb | HR < 1 indicates a clinical advantage for carbamazepine |
| *Illustrative risks in the topiramate and carbamazepine groups are calculated at the median time to first seizure or time to 12‐month remission (i.e. the time to 50% of participants experiencing a first seizure or 12‐months of remission) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'time to first seizure' or 'time to 12‐month remission' between the treatment groups. CI: confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect Moderate certainty: : further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low certainty: we are very uncertain about the estimate | ||||||
aPooled HR for all participants adjusted for seizure type. bDowngraded once for imprecision and applicability, limited information on generalised seizure types and most participants do not have a classified seizure type in this subgroup so the interpretation of this seizure type is unclear.
The second 'Summary of findings' table reports the secondary outcomes of 'time to first seizure' and 'time to 12‐month remission' in the subgroups of participants with focal onset epilepsy and generalised onset or unclassified epilepsy overall for all participants, adjusted by epilepsy type.
We determined the certainty of the evidence using the GRADE approach (Hultcrantz 2017; GRADEPro GDT 2015), whereby we downgraded evidence in the presence of a high risk of bias in at least one trial, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results or high probability of publication bias. We downgraded evidence by one level if we considered the limitation to be serious and two levels if we considered it to be very serious.
Results
Description of studies
Results of the search
We identified 136 records from the databases and search strategies outlined in Electronic searches. We found no additional records by handsearching and checking the reference lists of included studies. We removed 30 duplicate records and screened 106 records (title and abstract) for inclusion in the review. We excluded 85 records based on title and abstract, and assessed 16 records describing four full‐text articles for inclusion in the review. We excluded three records linked to a single study from the review (see Excluded studies below) and included three studies in the review described in 13 records (see Included studies below). See Figure 1 for a PRISMA study flow diagram.
1.
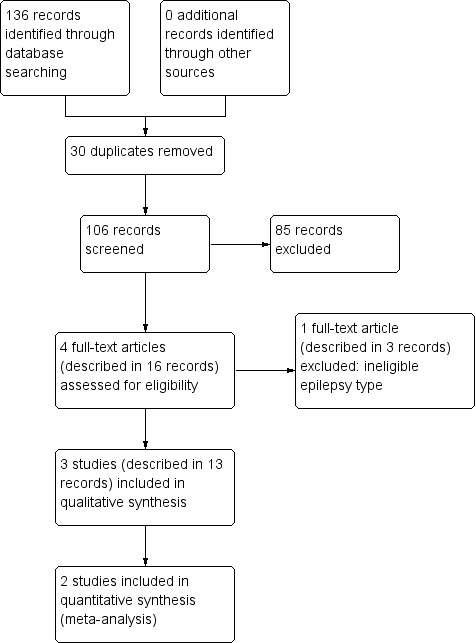
Study flow diagram.
Included studies
We included three studies in the review (Privitera 2003; Resendiz‐Aparicio 2004; SANAD A 2007).
One trial recruited individuals over the age of six years (Privitera 2003), and another trial recruited individuals over the age of four years (SANAD A 2007). The third trial recruited children between the ages of six and 18 years (Resendiz‐Aparicio 2004).
One trial recruited individuals with focal seizures (with or without secondary generalisation) (Resendiz‐Aparicio 2004). SANAD A 2007 was designed to recruit individuals with focal seizures only, but some individuals with generalised onset or unclassified seizures were recruited; we examine this seizure classification in subgroup analysis. Privitera 2003 was designed in two strata, based on whether the recommended treatment would be carbamazepine or sodium valproate. Within the two strata, participants were randomised to topiramate 100 mg/day or 200 mg/day, or carbamazepine/sodium valproate depending on the stratum. Only the carbamazepine stratum (participants randomised to carbamazepine or one of the two doses of topiramate) was eligible for the randomised comparison in this review. The majority of participants within this stratum had focal seizures but some individuals with generalised onset or unclassified seizures were also recruited; we examine this seizure classification in subgroup analysis.
Two trials recruited individuals with new‐onset seizures (Privitera 2003; Resendiz‐Aparicio 2004), and one trial recruited individuals with new‐onset, relapsed or recurrent seizures (failure of an antiepileptic drug (AED) not randomised in the trial) (SANAD A 2007).
All three trials were conducted in a multicentre setting; Resendiz‐Aparicio 2004 was conducted in Mexico, SANAD A 2007 was conducted in the UK and Privitera 2003 was conducted in centres across the USA, Canada, Europe and South America.
Individual participant data (IPD) were available for two trials randomising 1151 participants to carbamazepine or topiramate (Privitera 2003; SANAD A 2007). For the third trial, which recruited 88 participants, we were unable to contact the original authors and so IPD could not be included in this review (Resendiz‐Aparicio 2004). Overall, IPD were available for 93% of the total eligible 1239 participants.
Data were available for the following participant characteristics (percentage of 1151 participants with data available): drug randomised (100%), sex (98%, data missing for 18 participants in SANAD A 2007), age at randomisation (98%, data missing for 18 participants in SANAD A 2007), number of seizures in six months prior to randomisation (98%, missing for 21 participants in SANAD A 2007) and seizure type (90%, data missing for 22 participants in Privitera 2003 and 89 participants in SANAD A 2007).
Results of neurological examinations were available for 738 of 756 participants (98%) from SANAD A 2007 (data for 18 participants missing). This information was not available for Privitera 2003.
No information was available from either trial regarding electroencephalographic (EEG) or computerised tomography/magnetic resonance imaging (CT/MRI) results and time since first seizure to randomisation.
See the Characteristics of included studies and Table 3 for further details.
1. Demographic characteristics of trial participants (trials providing IPD).
| Characteristic | Privitera 2003 | SANAD A 2007 | ||||
| CBZ | TPM | Missing | CBZ | TPM | Missing | |
| Focal seizures n (%) | 91 (71) | 194 (73) | 22 | 333 (88) | 321 (85) | 89 |
|
Male gender n (%) |
68 (52) | 147 (55) | 0 | 204 (55) | 204 (55) | 18 |
| Abnormal neurological exam, n (%) | NA | NA | 395 | 87 (24) | 105 (28) | 18 |
| Age at entry (years), mean (SD), range | 35.4 (18.7), 6 to 80 | 33.9 (18.2), 6 to 75 | 0 | 39.3 (18.4), 5 to 82 | 38.7 (18.6), 5 to 86 | 18 |
| Number of seizures in prior 6 months: median (range) | 4 (0 to 2400) | 4 (0 to 1346) | 0 | 4 (0 to 467) | 4 (0 to 393) | 21 |
CBZ = carbamazepine, TPM = topiramate, NA = not available, SD = standard deviation
Excluded studies
We excluded one study described in three records (Kang 2007). This study recruited children with only benign rolandic epilepsy, which was an ineligible seizure type for this review.
Risk of bias in included studies
For further details, see the Characteristics of included studies and Figure 2.
2.
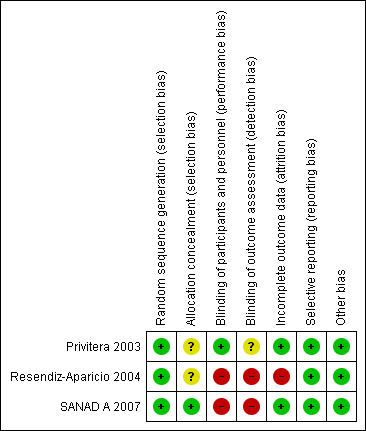
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
As all three trials described adequate methods of generation of a random list we judged them to be at low risk of bias; Privitera 2003 used computer‐generated block randomisation, Resendiz‐Aparicio 2004 used random number tables and SANAD A 2007 used minimisation.
SANAD A 2007 used telephone randomisation to a central allocation service, and so we judged the study to be at low risk of bias for allocation concealment. As the other two trials did not describe a method of allocation concealment we judged them to be at unclear risk of bias.
Blinding
We judged the two open‐label trials to be at high risk of performance and detection bias (Resendiz‐Aparicio 2004; SANAD A 2007). The third trial comprised a six‐month double‐blind phase followed by an open‐label extension phase; it was unclear if outcome assessors were blinded in this trial.
Incomplete outcome data
In theory, a review using IPD should overcome issues of attrition bias as unpublished data can be provided, unpublished outcomes calculated, and all randomised participants can be analysed by an intention‐to‐treat approach. Both trials providing IPD for all randomised individuals reported the extent of follow‐up for each individual (Privitera 2003; SANAD A 2007). We queried any missing data with the original trial authors. From the information provided by the authors, we deemed the small amount of missing data present (see Included studies) to be missing at random and considered that it did not affect our analysis.
For the trial for which no IPD were provided, we included only those participants who completed the trial in analyses; this is not an intention‐to‐treat approach so we judged this trial to be at high risk of attrition bias (Resendiz‐Aparicio 2004).
Selective reporting
In theory, a review using IPD should overcome issues of reporting biases as unpublished data can be provided and unpublished outcomes calculated. We requested trial protocols in all IPD requests and protocols were provided for Privitera 2003 and SANAD A 2007. We received sufficient IPD to calculate all outcomes for both trials.
For the trial for which no IPD were provided, no protocol was available and the trial publication was translated from Spanish by SJN. We judged seizure outcomes and adverse events to be well reported and to be at low risk of selective reporting bias (Resendiz‐Aparicio 2004).
Other potential sources of bias
We identified no other potential sources of bias in any of the trials.
Effects of interventions
Table 4 gives details regarding the number of individuals (with IPD) contributing to each analysis, Table 1 summarises the results for the primary outcome ‘time to treatment failure’ and Table 2 summarises the results for the secondary outcomes ‘time to first seizure’ and ‘time to 12‐month remission.’
2. Number of participants included in analyses (trials providing IPD).
| Privitera 2003 | SANAD A 2007a | Total | |||||||
| CBZ | TPM | Total | CBZ | TPM | Total | CBZ | TPM | Total | |
| Number randomised | 129 | 266 | 395 | 378 | 378 | 756 | 507 | 644 | 1151 |
| Time to treatment failure | 129 | 266 | 395 | 368 | 366 | 734 | 497 | 632 | 1129 |
| Time to first seizure | 129 | 266 | 395 | 362 | 358 | 720 | 491 | 624 | 1115 |
|
Time to 6 and 12‐month remission |
129 | 266 | 395 | 362 | 358 | 720 | 491 | 624 | 1115 |
CBZ = carbamazepine, TPM = topiramate
aWithdrawal time missing for 22 participants and seizure data after follow‐up missing for 36 participants in SANAD A 2007.
Survival curve (cumulative incidence) plots are shown in Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10, Figure 11, Figure 12, Figure 13 and Figure 14. We produced all cumulative incidence plots in Stata software version 14.1. We used Stata software version 14 to produce all survival curve plots using data from all trials providing IPD combined (Stata 2015).
3.
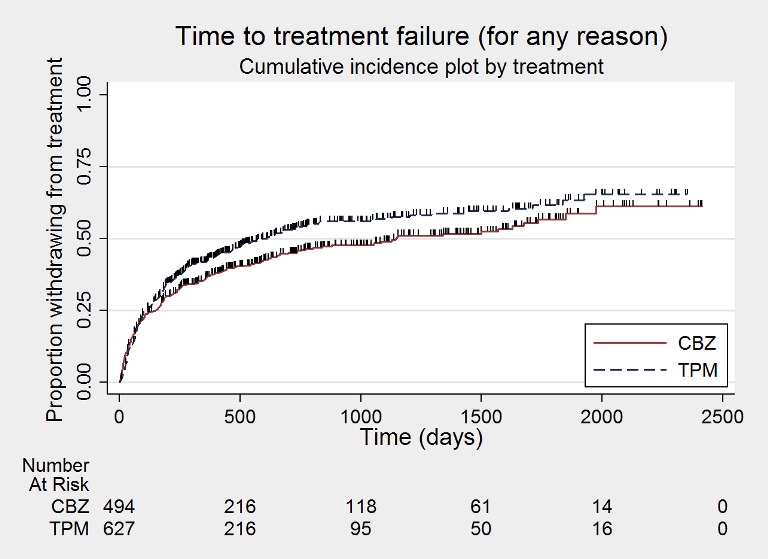
Time to treatment failure ‐ any reason related to the treatment (CBZ: carbamazepine; TPM: Topiramate)
4.
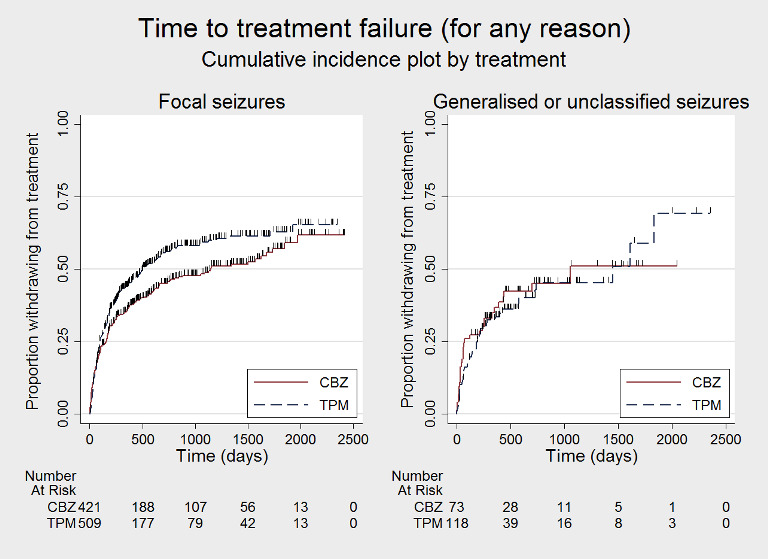
Time to treatment failure ‐ any reason related to the treatment, by seizure type (CBZ: carbamazepine; TPM: Topiramate)
5.
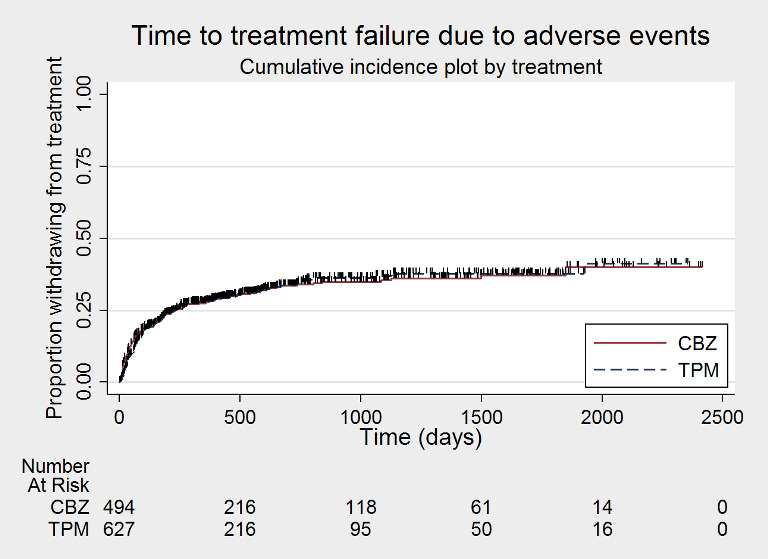
Time to treatment failure due to adverse events (CBZ: carbamazepine; TPM: Topiramate)
6.
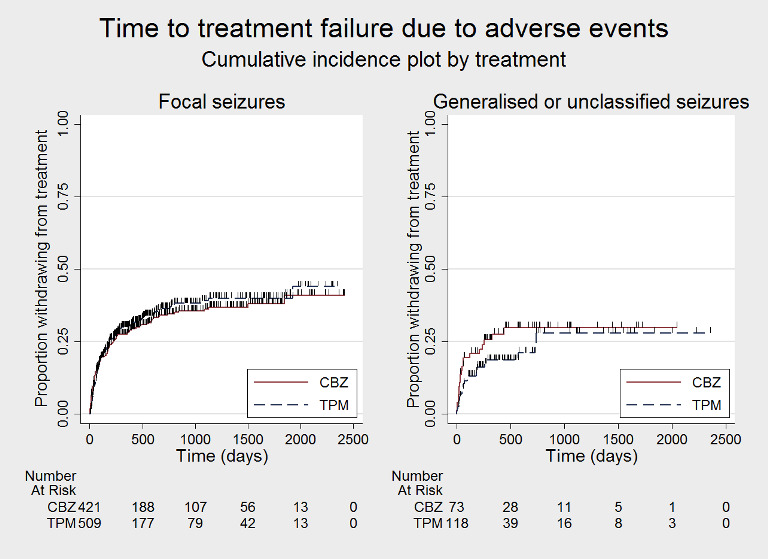
Time to treatment failure due to adverse events, by seizure type (CBZ: carbamazepine; TPM: Topiramate)
7.
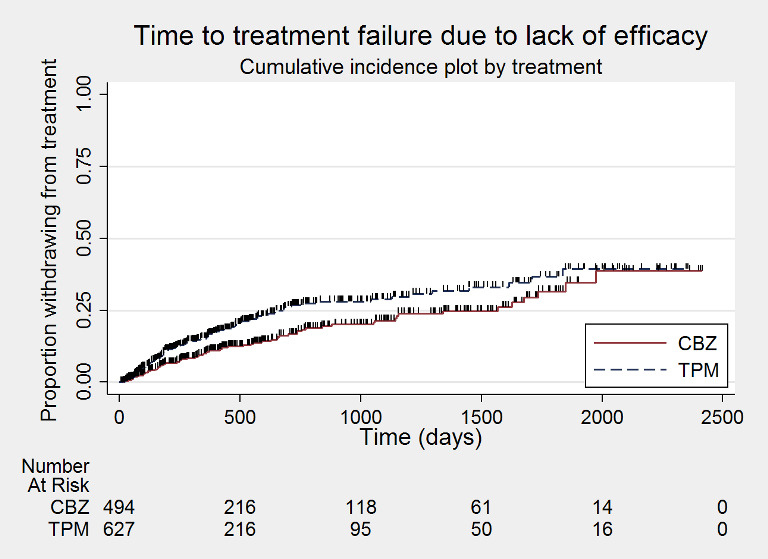
Time to treatment failure due to lack of efficacy (CBZ: carbamazepine; TPM: Topiramate)
8.
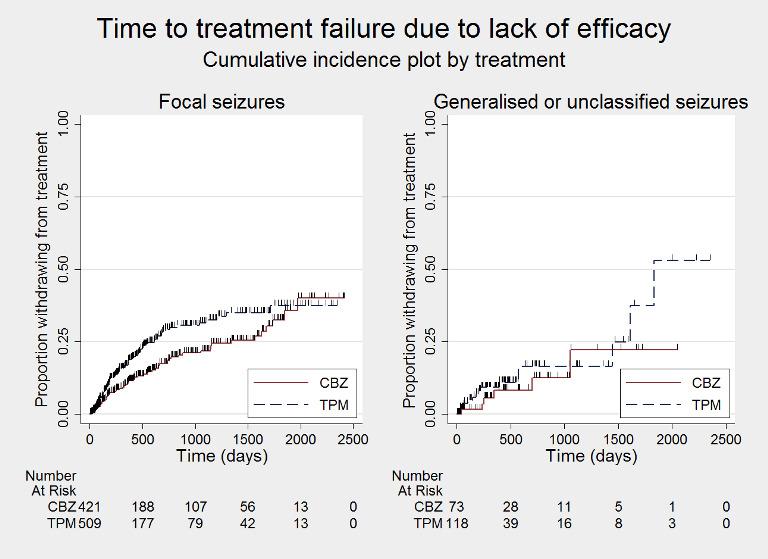
Time to treatment failure due to lack of efficacy, by seizure type (CBZ: carbamazepine; TPM: Topiramate)
9.
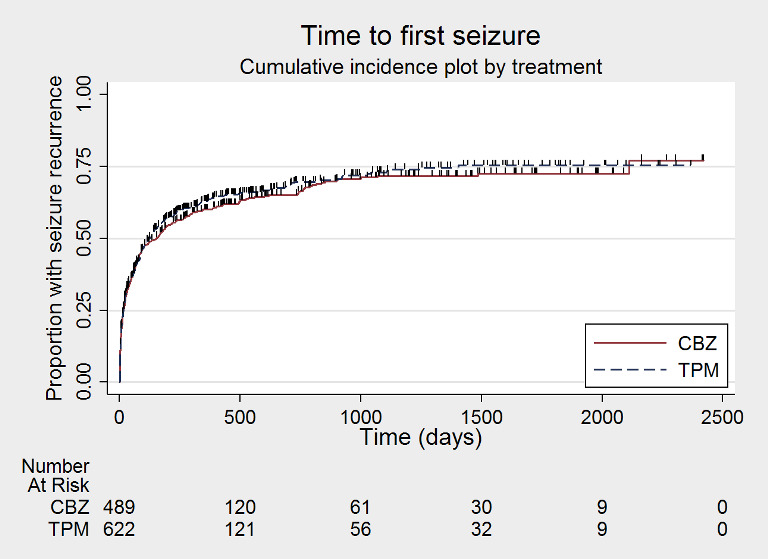
Time to first seizure after randomisation (CBZ: carbamazepine; TPM: Topiramate)
10.
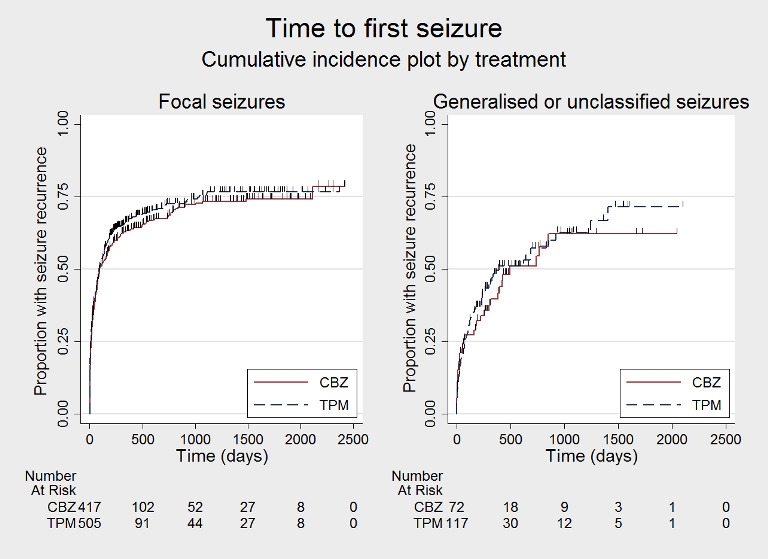
Time to first seizure after randomisation, by seizure type (CBZ: carbamazepine; TPM: Topiramate)
11.
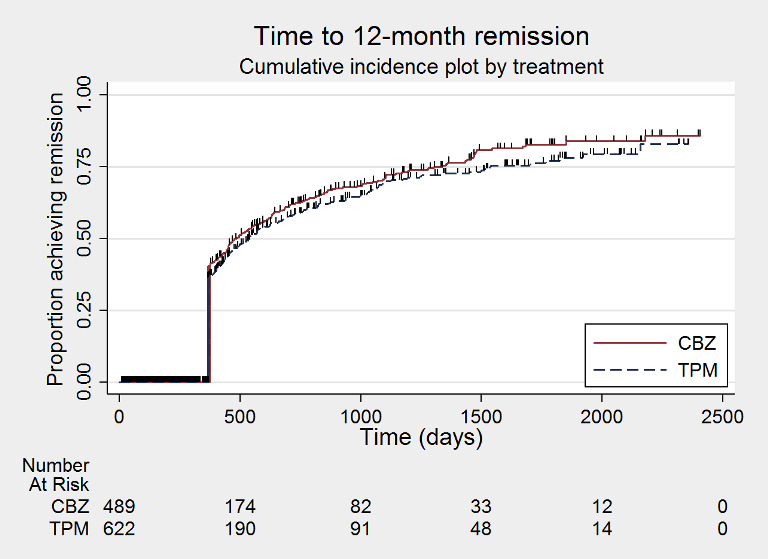
Time to 12‐month remission (CBZ: carbamazepine; TPM: Topiramate)
12.
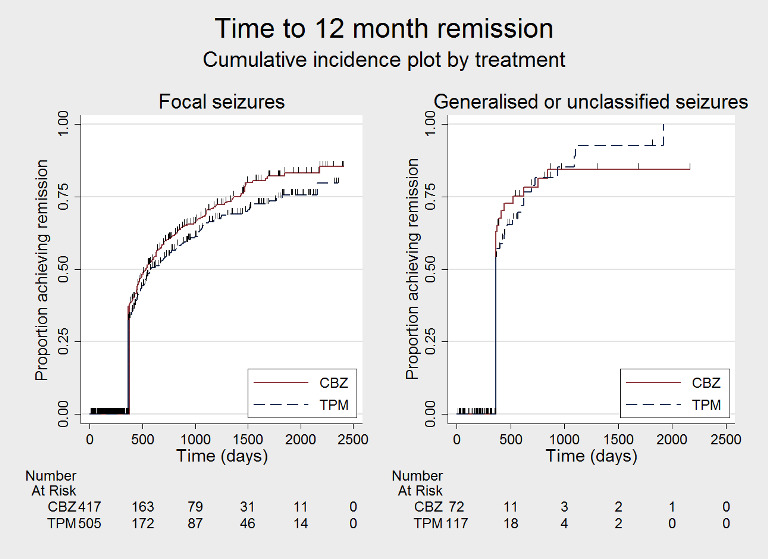
Time to 12‐month remission by seizure type (CBZ: carbamazepine; TPM: Topiramate)
13.
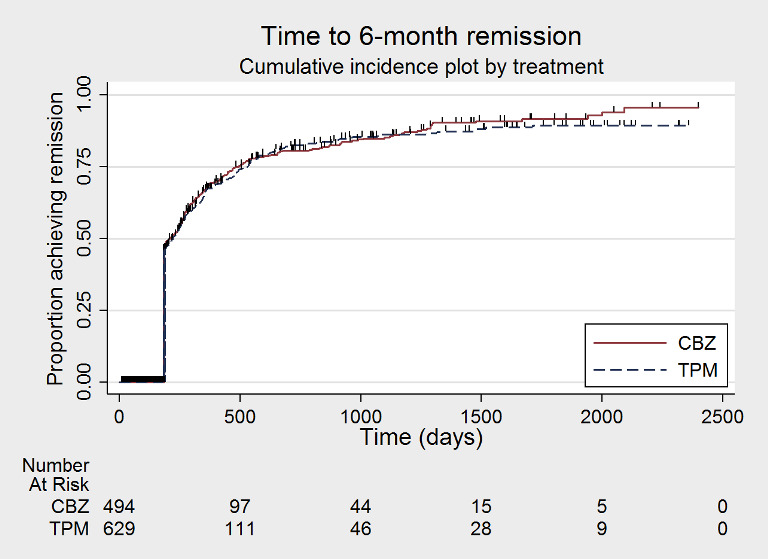
Time to 6‐month remission (CBZ: carbamazepine; TPM: Topiramate)
14.
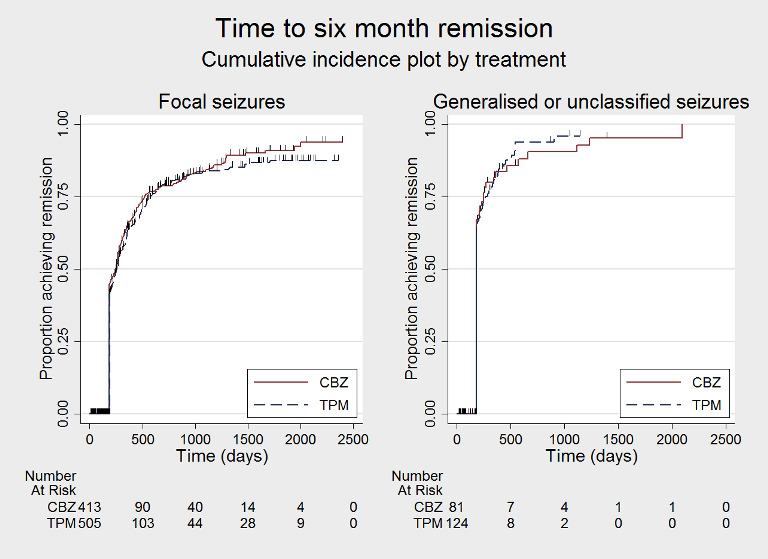
Time to 6‐month remission by seizure type (CBZ: carbamazepine; TPM: Topiramate)
We note that participants with event times of zero (i.e. those who experienced treatment failure or experienced seizure recurrence on the day of randomisation), are not included in the 'numbers at risk' on the graphs and that data are not stratified by trial within these survival curve plots. All figures are intended to provide a visual representation of outcomes, extent of follow‐up and visual differences between seizure types. These graphs are not intended to show statistical significance and numerical values may vary compared to the text due to differences in methodology.
We calculated all the hazard ratios (HRs) presented using generic inverse variance fixed‐effect meta‐analysis unless otherwise stated. All analyses met the assumption of proportional hazards (addition of time varying covariate into the model non‐significant) unless stated.
Primary outcome
Time to treatment failure (retention time)
For this outcome, a HR less than 1 indicates a clinical advantage for topiramate.
Table 5 shows the reasons for premature termination for 1151 participants in the two trials included in this analysis and how we classified these treatment failures or withdrawals in analysis of IPD. Times to treatment failure were available for 1129 participants in the two trials (98% of total 1151 participants included in analysis). Withdrawal times were missing for 22 participants in SANAD A 2007 (see Table 4); however, as all 22 participants withdrew for reasons which would have been censored in analysis, we consider the impact of these missing participants on the analysis to be negligible.
3. Reasons for premature discontinuation (treatment failure) in trials providing IPD.
| Study | Privitera 2003 | SANAD A 2007e |
Grand Total |
||||||
|
Reason (and classification in analysis) |
CBZ |
TPM 100 mg/day |
TPM 200 mg/day |
TPM (pooled) |
Total | CBZ | TPM | Total | |
| Completed study (Censored) | 63 | 64 | 66 | 130 | 193 | 151 | 137 | 288 | 481 |
| Adverse event (Event) | 32 | 26 | 30 | 56 | 88 | 104 | 103 | 207 | 295 |
| Ineffective treatment (Event) | 10 | 18 | 13 | 31 | 41 | 43 | 55 | 98 | 139 |
| Other reason (Event)b | 7 | 9 | 8 | 17 | 24 | 10 | 16 | 26 | 50 |
| Both ineffective treatment and adverse events (Event) |
0 | 0 | 0 | 0 | 0 | 20 | 28 | 48 | 48 |
| Remission (Censored) | 0 | 0 | 0 | 0 | 0 | 25 | 19 | 44 | 44 |
| Other reason (Censored)c | 3 | 4 | 2 | 6 | 9 | 19 | 12 | 31 | 40 |
| Participant choice (Event)d | 5 | 9 | 7 | 16 | 21 | 6 | 8 | 14 | 35 |
| Lost to follow‐up (Censored) | 9 | 6 | 4 | 10 | 19 | 0 | 0 | 0 | 19 |
| Total censored | 75 | 74 | 72 | 146 | 221 | 201 | 176 | 377 | 598 |
| Total events | 54 | 62 | 58 | 120 | 174 | 177 | 202 | 379 | 553 |
| Grand total | 129 | 136 | 130 | 266 | 395 | 378 | 378 | 756 | 1151 |
CBZ = carbamazepine, TPM = topiramate
aPrimary reason for discontinuation specified: participants may have withdrawn from allocated treatment for a combination of reasons. bOther treatment‐related failures: drug‐related death, pregnancy or perceived remission (SANAD A 2007). Specified only as 'other reason' in Privitera 2003. cOther withdrawals (not treatment‐related): epilepsy diagnosis changed and death not related to treatment (SANAD A 2007). Specified only as 'other reason' in Privitera 2003. dWithdrawal of consent/participant choice classified as an event in this review but censored in included trial (SANAD A 2007). Sensitivity analysis classifying withdrawal of consent as a censored observation did not change conclusions (results available on request). eWithdrawal reasons available for all participants in the two studies but withdrawal times missing for 22 participants in SANAD A 2007 (see Table 4). These 22 participants were not included in analysis of time to treatment failure, but all 22 withdrew for reasons which would have been censored in analysis, therefore the impact of these missing participants on the analysis is minor.
Of 1151 participants, 670 (58%) prematurely withdrew from treatment: 377 of 644 (59%) participants randomised to topiramate and 293 of 507 (58%) participants randomised to carbamazepine. We deemed 553 participants (83% of total treatment failures) to have failed treatment for reasons related to the allocated drug and classified these reasons as ’events’ in analysis ‐ 322 randomised to topiramate (85% of topiramate treatment failures) and 231 randomised to carbamazepine (79% of carbamazepine treatment failures). The most common treatment‐related reason for treatment failure was adverse events: 44% of total treatment failures, 159 participants randomised to topiramate (42% of total topiramate treatment failures) and 136 participants randomised to carbamazepine (46% of total carbamazepine treatment failures).
We classed the other 117 treatment failures or withdrawals (55 participants randomised to topiramate and 62 randomised to carbamazepine) to be not related to the allocated drug and censored these participants in analysis, in addition to the 481 participants (267 receiving topiramate and 214 receiving carbamazepine) who completed the trial without failing or withdrawing from the treatment.
Considering 'time to treatment failure for any reason related to the treatment', the overall pooled HR (for 1129 participants providing IPD from 2 trials) was 1.15 (95% confidence interval (CI) 0.97 to 1.37; P = 0.11, moderate‐certainty evidence; Analysis 1.1), indicating a potential advantage to carbamazepine that was not statistically significant; in other words, treatment failure may occur earlier on topiramate than carbamazepine but we cannot rule out a slight advantage to topiramate or no difference between the drugs. No heterogeneity was present between trials (I2 = 0%).
1.1. Analysis.
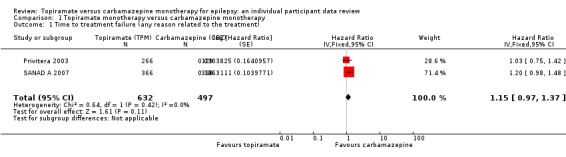
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 1 Time to treatment failure (any reason related to the treatment).
Considering 'time to treatment failure due to adverse events' (all other reasons for treatment failure or treatment withdrawal censored in analysis), the overall pooled HR (for 1129 participants providing IPD from 2 trials) was 1.01, 95% CI 0.82 to 1.26; P = 0.90, moderate‐certainty evidence; Analysis 1.2), indicating no clear differences between the drugs. No important heterogeneity was present between trials (I2 = 15%).
1.2. Analysis.
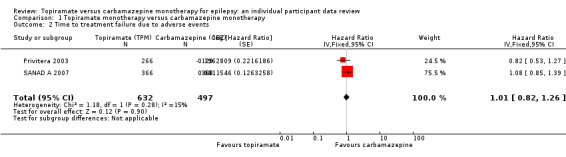
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 2 Time to treatment failure due to adverse events.
Considering 'time to treatment failure due to lack of efficacy' (all other reasons for treatment failure or treatment withdrawal censored in analysis), the overall pooled HR (for 1129 participants providing IPD from 2 trials) was 1.45, 95% CI 1.08 to 1.96; P = 0.01, moderate‐certainty evidence; Analysis 1.3), indicating a statistically significant advantage to carbamazepine; in other words, treatment failure due to adverse events occurred earlier on topiramate than carbamazepine in the two included trials. No heterogeneity was present between trials (I2 = 0%).
1.3. Analysis.
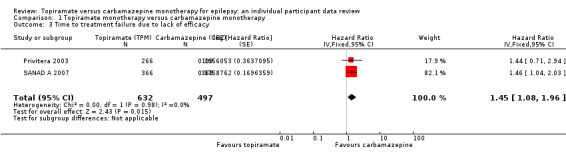
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 3 Time to treatment failure due to lack of efficacy.
Subgroup analyses: seizure type (focal onset versus generalised onset or unclassified epilepsy)
See Subgroup analysis and investigation of heterogeneity for more details regarding the definitions of subgroups.
Considering time to treatment failure for any reason related to the treatment, for participants with focal onset seizures (937 participants providing IPD), the pooled HR was 1.21 (95% CI 1.01 to 1.46; P = 0.04, moderate‐certainty evidence; Analysis 1.4), indicating a statistically significant advantage for carbamazepine; in other words, treatment failure due to adverse events occurred earlier on topiramate than carbamazepine in the two included trials. For participants with generalised onset or unclassified epilepsy (192 participants providing IPD), the pooled HR was 0.88 (95% CI 0.56 to 1.39; P = 0.59, low‐certainty evidence; Analysis 1.4), indicating that treatment failure may occur earlier on carbamazepine than topiramate but CIs are wide so we cannot rule out an advantage to carbamazepine or no differences between the drugs.
1.4. Analysis.
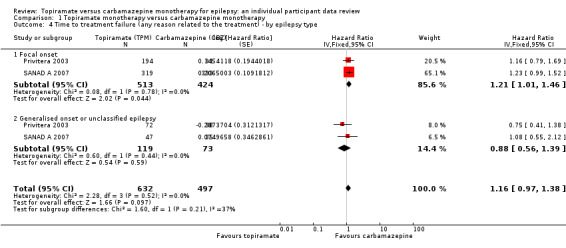
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 4 Time to treatment failure (any reason related to the treatment) ‐ by epilepsy type.
The overall pooled HR (adjusted for epilepsy type for 1129 participants) was HR 1.16 (95% CI 0.97 to 1.38, P = 0.10, moderate‐certainty evidence, Analysis 1.4), indicating (as above) a potential advantage to carbamazepine that is not statistically significant. No between‐trial heterogeneity was present overall or by subgroup (I2 = 0%) and there was no evidence of a difference between the subgroups (test for subgroup differences P = 0.21; Analysis 1.4).
Considering 'time to treatment failure due to adverse events' (all other reasons for treatment failure or treatment withdrawal censored in analysis), for participants with focal onset seizures (937 participants providing IPD), the pooled HR was 1.08 (95% CI 0.85 to 1.36; P = 0.53, moderate‐certainty evidence; Analysis 1.5), indicating no clear differences between the drugs. For participants with generalised onset or unclassified epilepsy (192 participants providing IPD), the pooled HR was 0.72 (95% CI 0.39 to 1.31; P = 0.28, low‐certainty evidence; Analysis 1.5), indicating that treatment failure may occur earlier on carbamazepine than topiramate but CIs are wide so we cannot rule out an advantage to carbamazepine or no differences between the drugs.
1.5. Analysis.
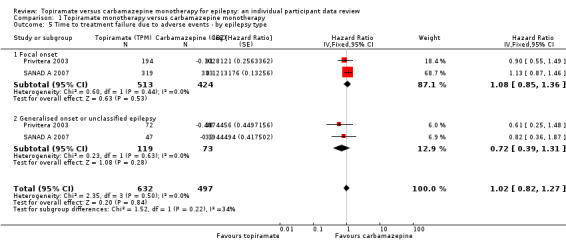
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 5 Time to treatment failure due to adverse events ‐ by epilepsy type.
The overall pooled HR (adjusted for epilepsy type for 1129 participants) was HR 1.02 (95% CI 0.82 to 1.27; P = 0.84, moderate‐certainty evidence; Analysis 1.5), indicating no clear differences between the drugs. No between‐trial heterogeneity was present overall or by subgroup (I2 = 0%) and there was no evidence of a difference between the subgroups (test for subgroup differences P = 0.22; Analysis 1.5).
Considering 'time to treatment failure due to lack of efficacy' (all other reasons for treatment failure or treatment withdrawal censored in analysis), for participants with focal onset seizures (937 participants providing IPD), the pooled HR was 1.47 (95% CI 1.07 to 2.02; P = 0.02, moderate‐certainty evidence; Analysis 1.6), indicating a statistically significant advantage to carbamazepine; in other words, for individuals with focal onset seizures, treatment failure due to adverse events occurred earlier on topiramate than carbamazepine in the two included trials. For participants with generalised onset or unclassified epilepsy (192 participants providing IPD), the pooled HR was 1.41 (95% CI 0.54 to 3.67; P = 0.48, low‐certainty evidence; Analysis 1.6), indicating that treatment failure may occur earlier on topiramate than carbamazepine but CIs are wide so we cannot rule out an advantage to topiramate or no differences between the drugs.
1.6. Analysis.
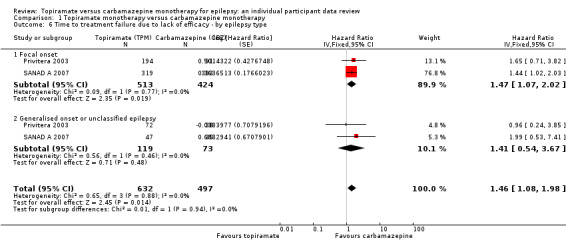
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 6 Time to treatment failure due to lack of efficacy ‐ by epilepsy type.
The overall pooled HR (adjusted for epilepsy type for 1129 participants) was HR 1.46 (95% CI 1.08 to 1.98; P = 0.01, moderate‐certainty evidence; Analysis 1.6), indicating a statistically significant advantage to carbamazepine; in other words, treatment failure due to adverse events occurred earlier on topiramate than carbamazepine in the two included trials. No between‐trial heterogeneity was present overall or by subgroup (I2 = 0%) and there was no evidence of a difference between the subgroups (test for subgroup differences P = 0.94; Analysis 1.6).
Sensitivity analysis
We performed a sensitivity analysis including only IPD from only the six‐month double‐blind period of Privitera 2003; participants who failed treatment after six months (9 receiving carbamazepine and 30 receiving topiramate) were censored at six months. When only treatment from the first six months of Privitera 2003 were combined with IPD from SANAD A 2007, numerical results were very similar and conclusions were unchanged (results available from authors on request).
One included trial allocated participants to three treatment arms, 100 mg/day topiramate, 200 mg/day topiramate or carbamazepine (Privitera 2003). Table 6 shows sensitivity analysis comparing the primary analysis (pooled topiramate arms versus carbamazepine), topiramate 100 mg/day versus carbamazepine and topiramate 200 mg/day versus carbamazepine. Results were similar across all three analyses and conclusions were unchanged.
4. Sensitivity analysis by topiramate dose ‐ Privitera 2003.
| Outcomea | Topiramate (both arms) | Carbamazepine | Topiramate 200 mg | Carbamazepine | Topiramate 100 mg | Carbamazepine |
| n = 226 | n = 129 | n = 130 | n = 129 | n = 136 | n = 129 | |
| Time to treatment failure (any reason) | HR 1.04 (95% CI 0.75 to 1.42), P = 0.85 | HR 1.03 (95% CI 0.71 to 1.48), P = 0.89 | HR 1.05 (95% CI 0.73 to 1.52), P = 0.79 | |||
| Time to treatment failure (adverse events) | HR 0.82 (95% CI 0.53 to 1.27), P = 0.38 | HR 0.92 (95% CI 0.56 to 1.52), P = 0.75 | HR 0.73 (95% CI 0.44 to 1.23), P = 0.23 | |||
| Time to treatment failure (lack of efficacy) | HR 1.44 (95% CI 0.71 to 2.94), P = 0.31 | HR 1.26 (95% CI 0.56 to 2.89), P = 0.57 | HR 1.60 (95% CI 0.74 to 3.44), P = 0.24 | |||
| Time to first seizure | HR 1.32 (95% CI 0.96 to 1.81), P = 0.09 | HR 1.34 (95% CI 0.94 to 1.91), P = 0.11 | HR 1.29 (95% CI 0.89 to 1.86), P = 0.18 | |||
| Time to 12‐month remission | HR 0.82 (95% CI 0.51 to 1.33), P = 0.42 | HR 0.83 (95% CI 0.48 to 1.44), P = 0.5 | HR 0.79 (95% CI 0.46 to 1.37), P = 0.41 | |||
| Time to 6‐month remission | HR 0.88 (95% CI 0.65 to 1.18), P = 0.39 | HR 0.84 (95% CI 0.59 to 1.18), P = 0.31 | HR 0.93 (95% CI 0.66 to 1.31), P = 0.66 | |||
HR = hazard ratio
The reason for treatment failure 'participant choice' was classified as an event in this review but censored in the included trial (SANAD A 2007). This was the primary reason for treatment failure specified in 14 participants (see Table 5). Sensitivity analysis classifying this reason as a censored observation for these 14 participants did not change our conclusions (results available from authors on request).
Secondary outcomes
Time to first seizure post‐randomisation
For this outcome, a HR less than 1 indicates a clinical advantage for topiramate.
No seizure recurrence data after randomisation were available for 36 participants in SANAD A 2007, therefore 1115 participants (97% of total 1151 participants) from the two trials were included in the analysis of 'time to first seizure after randomisation'.
A total of 720 participants (65% of participants included in analysis) experienced seizure recurrence, 403 of 624 (65%) receiving topiramate and 317 of 491 (65%) receiving carbamazepine.
The overall pooled HR (for 1115 participants providing IPD from two trials) was 1.09 (95% CI 0.94 to 1.27; P = 0.24, high‐certainty evidence; Analysis 1.7), indicating a potential advantage for carbamazepine that was not statistically significant; in other words, seizure recurrence may occur earlier on topiramate than carbamazepine but we cannot rule out a slight advantage to topiramate or no difference between the drugs. No important heterogeneity was present between trials (I2 = 39%).
1.7. Analysis.
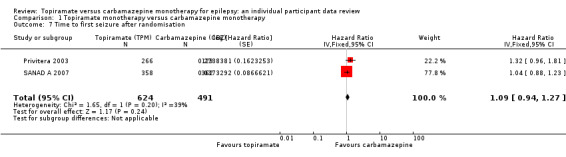
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 7 Time to first seizure after randomisation.
Subgroup analyses: seizure type (focal onset versus generalised onset or unclassified epilepsy)
See Subgroup analysis and investigation of heterogeneity for more details regarding the definitions of subgroups.
For participants with focal onset seizures (925 participants providing IPD), the pooled HR was 1.12 (95% CI 0.95 to 1.29; P = 0.17, high‐certainty evidence; Analysis 1.8) and for participants with generalised onset or unclassified epilepsy (190 participants providing IPD), the pooled HR was 1.08 (95% CI 0.70 to 1.66; P = 0.73, moderate‐certainty evidence; Analysis 1.8), both indicating a potential advantage to carbamazepine that was not statistically significant.
1.8. Analysis.
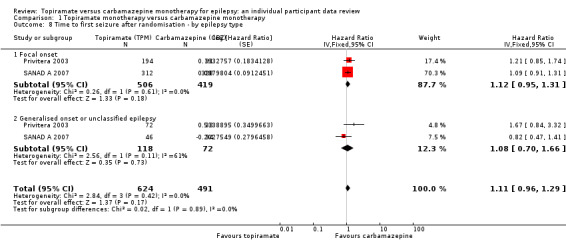
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 8 Time to first seizure after randomisation ‐ by epilepsy type.
There was no evidence of a difference between the subgroups (test for subgroup differences P = 0.89; Analysis 1.8). There was some heterogeneity between the two trials in the subgroup of generalised onset or unclassified epilepsy (I2 = 61%), which is likely due to the variability in the 'unclassifiable' nature of the epilepsy in many of the participants in this subgroup (i.e. the subgroup is likely to be comprised of some individuals experiencing focal epilepsy and others experiencing generalised epilepsy). No heterogeneity was present in the subgroup of participants classified as having focal epilepsy (I2 = 0%).
The overall pooled HR (adjusted for epilepsy type for 1115 participants) was HR 1.11 (95% CI 0.96 to 1.29; P = 0.17, high‐certainty evidence; Analysis 1.8), indicating an advantage for carbamazepine that was not statistically significant.
Sensitivity analysis
We performed a sensitivity analysis including only IPD from the six‐month double‐blind period of Privitera 2003; participants who experienced a first seizure recurrence after six months (8 receiving carbamazepine and 11 receiving topiramate) were censored at six months. When only seizure recurrences from the first six months of Privitera 2003 were combined with IPD from SANAD A 2007, the numerical results were very similar and our conclusions were unchanged (results available from authors on request).
One included trial allocated participants to three treatment arms, 100 mg/day topiramate, 200 mg/day topiramate or carbamazepine (Privitera 2003). Table 6 shows sensitivity analysis comparing the primary analysis (pooled topiramate arms versus carbamazepine), topiramate 100 mg/day versus carbamazepine and topiramate 200 mg/day versus carbamazepine. Results were similar across all three analyses and conclusions were unchanged.
Time to 12‐month remission of seizures
For this outcome, a HR less than 1 indicates a clinical advantage for carbamazepine.
No seizure recurrence data after randomisation were available for 36 participants in SANAD A 2007, therefore 1115 participants (97% of total 1151 participants) from the two trials were included in the analysis of time to 12‐month remission.
A total of 558 participants (50% of participants included in analysis) achieved 12‐month remission; 277 of 624 (44%) receiving topiramate and 281 of 491 (57%) receiving carbamazepine.
Of these 558 participants, 301 achieved immediate remission (54%), i.e. no seizure recurrence in the immediate 12 months following randomisation; 151 received topiramate and 150 received carbamazepine.
The overall pooled HR (for 1115 participants providing IPD from 2 trials) was 0.85 (95% CI 0.72 to 1.01; P = 0.07, high‐certainty evidence; Analysis 1.9), indicating a potential advantage for carbamazepine that was not statistically significant; in other words, a seizure‐free period of 12 months may occur earlier on carbamazepine than topiramate but we cannot rule out a slight advantage to topiramate or no difference between the drugs. No heterogeneity was present between trials (I2 = 0%).
1.9. Analysis.
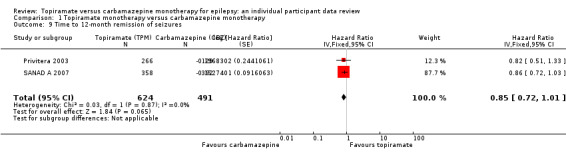
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 9 Time to 12‐month remission of seizures.
Subgroup analyses: seizure type (focal onset versus generalised onset or unclassified epilepsy)
See Subgroup analysis and investigation of heterogeneity for more details regarding the definition of subgroups.
For participants with focal onset seizures (925 participants providing IPD), the pooled HR was 0.82 (95% CI 0.69 to 0.99; P = 0.04, high‐certainty evidence; Analysis 1.10), indicating a statistically significant advantage for carbamazepine; in other words, for individuals with focal onset seizures, a seizure‐free period of 12 months occurs significantly earlier on carbamazepine compared to topiramate in the two included trials. For participants with generalised onset or unclassified epilepsy (190 participants providing IPD), the pooled HR was 0.92 (95% CI 0.61 to 1.41; P = 0.71, moderate‐certainty evidence; Analysis 1.10), indicating that a seizure‐free period of 12 months may occur earlier on carbamazepine but CIs are wide so we cannot rule out an advantage to topiramate or no differences between the drugs.
1.10. Analysis.
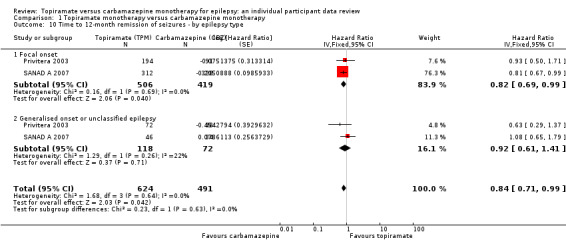
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 10 Time to 12‐month remission of seizures ‐ by epilepsy type.
The overall pooled HR (adjusted for epilepsy type for 1115 participants) was HR 0.84 (95% CI 0.71 to 0.99; P = 0.04, high‐certainty evidence; Analysis 1.10), indicating a statistically significant advantage for carbamazepine; in other words, a seizure‐free period of 12 months occurs significantly earlier on carbamazepine compared to topiramate in the two included trials. No important between‐trial heterogeneity was present overall or by subgroup (I2 < 25% for all analyses) and there was no evidence of a difference between the subgroups (test for subgroup differences P = 0.63; Analysis 1.10).
Sensitivity analysis
One included trial allocated participants to three treatment arms, 100 mg/day topiramate, 200 mg/day topiramate or carbamazepine (Privitera 2003). Table 6 shows sensitivity analysis comparing the primary analysis (pooled topiramate arms versus carbamazepine), topiramate 100 mg/day versus carbamazepine and topiramate 200 mg/day versus carbamazepine. Results were similar across all three analyses and conclusions were unchanged.
Time to six‐month remission of seizures
For this outcome, a HR less than 1 indicates a clinical advantage for carbamazepine.
No seizure recurrence data after randomisation were available for 36 participants in SANAD A 2007, therefore 1115 participants (97% of total 1151 participants) from the two trials were included in the analysis of time to six‐month remission.
A total of 790 participants (71% of participants included in analysis) achieved six‐month remission; 422 of 624 (68%) receiving topiramate and 368 of 491 (75%) receiving carbamazepine.
Of these 790 participants, 441 achieved immediate remission (56% of participants achieving remission), 240 receiving topiramate and 201 receiving carbamazepine.
The overall pooled HR (for 1115 participants providing IPD from 2 trials) was 0.88 (95% CI 0.76 to 1.01; P = 0.09, high‐certainty evidence; Analysis 1.11), indicating an advantage for carbamazepine that was not statistically significant; in other words, a seizure‐free period of six months may occur earlier on carbamazepine than topiramate but we cannot rule out a slight advantage to topiramate or no difference between the drugs. No heterogeneity was present between trials (I2 = 0%).
1.11. Analysis.
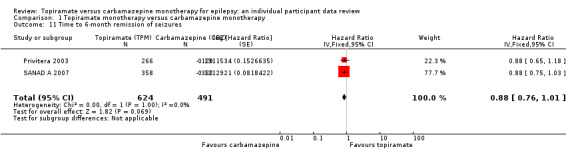
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 11 Time to 6‐month remission of seizures.
Subgroup analyses: seizure type (focal onset versus generalised onset or unclassified epilepsy)
See Subgroup analysis and investigation of heterogeneity for more details regarding the definitions of subgroups.
For participants with focal onset seizures (925 participants providing IPD), the pooled HR was 0.86 (95% CI 0.74 to 1.01; P = 0.06, high‐certainty evidence; Analysis 1.12) and for participants with generalised onset or unclassified epilepsy (190 participants providing IPD), the pooled HR was 0.93 (95% CI 0.66 to 1.30; P = 0.67, moderate‐certainty evidence; Analysis 1.12), both indicating a potential advantage to carbamazepine that was not statistically significant.
1.12. Analysis.
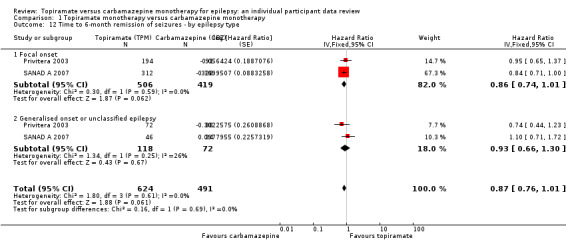
Comparison 1 Topiramate monotherapy versus carbamazepine monotherapy, Outcome 12 Time to 6‐month remission of seizures ‐ by epilepsy type.
The overall pooled HR (adjusted for epilepsy type for 1115 participants) was HR 0.87 (95% CI 0.76 to 1.01; P = 0.06, high‐certainty evidence; Analysis 1.12), indicating a potential advantage for carbamazepine that was not statistically significant; in other words, a seizure‐free period of 12 months may occur earlier on carbamazepine than topiramate but we cannot rule out a slight advantage to topiramate or no difference between the drugs. No important between‐trial heterogeneity was present overall or by subgroup (I2 < 30% for all analyses) and there was no evidence of a difference between the subgroups (test for subgroup differences P = 0.69; Analysis 1.12).
Sensitivity analysis
We performed a sensitivity analysis including only IPD from the six‐month double‐blind period of Privitera 2003; those who achieved six months of remission after six months (i.e. those who did not experience immediate six‐month remission, 11 receiving carbamazepine and 41 receiving topiramate) were censored at six months. When only immediate six‐month remission data from Privitera 2003 were combined with IPD from SANAD A 2007, the pooled HR was 0.86 (0.75 to 1.00, P = 0.05), indicating a statistically significant advantage for carbamazepine over topiramate. We note that this analysis combines immediate six‐month remission in Privitera 2003 with six‐month remission at any time in SANAD A 2007. When analysing only immediate six‐month remission in SANAD A 2007, the pooled HR was 0.88 (0.73 to 1.07, P = 0.19) indicating no significant difference between the drugs.
One included trial allocated participants to three treatment arms, 100 mg/day topiramate, 200 mg/day topiramate or carbamazepine (Privitera 2003). Table 6 shows sensitivity analysis comparing the primary analysis (pooled topiramate arms versus carbamazepine), topiramate 100 mg/day versus carbamazepine and topiramate 200 mg/day versus carbamazepine. Results were similar across all three analyses and conclusions were unchanged.
Incidence of adverse events
We were provided with IPD for adverse events experienced during the trial for two trials (Privitera 2003; SANAD A 2007).
Due to the wide range of events reported in the trials and the differences in adverse‐event profiles of the two drugs, we have not analysed adverse event data in meta‐analysis and provide a narrative report. This information is summarised in Table 7 and Table 8. All adverse events are reported according to the definitions within the data provided to us.
5. Summary of adverse events experienced.
| Study and drug | Privitera 2003 | SANAD A 2007 | |||||
| TPM 100 | TPM 200 | CBZ | Total | TPM | CBZ | Total | |
| Number experiencing adverse events | 120 | 114 | 111 | 345 | 283 | 260 | 543 |
| Number of adverse events | 1063 | 1035 | 970 | 3068 | 2503 | 1339 | 3842 |
| Number of adverse events per person (range) | 1 to 40 | 1 to 30 | 1 to 37 | NA | 1 to 35 | 1 to 37 | NA |
| Number of drug related adverse eventsa | 578 | 613 | 537 | 1728 | NA | NA | NA |
| Number of adverse events requiring action/treatment changeb | 76 | 90 | 72 | 238 | 705 | 529 | 1234 |
| Number of participants needing a treatment change/dose changeb | 27 | 31 | 32 | 90 | 185 | 173 | 358 |
CBZ = carbamazepine, NA = not available, TPM = topiramate, TPM 100 = topiramate 100 mg/day, TPM 200 = topiramate 200 mg/day (Privitera 2003)
aDefined as events which are 'very likely,' 'probably' or 'possibly' related in Privitera 2003. Not stated if events were drug‐related in SANAD A 2007. bInformation given only for drug discontinuation in Privitera 2003. Information on drug discontinuation and dose change in SANAD A 2007.
6. Most commonly reported adverse events.
|
Most commonly reported adverse eventsa |
Privitera 2003 | SANAD A 2007 | Total | |||||||
| CBZ | TPM 100 | TPM 200 | Total | CBZ | TPM | Total | CBZ | TPM | Total | |
| Aggression | 0 (0) | 8 (6) | 5 (2) | 13 (8) | 41 (25) | 75 (50) | 116 (75) | 41 (25) | 88 (58) | 129 (83) |
| Anorexia/weight loss | 32 (16) | 45 (26) | 54 (34) | 131 (76) | 16 (14) | 126 (82) | 142 (96) | 48 (30) | 225 (142) | 273 (172) |
| Anxiety/depression | 24 (15) | 48 (27) | 60 (39) | 132 (81) | 46 (35) | 107 (71) | 153 (106) | 70 (50) | 215 (137) | 285 (187) |
| Aphasia | 18 (10) | 10 (7) | 34 (14) | 62 (31) | 11 (10) | 16 (16) | 27 (26) | 29 (20) | 60 (37) | 89 (57) |
| Ataxia | 7 (4) | 11 (6) | 9 (6) | 27 (16) | 30 (23) | 21 (14) | 51 (37) | 37 (27) | 41 (26) | 78 (53) |
| Chest infection/bronchitis | 36 (23) | 41 (25) | 54 (26) | 131 (74) | 6 (6) | 3 (3) | 9 (9) | 42 (29) | 98 (54) | 140 (83) |
| Cold/fever/influenza | 14 (13) | 20 (11) | 15 (15) | 49 (39) | 3 (3) | 4 (4) | 7 (7) | 17 (16) | 39 (30) | 56 (46) |
| Concentration | 6 (5) | 15 (7) | 28 (11) | 49 (23) | 11 (11) | 8 (7) | 19 (18) | 17 (16) | 51 (25) | 68 (41) |
| Confusion | 6 (4) | 5 (4) | 10 (6) | 21 (14) | 33 (25) | 45 (34) | 78 (59) | 39 (29) | 60 (44) | 99 (73) |
| Dental | 6 (3) | 10 (7) | 9 (5) | 25 (15) | 17 (14) | 13 (13) | 30 (27) | 23 (17) | 32 (25) | 55 (42) |
| Dizzy/faint | 49 (30) | 44 (24) | 35 (23) | 128 (77) | 64 (51) | 76 (49) | 140 (100) | 113 (81) | 155 (96) | 268 (177) |
| Drowsy/tired | 130 (60) | 97 (51) | 79 (45) | 306 (156) | 267 (187) | 188 (139) | 455 (326) | 397 (247) | 364 (235) | 761 (482) |
| Gastrointestinal disturbances | 88 (51) | 50 (32) | 53 (28) | 191 (111) | 49 (41) | 48 (32) | 97 (73) | 137 (92) | 151 (92) | 288 (184) |
| Headache | 75 (39) | 84 (38) | 40 (24) | 199 (101) | 97 (65) | 76 (44) | 173 (109) | 172 (104) | 200 (106) | 372 (210) |
| Increased/worsened seizures | 2 (2) | 5 (4) | 0 (0) | 7 (6) | 41 (30) | 30 (24) | 71 (54) | 43 (32) | 35 (28) | 78 (60) |
| Kidney/urinary problems | 11 (6) | 15 (7) | 22 (12) | 48 (25) | 10 (10) | 21 (15) | 31 (25) | 21 (16) | 58 (34) | 79 (50) |
| Memory | 8 (6) | 19 (10) | 26 (12) | 53 (28) | 71 (48) | 92 (62) | 163 (110) | 79 (54) | 137 (84) | 216 (138) |
| Mood/behavioural change | 19 (10) | 22 (14) | 29 (15) | 70 (39) | 56 (42) | 97 (76) | 153 (118) | 75 (52) | 148 (105) | 223 (157) |
| Nausea/vomiting | 57 (35) | 21 (19) | 27 (23) | 105 (77) | 54 (49) | 32 (29) | 86 (78) | 111 (84) | 80 (71) | 191 (155) |
| Pain | 26 (19) | 14 (9) | 39 (19) | 79 (47) | 15 (13) | 20 (17) | 35 (30) | 41 (32) | 73 (45) | 114 (77) |
| Pins and needles | 17 (5) | 116 (38) | 135 (45) | 268 (88) | 23 (17) | 205 (148) | 228 (165) | 40 (22) | 456 (231) | 496 (253) |
| Rash | 61 (35) | 42 (22) | 25 (17) | 128 (74) | 99 (81) | 54 (44) | 153 (125) | 160 (116) | 121 (83) | 281 (199) |
| Sleep problems/nightmares | 14 (6) | 24 (14) | 23 (12) | 61 (32) | 24 (16) | 40 (30) | 64 (46) | 38 (22) | 87 (56) | 125 (78) |
| Vision | 7 (5) | 8 (5) | 3 (3) | 18 (13) | 33 (28) | 24 (23) | 57 (51) | 40 (33) | 35 (31) | 75 (64) |
| Weight gain | 8 (3) | 5 (4) | 0 (0) | 13 (7) | 42 (27) | 25 (15) | 67 (42) | 50 (30) | 30 (19) | 80 (49) |
CBZ = carbamazepine, TPM = topiramate, TPM 100 = topiramate 100 mg/day, TPM 200 = topiramate 200 mg/day (Privitera 2003)
a. Results are expressed as Number of events (Number of Participants), where events = number of adverse events reported; participants = number of participants reporting the adverse event (a participant could report the same type of adverse event multiple times).
Less commonly reported adverse events are not summarised in this table but details are available on request from the review authors. General terminology for the type of adverse events was defined by the review authors based on the individual participant data provided.
The five most commonly reported adverse events with the two drugs were drowsiness or fatigue, ‘pins and needles’ (tingling sensation), headache, gastrointestinal disturbance and anxiety or depression. Rash and dizziness (feeling faint) were also commonly reported with carbamazepine, and anorexia or weight loss was commonly reported with topiramate.
In Privitera 2003, 58 serious adverse events were reported in 29 individuals.
With topiramate 100 mg, there were 12 serious adverse events in 10 participants. One event of renal calculus in one participant, and one event of grand mal convulsions in one participant, were possibly related to treatment. All other events were unlikely to be related to treatment: two events of grand mal convulsions in two participants; and one event of 'regression', one event of hypotension, one event of thrombophlebitis, one event of worsened convulsions, one event of abnormal hepatic function, one event of oedema, one event of asthenia and one event of aggravated depression (resulting in withdrawal of the drug), all in one participant each.
With topiramate 200 mg, there were 29 serious adverse events in 11 participants. One event of renal calculus was very likely to be related to the treatment and another event of renal calculus was probably related to treatment. Nine events of confusion and aggravated depression in one participant were also probably related to treatment. The drug was withdrawn from these three participants. All other events were unlikely to be related to treatment: two events of adenocarcinoma in one participant; three events of dizziness, nausea and palpitations in one participant; eight events of headache, back pain, confusion, fever and upper respiratory tract infection in one participant; and one injury, one event of asthma, one event of migraine, one event of ileus, one event of chest pain and one event of foetal death, all in one participant each. None of the unrelated adverse events resulted in withdrawal of the drug.
On carbamazepine, there were 17 serious adverse events in eight participants. One event of grand mal convulsions was very likely to be related to the treatment. All other events were unlikely to be related to treatment: seven events of diverticulitis and hypertension in one participant; two events of enteritis in one participant; three events of dyspnoea and chest pain in one participant; and one event of syncope, one abscess, one injury and one case of abdominal pain, all in one participant each. None of these events resulted in withdrawal of treatments.
In SANAD A 2007, 179 events resulting in hospitalisation were reported in 101 participants (not stated whether events were related to treatment).
On topiramate, there were 88 hospitalisation events in 55 participants: 21 events of worsening seizures or status epilepticus in 13 participants (resulting in withdrawal of the drug in 2 participants); accidental injuries in four participants; six events of headache in three participants (resulting in withdrawal of the drug in 1 participant); accidental drug overdoses in three participants; brain tumours in three participants (resulting in withdrawal of the drug in 1 participant); abdominal pain in three participants (resulting in withdrawal of the drug in 1 participant); three coronary artery bypass grafts in two participants; chest pain in two participants; renal malignancy in two participants; depression in two participants; visual disturbances in two participants (resulting in withdrawal of the drug in 1 participant); self‐harm/suicide attempt in two participants (resulting in withdrawal of the drug in one participant); urinary tract infections in two participants; thrombosis in two participants; three events of ataxia in one participant; three events of Crohn's disease in one participant; two events of dizziness in one participant; and one cataract operation, one event of hypertension, one event of sarcoidosis, one testicular lump, one event of urinary incontinence, one miscarriage, one event of Henoch‐Schonlein purpura, one event of Steven Johnsons syndrome (resulting in withdrawal of the drug), and one collapsed lung, all in one participant each.
On carbamazepine, there were 91 hospitalisation events in 46 participants: worsening of seizures in 12 participants; cardiovascular events in five participants; attempted suicide in three participants; seizure‐related injury in three participants; allergic rash in two participants; pneumonia in two participants; and antiphospholipid syndrome, arthritis, stomach cancer, urinary tract infection, disorientation, psychotic illness (resulting in withdrawal of the drug), exacerbation of chronic obstructive pulmonary disease, hysterectomy (resulting in withdrawal of the drug), torsion of testis, myringotomy, infection, worsening of seizures and visual disturbance (resulting in withdrawal of the drug), constipation (resulting in withdrawal of the drug), low serum, breast cancer, abdominal pain, ataxia, childbirth, and headache, all in one participant each.
Summary of aggregate results reported in Resendiz‐Aparicio 2004
IPD were not available for the 88 participants randomised in Resendiz‐Aparicio 2004.
Forty‐six participants were randomised to topiramate and 42 were randomised to carbamazepine; 23 participants dropped out due to adverse events, lack of efficacy or loss to follow‐up (13 randomised to topiramate and 10 randomised to carbamazepine). Results were presented only for the 33 participants randomised to topiramate and 32 randomised to carbamazepine who did not drop out of the study.
Thirty participants on topiramate and 26 on carbamazepine achieved six months of freedom from seizures after 12 months of treatment and 32 participants receiving topiramate and 27 receiving carbamazepine achieved a 50% or more reduction in seizures during the same time frame. The average number of seizures was significantly lower in the topiramate group than in the carbamazepine group at six and nine months (P value of t‐test = 0.01).
No clinically significant changes were observed in clinical or physical examinations in either group. Adverse event experiences were mild and similar between groups: somnolence (in 3 receiving topiramate and 6 receiving carbamazepine); dizziness (1 receiving topiramate and 2 receiving carbamazepine); weight loss or anorexia (5 receiving topiramate); gastritis (1 receiving topiramate); nausea (1 receiving topiramate); rash (1 receiving carbamazepine); headache (1 receiving carbamazepine); uncontrolled seizures (1 receiving carbamazepine).
Discussion
Summary of main results
Individual participant data (IPD) were available for two trials recruiting 1151 participants to carbamazepine or topiramate (Privitera 2003; SANAD A 2007). For the third trial, which recruited 88 participants, we could not contact the original authors, and so we could not include IPD from this trial (Resendiz‐Aparicio 2004). Overall, IPD were available for 93% of the total eligible 1239 participants.
This review provides moderate‐certainty evidence showing no statistically significant difference between topiramate and carbamazepine for our primary global efficacy outcome 'time to treatment failure for any reason related to treatment' (pooled hazard ratio (HR) 1.16, 95% confidence interval (CI) 0.97 to 1.38) and for 'time to treatment failure due to adverse events' (pooled HR 1.02, 95% CI 0.82 to 1.27) and a statistically significant advantage for carbamazepine over topiramate for 'time to treatment failure lack of efficacy' (pooled HR 1.46, 95% CI 1.08 to 1.98) for all participants including those with focal onset seizures, generalised tonic‐clonic seizures (with or without other seizure types) and unclassified seizure types. A statistically significant advantage (high‐certainty evidence) for carbamazepine over topiramate was also observed for secondary outcome 'time to 12‐month remission' (pooled HR 0.84, 95% CI 0.71 to 0.99) for all participants.
Considering only individuals with focal onset seizures (81% of participants contributing to the analysis) results of this review provide high to moderate‐certainty evidence. A statistically significant advantage for carbamazepine over topiramate was observed for our primary outcome 'time to treatment failure for any reason related to treatment' (pooled HR 1.21, 95% CI 1.01 to 1.46) and for 'time to treatment failure lack of efficacy' (pooled HR 1.47, 95% CI 1.07 to 2.02) but no differences were found between the drugs in terms of 'time to treatment failure due to adverse events' (pooled HR 1.08, 95% CI 0.85 to 1.36). As also observed for all participants, for individuals with focal onset seizures, a statistically significant advantage (high‐certainty evidence) for carbamazepine over topiramate was also observed for secondary outcome 'time to 12‐month remission' (pooled HR 0.82, 95% CI 0.69 to 0.99).
The results of this review (high‐ to moderate‐certainty evidence) also show no statistically significant difference between topiramate and carbamazepine for our secondary outcomes of 'time to first seizure' and 'time to six‐month remission' for all individuals and by seizure type.
Evidence for individuals with generalised tonic‐clonic seizures (with or without other seizure types) (9% of participants contributing to the analysis), and unclassified seizure types (10% of participants contributing to the analysis) was very limited and of moderate to low certainty for all outcomes; no statistically significant differences were found but CIs were wide, therefore we cannot exclude an advantage to either drug, or no difference between drugs for generalised onset seizures and unclassified epilepsy.
The five most commonly reported adverse events with the two drugs were drowsiness or fatigue, ‘pins and needles’ (tingling sensation), headache, gastrointestinal disturbance and anxiety or depression. Rash and dizziness (feeling faint) were also commonly reported with carbamazepine, and anorexia or weight loss was commonly reported with topiramate. The rates of adverse events and serious adverse events were similar across the two drugs.
Overall completeness and applicability of evidence
We believe our systematic electronic searches identified all relevant evidence for this review. We gratefully received IPD for 1151 individuals (93% of 1239 individuals from all eligible trials) from the authors or sponsors of two trials (Privitera 2003; SANAD A 2007), that included a comparison of topiramate with carbamazepine for the treatment of epilepsy.
At the time the review was conducted, we were unable to obtain IPD for the remaining trial (Resendiz‐Aparicio 2004), which randomised a total of 88 participants. We were not able to make contact with a study author. If we receive IPD from this trial, we will include them in future review updates. We do not believe that our failure to obtain IPD from 7% of eligible participants from this single trial has had a large impact on the applicability of the results of the review.
Eligible seizure types included in this review were focal onset and generalised tonic‐clonic (with or without other generalised types). Due to the design of the two studies contributing to analysis, a majority of participants recruited into these trials experienced focal onset seizures (82% of participants contributing to the analysis) and the majority of the remaining participants had an unclassified seizure type.
As a result, the results of this review are primarily applicable to participants with focal onset seizures and we encourage caution in the interpretation of results for the small subgroup of participants who had generalised onset or unclassified epilepsy.
Quality of the evidence
The two trials for which IPD were made available (as well as additional trial design information from trial authors/sponsors) were generally of good quality. One of the trials was double‐blind (Privitera 2003), and one was open‐label (SANAD A 2007). While it is argued that an open‐label design is more pragmatic and reflective of the 'real world' treatment of a chronic condition, such as epilepsy, where treatments are likely to be taken long term by participants (SANAD A 2007), significantly more participants withdrew from treatment in the open‐label study than in the double‐blind study (51% versus 44%, Chi2 P = 0.03). Both of the trials contributing to analysis in this review compared a 'new' intervention with a 'standard' intervention, and knowledge of the treatment allocation may have influenced the choice of the participant or clinician to continue taking the treatment. This, in turn, may have influenced the perceived effectiveness of the two drugs under comparison. We have, therefore, considered an open‐label design to potentially introduce bias into the results for the subjective outcome of time to treatment failure, but not for the objective secondary outcomes of time to first seizure and remission.
Due to the potential risk of bias from an open‐label design, we have rated the evidence provided in this review, according to GRADE criteria, for our primary outcome of time to treatment failure as ‘moderate certainty’ for all participants and the subgroup of participants with focal onset seizures. Due to the limited number of participants with generalised onset seizures (and, hence, the potential misclassification of seizure type), we have rated this evidence as low certainty for the primary outcome, see Table 1. For our secondary (objective) outcomes of time to first seizure and remission, we have rated evidence as high certainty (moderate certainty in the subgroup of participants with generalised onset seizures for the reasons stated above) (see Table 2).
Potential biases in the review process
We were able to include IPD for 1151 of 1239 eligible participants (93%) from two of three trials in this review and were able to analyse all outcomes using IPD. Such an approach has many advantages, such as allowing the standardisation of definitions of outcomes across trials. In addition, attrition and reporting biases are reduced as we can perform additional analyses and calculate additional outcomes from unpublished data. For the outcomes we used in this review that are of a time‐to‐event nature, an IPD approach is considered to be the ’gold standard’ approach to analysis (Parmar 1998).
For reasons outside of our control, we were unable to obtain IPD for 88 participants from one trial for inclusion in this review. However, we do not believe that the exclusion of 7% of eligible participants is likely to have impacted on the conclusions of this review (see Overall completeness and applicability of evidence).
Finally, we made some assumptions in the statistical methodology used in this review. First, when we received only follow‐up dates and seizure frequencies from the authors of the included studies, we used linear interpolation to estimate approximate seizure dates. We are aware that an individual’s seizure patterns may be non‐linear; therefore, we recommend caution when interpreting the numerical results of the seizure‐related outcomes.
We also made an assumption that treatment effect for each outcome did not change over time (proportional hazards assumption, see Data synthesis). We are aware that in trials of long duration (e.g. SANAD A 2007, which was of over one year in duration), the assumption that treatment effect remains constant over time is unlikely to be appropriate; for example, there is likely to be a difference between participants who achieve immediate remission compared with participants who achieve later remission. Therefore, if future updates of this review include more trials of long duration, we would like to perform statistical analyses that allow for treatment effects to vary over time.
Agreements and disagreements with other studies or reviews
To our knowledge, this is the only systematic review and meta‐analysis that compares topiramate and carbamazepine monotherapy for focal onset seizures and generalised onset tonic‐clonic seizures. A network meta‐analysis has been published (Nevitt 2017a), comparing all direct and indirect evidence from topiramate, carbamazepine, and other standard and new antiepileptic drugs (AEDs) licensed for monotherapy. The results of this review generally agree with the results of the network meta‐analysis; results of this network meta‐analysis showed a potential advantage for carbamazepine compared with topiramate which was not statistically significant for all outcomes (time to treatment failure, time to first seizure and time to six‐month and 12‐month remission). No statistically significant differences were found between the drugs for all outcomes for participants with generalised onset seizures.
Authors' conclusions
Implications for practice.
Current UK guidelines recommend carbamazepine or lamotrigine as first‐line treatment for adults and children with new‐onset focal seizures, and sodium valproate for adults and children with new‐onset generalised seizures. Topiramate is not currently recommended as a first‐ or second‐line treatment for use in new‐onset focal or generalised seizures (NICE 2012). The results of this review do not provide any conclusive evidence for or against these guidelines.
There is some suggestion from the moderate‐certainty evidence provided by the results of this review that carbamazepine may be a more effective drug for individuals with new‐onset focal seizures in terms of treatment retention (treatment failure due to lack of efficacy, or adverse events, or both occurred later with carbamazepine) and that these individuals may achieve a year of remission from seizures earlier with carbamazepine than with topiramate. However, this difference was not observed for recurrence of a first seizure and for remission of a shorter period.
For individuals with new‐onset generalised tonic‐clonic seizures (with or without other generalised seizure types), the evidence in the review is limited due to the small numbers of participants with generalised seizure types recruited into the included trials. Furthermore, an important proportion of individuals had unclassified seizure types, and evidence is limited and inconclusive for these participants.
There is evidence that carbamazepine may exacerbate some generalised seizure types, and so should be used with caution in individuals with this seizure type (Liporace 1994; Shields 1983; Snead 1985). Topiramate may be an effective alternative treatment option to sodium valproate for new‐onset generalised seizures, but more evidence is required to confirm this (NICE 2012). Newer antiepileptic drugs (AEDs), such as topiramate, may be associated with less intolerable side effects than older drugs, such as carbamazepine (French 2007); however, the results of the review do not suggest that topiramate is better‐ or worse‐tolerated than carbamazepine.
Implications for research.
Results of this review are taken from the synthesis of 1151 of 1239 eligible participants from two of three eligible trials. Some of the pooled results from the two studies included in this review suggested a potential advantage for carbamazepine over topiramate but these results did not reach statistical significance; we therefore do not rule out that important differences may exist between the drugs which may come to light if more evidence can be incorporated into the review during future updates.
This review highlights the need for the design of future AED monotherapy trials that recruit individuals with specific epilepsy syndromes to be powered to detect a difference between particular AEDs. An approach likely to reflect and inform clinical practice, as well as being statistically powerful, would be to recruit heterogeneous populations for whom epilepsy syndromes have been adequately defined, with testing for interactions between treatments and epilepsy syndromes.
In view of potential problems arising from unclassified seizures and the misclassification of seizure type, it is important that epilepsy syndromes should be well defined in the inclusion criteria of future trials, with adequate checking mechanisms to ensure that classifications are accurate and a system to recognise uncertainty surrounding epilepsy syndromes in individuals within trials. This most commonly applies to tonic‐clonic seizures that may be generalised at onset, or which may be secondarily generalised. In any trial, such unclassified individuals need to be clearly identified, because if they are not they may confound the interpretation of the results for well‐classified individuals. We need to know how to manage participants whose classification we find more difficult.
It is also important that future trials are of a sufficient duration to measure the long‐term effectiveness of AEDs ‐ treatments that will be life‐long for many individuals with epilepsy ‐ as well as psychosocial, quality of life and health economic outcomes. Consideration is also required in the design of a trial regarding whether to blind participants and outcome assessors to treatment allocation. While an open‐label design is a more pragmatic and practical approach for large long‐term trials, when trials compare a new intervention with an established 'standard' intervention, masking of treatment may be important to avoid preconceptions over the relative effectiveness of the drugs.
The choice of outcomes at the design stage of a trial and the presentation of the results of outcomes, particularly of a time‐to‐event nature, require very careful consideration. While the majority of trials of a monotherapy design record an outcome measuring efficacy (seizure control) and an outcome measuring tolerability (adverse events), there is little uniformity between the definition of the outcomes and the reporting of the summary statistics related to the outcomes (Nolan 2013a), making an aggregate data approach to meta‐analysis in reviews of monotherapy trials impossible. Where trial authors cannot or will not make individual participant data (IPD) available for analysis, we are left with no choice but to exclude a proportion of relevant evidence from the review, which may impact upon the interpretation of the results of the review and the applicability of the evidence and conclusions. The International League Against Epilepsy recommends that trials of a monotherapy design should adopt a primary effectiveness outcome of 'time to treatment failure (retention time)' and should be of a duration of at least 48 weeks to allow for the assessment of longer‐term outcomes, such as remission (ILAE 1998; ILAE 2006). If trials followed these recommendations, an aggregate data approach to meta‐analysis could be feasible, reducing the resources and time required by an IPD approach.
A network meta‐analysis has also been published (Nevitt 2017a), comparing all direct and indirect evidence from topiramate, carbamazepine, and other standard and new AEDs licensed for monotherapy. This network meta‐analysis will be updated as more information becomes available; however, we acknowledge that as topiramate is not considered to be a first‐line agent for individuals with a new diagnosis of the seizure types within this review, it is unlikely that a substantial amount of new evidence will become available for this review.
What's new
| Date | Event | Description |
|---|---|---|
| 22 May 2018 | New search has been performed | We updated the searches on 22 May 2018; we have not included any new trials. We replaced the term 'partial' by 'focal', in accordance with the most recent classification of epilepsies of the International League Against Epilepsy (Scheffer 2017). |
| 22 May 2018 | New citation required but conclusions have not changed | Conclusions are unchanged |
History
Protocol first published: Issue 1, 2016 Review first published: Issue 12, 2016
| Date | Event | Description |
|---|---|---|
| 26 April 2017 | Amended | Declarations of interest section updated. |
Acknowledgements
We are grateful to the Cochrane Epilepsy Group Information Specialist, Graham Chan, for producing and performing all electronic searches.
We are greatly indebted to the original trial investigators and sponsors for their time and efforts in making individual participant data (IPD) available for this review.
This review update was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to the Epilepsy Group. The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service or the Department of Health.
Appendices
Appendix 1. Cochrane Register of Studies (CRS Web) search strategy
1. Topiram* or Tipiram* or Topamax or TPM or qudexy AND CENTRAL:TARGET
2. MeSH DESCRIPTOR Carbamazepine Explode All AND CENTRAL:TARGET
3. Carbamezepin* or CBZ or SPD417 or Amizepine or "Apo‐Carbamazepine" or Atretol or Biston or Calepsin or Carbagen or Carbamazepen* or Carbatrol or Carbazepin* or Carbelan or Epitol or Equetro or Finlepsin or Karbamazepin or Lexin or Neurotol or "Novo‐Carbamaz" or "Nu‐Carbamazepine" or Sirtal or Stazepin or Stazepine or "Taro‐Carbamazepine" or Tegretal or Tegretol or Telesmin or Teril or Timonil AND CENTRAL:TARGET
4. #2 OR #3 AND CENTRAL:TARGET
5. #1 AND #4 AND CENTRAL:TARGET
6. ((adjunct* or "add‐on" or "add on" or adjuvant* or combination* or polytherap*) not (monotherap* or alone or singl*)):TI AND CENTRAL:TARGET
7. #5 NOT #6 AND CENTRAL:TARGET
8. MESH DESCRIPTOR Epilepsy EXPLODE ALL AND CENTRAL:TARGET
9. MESH DESCRIPTOR Seizures EXPLODE ALL AND CENTRAL:TARGET
10. (epilep* OR seizure* OR convuls*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
11. #8 OR #9 OR #10 AND CENTRAL:TARGET
12. #7 AND #11
13. >14/04/2016:CRSINCENTRAL AND CENTRAL:TARGET
14. #12 AND #13
Appendix 2. MEDLINE (Ovid) search strategy
This strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomized trials (Lefebvre 2011).
1. (Topiram$ or Tipiramate or Topamax).mp.
2. exp Carbamazepine/
3. (Carbamezepin$ or CBZ or SPD417 or Amizepine or "Apo‐Carbamazepine" or Atretol or Biston or Calepsin or Carbagen or Carbamazepen$ or Carbatrol or Carbazepin$ or Carbelan or Epitol or Equetro or Finlepsin or Karbamazepin or Lexin or Neurotol or "Novo‐Carbamaz" or "Nu‐Carbamazepine" or Sirtal or Stazepin or Stazepine or "Taro‐Carbamazepine" or Tegretal or Tegretol or Telesmin or Teril or Timonil).mp.
4. 2 or 3
5. exp Epilepsy/
6. exp Seizures/
7. (epilep$ or seizure$ or convuls$).tw.
8. 5 or 6 or 7
9. exp *Pre‐Eclampsia/ or exp *Eclampsia/
10. 8 not 9
11. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
12. clinical trials as topic.sh.
13. trial.ti.
14. 11 or 12 or 13
15. exp animals/ not humans.sh.
16. 14 not 15
17. 1 and 4 and 10 and 16
18. ((adjunct$ or "add‐on" or "add on" or adjuvant$ or combination$ or polytherap$) not (monotherap$ or alone or singl$)).ti.
19. 17 not 18
20. limit 19 to ed=20160414‐20180522
21. 19 not (1$ or 2$).ed.
22. 21 and (2016$ or 2017$ or 2018$).dt.
23. 20 or 22
24. remove duplicates from 23
Appendix 3. ClinicalTrials.gov search strategy
Interventional Studies | Epilepsy | Topiramate AND Carbamazepine | First posted on or after 04/14/2016
Appendix 4. ICTRP search strategy
Condition: epilepsy
Intervention: Topiramate AND Carbamazepine
Date of registration between 14/04/2016 and 22/05/2018
Data and analyses
Comparison 1. Topiramate monotherapy versus carbamazepine monotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Time to treatment failure (any reason related to the treatment) | 2 | 1129 | Hazard Ratio (Fixed, 95% CI) | 1.15 [0.97, 1.37] |
| 2 Time to treatment failure due to adverse events | 2 | 1129 | Hazard Ratio (Fixed, 95% CI) | 1.01 [0.82, 1.26] |
| 3 Time to treatment failure due to lack of efficacy | 2 | 1129 | Hazard Ratio (Fixed, 95% CI) | 1.45 [1.08, 1.96] |
| 4 Time to treatment failure (any reason related to the treatment) ‐ by epilepsy type | 2 | 1129 | Hazard Ratio (Fixed, 95% CI) | 1.16 [0.97, 1.38] |
| 4.1 Focal onset | 2 | 937 | Hazard Ratio (Fixed, 95% CI) | 1.21 [1.01, 1.46] |
| 4.2 Generalised onset or unclassified epilepsy | 2 | 192 | Hazard Ratio (Fixed, 95% CI) | 0.88 [0.56, 1.39] |
| 5 Time to treatment failure due to adverse events ‐ by epilepsy type | 2 | 1129 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.82, 1.27] |
| 5.1 Focal onset | 2 | 937 | Hazard Ratio (Fixed, 95% CI) | 1.08 [0.85, 1.36] |
| 5.2 Generalised onset or unclassified epilepsy | 2 | 192 | Hazard Ratio (Fixed, 95% CI) | 0.72 [0.39, 1.31] |
| 6 Time to treatment failure due to lack of efficacy ‐ by epilepsy type | 2 | 1129 | Hazard Ratio (Fixed, 95% CI) | 1.46 [1.08, 1.98] |
| 6.1 Focal onset | 2 | 937 | Hazard Ratio (Fixed, 95% CI) | 1.47 [1.07, 2.02] |
| 6.2 Generalised onset or unclassified epilepsy | 2 | 192 | Hazard Ratio (Fixed, 95% CI) | 1.41 [0.54, 3.67] |
| 7 Time to first seizure after randomisation | 2 | 1115 | Hazard Ratio (Fixed, 95% CI) | 1.09 [0.94, 1.27] |
| 8 Time to first seizure after randomisation ‐ by epilepsy type | 2 | 1115 | Hazard Ratio (Fixed, 95% CI) | 1.11 [0.96, 1.29] |
| 8.1 Focal onset | 2 | 925 | Hazard Ratio (Fixed, 95% CI) | 1.12 [0.95, 1.31] |
| 8.2 Generalised onset or unclassified epilepsy | 2 | 190 | Hazard Ratio (Fixed, 95% CI) | 1.08 [0.70, 1.66] |
| 9 Time to 12‐month remission of seizures | 2 | 1115 | Hazard Ratio (Fixed, 95% CI) | 0.85 [0.72, 1.01] |
| 10 Time to 12‐month remission of seizures ‐ by epilepsy type | 2 | 1115 | Hazard Ratio (Fixed, 95% CI) | 0.84 [0.71, 0.99] |
| 10.1 Focal onset | 2 | 925 | Hazard Ratio (Fixed, 95% CI) | 0.82 [0.69, 0.99] |
| 10.2 Generalised onset or unclassified epilepsy | 2 | 190 | Hazard Ratio (Fixed, 95% CI) | 0.92 [0.61, 1.41] |
| 11 Time to 6‐month remission of seizures | 2 | 1115 | Hazard Ratio (Fixed, 95% CI) | 0.88 [0.76, 1.01] |
| 12 Time to 6‐month remission of seizures ‐ by epilepsy type | 2 | 1115 | Hazard Ratio (Fixed, 95% CI) | 0.87 [0.76, 1.01] |
| 12.1 Focal onset | 2 | 925 | Hazard Ratio (Fixed, 95% CI) | 0.86 [0.74, 1.01] |
| 12.2 Generalised onset or unclassified epilepsy | 2 | 190 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.66, 1.30] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Privitera 2003.
| Methods | Multinational, randomised, double‐blind trial conducted at 115 centres across the USA, Canada, Europe and South America Four treatments: CBZ, SV and TPM (2 arms, 100 mg/day and 200 mg/day) (see Notes) |
|
| Participants | Participants over the age of 6 years and over 30 kg in weight, with a diagnosis of epilepsy within the three months before trial entry and no previous AED treatment except emergency treatment Number randomised (ITT population): CBZ = 129, TPM = 266 (CBZ branch) 215 male participants (54%) 322 participants with focal epilepsy (82%) Mean age (range): 34 (6 to 80 years) |
|
| Interventions | Monotherapy with CBZ or TPM Starting doses: CBZ = 200 mg/day, TPM = 25 mg/day Target doses (after 4 week titration): CBZ = 600 mg/day, TPM = 100 or 200 mg/day (see Notes) Range of follow‐up: 0 to 29 months |
|
| Outcomes | Time to exit from the study Time to first seizure Proportion of seizure‐free participants during the last 6 months of double‐blind treatment Safety assessment: most commonly occurring adverse events |
|
| Notes | IPD provided for all outcomes of this review by trial sponsor Johnson & Johnson. Trial designed in two strata based on whether recommended treatment would be CBZ or SV. Within the two strata, participants were randomised to 100 mg/day TPM, 200 mg/day TPM or CBZ/SV depending on the strata. Data analysed according to the separate strata in this review with the two TPM doses analysed together; separate doses of TPM are considered in sensitivity analysis (see Data extraction and management) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was balanced using permuted blocks of size three and stratified by trial centre, according to a computer‐generated randomisation schedule prepared by the trial sponsor |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial was double‐blinded for the first 6 months, followed by an open‐label phase |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants from the ITT population analysed from IPD provided (see footnote 2). Eight participants with no follow‐up data were excluded from ITT population |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Resendiz‐Aparicio 2004.
| Methods | Randomised open‐label trial conducted in several hospitals in Mexico Two treatment arms: CBZ and TPM |
|
| Participants | Participants between 2 and 18 years with newly diagnosed focal epilepsy (with or without secondary generalisation) with at least two unprovoked seizures more than 24 hours apart and at least one seizure in the last 6 months. Participants must have no established treatment and have received no antiepileptic treatment within the past 30 days Number randomised: CBZ = 42, TPM = 46. Number included in analysis CBZ = 32, TPM = 33 100% focal epilepsy 33 male participants (60%) included in analysis Mean age (range): CBZ = 10 (5 to 17) years, TPM = 8 (2 to 16) years for participants included in analysis |
|
| Interventions | Monotherapy with CBZ or TPM Treatments titrated to a maximum of CBZ = 20 to 25 mg/kg/day, TPM = 9 mg/kg/day Follow‐up assessments at 6 and 9 months, range of follow‐up not stated |
|
| Outcomes | Seizure freedom and frequency of seizures during the trial Adverse events during the trial Laboratory results |
|
| Notes | The trial was published in Spanish; the characteristics and outcomes were translated. Outcomes chosen for this review were not reported; contact could not be made with trial author to provide IPD Results presented only for those who completed the trial. Those with less than 35% reduction of seizures were excluded from analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number tables used to assign participants to treatment groups |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rates reported (23 drops outs, 10 for CBZ and 13 for TPM). Only those who completed the trial were included in analysis (non responders to treatment excluded), this is not an ITT approach |
| Selective reporting (reporting bias) | Low risk | No protocol available. Seizure outcomes and adverse events well reported |
| Other bias | Low risk | None identified |
SANAD A 2007.
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in the UK Five treatment arms: LTG, CBZ, GBP, TPM and OXC |
|
| Participants | Adults and children over the age of 4 years with newly diagnosed focal epilepsy, relapsed focal epilepsy or failed treatment with a previous drug not used in this trial. Number randomised: CBZ = 378, TPM = 378 408 male participants (54%) 654 focal epilepsy (97%) 139 had received previous AED treatment (18%) Mean age (range): 39 (5 to 86) years |
|
| Interventions | Monotherapy for CBZ or TPM Titration doses and maintenance doses decided by treating clinician. Range of follow‐up: 0 to 86 months |
|
| Outcomes | Time to treatment failure Time to 1 year (12 month) remission Time to 2 year remission Time to first seizure Health‐related quality of life via the NEWQOL (Newly Diagnosed Epilepsy Quality of Life Battery) Health economic assessment and cost‐effectiveness of the drugs (cost per QALY gained and cost per seizure avoided) Frequency of clinically important adverse events |
|
| Notes | IPD provided for time to treatment failure, time to first seizure, time to 6‐month remission, time to 12‐month remission (trial co‐ordinated at our site) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer minimisation programme stratified by centre, sex and treatment history |
| Allocation concealment (selection bias) | Low risk | Telephone randomisation to a central randomisation allocation service |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Abbreviations AED: antiepileptic drug; CBZ: carbamazepine; GBP: gabapentin; IPD: individual participant data, ITT: intention‐to‐treat; LTG: lamotrigine; OXC: oxcarbazepine; QALY: quality‐adjusted life‐year; SV: sodium valproate; TPM: topiramate
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Kang 2007 | Ineligible epilepsy type |
Contributions of authors
SJ Nevitt wrote the text of the protocol with the input of M Sudell, C Tudur Smith and AG Marson.
SJ Nevitt requested all individual participant data (IPD), under the supervision of C Tudur Smith and AG Marson.
SJ Nevitt and M Sudell prepared IPD for analysis, conducted analyses of the review and interpreted results under the supervision of C Tudur Smith (statistical interpretation) and AG Marson (clinical interpretation).
SJ Nevitt wrote the text of the review with the input of M Sudell, C Tudur Smith and AG Marson.
Sources of support
Internal sources
No sources of support supplied
External sources
National Institute of Health Research (NIHR), UK.
Declarations of interest
SJN: none known MS: none known CTS: none known AGM: a consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. Professor Tony Marson is part funded by National Institute for Health Research Collaboration for Leadership in Applied Health Research and Care North West Coast (NIHR CLAHRC NWC).
Unchanged
References
References to studies included in this review
Privitera 2003 {published data only}
- Ben‐Menachem E, Privitera MD, Neto W, Wu SC. Response to topiramate (TPM), carbamazepine (CBZ) or valproate (VPA) by seizure type in newly diagnosed epilepsy. Epilepsia 2005;46(Suppl 6):274‐5, Abstract no: p843. [Google Scholar]
- Edwards JC, Privitera MD, Neto W, Wu S. Topiramate (TPM) vs carbamazepine (CBZ) and valproate (VPA) in newly diagnosed epilepsy: effectiveness according to seizure type. Epilepsia 2004;45(Suppl 7):139‐40, Abstract no: 1.369. [Google Scholar]
- Privitera MD, Arroyo S, Squires L, Wang S, Twyman R. 100 mg/day topiramate as the initial target dose in newly diagnosed epilepsy. Epilepsia 2003;44(Suppl 9):270, Abstract no: 2.5. [Google Scholar]
- Privitera MD, Brodie MJ, Mattson RH, Chadwick DW, Neto W, Wang S. Topiramate, carbamazepine and valproate monotherapy: double‐blind comparison in newly diagnosed epilepsy. Acta Neurologica Scandinavica 2003;107(3):165‐75. [DOI] [PubMed] [Google Scholar]
- Privitera MD, Brodie MJ, Neto W, Wang S. Topiramate vs. investigator choice of carbamazepine or valproate as monotherapy in newly diagnosed epilepsy. Epilepsia 2000;41(Suppl 7):93‐4, Abstract no: 2.019. [Google Scholar]
- Privitera MD, Brodie MJ, Neto W, Wang S, Topiramate EPMN‐105 Study Group. Monotherapy in newly diagnosed epilepsy: topiramate vs investigator choice of carbamazepine or valproate [abstract]. Epilepsia 2000;41(Suppl Florence):138. [Google Scholar]
- Sander J, Arroyo S, Privitera M, Squires L, Wang S, Twyman R. Topiramate in newly diagnosed epilepsy: 100 mg/day as initial target dose. Epilepsia 2003;44(Suppl 8):82, Abstract no: P184. [Google Scholar]
- Wheless JW, Neto W, Wang S. Topiramate, carbamazepine, and valproate in children with newly diagnosed epilepsy: a unique trial design. Neurology 2001;56(8 Suppl 3):A234‐5, Abstract no: S32.005. [DOI] [PubMed] [Google Scholar]
- Wheless JW, Neto W, Wang S, EPMN‐105 Study Group. Topiramate, carbamazepine, and valproate monotherapy: double‐blind comparison in children with newly diagnosed epilepsy. Journal of Child Neurology 2004;19(2):135‐41. [DOI] [PubMed] [Google Scholar]
Resendiz‐Aparicio 2004 {published data only}
- Resendiz‐Aparicio JC, Rodriguez‐Rodriguez E, Contreras‐Bernal J, Ceja‐Moreno H, Barragan‐Perez E, Garza‐Morales S, et al. A randomised open trial comparing monotherapy with topiramate versus carbamazepine in the treatment of paediatric patients with recently diagnosed epilepsy. Revista de Neurologia 2004;39(3):201‐4. [PubMed] [Google Scholar]
SANAD A 2007 {published data only}
- Marson A, Smith D, Tudur Smith C, Williamson P, Jacoby A, Chadwick D. Carbamazepine versus gabapentin, lamotrigine, oxcarbazepine and topiramate for epilepsy: results from arm A of the SANAD trial. Epilepsia 2006;47(Suppl 4):272. [Google Scholar]
- Marson AG, Al‐Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet 2007;369(9566):1000‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson AG, Appleton R, Baker GA, Chadwick DW, Doughty J, Eaton B, et al. A randomised controlled trial examining the longer‐term outcomes of standard versus new antiepileptic drugs. The SANAD trial. Health Technology Assessment 2007;11(37):iii‐iv, ix‐x, 1‐134. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Kang 2007 {published data only}
- Kang HC, Eun BL, Lee CW, Moon HK, Kim JS, Kim DW, et al. A multicenter, randomized, open‐labeled, clinical study to evaluate the effect on cognitive and behavioral function of topiramate compared with carbamazepine as monotherapy in children with benign rolandic epilepsy. Epilepsia 2006;47(Suppl 4):138, Abstract no: 2.057. [Google Scholar]
- Kang HC, Eun BL, Wu Lee C, Ku Moon H, Kim JS, Wook Kim D, et al. The effects on cognitive function and behavioural problems of topiramate compared to carbamazepine as monotherapy for children with benign rolandic epilepsy. Epilepsia 2007;48(9):1716‐23. [DOI] [PubMed] [Google Scholar]
- Lim K, Kim HD, Korean BRE Study Group. Low‐dose topiramate compared with carbamazepine in treating benign rolandic epilepsy. Epilepsia 2004;45(Suppl 7):322‐3, Abstract no: 2.390. [Google Scholar]
Additional references
Annegers 1999
- Annegers JF, Dubinsky S, Coan SP, Newmark ME, Roht L. The incidence of epilepsy and unprovoked seizures in multiethnic, urban health maintenance organizations. Epilepsia 1999;40(4):502‐6. [DOI] [PubMed] [Google Scholar]
Bell 2014
- Bell GS, Neligan A, Sander JW. An unknown quantity—the worldwide prevalence of epilepsy. Epilepsia 2014;55(7):958‐62. [DOI] [PubMed] [Google Scholar]
Brodie 1996
- Brodie MJ, Dichter MA. Antiepileptic drugs. New England Journal of Medicine 1996;334(3):168‐75. [DOI] [PubMed] [Google Scholar]
Bromley 2014
- Bromley R, Weston J, Adab N, Greenhalgh J, Sanniti A, McKay AJ, et al. Treatment for epilepsy in pregnancy: neurodevelopmental outcomes in the child. Cochrane Database of Systematic Reviews 2014, Issue 10. [DOI: 10.1002/14651858.CD010236.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cockerell 1995
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the National General Practice Study of Epilepsy. Lancet 1995;346(8968):140‐4. [DOI] [PubMed] [Google Scholar]
Coulter 1993
- Coulter DA, Sombati S, DeLorenzo RJ. Selective effects of topiramate on sustained repetitive firing and spontaneous bursting in cultured hippocampal neurons. Epilepsia 1993;34(Suppl 2):123. [DOI] [PubMed] [Google Scholar]
Excel 2010 [Computer program]
- Microsoft. Microsoft Excel. Version accessed 13 May 2016. Redmond (WA): Microsoft, 2010.
French 2007
- French JA, Kanner AM, Bautista J, Abou‐Khalil B, Browne T, Harden CL, et al. Appendix C: Efficacy and tolerability of the new antiepileptic drugs I: Treatment of new onset epilepsy: Report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. CONTINUUM Lifelong Learning in Neurology 2007;13(4):203‐11. [DOI] [PubMed] [Google Scholar]
Gilliam 2003
- Gilliam FG, Veloso F, Bomhof MA, Gazda SK, Biton V, Ter Bruggen JP, et al. A dose‐comparison trial of topiramate as monotherapy in recently diagnosed partial epilepsy. Neurology 2003;60(2):196‐202. [DOI] [PubMed] [Google Scholar]
Global Burden of Disease Study 2013
- Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015;386(9995):743‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEPro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Version accessed 13 May 2016. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Hauser 1993
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota 1935‐1984. Epilepsia 1993;34(3):453‐68. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2017
- Higgins JP, Altman DG, Sterne JA, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org. Cochrane.
Hirtz 2007
- Hirtz D, Thurman DJ, Gwinn‐Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the "common" neurologic disorders?. Neurology 2007;68(5):326‐37. [DOI] [PubMed] [Google Scholar]
Hultcrantz 2017
- Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, et al. The GRADE Working Group clarifies the construct of certainty of evidence. Journal of clinical epidemiology 2017 Jul 1;87:4‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hunt 2008
- Hunt S, Russell A, Smithson WH, Parsons L, Robertson I, Waddell R, et al. Topiramate in pregnancy: preliminary experience from the UK Epilepsy and Pregnancy Register. Neurology 2008;71(4):272‐6. [DOI] [PubMed] [Google Scholar]
ILAE 1998
- ILAE Commission on Antiepileptic Drugs. Considerations on designing clinical trials to evaluate the place of new antiepileptic drugs in the treatment of newly diagnosed and chronic patients with epilepsy. Epilepsia 1998;39(7):799‐803. [DOI] [PubMed] [Google Scholar]
ILAE 2006
- Glauser T, Ben‐Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C, et al. ILAE treatment guidelines: evidence based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 2006;47(7):1094‐120. [DOI] [PubMed] [Google Scholar]
Juul‐Jenson 1983
- Juul‐Jenson P, Foldspang A. Natural history of epileptic seizures. Epilepsia 1983;24(3):297‐312. [DOI] [PubMed] [Google Scholar]
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [DOI] [PubMed] [Google Scholar]
Kwan 2000
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. New England Journal of Medicine 2000;342(5):314‐9. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Liporace 1994
- Liporace JD, Sperling MR, Dichter MA. Absence seizures and carbamazepine in adults. Epilepsia 1994;35(5):1026‐8. [DOI] [PubMed] [Google Scholar]
MacDonald 1995
- MacDonald RL, Kelly KM. Antiepileptic drug mechanisms of action. Epilepsia 1995;36(Suppl 2):S2‐12. [DOI] [PubMed] [Google Scholar]
MacDonald 2000
- MacDonald BK, Johnson AL, Goodridge DM, Cockerell OC, Sander JWA, Shorvon SD. Factors predicting prognosis of epilepsy after presentation with seizures. Annals of Neurology 2000;48(6):833‐41. [PubMed] [Google Scholar]
Malafosse 1994
- Malfosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R. Idiopathic Generalised Epilepsies: Clinical, Experimental and Genetic. Eastleigh: John Libbey and Company, 1994. [Google Scholar]
Marson 2000
- Marson AG, Williamson PR, Hutton JL, Clough HE, Chadwick DW. Carbamazepine versus valproate monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2000, Issue 3. [DOI: 10.1002/14651858.CD001030] [DOI] [PMC free article] [PubMed] [Google Scholar]
Matlow 2012
- Matlow J, Koren G. Is carbamazepine safe to take during pregnancy?. Canadian Family Physician 2012;58(2):163‐4. [PMC free article] [PubMed] [Google Scholar]
Meador 2008
- Meador K, Reynolds M, Crean S, Fahrbach K, Probst C. Pregnancy outcomes in women with epilepsy: a systematic review and meta‐analysis of published pregnancy registries and cohorts. Epilepsy Research 2008;81(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Morrow 2006
- Morrow J, Russel A, Guthrie E, Parsons L, Robertson I, Waddell R, et al. Malformation risks of antiepileptic drugs in pregnancy: a prospective study from the UK Epilepsy and Pregnancy Register. Journal of Neurology, Neurosurgery, and Neuropsychiatry 2006;77(2):193‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Murray 1994
- Murray CJ, Lopez AD, World Health Organization. Global Comparative Assessments in the Health Sector: Disease Burden, Expenditures and Intervention Packages. Geneva: World Health Organization, 1994. [Google Scholar]
Nevitt 2017a
- Nevitt SJ, Sudell M, Weston J, Tudur Smith C, Marson A. Antiepileptic drug monotherapy for epilepsy: a network meta‐analysis of individual participant data. Cochrane Database of Systematic Reviews 2017, Issue 12. [DOI: 10.1002/14651858.CD011412.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2017b
- Nevitt SJ, Marson AG, Weston J, Tudur‐Smith C. Carbamazepine versus phenytoin monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.CD001911.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2018a
- Nevitt SJ, Tudur Smith C, Weston J, Marson AG. Lamotrigine versus carbamazepine monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2018, Issue 6. [DOI: 10.1002/14651858.CD001031.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2018b
- Nevitt SJ, Marson AG, Weston J, Tudur Smith C. Sodium valproate versus phenytoin monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2018, Issue 8. [DOI: 10.1002/14651858.CD001769.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2018c
- Nevitt SJ, Tudur Smith C, Marson AG. Oxcarbazepine versus phenytoin monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2018, Issue 10. [DOI: 10.1002/14651858.CD003615.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2018d
- Nevitt SJ, Marson AG, Tudur‐Smith C. Carbamazepine versus phenobarbitone monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2018, Issue 10. [DOI: 10.1002/14651858.CD001904.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ngugi 2010
- Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Estimation of the burden of active and life‐time epilepsy: a meta‐analytic approach. Epilepsia 2010;51(5):883‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2012
- National Institute for Health and Care Excellence. Epilepsies: diagnosis and management. Clinical guideline [CG137]. www.nice.org.uk/guidance/cg1373 (accessed 10 November 2016). London: National Institute for Health and Care Excellence, 2012.
Nolan 2013a
- Nolan SJ, Sutton L, Marson A, Tudur Smith C. Consistency of outcome and statistical reporting of time‐to‐event data: the impact on Cochrane Reviews and meta‐analyses in epilepsy. Better knowledge for better health. 21st Cochrane Colloquium; 19‐23 September 2013; Quebec City, Canada. Quebec City, Canada: The Cochrane Collaboration, 2013:114‐5. [Google Scholar]
Nolan 2013b
- Nolan SJ, Tudur Smith C, Pulman J, Marson AG. Phenobarbitone versus phenytoin monotherapy for partial onset seizures and generalised onset tonic‐clonic seizures. Cochrane Database of Systematic Reviews 2013, Issue 1. [DOI: 10.1002/14651858.CD002217.pub2] [DOI] [PubMed] [Google Scholar]
Olafsson 2005
- Olafsson E, Ludvigsson P, Gudmundsson G, Hesdorfer D, Kjartansson O, Hauser WA. Incidence of unprovoked seizures and epilepsy in Iceland and assessment of the epilepsy syndrome classification: a prospective study. Lancet Neurology 2005;4:627‐34. [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Ragsdale 1991
- Ragsdale DS, Scheuer T, Catterall WA. Frequency and voltage‐dependent inhibition of type IIA Na+ channels,expressed in a mammalian cell line, by local anaesthetic,antiarrhythmic, and anticonvulsant drugs. Molecular Pharmacology 1991;40(5):756‐65. [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
SANAD B 2007
- Marson AG, Al‐Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, et al. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet 2007;369(9566):1016‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sander 1996
- Sander JW, Shorvon SD. Epidemiology of the epilepsies. Journal of Neurology, Neurosurgery, and Psychiatry 1996;61(5):433‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sander 2004
- Sander JW. The use of anti‐epileptic drugs ‐ principles and practice. Epilepsia 2004;45(Suppl 6):28‐34. [DOI] [PubMed] [Google Scholar]
Scheffer 2017
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shakir 1980
- Shakir RA. Sodium valproate, phenytoin and carbamazepine as sole anticonvulsants. The Place of Sodium Valproate in the Treatment of Epilepsy. London: Academic Press Inc and the Royal Society of Medicine, 1980:7‐16. [Google Scholar]
Shields 1983
- Shields WD, Saslow E. Myoclonic, atonic, and absence seizures following institution of carbamazepine therapy in children. Neurology 1983;33(11):1487‐9. [DOI] [PubMed] [Google Scholar]
Snead 1985
- Snead OC, Hosey LC. Exacerbation of seizures in children by carbamazepine. New England Journal of Medicine 1985;313(15):916‐21. [DOI] [PubMed] [Google Scholar]
Stata 2015 [Computer program]
- StataCorp. Stata. Version 14. College Station, TX, USA: StataCorp, 2015.
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials 2007;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tudur Smith 2007
- Tudur Smith C, Marson AG, Chadwick DW, Williamson PR. Multiple treatment comparisons in epilepsy monotherapy trials. Trials 2007;5(8):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weston 2016
- Weston J, Bromley R, Jackson CF, Adab N, Clayton‐Smith J, Greenhalgh J, et al. Monotherapy treatment of epilepsy in pregnancy: congenital malformation outcomes in the child. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD010224.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
White 1997
- White HS, Brown SD, Woodhead JH, Skeen GA, Wolf HH. Topiramate enhances GABA‐mediated chloride flux and GABA‐evoked chloride currents in murine brain neurons and increases seizure threshold. Epilepsy Research 1997;28(3):167‐79. [DOI] [PubMed] [Google Scholar]
Williamson 2002
- Williamson PR, Tudur Smith C, Hutton JL, Marson AG. Aggregate data meta‐analysis with time‐to‐event outcomes. Statistics in Medicine 2002;21(11):3337‐51. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Nolan 2016a
- Nolan SJ, Sudell M, Tudur Smith C, Marson AG. Topiramate versus carbamazepine monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 1. [DOI: 10.1002/14651858.CD012065] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nolan 2016b
- Nolan SJ, Sudell M, Tudur Smith C, Marson AG. Topiramate versus carbamazepine monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD012065.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]