

Abstract
Anthracycline‐induced cardiotoxicity (ACT) is a severe adverse drug reaction for a subset of children treated with anthracyclines as part of chemotherapy protocols. The identification of genetic markers associated with increased ACT susceptibility has clinical significance toward improving patient care and our understanding of the molecular mechanisms involved in ACT. Human‐induced pluripotent stem cell–derived cardiomyocytes represent a novel approach to determine the pharmacogenomics of ACT and guide the development of genetic screening tests.
The anthracyclines
The anthracyclines are a potent class of chemotherapy agent used widely in the treatment of childhood cancers. Following their introduction to clinical practice in the early 1960s and the increased international collaboration among clinical trial centers since, major advances in survival outcomes for children with cancer have been achieved. According to survivorship statistics from the United States in 2016, overall 5‐year survival rates for all childhood cancers have risen from 58% for those diagnosed between 1975 and 1977 to 83% for children diagnosed between 2005 and 2011.1 Similarly, in Australia, 5‐year survival rates for all childhood cancers have risen from 72% for those diagnosed between 1983 and 1992 to 84% for children diagnosed between 2003 and 2012.2 Owing to their effectiveness, anthracyclines are now included in over 50% of all paediatric chemotherapy regimens.3
Today, there are five different anthracyclines approved for use as chemotherapeutics. They are used in pediatric settings for the treatment of hematopoietic malignancies and solid tumors, including neuroblastoma, nephroblastoma, osteosarcoma, and Ewing's sarcoma. Their chemical structures are shown in Figure 1. Daunorubicin and doxorubicin are very similar compounds, the only difference being the primary chain of doxorubicin terminates with a hydroxyl group rather than the methyl group in daunorubicin. The second generation analog epirubicin is an epimer of doxorubicin with a shorter half‐life and reported reduced cardiotoxic effects.4, 5 Idarubicin is derived from daunorubicin and lacks the 4‐methoxy group on ring D making it more lipophilic than its parent compound. Structurally related to the anthracyclines, mitoxantrone is an anthracenedione that is used in combination therapy for the treatment of acute myeloid leukemia in pediatric patients. All types of anthracyclines have been linked to the development of anthracycline‐induced cardiotoxicity (ACT). Although the precise molecular mechanism for ACT remains unknown, several theories exist to describe its pathogenesis.
Figure 1.
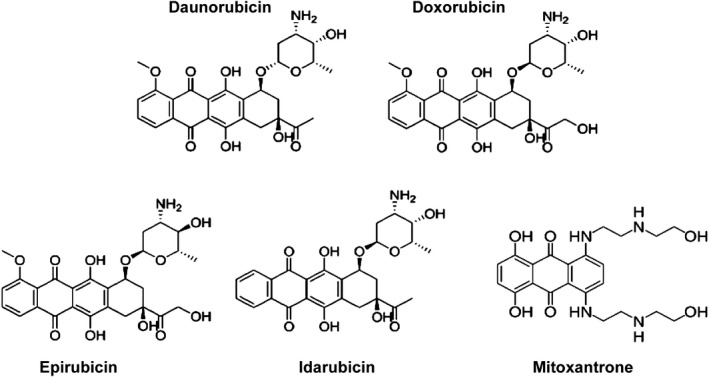
Chemical structures of clinically approved anthracyclines.
Act
The therapeutic use of anthracyclines is complicated by the adverse drug reaction of cardiotoxicity. The severity of ACT is variable, ranging from asymptomatic cardiac dysfunction evidenced by echocardiogram to the development of cardiomyopathy and congestive heart failure, which affects 57% and 16% of children, respectively, postanthracycline exposure.6 Risk factors for ACT include younger age, female sex, higher cumulative anthracycline dose, and concomitant radiotherapy to the mediastinum.7 One in ten children exposed to cumulative anthracycline doses > 300 mg/m2 develop anthracycline‐induced congestive heart failure,8 which is often refractive to medical therapy and associated with a poor prognosis.9 ACT can have an early onset, defined as developing within 1 year of treatment, or a late onset, with increasing cumulative incidence up to 30 years posttreatment.10 Susceptibility for the development of ACT has been suggested to have a genetic basis. Many studies have identified associations between ACT and polymorphisms of genes that can be grouped into various mechanistic pathways. Among these genes are RARG, encoding the retinoic acid receptor‐γ involved in DNA damage11; ABCC5, an ATP cassette binding transporter12; SLC28A3, a sodium‐coupled nucleoside transporter involved in transport of antineoplastic drugs13; and CBR3, a carbonyl reductase that metabolizes the anthracycline doxorubicin.14 Currently, predicting which patients will develop ACT is not possible. However, in 2016, a breakthrough study identified that human‐induced pluripotent stem cells‐derived cardiomyocytes (hiPSC‐CMs) from patients with breast cancer exposed to anthracyclines were able to recapitulate individual patients' sensitivity to ACT at a cellular level.15 Exposure to anthracycline hiPSC‐CMs from patients with ACT displayed decreased cell viability, increased reactive oxygen species (ROS) production, impaired calcium handling, and mitochondrial dysfunction. Thus, hiPSC‐CMs represent a novel platform for further characterization of the genetic basis of ACT, which is an area of great research interest due to its potential translation into clinical practice.
The scope of this review is to briefly discuss the current prevailing theories for the molecular mechanism of ACT, which include the inhibition of topoisomerase 2B (TOP2B), the generation of ROS, and impaired calcium handling due to the C‐13 alcohol metabolites of anthracyclines. The key pharmacogenetic variants identified from pediatric cohorts to be associated with either ACT sensitivity or resistance will then be discussed in detail according to the mechanistic pathways they influence. These include DNA damage pathways, drug transport, oxidative stress defenses, iron metabolism, and sarcomere structure and function.
Genomic variants associated with ACT sensitivity and resistance in paediatric oncology cohorts
Several studies have been conducted in pediatric oncology cohorts to identify genetic variants associated with ACT (Table 1). Both candidate gene approaches and genomewide association studies (GWAS) have yielded significant single nucleotide polymorphisms (SNPs) in 18 genes, which are discussed herein. Identified genetic variants associated with ACT have roles in DNA damage pathways, drug transport, oxidative stress defenses, iron metabolism, and sarcomere structure and function, as summarized in Figure 2. To date, the genetic studies presented are limited by small sample sizes, and follow‐up studies aimed at functional validation of reported gene associations are only beginning to emerge.16 Importantly, among all the studies identified, a consensus does not exist upon clear clinical and echocardiographic research criteria for an ACT diagnosis, which would allow more accurate comparisons to be drawn between cohorts regarding the timing of onset and severity of ACT.
Table 1.
Studies identifying genetic variants associated with ACT in pediatric cancer cohorts according to proposed mechanistic pathway
| Authors | Year | Cohort (n) | Cases (n) | Controls (n) | Study approach | Genes with significant SNPs | SNP affect upon ACT |
|---|---|---|---|---|---|---|---|
| A. DNA damage | |||||||
| Aminkeng et al. | 2015 |
280 96 80 |
32 22 19 |
248 74 61 |
GWAS | RARG rs2229774 | Sensitizing |
| B. Anthracycline metabolism and transport | |||||||
| Visscher et al. | 2012 |
156 188 96 |
38 40 43 |
118 148 53 |
SNP array | SLC28A3 rs78537585 | Protective |
| Visscher et al. | 2013 | 177 | 46 | 131 | SNP array |
SLC28A3 rs78537585
UGT1A6 rs17863783 SULT2B1 rs10426377 |
Protective Sensitizing Sensitizing |
| Semsei et al. | 2012 | 234 | ‐ | ‐ | Candidate gene approach | ABCC1 rs3743527, rs246221, rs3743527 | Sensitizing |
| Armenian et al. | 2013 | 255 | 77 | 178 | SNP array |
ABCC2 rs8187710
RAC2 rs1305833 ** HFE rs1799945 ** |
Sensitizing Sensitizing Sensitizing |
| C. Oxidative stress capacity | |||||||
| Krajinovic et al. | 2016 | 251 | ‐ | ‐ | GWAS |
ABCC5 rs7627754
NOS3 rs1799983 |
Sensitizing Protective |
| Blanco et al. | 2012 | 487 | 170 | 317 | Candidate gene approach | CBR3 rs1056892 | Sensitizing |
| Ruiz‐Pinto et al. | 2017 | 93 | 58 | 35 | SNP array | GPR35 rs12468485 | Sensitizing |
| Wang et al. | 2014 | 287 | 93 | 194 | SNP array | HAS3 rs2232228 | Sensitizing |
| Windsor et al. | 2012 | 58 | 41 | 17 | Candidate gene approach | GSTP1 rs1695 | Sensitizing |
| Rajic´ et al. | 2009 | 76 | 43 | 33 | Candidate gene approach | CAT rs10836235 | Protective |
| Hildebrandt et al. | 2017 | 108 | 46 | 62 | Candidate gene approach |
PLCE1 rs932764
ATP2B1 rs17249754 |
Protective Protective |
| D. Impaired iron metabolism | |||||||
| Lipshultz et al. | 2013 | 184 | – | – | Candidate gene approach | HFE rs1800562 | Sensitizing |
| E. Sarcomere dysfunction | |||||||
| Wang et al. | 2016 |
331 54 |
112 54 |
219 0 |
GWAS | CELF4 rs1786814 | Sensitizing |
The RAC2 and HFE variants identified in Armenians’ study are proposed to mediate ACT through mechanisms involving reactive oxygen species and impaired iron metabolism, respectively. Variant identification and sequence references all obtained from https://varsome.com.
ACT, anthracycline‐induced cardiotoxicity; ATP, adenosine‐5’‐triphosphate; CAT, cationic amino acid transporter; CBR, carbonyl reductase; CELF, CUGBP Elav‐like family member; GPR, G‐protein‐coupled receptor; GST, glutathione s‐transferase; GWAS, genomewide association study; HAS, hyaluronan synthase; NOS, nitric oxide synthase; PLCE, phospholipase C epsilon; RARG, retinoic acid receptor‐gamma; SLC, solute carrier; SNP, single nucleotide polymorphism; SULT, sulfotransferase; UGT, UDP‐glucuronosyltransferase.
Figure 2.
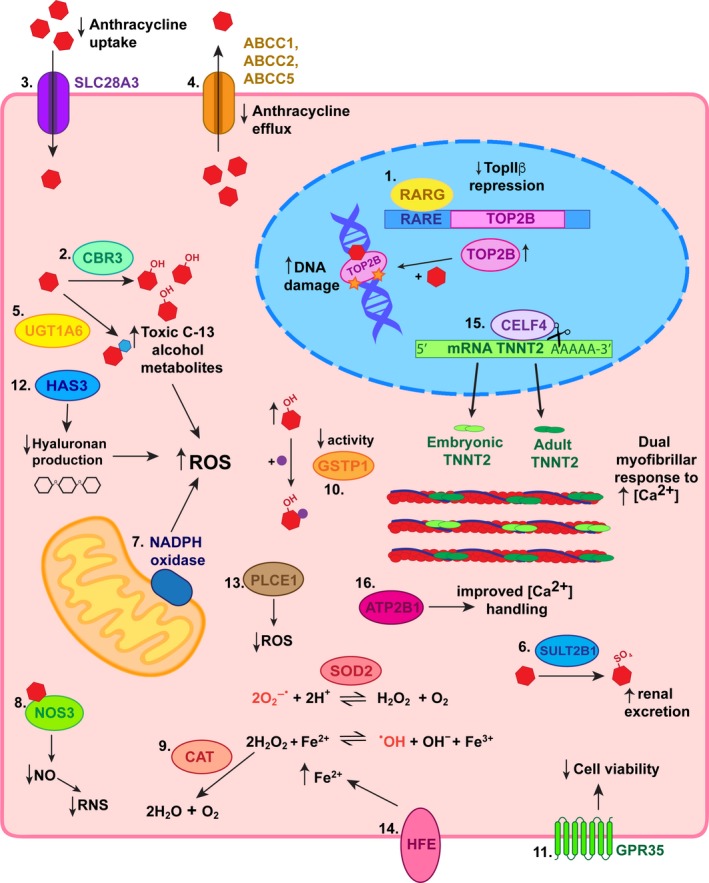
Summary of proposed mechanisms for identified genetic variants associated with anthracycline‐induced cardiotoxicity (ACT) in children. 1. The retinoic acid receptor‐gamma (RARG) variant decreases repression of topoisomerase (TOP)2B such that its levels increase enabling greater amounts of doxorubicin to stabilize its complex with double‐stranded DNA breaks. This leads to increased DNA damage and signals for apoptosis. 2. Carbonyl reductase (CBR)3 variants with increased catalytic activity result in accumulation of toxic C‐13 alcohol metabolites. 3. Decreased anthracycline uptake into cardiomyocytes due to a variant in transporter solute carrier (SLC)28A3 is protective against ACT. 4. The UDP‐glucuronosyltransferase (UGT)1A6 variant increases glucuronidation of anthracyclines resulting in higher levels of toxic metabolites, which predispose patients to ACT. 5. Decreased anthracycline efflux due to polymorphisms in ABC transporters leads to an accumulation of anthracyclines in cardiomyocytes sensitizing them to ACT. 6. Sulfotransferase (SULT)2B1 variant increases sulfonation and therefore renal excretion of anthracyclines and are thus protective against ACT. 7. Mutations in the RAC2 subunit of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase result in increased reactive oxygen species (ROS), which predisposes to ACT. 8. Nitric oxide synthase (NOS)3 variant decreases production of nitric oxide (NO) and consequential reactive nitrogen species (RNS), which culminates in resistance to ACT. 9. A variant resulting in increased cationic amino acid transporter (CAT) expression protects against ACT as hydrogen peroxide is diverted away from conversion to the hydroxyl radical. 10. Conversely, variation causing decreased activity of glutathione s‐transferase p (GSTP)1 results in reduced protection against ROS and therefore increased susceptibility to ACT. 11. A variant in G‐protein‐coupled receptor (GPR)35 has been associated with decreased cell viability upon exposure to anthracyclines. 12. Decreased production of hyaluronan, an antioxidant, due to a polymorphism in hyaluronan synthase (HAS)3 predisposes toward ACT via increased ROS. 13. The phospholipase C epsilon (PLCE)1 variant protects against ACT by reducing the oxidative stress anthracyclines cause. 14. HFE mutations increasing intracellular Fe2+ concentration drive production of hydroxyl radicals. 15. CUGBP Elav‐like family member (CELF)4 variant results in the persistence of alternative splice variants of cardiac troponin T, which are developmentally inappropriate. Co expression of embryonic and adult cardiac troponin T results in a dual capacity of cardiomyocytes to respond to the increasing intracellular calcium levels that occur when anthracyclines are present. 16. An ATP2B1 variant has been associated with ACT resistance potentially as a result of improved calcium handling that optimises sarcomere function during challenge. RARE, retinoic acid receptor element; SOD2, superoxide dismutase II.
Dna damage
DNA topoisomerase I (TOP1) and II (TOP2) relieve tension in overwound DNA by introducing transient single‐stranded or double‐stranded DNA breaks, respectively. Anthracyclines target and stabilize the intermediate TOP2‐cleaved DNA complex, preventing reannealing and causing an accumulation of double‐stranded DNA breaks17 leading to activation of DNA damage pathways ultimately resulting in programmed cell death. Two isoenzymes of TOP2 exist in mammals and both interact with anthracyclines. TOP2A is overexpressed in tumor cells and proposed to mediate the antineoplastic effects of anthracyclines; however, it is not expressed in the adult heart. TOP2B is expressed ubiquitously and has been implicated in the mechanism for the cardiotoxic effects of anthracyclines. Selective deletion of TOP2B from cardiomyocytes protects mice from doxorubicin‐induced heart failure.18 In addition, clustered regularly interspaced short palindromic repeats and associated protein 9 (CRISPR/Cas9) disruption of TOP2B from human embryonic stem cell–derived cardiomyocytes reduces the levels of doxorubicin‐induced double‐stranded DNA breaks and cell death.19
Further support for the role of TOP2B in ACT comes from studies using dexrazoxane, a clinically used cardioprotective agent that inhibits the catalytic activity of TOP2B and reduces ROS production by chelating iron. Preincubation with dexrazoxane prior to doxorubicin exposure reduced expression of phosphorylated H2A histone, a DNA damage signal, in rat cardiomyocytes.20 Furthermore, dexrazoxane protects neonatal ventricular cardiomyocytes from doxorubicin and daunorubicin‐induced cytotoxicity, as measured by lactate dehydrogenase levels.21 Dexrazoxane did not protect neonatal ventricular cardiomyocytes from oxidative stress‐induced cytotoxicity suggesting its actions to reduce ROS are not essential to its cardioprotective function. In contrast, dexrazoxane did not improve hiPSC‐CM viability following doxorubicin exposure.15 However, the antioxidant N‐acetyl‐l‐cysteine was shown to protect hiPSC‐CMs, which would indicate ROS‐based toxicity does contribute to ACT.
RARG variant results in sensitivity to ACT
A GWAS conducted in 280 pediatric oncology patients and replicated in two further independent cohorts of 96 and 80 patients identified SNP rs2229774 in RARG to be strongly associated with ACT (P = 5.9 × 10−8; odds ratio (OR): 4.7).11 RARG binds DNA regulatory sequences called retinoic acid receptor elements (RAREs) and regulates downstream gene expression in response to its agonist all‐trans retinoic acid. Luciferase reporter assays in HEK293T cells showed RARG rs2229774 (p.Ser427Leu) reduced expression of a RARE reporter gene compared to wild‐type RARG, suggesting that deregulated expression of a downstream gene could contribute to ACT. RARG is known to bind the TOP2B promoter. Rat cardiomyocytes (H9c2 cells) expressing RARG rs2229774 (p.Ser427Leu) do not repress TOP2B expression to the extent that wild‐type RARG does. This suggests that in cardiomyocytes with the RARG rs2229774 (p.Ser427Leu) variant, higher levels of TOP2B are present, which leads to a greater accumulation of DNA damage and subsequent cardiomyocyte death when anthracyclines are present.
Anthracycline metabolism and transport
An alternative hypothesis for the mechanism of ACT involves the C‐13 alcohol metabolites of anthracyclines. Carbonyl reductases convert doxorubicin to doxorubicinol, and the accumulation of doxorubicinol in cardiomyocytes is associated with impaired cardiac relaxation.22 With increasing amounts of doxorubicinol there are also greater amounts of cellular injury and cardiomyocyte death.23 Doxorubicinol is able to directly modulate the calcium handling mechanisms of cardiomyocytes. It binds and inhibits both the Ca2+‐ATPase (SERCA2A) and the Ryanodine receptor (RyR2), which replenish the sarcoplasmic reticulum calcium stores and enable sarcoplasmic reticulum calcium release, respectively.24 This results in impaired cardiomyocyte contraction through reduced calcium release into the cytosol. Furthermore, doxorubicinol can bind and inhibit the F1F0 proton pump, leading to reduced energy production that fails to meet the high‐energy requirements of cardiomyocytes. Heart‐specific overexpression of carbonyl reductases in mice results in the accelerated development of acute and chronic forms of ACT due to increased doxorubicinol levels.25 To further support a role for the carbonyl reductases in the mechanism of ACT, a polymorphism in carbonyl reductase 3 (CBR3) rs1056892, which results in p.Val244Met, was identified to be associated with a significant increase in the risk of cardiomyopathy following exposure to low doses of anthracyclines in children with cancer.14
CBR3 polymorphism predisposes to the development of ACT
Carbonyl reductases metabolize anthracyclines to their toxic C‐13 alcohol metabolites. SNPs in CBR1 rs894010988 (c.*246G>A; within 3′UTR) and rs1056892 in CBR3 (p.Val244Met) are known to affect their catalytic activity and, therefore, the metabolism of anthracyclines. Genotyping a case‐controlled cohort of childhood cancer survivors (n = 30 cases, n = 115 controls) for CBR3 rs1056892 (p.Val244Met) indicated a trend toward significance for an association with ACT (P = 0.056). Furthermore, recombinant CBR3V244 produced 2.6‐fold greater amounts of toxic metabolite doxorubicinol compared to CBR3M244.26 In a replicate study, in a larger cohort (n = 170 cases, n = 317 controls) for ACT‐associated SNPs in CBRs, no significant association was determined for CBR1.26 However, the homozygous G genotype at polymorphism rs1056892 in CBR3 (p.Val244) was found to correlate with a 3.3‐fold increased risk of cardiomyopathy following exposure to anthracyclines (1–250 mg/m2) compared with those with a GA or AA genotype.14
SLC28A3 variant confers resistance to ACT, whereas UGT1A6 variant predisposes to ACT
SLC28A3 encodes a sodium‐coupled nucleoside transporter that is expressed in the human heart. It is able to carry both purines and pyrimidines, as well as drugs, including anthracyclines. A study of 2,977 SNPs in 220 genes involved in drug metabolism compared the genetic profiles of children who developed ACT (n = 38 cases) with those who did not (n = 118 controls) following treatment with anthracyclines using a customized Illumina GoldenGate array.27 A synonymous variant of SLC28A3, rs78537585, (which results in a different codon encoding amino acid 461 in SLC28A3, i.e., p.Leu461Leu) was strongly associated with resistance to ACT (P = 1.8 × 10−5; OR: 0.35). The identification of this variant has been replicated in two further independent paediatric cohorts from Canada (n = 188) and The Netherlands (n = 96). Although a mechanism for SLC28A3 protecting against ACT was not proposed, by describing its activity in transporting anthracyclines, the suggestion is that a variant may reduce the uptake of anthracycline into cardiomyocytes. However, given the variant is synonymous and does not alter the structure of the transporter it is also unlikely to change its activity. Nevertheless, a strength of this study is the endeavor to generate a risk‐stratification model: high, intermediate, and low risk for predicting ACT according to a genetic profile of several genes. Combining genetic risk factors with clinical risk factors was superior to predicting ACT than either set of factors alone. In 2013, Visscher and colleagues27 were able to reproduce their findings in a third independent cohort of children. The association between ACT and the SLC28A3 variant remains to be validated in functional assays. In this study, however, a previously identified variant in UDP‐glucoronyl transferase (UGT1A6) was found to be significant for an association with ACT (P = 6.2 × 10−3; OR: 7.98) UGT1A6 glucoronidates various substrates, including anthracycline metabolites. It is speculated that altered activity in the variant rs17863783 UGT1A6 (which is a synonymous variant; i.e., p.Val209Val) may lead to an accumulation of toxic anthracycline metabolites, which predispose to ACT. However, synonymous variants do not alter proteins structure and are, therefore, unlikely to change function. Furthermore, SLC28A3 is not expressed in human cardiac or liver tissues, whereas UGT1A6 is found in the liver and may result in impaired anthracycline metabolism. Nevertheless, the functional consequences of these genetic associations are difficult to discern.
ABCC1, ABCC2, and ABCC5 polymorphisms sensitize to ACT
ATP‐binding cassette proteins are membrane‐bound transporters involved in xenobiotic clearance from cells using energy derived from ATP hydrolysis. Their substrates include numerous chemotherapeutic agents, including anthracyclines.28 ABCC1 (ATP‐binding cassette, subfamily C, member 1) is highly expressed in the human heart, and mouse studies have revealed its expression levels are upregulated in the heart following exposure to doxorubicin.29 Several SNPs are known to modulate the activity of ABCC1 and, therefore, potentially influence the ability of cardiomyocytes to clear anthracyclines and limit oxidative stress. A cohort of pediatric patients (n = 234) diagnosed with acute lymphoblastic leukemia (ALL) were genotyped for nine SNPs in ABCC1.30 The TT genotype at SNP rs3743527 (3′UTR; c.*543C>T) and its combination with either the TT or TC genotype at SNP rs246221 (synonymous variant, p.Val275Val) were significantly associated with a decreased left ventricular fractional shortening, as measured by echocardiography, following treatment with anthracycline.30 SNP rs246221 is a synonymous variant in ABCC1 (p.Val275Val) and, therefore, does not alter its structure, which decreases the likelihood of it having an effect on the activity of the transporter. In comparison, SNP rs3743527 occurs within the 3′UTR of ABCC1 mRNA and perhaps exerts its influence by decreasing gene expression levels posttranscriptionally. Polymorphisms in additional ABC transporter family members, ABCC2 and ABCC5, have also been reported. A retrospective case‐controlled study identified SNP rs8187710 (p.Cys1515Tyr) in ABCC2 to be associated with a 4.3‐fold risk of ACT (P < 0.01) upon genetic profiling of a mixed cohort of child and adult patients exposed to anthracyclines prior to hematopoietic cell transplant.31 Polymorphisms in 16 candidate genes with roles in iron homeostasis, antioxidant stress, cardiac remodeling, and anthracycline metabolism were screened for and also identified two further SNPs with significant correlations to ACT; SNP rs1799945 (c.187C>G; p.His63Asp) in HFE, which is consistent with evidence discussed below and SNP rs13058338 (intronic, c.108‐3812A>T) in the RAC2, which encodes a subunit of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Furthermore, ABCC5 has been implicated in ACT with the TT genotype at SNP rs7627754 (in promoter region) being associated with reduced ejection fraction and fractional shortening.12
Sulfotransferase family cytosolic member 2B1 is sensitizing to ACT
Sulfotransferase family cytosolic member 2B1 (SULT2B1) is an enzyme that conjugates sulfate groups to drugs, increasing solubility in water and promoting renal excretion. The SULT2B1 variant rs10426377 (intronic c.423 + 1540C>A) was initially identified as having a weak association with ACT in pediatric patients.13 However, only upon replication in a second study did the association with ACT reach significance (P = 0.054).27 Interestingly, when stratified by sex, this SULT2B1 variant only exerted a sensitizing effect in men and not women.27 It is possible the SULT2B1 variant may result in a loss of catalytic function preventing anthracycline metabolites from becoming sulfonated and renally excreted. Anthracycline metabolite levels would, therefore, accumulate and be able to mediate their toxic effects for greater periods of time. However, additional work is required to establish if the variant rs10426377 alters SULT2B1 expression levels. Furthermore, it is important to note that SULT2B1 is not highly expressed in human cardiac tissue and, therefore, would mediate its effect upon ACT indirectly.
Oxidative stress capacity
The cardiotoxic effects of anthracyclines have also been attributed to their ability to induce oxidative stress. Cardiomyocytes are particularly vulnerable to ROS‐induced damage having poorer anti‐oxidant defenses compared with hepatocytes.32 Anthracyclines generate ROS by two mechanisms: an enzymatic pathway involving the electron transport chain of mitochondria33 and a nonenzymatic pathway involving redox cycling of iron‐anthracycline complexes.34 Both result in the formation of superoxide anions (O2 −•), which can either directly cause subcellular damage or become converted to the highly reactive hydroxyl radical (•OH) via a series of reactions with superoxide dismutase and further reduction by Fe2+ shown in Figure 3.35 Superoxide anions and hydroxyl radicals cause lipid peroxidation of cardiomyocyte membranes resulting in disruption of membrane integrity allowing intracellular proteins, including lactate dehydrogenase and cardiac troponin to be released.36 Detailed examination of anthracycline‐induced cellular morphology changes in cardiomyocytes by electron microscopy of endomyocardial biopsies demonstrated that myocyte vacuolization and myofibrillar lysis are the characteristic morphological features of ACT.37, 38
Figure 3.
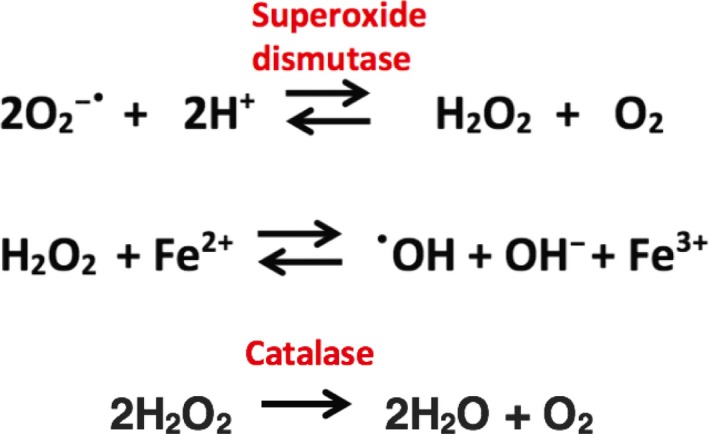
Superoxide anion conversion pathway to hydroxyl radical and catalase defense pathway.
The role of cardiolipin and NADPH oxidase in ACT
Cardiolipin is a phospholipid enriched in the inner mitochondrial membrane for which anthracyclines have a high affinity.33 Being lipophilic molecules, anthracyclines are able to diffuse passively across cell membranes and into mitochondria. Once bound to cardiolipin, anthracycline is reduced from its quinone form to a semiquinone by nicotinamide adenine dinucleotide. The anthracycline semiquinone then converts back to its quinone form by transferring the electron to an oxygen molecule and forming a superoxide anion.22 Thus, the electron transport chain is short‐circuited and ROS are generated. Alternatively, the NADPH oxidase complex has also been implicated in the mechanism of ACT. It is expressed in cardiomyocyte mitochondria and comprised of a catalytic core situated in the membrane with four cytosolic subunits that are recruited upon its activation. NADPH oxidase generates ROS, which are known to induce a hypertrophic response that serves as an initial compensation mechanism by the failing heart, which then becomes pathological.39 The SNP rs13058338 (intronic; c.108‐3812A>T) in RAC2, which encodes an Rho‐GTPase that regulates the NADPH oxidase, has been correlated with susceptibility to ACT.31 It is possible this variant accelerates ROS production and drives the hypertrophic response to become pathological. Cardiac‐specific RAC1 overexpression in mice results in the development of cardiomyopathy.39 Furthermore, NADPH oxidase (NOX2) knockout mice are protected against doxorubicin‐induced heart failure.40 Inhibitors of NADPH oxidase have also been shown to reduce the damage to cardiomyocytes upon anthracycline exposure.41
NOS3 mutation protective against ACT
Krajinovic et al.12 investigated the influence of polymorphisms of genes involved in doxorubicin metabolism upon late‐onset ACT. A cohort of 251 children with ALL exposed to doxorubicin and monitored by echocardiography were genotyped for 33 common polymorphisms of 12 genes involved in drug metabolism. Nitric oxide synthase (NOS3) variant rs1799983 (p.Asp298Glu) was identified to have a protective effect upon the ejection fraction (EF) of high‐risk patients. The variant is known to decrease endothelial nitric oxide (NO) production. Anthracyclines are able to bind NOS3 and inhibit its activity, thus reducing the amount of vasodilator NO produced; excess levels of NO increase the generation of reactive nitrogen species. Expediting the decomposition of the reactive nitrogen species using a catalyst has been shown to reduce doxorubicin‐induced cardiomyocyte injury and cell death.42 The NOS3 −/− mice are protected against cardiotoxicity following doxorubicin exposure compared with mice with cardiomyocyte‐specific overexpression of wild‐type NOS3, which were shown to be more sensitive to injury and cell death.43 Taking together the above studies, NOS3 has been implicated in the pathogenesis of ACT.
Catalase variant protects against ACT, whereas GSTP1 variant increases susceptibility
Variants inhibiting the activity of enzymes involved in oxidative stress are candidates for an association with ACT because ROS generation is a well‐described potential theory for its pathogenesis. During oxidative stress, superoxide dismutase II (SOD2) converts superoxide to hydrogen peroxide, catalase (CAT) converts hydrogen peroxide to water, which prevents its reduction by Fe2+ to the damaging hydroxyl radical (Figure 2). Glutathione‐S‐transferase θ and μ (GSTT1 and GSTM1) conjugate free glutathione to drug metabolites, including chemotherapeutics to prevent their damaging interactions with lipids, protein, and DNA. A cohort of childhood cancer survivors (n = 76) diagnosed with ALL had their genotypes at known inactivating SNPs in SOD2, CAT, GSTT1, and GSTM1 determined from archived bone marrow smears. The homozygous CC allele for SNP rs10836235 (intronic, c.66 + 78C>G) in CAT reached statistical significance (P = 0.02) for an association with resistance to ACT.44 Rajić et al.44 speculate that this SNP in intron 1 interferes with binding of AP‐2, a negative regulator of CAT transcription and, therefore, there are higher levels of CAT expressed in these patients protecting them from ROS and subsequent ACT. No variants from the other genes were found to have significant associations with ACT. However, a variant in GSTP1 rs1695 (c.313A>G; Ile105Val) was associated with cardiotoxicity (P = 0.008) in a cohort of children (n = 60) treated with doxorubicin for osteosarcoma.45 Already vulnerable to oxidative stress, cardiomyocytes are proposed to be rendered further unprotected from ROS in patients with reduced activity of GSTP1. In this retrospective study, an initial drop in EF on echocardiography from that recorded at diagnosis was seen in 16 patients (29%) and considered to represent early ACT. By the end of treatment, 25 patients (45%) had decreased cardiac EF. The need for future prospective studies monitoring the EF throughout treatment in large cohorts is highlighted by this study as well as the need to validate gene associations with ACT.
GPR35 missense mutation increases susceptibility to and severity of ACT
The genetic association studies discussed so far have focused on common SNPs with a minor allele frequency (MAF) > 5%. Alternatively, Ruiz‐Pinto et al.46 hypothesize that low frequency alleles (MAF < 5%) also have a role to play in complex pathophysiological processes, such as ACT. An Illumina HumanExome BeadChip array, enriched for low‐frequency alleles (MAF < 1%) was used to profile case‐control matched pediatric patients with cancer (n = 35 cases, n = 58 controls). Cases of ACT were defined as chronic ACT if the onset occurred > 1 year following anthracycline treatment. Upon gene‐based testing, as opposed to single‐variant testing performed in other studies, G‐protein coupled receptor 35 (GPR35) was detected to have a significant association with chronic ACT.46 The missense mutation rs12468485 in GPR35 (c.758C>T p.T253M) carried the strongest association with chronic ACT, and the T allele conferred increased risk of chronic ACT and increased severity of ACT at low doses of anthracycline. GPR35 has been found to be upregulated in patients with severe congestive heart failure.47 An in vitro study has shown its expression levels in cardiomyocytes are exquisitely sensitive to hypoxic conditions upon which they are upregulated. In turn, overexpression of GPR35 in cardiomyocytes results in reduced cell viability.48 It is possible that the ROS generated by anthracyclines mimic the hypoxic conditions in the failing heart, leading to increased GPR35 expression, which is correlated with reduced cell viability. Functional validation of the observed genotype‐phenotype associations have yet to be performed.
Hyaluronan synthase‐3 variant modifies risk of developing ACT
Hyaluronan is a glycosaminoglycan found in the extracellular matrix. It is synthesized by hyaluronan synthase‐3 (HAS3) and forms polymers that are known to serve as scaffolds during tissue remodeling following injury. Wang et al.49 identified an SNP rs2232228 (synonymous variant; p.Ala93Ala) in HAS3 to be associated with altering the risk of developing ACT. An SNP array (ITMAT/Broad CARe) of 34,912 common variants in 2,100 genes associated with cardiovascular disease was used to profile a case‐controlled cohort of childhood cancer survivors with and without cardiomyopathy postanthracycline exposure (n = 93 cases, n = 194 controls). Patients with the AA genotype in SNP rs2232228 HAS3 exposed to high doses of anthracycline > 250 mg/m2 were at 8.9‐fold greater risk of developing ACT compared with those with the GG genotype.49 This dose‐dependent increase in the risk of developing ACT among patients with the AA genotype was reproduced in an independent replication cohort of patients (n = 76) with diagnosed ACT. Given hyaluronan has antioxidant properties, it is suggested that patients with the AA genotype in HAS3 have decreased production of hyaluronan leaving them susceptible to ROS‐induced damage generated by anthracyclines. In addition, hyaluronan has been found to promote rat cardiomyocyte survival during ROS damage by directly binding its receptor CD44 and stimulating cell proliferation.50 Given rs2232228 does not change the protein sequence, one proposed mechanism was that the cardiac HAS3 expression levels of the AA genotype would be lower; however, no statistically significant difference between genotypes was observed.49 Thus, the mechanistic basis by which this synonymous variant leads to ACT remains to be discovered.
Phospholipase C ε variants confers protection against ACT
Long‐term survivors of childhood cancer with hypertension have an increased risk of developing ACT. It is possible the increased workload on the heart due to hypertension exacerbates any underlying, perhaps asymptomatic, dysfunction to produce symptomatic ACT. Genetic analysis of 108 childhood cancer survivors was performed for a panel of 12 known hypertension susceptibility loci.51 Two variants, PLCE1 rs932764 (intronic variant; c.1492 + 3724A>G) and ATP2B1 rs17249754 (discussed in E. Sarcomere dysfunction; intronic variant; c.‐221‐10702C>T) were associated with protection against the development of ACT, and both are expressed in human cardiac tissue.51 Phospholipase C ε (PLCE1) is a second messenger, able to induce Ca2+ signalling and activate protein kinase C and D. Using hiPSC‐CMs that were exposed to doxorubicin for 48 hours, expression of PLCE1 was found to be downregulated. PLCE1 is known to reduce ROS generation through the activation of protein kinase D. Therefore, this PLCE1 variant potentially protects against the development of ACT by having increased capacity to mitigate the toxic effects of ROS. PLCE1‐deficient mice have decreased cardiac function following exposure to hypertrophic stress signals both acutely and chronically.52
Impaired iron metabolism
HFE deficiency increases sensitivity to ACT
Given iron chelators such as dexrazoxane have cardioprotective effects, it was hypothesized by Miranda et al.53 that genes responsible for hereditary hemochromatosis, a condition marked by iron overload, could play a role in sensitizing patients to ACT. In their study, HFE −/− mice and wild‐type mice (n = 7) were treated with intraperitoneal injections of doxorubicin (20 mg/kg) and the HFE‐deficient mice were found at day 4 posttreatment to have higher levels of serum creatine kinase, a biomarker of cardiac damage compared to wild‐type mice.53 This study generated interest for profiling childhood cancer survivors for the two SNPs in HFE rs1800562 (p.C282Y) and rs1799945 (p.H63D) that are linked to hereditary haemochromatosis. HFE encodes a major histocompatibility complex class 1–like protein that is able to bind transferrin, an iron transport molecule, and regulate the production of the master regulator of iron storage, hepcidin. Lipshultz et al.54 recruited 184 pediatric patients with cancer between 2005 and 2007 and identified their genotype at the above two SNPs in HFE. Of this cohort, 10% carried the SNP rs1800562 (p.C282Y) and the heterozygous rs1800562 (p.C282Y) genotype correlated with multiple increases in serum cardiac troponin T during chemotherapy and reduced left ventricular function on echocardiography at 2.2‐year follow‐up. These findings suggest that carriers of HFE SNPs are more sensitive to developing ACT and would potentially benefit from receiving iron chelation therapy with dexrazoxane prior to commencing treatment. Although Lipshultz et al.54 suggest HFE genotyping prior to anthracycline exposure, there are implications for patients and their families with regard to the rare finding of a homozygous.
Sarcomere dysfunction
CUGBP Elav‐like family member‐4 variant modifies risk of developing ACT
In a follow‐up study, Wang et al.55 identified an SNP in CUGBP Elav‐like family member 4 (CELF4) associated with altering the risk of developing ACT. CELF4 is an RNA binding protein involved in tissue‐specific, developmentally regulated pre‐mRNA splicing. It is known to mediate the alternative splicing of the gene TNNT2, which encodes cardiac troponin T, a component of thin filaments in sarcomeres. Splice variants of cardiac troponin T bearing an alternative exon 5 are predominately expressed in the embryonic heart and significantly downregulated during development into the adult heart.56 CELF4 variants with reduced activity to target TNNT2 pre‐mRNA enable the continued expression of embryonic cardiac troponin T in the adult heart. This results in a dual capacity for thin myofilaments to handle increasing calcium concentrations and potentially compromises their contractility and ultimately left ventricular ejection fraction. In favor of this proposed mechanism are findings of multiple TNNT2 splice variants present in patients with cardiomyopathies.57 In contrast to their previous approach using an SNP array of common variants associated with cardiovascular disease, Wang et al.55 used a case‐controlled GWAS performed in childhood cancer survivors comparing those who had developed cardiomyopathy postanthracycline with healthy controls (n = 112 cases, n = 219 controls) and identified an association with SNP rs1786814 (intronic, c.287‐11458C>T) in CELF4. Patients exposed to doses of anthracycline > 300 mg/m2 with the CC genotype (publication 55 used nomenclature GG, GA, and AA) had a 10.2‐fold increased risk of developing ACT. However, the CT and TT genotypes for CELF4 rs1786814 mitigated the risk of ACT. These findings were repeated in an independent replication cohort (n = 54) of childhood cancer survivors with cardiomyopathy. To examine whether the high‐risk CC genotype for the CELF4 SNP was associated with the co‐expression of multiple TNNT2 splice variants, DNA was isolated from 33 healthy heart samples. The CC genotype was detected in 21 of 33 samples with embryonic and adult TNNT2 splice variants being found more commonly in these samples.
ATP2B1 variant confers protection against ACT
ATP2B1 encodes a plasma membrane ATPase, which is the calcium pump responsible for maintaining low cytosolic calcium during cardiomyocyte relaxation. SNP rs17249754 (intronic; c.‐221‐10702C>T) in ATP2B1 confers resistance to ACT.51 Following doxorubicin exposure, hiPSC‐CMs had increased expression levels of ATP2B1 and were less susceptible to ACT.51 Because doxorubicin is known to impair calcium handling, expression of ATP2B1 may become upregulated in response to rising cytosolic calcium levels and help reduce excess cytosolic calcium. Efficient cytosolic calcium clearance is necessary for sarcomere relaxation and maintenance of calcium‐coupled contraction.
Discussion
The advent of hiPSC‐CMs has provided a relevant human cardiomyocyte model cell type in which to perform functional validation studies to demonstrate that a given genetic variant may predispose an individual to ACT (Figure 4). Importantly, functional validation studies of ACT‐associated genomic variants remain to be done. Clinical practice recommendations developed by the Canadian Pharmacogenomic Network for Drug Safety suggest pharmacogenomic testing of RARG rs2229774 (p.S427L), SLC28A3 rs7853758 (p.L461L), and UGT1A6 rs17863783 (p.V209V).58 However, the clinical significance of the latter two variants must be interpreted with care because they are silent mutations that do not alter the protein structure or function, nor do they create alternative splice transcripts. Furthermore, SLC28A3 and UGT1A6 are not expressed in cardiac tissue, suggesting their influence is mediated through an indirect mechanism.59 These guidelines, although published, have not been incorporated broadly into pediatric oncology practice because of the low level of evidence and lack of robust functional validation or prospective trials. Studies demonstrating the functional validity in relevant disease models, for example, patient‐specific hiPSC‐CMs (i.e., iPSCs from pediatric oncology patients exposed to anthracyclines that have been differentiated into cardiomyocytes), are still required in order to provide robust evidence to guide the development of genetic screening tests that are applicable to the clinic.
Figure 4.
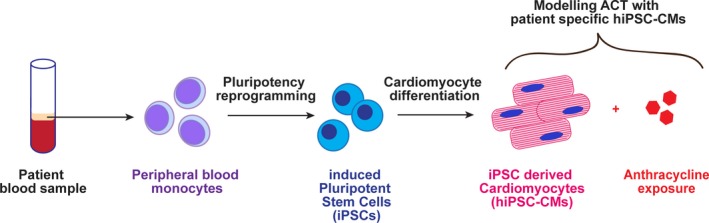
Disease modeling anthracycline‐induced cardiotoxicity (ACT) with patient‐specific human‐induced pluripotent stem cells‐derived cardiomyocytes (hiPSC‐CMs).
Broadly, however, hiPSC‐CMs are a useful model to study the molecular mechanism of ACT. Holmgren et al.60 examined the effect of doxorubicin on global protein expression in hiPSC‐CMs and found a dose‐dependent increase in the number of differentially expressed proteins that was maximal at day 14 postexposure. Interestingly, proteins involved in sarcomere function, including myosin light and heavy chains, troponins and tropomyosins were downregulated following doxorubicin treatment, which is in keeping with the reduced contractility of cardiomyocytes and a potential mechanism for ACT. Additionally, changes to gene expression following doxorubicin exposure have been identified in hiPSC‐CMs using RNASeq studies.15, 60, 61, 62 For example, genes involved in the DNA damage repair response pathway and cell cycle regulation are significantly downregulated following doxorubicin exposure.61 Furthermore, this approach has highlighted interesting linkages, such variation at rs885004 (intronic, c.862‐360C>T) altering SLC28A3 expression levels in hiPSCs.62
The complex nature of the pharmacogenomics of ACT is only beginning to be unravelled and there is still much to be determined. Current knowledge implicates TOP2B inhibition, ROS generation, and the C‐13 anthracycline alcohol metabolites in the development of ACT. The GWAS and candidate gene approach studies to date, to determine genomic variants associated with ACT, have been relatively underpowered and await further independent replication studies to confirm the original findings. However, the recent advent of rapid hiPSC‐CM generation from larger cohorts provides a tool to further dissect the molecular response to anthracycline to identify quantitative trait loci associated with ACT.62 Functional validation of identified genetic variants will improve our understanding of how identified variants contribute to ACT susceptibility. The hiPSC‐CMs generated from ACT‐sensitive and ACT‐resistant pediatric oncology patients provide an experimentally tractable human model system15 to validate genetic variants and further characterize the molecular mechanisms of ACT. These studies will inform the incorporation of genetic screening for ACT sensitivity into clinical practice.
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
References
- 1. Miller, K. et al Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 66, 271–289 (2016). [DOI] [PubMed] [Google Scholar]
- 2. Australian Childhood Cancer Registry . A summary of childhood cancer statistics 1983–2014. Queensland: Cancer Council (2014). <https://cancerqld.blob.core.windows.net/site/content/uploads/2017/12/A-summary-of-childhood-cancer-in-Australia-1983-2014.pdf>. Accessed February 27, 2018.
- 3. Smith, L. et al Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta‐analysis of randomised controlled trials. BMC Cancer 10, 1–14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Minotti, G. , Menna, P. , Salvatorelli, E. , Cairo, G. & Gianni, L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 56, 185–229 (2004). [DOI] [PubMed] [Google Scholar]
- 5. Jain, K. et al A prospective randomized comparison of epirubicin and doxorubicin in patients with advanced breast cancer. J. Clin. Oncol. 3, 818–826 (1985). [DOI] [PubMed] [Google Scholar]
- 6. Van der Pal, H. et al Cardiac function in 5‐year survivors of childhood cancer a long‐term follow‐up study. Arch. Intern. Med. 170, 1247–1255 (2010). [DOI] [PubMed] [Google Scholar]
- 7. Lipshultz, S. , Alvarez, J. & Scully, R. Anthracycline associated cardiotoxicity in survivors of childhood cancer. Heart 94, 525–533 (2008). [DOI] [PubMed] [Google Scholar]
- 8. Van Dalen, E. , van der Pal, H. , Kok, W. , Caron, H. & Kremer, L. Clinical heart failure in a cohort of children treated with anthracyclines: a long‐term follow‐up study. Eur. J. Cancer 42, 3191–3198 (2006). [DOI] [PubMed] [Google Scholar]
- 9. Felker, G.M. et al Underlying causes and long term survival in patients with initially unexplained cardiomyopathy. N. Engl. J. Med. 342, 1077–1084 (2000). [DOI] [PubMed] [Google Scholar]
- 10. Mulrooney, D.A. et al Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ 339, b4606 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aminkeng, F. et al A coding variant in RARG confers susceptibility to anthracycline‐induced cardiotoxicity in childhood cancer. Nat. Genet. 47, 1079–1084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krajinovic, M. et al Polymorphisms of ABCC5 and NOS3 genes influence doxorubicin cardiotoxicity in survivors of childhood acute lymphoblastic leukemia. Pharmacogenomics J. 16, 530–535 (2016). [DOI] [PubMed] [Google Scholar]
- 13. Visscher, H. et al Pharmacogenomic prediction of anthracycline‐induced cardiotoxicity in children. J. Clin. Oncol. 30, 1422–1428 (2012). [DOI] [PubMed] [Google Scholar]
- 14. Blanco, J. et al Anthracycline‐related cardiomyopathy after childhood cancer: role of polymorphisms in carbonyl reductase genes—a report from the Children's Oncology Group. J. Clin. Oncol. 30, 1415–1421 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burridge, P.W. et al Human induced pluripotent stem cell‐derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin‐induced cardiotoxicity. Nat. Med. 22, 547–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Linschoten, M. , Teske, A.J. , Cramer, M.J. , van der Wall, E. & Asselbergs, F.W. Chemotherapy‐related cardiac dysfunction: a systematic review of genetic variants modulating individual risk. Circ. Genom. Precis. Med. 11, e001753 (2018). [DOI] [PubMed] [Google Scholar]
- 17. Tewey, K.M. , Rowe, T.C. , Yang, L. , Halligan, B.D. & Liu, L.F. Adriamycin‐induced DNA damage mediated by mammalian DNA topoisomerase II. Science 226, 466 (1984). [DOI] [PubMed] [Google Scholar]
- 18. Zhang, S. et al Identification of the molecular basis of doxorubicin‐induced cardiotoxicity. Nat. Med. 18, 1639–1642 (2012). [DOI] [PubMed] [Google Scholar]
- 19. Maillet, A. et al Modeling doxorubicin‐induced cardiotoxicity in human pluripotent stem cell derived‐cardiomyocytes. Sci. Rep. 6, srep25333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lyu, Y.L. et al Topoisomerase IIbeta mediated DNA double‐strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 67, 8839–8846 (2007). [DOI] [PubMed] [Google Scholar]
- 21. Vavrova, A. et al Catalytic inhibitors of topoisomerase II differently modulate the toxicity of anthracyclines in cardiac and cancer cells. PLoS One 8, e76676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mordente, A. , Meucci, E. , Martorana, G. , Giardina, B. & Minotti, G. Human heart cytosolic reductases and anthracycline cardiotoxicity. IUBMB Life 52, 83–88 (2001). [DOI] [PubMed] [Google Scholar]
- 23. Mushlin, P. , Cusack, B. , Boucek, R. , Andrejuk, T. , Li, X. & Olson, R. Time‐related increases in cardiac concentrations of doxorubicinol could interact with doxorubicin to depress myocardial contractile function. Br. J. Pharmacol. 110, 975–982 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanna, A. , Lam, A. , Tham, S. , Dulhunty, A. & Beard, N. Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol. Pharmacol. 86, 438–449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Forrest, G.L. , Gonzalez, B. , Tseng, W. , Li, X. & Mann, J. Human carbonyl reductase overexpression in the heart advances the development of doxorubicin‐induced cardiotoxicity in transgenic mice. Cancer Res. 60, 5158–5164 (2000). [PubMed] [Google Scholar]
- 26. Blanco, J. et al Genetic polymorphisms in the carbonyl reductase 3 gene CBR3 and the NAD(P)H:quinone oxidoreductase 1 gene NQO1 in patients who developed anthracycline‐related congestive heart failure after childhood cancer. Cancer 112, 2789–2795 (2008). [DOI] [PubMed] [Google Scholar]
- 27. Visscher, H. et al Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline induced cardiotoxicity in children. Pediatr. Blood Cancer 60, 1375–1381 (2013). [DOI] [PubMed] [Google Scholar]
- 28. Couture, L. , Nash, J. & Turgeon, J. Role of ATP‐binding cassette transporters in drug distribution to the heart and protection from toxic compounds. Heart Metab. 35, 1–6 (2007). [Google Scholar]
- 29. Jungsuwadee, P. et al Mrp1 localization and function in cardiac mitochondria after doxorubicin. Mol. Pharmacol. 75, 111726 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Semsei, A. et al ABCC1 polymorphisms in anthracycline‐induced cardiotoxicity in childhood acute lymphoblastic leukaemia. Cell Biol. Int. 36, 79–86 (2012). [DOI] [PubMed] [Google Scholar]
- 31. Armenian, S.H. et al Genetic susceptibility to anthracycline‐related congestive heart failure in survivors of haematopoietic cell transplantation. Br. J. Haematol. 163, 205–213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Doroshow, J. , Locker, G. & Myers, C. Enzymatic defenses of the mouse heart against reactive oxygen metabolites. J. Clin. Invest. 65, 128–135 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goormaghtigh, E. , Huart, P. , Praet, M. , Brasseur, R. & Ruysschaert, J.M. Structure of the Adriamycin‐cardiolipin complex role in mitochondrial toxicity. Biophys. Chem. 35, 247–257 (1990). [DOI] [PubMed] [Google Scholar]
- 34. Gianni, L. , Zweier, J. , Levy, A. & Myers, C. Characterisation of the cycle of iron‐mediated electron transfer from Adriamycin to oxygen. J. Biol. Chem. 250, 6820–6826 (1984). [PubMed] [Google Scholar]
- 35. Horenstein, M.S. , Heide, R. & L'Ecuyer, T. Molecular basis of anthracycline‐induced cardiotoxicity and its prevention. Mol. Genet. Metab. 71, 436–444 (2000). [DOI] [PubMed] [Google Scholar]
- 36. Singal, P.K. , Iliskovic, N. , Li, T. & Kumar, D. Adriamycin cardiomyopathy: pathophysiology and prevention. FASEB J. 11, 931–936 (1997). [DOI] [PubMed] [Google Scholar]
- 37. Billingham, M. , Mason, J. , Bristow, M. & Daniels, J. Anthracycline cardiomyopathy monitored by morphologic changes. Cancer Treat. Rep. 62, 865–872 (1978). [PubMed] [Google Scholar]
- 38. Glass, C. & Mitchell, R. Winning the battle, but losing the war: mechanisms and morphology of cancer‐therapy‐associated cardiovascular toxicity. Cardiovasc. Pathol. 30, 55–63 (2017). [DOI] [PubMed] [Google Scholar]
- 39. Akki, A. , Zhang, M. , Murdoch, C. , Brewer, A. & Shah, A. NADPH oxidase signalling and cardiac myocyte function. J. Mol. Cell. Cardiol. 47, 15–22 (2009). [DOI] [PubMed] [Google Scholar]
- 40. Zhao, Y. et al Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with doxorubicin chemotherapy. Cancer Res. 70, 9287–9297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Garner, A.P. et al Nitric oxide synthases catalyse the activation of redox cycling and bioreductive anticancer agents. Cancer Res. 59, 1929–19234 (1999). [PubMed] [Google Scholar]
- 42. Pacher, P. et al Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin‐induced cardiac dysfunction. Circulation 107, 896–904 (2003). [DOI] [PubMed] [Google Scholar]
- 43. Neilan, T.G. et al Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation 2, 506–514 (2007). [DOI] [PubMed] [Google Scholar]
- 44. Rajic´, V. et al Influence of the polymorphism in candidate genes on late cardiac damage in patients treated due to acute leukemia in childhood. Leuk. Lymphoma 50, 1693–1698 (2009). [DOI] [PubMed] [Google Scholar]
- 45. Windsor, R. , Strauss, S. , Kallis, C. , Wood, N. & Whelan, J. Germline genetic polymorphisms may influence chemotherapy response and disease outcome in osteosarcoma. Cancer 118, 1856–1867 (2012). [DOI] [PubMed] [Google Scholar]
- 46. Ruiz‐Pinto, S. et al Exome array analysis identifies GPR35 as a novel susceptibility gene for anthracycline‐induced cardiotoxicity in childhood cancer. Pharmacogenet. Genomics 27, 445–453 (2017). [DOI] [PubMed] [Google Scholar]
- 47. Min, K.D. et al Identification of genes related to heart failure using global gene expression profiling of human failing myocardium. Biochem. Biophys. Res. Commun. 393, 55–60 (2010). [DOI] [PubMed] [Google Scholar]
- 48. Ronkainen, V.P. et al Hypoxia inducible factor 1‐induced G protein‐coupled receptor 35 expression is an early marker of progressive cardiac remodelling. Cardiovasc. Res. 101, 69–77 (2014). [DOI] [PubMed] [Google Scholar]
- 49. Wang, X. et al Hyaluronan synthase 3 variant and anthracycline‐related cardiomyopathy: a report from the Children's Oncology Group. J. Clin. Oncol. 32, 647–653 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Law, C. , Li, J. , Chou, H. , Chen, Y. & Chan, H. Hyaluronic acid‐dependent protection in H9C2 cardiomyocytes: a cell model of heart ischemia‐reperfusion injury and treatment. Toxicology 303, 54–71 (2013). [DOI] [PubMed] [Google Scholar]
- 51. Hildebrandt, M. et al Hypertension susceptibility loci are associated with anthracycline‐related cardiotoxicity in long‐term childhood cancer survivors. Sci. Rep. 7, 9698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang, H. et al Phospholipase C epsilon modulates beta‐adrenergic receptor‐dependent cardiac contraction and inhibits cardiac hypertrophy. Circ. Res. 97, 1305–1313 (2005). [DOI] [PubMed] [Google Scholar]
- 53. Miranda, C. et al HFE deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood 102, 2574–2580 (2003). [DOI] [PubMed] [Google Scholar]
- 54. Lipshultz, S. et al Impact of hemochromatosis gene mutations on cardiac status in doxorubicin‐treated survivors of childhood high‐risk leukemia. Cancer 119, 3555–3562 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang, X. et al CELF4 variant and anthracycline‐related cardiomyopathy: a Children's Oncology Group genome‐wide association study. J. Clin. Oncol. 34, 863–870 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cooper, T. & Ordahl, C. A single cardiac troponin T gene generates embryonic and adult isoforms via developmentally regulated alternate splicing. J. Biol. Chem. 260, 11140–11148 (1985). [PubMed] [Google Scholar]
- 57. Anderson, P. et al Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circ. Res. 76, 681–686 (1995). [DOI] [PubMed] [Google Scholar]
- 58. Aminkeng, F. et al Recommendations for genetic testing to reduce the incidence of anthracycline‐induced cardiotoxicity. Br. J. Clin. Pharmacol. 82, 683–695 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Craig, L. , Ekert, P. , Conyers, R. & Elliott, D. Genetic determinants of anthracycline cardiotoxicity – ready for the clinic? Br. J. Clin. Pharmacol. 83, 1141–1142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Holmgren, G. , Sartipy, P. , Andersson, C. , Lindahl, A. & Synnergren, J. Expression profiling of human pluripotent stem cell‐derived cardiomyocytes exposed to doxorubicin – integration and visualization of multi omics data. Toxicol. Sci. 163, 182–195 (2018). [DOI] [PubMed] [Google Scholar]
- 61. Reyes, M. , Ma, J. , Grove, M. , Ater, J. , Morrison, A. & Hildebrandt, M. RNA sequence analysis of inducible pluripotent stem cell‐derived cardiomyocytes reveals altered expression of DNA damage and cell cycle genes in response to doxorubicin. Toxicol. Appl. Pharmacol. 356, 44–53 (2018). [DOI] [PubMed] [Google Scholar]
- 62. Knowles, D.A. et al Determining the genetic basis of anthracycline‐cardiotoxicity by molecular response QTL mapping in induced cardiomyocytes. Elife 7, pii: e33480 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]