

Abstract
Background and purpose
To predict disability and cognition in multiple sclerosis (MS) after 6 and 12 years, using early clinical and imaging measures.
Methods
A total of 115 patients with MS were selected and followed up after 2 and 6 years, with 79 patients also being followed up after 12 years. Disability was measured using the Expanded Disability Status Scale (EDSS); cognition was measured only at follow‐up using neuropsychological testing. Predictors of interest included EDSS score, baseline brain and lesion volumes and their changes over 2 years, baseline age, clinical phenotype, sex and educational level.
Results
Higher 6‐year EDSS score was predicted by early EDSS score and whole‐brain volume changes and baseline diagnosis of primary progressive MS (adjusted R 2 = 0.56). Predictors for 12‐year EDSS score included larger EDSS score changes and higher T1‐hypointense lesion volumes (adjusted R 2 = 0.38). Year 6 cognition was predicted by primary progressive MS phenotype, lower educational level, male sex and early whole‐brain atrophy (adjusted R 2 = 0.26); year 12 predictors included male sex, lower educational level and higher baseline T1‐hypointense lesion volumes (adjusted R 2 = 0.14).
Conclusions
Patients with early signs of neurodegeneration and a progressive disease onset were more prone to develop both disability progression and cognitive dysfunction. Male sex and lower educational level only affected cognitive dysfunction, which remains difficult to predict and probably needs more advanced imaging measures.
Keywords: atrophy, cognition, disability, multiple sclerosis, prediction
Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system leading to neurodegeneration and chronic disability 1. Predicting clinical progression with only inflammatory magnetic resonance imaging (MRI) markers, such as lesion load, remains difficult 2, 3. Neurodegenerative markers, such as atrophy, have stronger correlations with disability and are present early in all disease phenotypes 4, 5. Atrophy is thought to be a promising predictor of disability progression on relatively short follow‐up (FU) periods 6, 7, 8, 9, although results are less consistent on longer‐term FU and longitudinal studies remain scarce 8, 10, 11.
In addition to physical disability, cognitive impairment is also commonly present in MS 12 and cognitive decline cannot be sufficiently explained by white matter lesion measures 13. Cross‐sectional studies have shown a stronger relation between cognition and atrophy measures 14, 15, 16, 17 and longitudinal studies indicated that atrophy may be able to predict cognitive impairment 18, 19. However, as with disability studies, most studies included relatively short FU periods and rather small sample sizes 14, 15, 16, 17, 18, 19. Furthermore, it remains unclear whether important predictors of physical disability, such as early imaging changes, are also predictive of cognitive decline.
Consequently, additional longitudinal studies that can confirm the promising prognostic value of early atrophy rates could help to identify patients with an especially unfavourable prognosis for disability as well as cognition. Therefore, the aim of this study was to determine the predictive value of early changes in brain, ventricular and lesion volumes for physical disability and cognitive function, after 6 and 12 years of MS.
Methods
Participants and clinical assessments
The study was approved by the institutional ethics review board and all subjects gave written informed consent prior to participation. Patients were selected from the prospectively included Amsterdam MS early inception cohort (n = 293) 20. Patients with an MRI scan at baseline and year 2 and clinical measurements at year 6 were retrospectively selected and analysed for the present study. Moreover, patients selected for the present study also required a diagnosis of MS at the latest at 6‐year FU, using the 2005 McDonald criteria 21. Of all 293 patients in the early inception cohort, our criteria resulted in the selection of 115 patients, with a mean age of 35.3 (SD 9.1) years of whom 76 (66.1%) were female. A subset of patients (n = 79) was also clinically assessed 12 years after baseline [mean age 34.9 (SD 8.9) years, 54 (68.4%) female]. Figure 1 shows an overview of all baseline and FU measurements.
Figure 1.

Baseline and follow‐up (FU) measurements. 9‐HPT, 9‐hole peg test; 25‐FWT, timed 25‐feet walk test; BL, baseline; EDSS, Expanded Disability Status Scale. Timeline of the baseline and FU measurements that were obtained (baseline and year 2) and the outcome measures (years 6 and 12) that were used. [Colour figure can be viewed at wileyonlinelibrary.com]
Previous MRI studies in the same cohort used different selection criteria and, for example, focused on only disability as an outcome measure 10, 20 with shorter FU periods.
Disease‐modifying treatment (DMT) strategies for all patients remained at the discretion of the individual treating physician. Educational level was measured using a scale from 1 to 7, with 1 representing unfinished primary school and 7 representing a university degree or higher and subdivided into low (level 1–3), middle (4 and 5) and high (6 and 7) levels of education 22.
Physical disability was measured using the Expanded Disability Status Scale (EDSS) 23 by certified EDSS raters at baseline, year 2, year 6 and year 12. Disability progression was used for descriptive purposes and defined as an increase in EDSS score of 1.5, 1.0 or 0.5 in the case of a baseline EDSS score of 0, 1.0–5.5 or ≥6, respectively 24. In addition, disability progression was also described using the EDSS‐plus, defined as progression on EDSS score (using the aforementioned criteria) or worsening of ≥20% on the 25‐feet walk test or 9‐hole peg test at FU 25.
Cognition was assessed at 6‐ and 12‐year FU (but not at baseline or year 2), using a comprehensive set of neuropsychological tests, which included a previously described expanded Brief Repeatable Battery of Neuropsychological tests 26. Cognitive domains included executive functioning (concept shifting test), verbal memory (selective reminding test), information processing speed (symbol digit modalities test), verbal fluency (word list generation), visuospatial memory (spatial recall test), working memory (memory comparison test) and attention (Stroop colour and word test). As previously published 26, the results of the different tests at years 6 and 12 were corrected for normal effects of age, sex and education. In addition, all cognitive data were corrected for normal changes in cognition over time, which were quantified using longitudinal data of 60 matched healthy controls, measured at the time of 6‐ and 12‐year FU. The Z‐scores of the different cognitive domains were averaged to derive a summary statistic of average cognition that was used for the statistical analyses. For descriptive purposes, patients were classified as mildly cognitively impaired when two of the domains had a Z‐score <1.5 SD compared with healthy controls and as cognitively impaired when Z‐scores on at least two domains were ≥2 SD below the Z‐scores of the healthy controls, based on previous work on the same cohort 26. Otherwise, patients were classified as cognitively preserved.
Magnetic resonance imaging
All patients underwent a brain MRI scan on a 1.0‐T magnetic resonance system at baseline and year 2 (Magnetom Impact Expert, Siemens AG, Erlingen, Germany) between December 2000 and September 2007, using a standard MS scan protocol including axial two‐dimensional dual‐echo proton density (PD)/T2 spin echo images for T2‐lesion volumes (repetition time, 2700 ms; echo time, 45/90 ms; flip angle, 90) and axial two‐dimensional T1‐weighted spin echo images (repetition time, 700 ms; echo time, 15 ms) (both with 5‐mm axial slices with a 0.5‐mm gap, in‐plane resolution 1 × 1 mm2). Pseudo‐T1 images (see below) were created using the PD/T2 images and used for atrophy measurements, in order to cancel out the effects of contrast administration, as pre‐gadolinium two‐dimensional‐T1‐weighted sequences were often lacking.
Magnetic resonance imaging processing
The T2‐hyperintense and T1‐hypointense lesion volumes were identified by experienced readers (M.M.S. and V.P.) with over 10 years of experience and outlined using a local thresholding technique, as described previously 20. T1‐hypointense lesions were classified as ‘black holes’ when their intensity was equal to or lower than cortical grey matter. At baseline and year 2, the PD/T2‐weighted images were used to create pseudo‐T1 images for longitudinal atrophy analyses 27, 28. The annualized percentage brain volume change (PBVC) and annualized percentage ventricular volume change (PVVC) were calculated between baseline and year 2 using SIENA 29 and VIENA 30 (FSL 4.1, http://www.fmrib.ox.ac.uk/fsl) on the pseudo‐T1 images, after lesion filling. In short, SIENA performs halfway registration between the images from the two time points and calculates the PBVC from the mean brain edge displacement between the two scans. For PVVC, only brain/non‐brain edge points on the ventricular edges were selected and their mean edge displacement was calculated. PBVC and PVVC were divided by the time interval between the visits to obtain annualized measures.
Statistical analyses
Normality of the data was checked by visual inspection of the histograms together with Kolmogorov–Smirnov testing. T1‐ and T2‐lesion volumes were log‐transformed. Multiple linear regression models were used to predict physical disability (EDSS score) and average cognitive performance (average Z‐score) at 6‐ and 12‐year FU. A backward selection procedure with a removal P = 0.10 was used; all reported β values were standardized β coefficients. Predictors of interest were baseline age, highest level of education attained, disease phenotype, EDSS score, T1‐ and T2‐lesion volumes and sex, as well as early changes (i.e. between baseline and year 2) in EDSS score, lesion volumes and annualized brain volume changes (PBVC and PVVC). In order to reduce the number of variables, baseline prognostic variables were only selected for multiple linear regression if univariate linear regression P < 0.10. Regression analysis for disability and cognition at year 6 was also performed including only the patients with 12‐year FU (n = 79).
Baseline characteristics of patients with and without 12‐year FU were compared using an independent‐samples t‐test, chi‐squared test or Mann–Whitney U‐test as appropriate. All statistical analyses were performed in SPSS 22.0 (IBM, SPSS, Chicago, IL, USA) (level of significance was set at P = 0.05).
Results
A total of 115 patients of the Amsterdam MS cohort were included. Patient characteristics at baseline, 6 years (n = 115) and 12 years (n = 79) are summarized in Table 1.
Table 1.
Patient characteristics
| All patients with 6 years FU | All patients with 12 years FU | |
|---|---|---|
| Clinical data | ||
| No. of patients | 115 | 79 |
| Disease type BL | ||
| CIS | 36/115 (31.3%) | 27/79 (34.2%) |
| RRMS | 68/115 (59.1%) | 50/79 (63.3%) |
| PPMS | 11/115 (9.6%) | 2/79 (2.5%) |
| Sex (female) | 76/115 (66.1%) | 54/79 (68.4%) |
| Age at BL (years) | 35.3 (±9.1) | 34.9 (±8.9) |
| Disease duration BL (years) | 0.92 (0.61–1.62) | 0.84 (0.61–1.60) |
| Level of education (range 1–7) | 5 (4–6) | 5 (4–6) |
| FU length (years) | 5.9 (5.3–6.5) | 11.8 (10.7–12.8) |
| Patients on DMT during FU | 64/115 (55.7%) | 50/79 (63.3%) |
| Disability | ||
| EDSS score BL | 2.0 (1.5–3.0) | 2.0 (1.5–3.0) |
| EDSS score at year 6 | 2.5 (2.0–3.5) | 2.0 (1.5–3.0) |
| EDSS score at year 12 | – | 3.0 (2.0–4.0) |
| EDSS score difference 0–2 years | 0.0 (−0.5 to 0.5) | 0.0 (−0.5 to 1.0) |
| EDSS score progression at year 6 | 40/115 (34.8%) | 24/79 (30.4%) |
| EDSS‐plus score progression at year 6 | 54/112 (48.2%) | 33/70 (42.3%) |
| EDSS score progression at year 12 | – | 40/79 (50.6%) |
| EDSS‐plus score progression at year 12 | – | 46/70 (65.7%) |
| Cognition | ||
| SDMT at year 6 | 54 (49–62) | 55 (49–62) |
| SDMT at year 12 | – | 53 (45–62) |
| Average cognition at year 6 (Z‐score) | −0.76 (±0.94) | −0.66 (±0.73) |
| Average cognition at year 12 (Z‐score) | – | −0.84 (±0.77) |
| MCI at year 6 | 21/115 (18.3%) | 11/79 (13.9%) |
| CI at year 6 | 28/115 (24.3%) | 18/79 (22.8%) |
| MCI at year 12 | – | 14/79 (17.7%) |
| CI at year 12 | – | 23/79 (29.1%) |
| MRI measures | ||
| PBVC (%/year) | −0.54 (±0.55) | −0.53 (±0.57) |
| PVVC (%/year) | 2.58 (±3.86) | 2.72 (±4.31) |
| T1‐hypointense lesion volume BL (mL) | 0.21 (±0.32) | 0.22 (±0.34) |
| T1‐Gd+ lesion volumes BL (mL) | 0.13 (± 0.51) | 0.13 (±0.23) |
| T2‐hyperintense lesion volume BL (mL) | 2.51 (±2.74) | 2.61 (±3.06) |
| T1‐hypointense lesion volume change 0–2 years (mL) | 0.04 (±0.22) | 0.03 (±0.22) |
| T1‐Gd+ lesion volumes at year 2 (mL) | 0.02 (±0.05) | 0.07 (±0.15) |
| T2‐hyperintense lesion volume change 0–2 years (mL) | 0.18 (±0.97) | 0.19 (±1.08) |
Patient characteristics of the 115 patients followed until 6 years after baseline and the 79 patients who were also seen 12 years after BL are given. Demographic, clinical and imaging information of both BL and FU are included. Data are given as mean ± SD, median (IQR) and n (%). BL, baseline; CI, cognitively impaired; CIS, clinically isolated syndrome; DMT, disease‐modifying treatment; EDSS, Expanded Disability Status Scale; FU, follow‐up; Gd+, gadolinium enhancing; IQR, interquartile range; MCI, mildly cognitively impaired; MRI, magnetic resonance imaging; PBVC, percentage brain volume change; PPMS, primary progressive multiple sclerosis; PVVC, percentage ventricular volume change; RRMS, relapsing‐remitting multiple sclerosis; SDMT, symbol digit modalities test.
At baseline, 36 patients (31.3%) were diagnosed with a clinically isolated syndrome, 68 (59.1%) with relapsing‐remitting MS and 11 (9.6%) with primary progressive MS (PPMS) using the 2005 McDonald criteria 21. At baseline, median disease duration based on the first symptom(s) was 0.92 [interquartile range (IQR), 0.61–1.62] years and the median EDSS score was 2.0 (IQR, 1.5–3.0). All baseline lesion volumes are also shown in Table 1. Patients with PPMS had a median EDSS score of 3.5 (IQR, 2.5–3.5) at baseline.
After 2 years of FU, median EDSS score remained 2.0 (IQR, 1.5–3.0), leading to an EDSS score change between baseline and year 2 of 0.0 (IQR, −0.5 to 0.5). Patients with PPMS had a median EDSS score of 4.0 (IQR, 4.0–5.0), which was a median of 0.5 (IQR, 0–1.5) change in EDSS score compared with baseline. Lesion and atrophy measurements are shown in Table 1.
After 6 years, 96 patients (83.5%) had relapsing‐remitting MS, 8 patients (7.0%) converted to secondary progressive MS and 11 (9.6%) had PPMS. Disability was still minimal with a median EDSS score of 2.5 (IQR, 2.0–3.5). About one‐third of the patients showed EDSS score progression compared with baseline (40/115 patients, 34.8%) and about half of the patients showed EDSS‐plus score progression (54/112 patients, 48.2%). Average cognitive performance at year 6 was Z = −0.76 (SD 0.94) below controls; 49/115 patients (42.6%) were classified as either mildly cognitively impaired (18.3%) or cognitively impaired (24.3%). A total of 54 patients (47.0%) used DMT at some point during the 6 years of FU.
Of the 79 patients who were evaluated after 12 years, 68 patients (86.1%) had relapsing‐remitting MS, 9 (11.4%) had secondary progressive MS (of whom five converted after the 6‐year visit) and two (2.5%) had PPMS. The median EDSS score at year 12 was 3.0 (IQR, 2.0–4.0), with EDSS score progression in 40/79 of the patients (50.6%) and EDSS‐plus score progression in 46/70 patients (65.7%). The average cognitive Z‐score was −0.84 (SD 0.77) and 37/79 (46.8%) patients were classified as either mildly cognitively impaired (17.7%) or cognitively impaired (29.1%). A total of 42 patients (53.2%) used DMT at some point during the 12 years of FU. The subset of patients with 12‐year FU showed significantly lower EDSS scores at baseline (P = 0.014), year 2 (P = 0.008) and year 6 (P = 0.001) than those who dropped out of the study, but not on the EDSS score change between baseline and year 2 (P = 0.280). Furthermore, a significantly lower number of patients with 12 years of FU had a baseline diagnosis of PPMS (only 2/11 patients with PPMS were followed for 12 years, P < 0.001). Results of the subgroup followed over 12 years are presented separately in Table 1.
Prediction of physical disability
The univariate models for disability are shown in Table 2 and Fig. 2. Variables with P < 0.10 were included for multivariate analyses as summarized in Table 3.
Table 2.
Univariate analyses of potential predictors
| Univariate linear regression | Disability year 6 | Disability year 12 | Cognition year 6 | Cognition year 12 | ||||
|---|---|---|---|---|---|---|---|---|
| Standardized β | P‐value | Standardized β | P‐value | Standardized β | P‐value | Standardized β | P‐value | |
| Age at BL | 0.296 | 0.001 | 0.325 | 0.003 | −0.072 | 0.445 | −0.136 | 0.232 |
| Sex (male versus female) | 0.056 | 0.552 | 0.021 | 0.854 | −0.268 | 0.004 | −0.244 | 0.030 |
| Education: medium versus low | −0.311 | 0.020 | −0.222 | 0.189 | 0.503 | <0.001 | 0.295 | 0.071 |
| Education: high versus low | −0.265 | 0.045 | −0.199 | 0.238 | 0.543 | <0.001 | 0.438 | 0.008 |
| Disease duration at BL | −0.039 | 0.679 | 0.038 | 0.737 | −0.070 | 0.456 | −0.064 | 0.573 |
| RRMS at BL | −0.122 | 0.194 | −0.092 | 0.419 | 0.152 | 0.106 | 0.096 | 0.402 |
| PPMS at BL | 0.450 | <0.001 | 0.173 | 0.127 | −0.274 | 0.003 | −0.147 | 0.195 |
| EDSS score at BL | 0.491 | <0.001 | 0.364 | 0.001 | −0.262 | 0.005 | −0.109 | 0.339 |
| EDSS score difference (BL–2 years) | 0.364 | <0.001 | 0.332 | 0.003 | −0.025 | 0.794 | −0.001 | 0.990 |
| DMT during FU | 0.179 | 0.055 | 0.132 | 0.244 | −0.071 | 0.451 | −0.113 | 0.323 |
| T1‐hypointense lesions BL | 0.016 | 0.869 | 0.281 | 0.012 | −0.070 | 0.456 | −0.263 | 0.019 |
| T1‐Gd+ lesions BL | −0.006 | 0.951 | 0.146 | 0.201 | −0.075 | 0.425 | 0.042 | 0.710 |
| T2‐hyperintense lesions BL | −0.028 | 0.609 | 0.086 | 0.576 | −0.117 | 0.211 | −0.198 | 0.339 |
| T1‐hypointense lesions; early change | 0.106 | 0.260 | 0.025 | 0.829 | −0.059 | 0.531 | −0.067 | 0.558 |
| T1‐Gd+ lesions 2 years | −0.110 | 0.286 | −0.101 | 0.427 | −0.037 | 0.721 | −0.107 | 0.346 |
| T2‐hyperintense lesions; early change | 0.085 | 0.367 | −0.061 | 0.592 | 0.029 | 0.760 | 0.016 | 0.889 |
| PBVC; early change | −0.158 | 0.091 | −0.037 | 0.743 | 0.222 | 0.017 | −0.010 | 0.931 |
| PVVC; early change | 0.048 | 0.609 | 0.064 | 0.576 | −0.145 | 0.122 | −0.109 | 0.339 |
Univariate analyses of the potential variables. Variables with a P < 0.10 (bold) were selected for the prediction models shown in Table 3 (disability) and Table 4 (cognition).
BL, baseline; DMT, disease‐modifying treatment; EDSS, Expanded Disability Status Scale; FU, follow‐up; Gd+, gadolinium enhancing; PBVC, percentage brain volume change; PPMS, primary progressive multiple sclerosis; PVVC, percentage ventricular volume change; RRMS, relapsing‐remitting multiple sclerosis.
Figure 2.
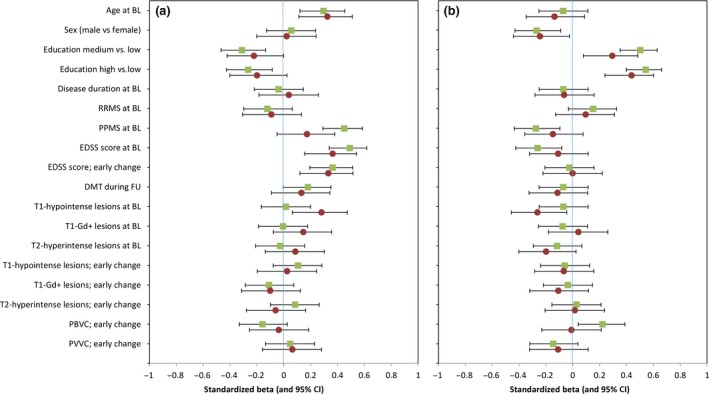
Univariate analyses of predictors for disability and cognition after 6 and 12 years of follow‐up (FU). Visualization of univariate analyses of potential predictors for disability (a) and cognition (b). Standardized β and 95% confidence interval (CI) of all variables for the 6‐year FU (squares) and 12‐year FU (circles). BL, baseline; DMT disease‐modifying treatment; EDSS, Expanded Disability Status Scale; Gd+, gadolinium enhancing; PBVC, percentage brain volume change; PPMS, primary progressive multiple sclerosis; PVVC, percentage ventricular volume change; RRMS, relapsing‐remitting multiple sclerosis. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 3.
Prediction models for disability
| Prediction model disability (EDSS score) at 6‐year FU | ||
|---|---|---|
| Adjusted R 2 = 0.559 | Standardized β | P‐value |
| EDSS score (BL–2 years) | 0.465 | <0.001 |
| PPMS at BL | 0.214 | 0.004 |
| Early whole‐brain atrophy rate (PBVC) | −0.143 | 0.026 |
| Prediction model disability (EDSS score) at 12‐year FU | ||
|---|---|---|
| Adjusted R 2 = 0.375 | Standardized β | P‐value |
| EDSS score (BL–2 years) | 0.443 | <0.001 |
| T1‐hypointense lesion volume at BL | 0.245 | 0.008 |
Prediction models for physical disability 6 and 12 years after BL. Both prediction models are corrected for baseline EDSS and the model for year 6 was also corrected for disease‐modifying treatment use during FU. Standardized β and corresponding P‐value are given per included variable. BL, baseline; EDSS, Expanded Disability Status Scale; FU, follow‐up; PBVC, percentage brain volume change per year; PPMS, primary progressive multiple sclerosis.
At year 6, the prediction model for disability explained 55.9% of the variance and included a higher change in EDSS score between baseline and year 2 (standardized β = 0.465, P < 0.001), a diagnosis of PPMS at baseline (standardized β = 0.214, P = 0.004) and early whole‐brain atrophy rate (PBVC, standardized β = −0.143, P = 0.026).
The prediction model for disability at year 12 explained 37.5% of the variability in disability by a worsening in EDSS score between baseline and year 2 (standardized β = 0.443, P < 0.001) and higher T1‐hypointense lesion volumes at baseline (standardized β = 0.245, P = 0.008). Both models were corrected for baseline EDSS score and the 6‐year model also for the use of DMT during FU.
Prediction of cognition
Prediction models for cognition included predictors selected by univariate analyses (see Table 2 and Fig. 2). Baseline EDSS score and DMT were not predictive of cognition and were therefore not included in the models. The multivariate analyses for cognition (Table 4) showed that lower cognitive functioning after 6 years was explained for 25.5% by a model containing a lower educational level (middle versus low: standardized β = 0.364, P = 0.004; high versus low: standardized β = 0.503, P < 0.001), male sex (standardized β = −0.213, P = 0.013), a diagnosis of PPMS at baseline (standardized β = −0.201, P = 0.019) and early whole‐brain atrophy rate (PBVC, standardized β = 0.181, P = 0.039).
Table 4.
Prediction models for cognitive decline
| Prediction model cognition at 6‐year FU | ||
|---|---|---|
| Adjusted R 2 = 0.255 | Standardized β | P‐value |
| High versus low level of education | 0.503 | <0.001 |
| Middle versus low level of education | 0.364 | 0.004 |
| Male sex | −0.213 | 0.013 |
| PPMS at BL | −0.201 | 0.019 |
| Early whole‐brain atrophy rate (PBVC) | 0.181 | 0.039 |
| Prediction model cognition at 12‐year FU | ||
|---|---|---|
| Adjusted R 2 = 0.141 | Standardized β | P‐value |
| Male sex | −0.256 | 0.019 |
| High versus low level of education | 0.242 | 0.026 |
| T1‐hypointense lesion volume at BL | −0.220 | 0.041 |
Prediction models for cognitive decline 6 and 12 years after BL. Standardized β and corresponding P‐value are given per included variable.
BL, baseline; FU, follow‐up; PBVC, percentage brain volume change per year; PPMS, primary progressive multiple sclerosis.
After 12 years, 14.1% of the variance in cognitive functioning was explained by a model consisting of male sex (standardized β = −0.256, P = 0.019), lower educational levels (high versus low: standardized β = 0.242, P = 0.026) and higher T1‐hypointense lesion volumes at baseline (standardized β = −0.220, P = 0.041).
When including only the 79 patients seen for the 12‐year FU, prediction models for 6‐year disability and cognition remained generally similar (adj. R 2 = 0.49 for disability, 0.20 for cognition), although the models did not include PPMS and PBVC anymore.
Discussion
In this longitudinal cohort of patients with MS, we determined early predictors for both physical disability and cognitive impairment at medium‐ and long‐term FU. The results suggest that a diagnosis of PPMS at baseline as well as faster early decreases in whole‐brain volume (PBVC) predict both worse physical disability and worse cognitive functioning 6 years after onset. However, predicting cognitive dysfunction remained more difficult with the set of measures available for this study. Interestingly, T1‐hypointense lesion volumes only became significant in the long‐term prediction model for physical disability as well as cognition. Early increases in EDSS score were predictive of physical disability only and a low educational level and male sex seemed specific for cognitive dysfunction.
Predictive value of early whole‐brain atrophy
In our study, faster whole‐brain atrophy rate (PBVC) was a predictor for both disability as well as lower cognitive functioning after 6 years, but not for year 12. This seems to be in contrast to previous data in which whole‐brain atrophy, T2‐lesion volumes at year 1 and central atrophy (measured with PVVC) predict disability progression over 10 years 10. Interestingly, correction for baseline EDSS score and the use of DMT, as was also performed in this study, excluded whole‐brain atrophy from their model. This seems to support another study on brain atrophy and disability progression in patients with a longer disease duration, which found a relation between atrophy and disability at 5 years, but not at 10 years of FU 8. A small 2‐year FU study also showed that both disability and cognitive impairment were correlated to loss of brain parenchyma 18. A possible explanation for the relative lack of predictive power for atrophy at longer periods may be the drop‐out of the more severe clinical cases after longer periods of time. This was also seen in the regression models including only the patients with a 12‐year FU in our cohort, which resulted in prediction models after 6 years no longer including patients with PPMS and PBVC as significant predictors. Furthermore, the lack of predictive power may also be caused by the need for more specific regional atrophy markers of neurodegeneration that require higher field strengths.
Predictive value of early central atrophy and lesions
Ventricular enlargement (PVVC) was not included in any of the prediction models and did not show direct correlations with cognition or disability at either time point, whereas previous work found early ventricular enlargement related to cognitive functioning 14, 15 and also disability 7, 10. Ventricular volume changes are thought to closely reflect thalamic volume changes 31, a structure known to closely relate to both cognitive and physical disability. Similar to our findings, another study on brain atrophy did not find PVVC as a predictor for disability. In that study, brain volumes were measured at baseline, 5‐year FU (81 patients) and 10‐year FU (50 patients) using a 1.5‐T magnetic resonance system 8, which could indicate the need for longitudinal imaging at higher field strengths.
Lesion volumes, especially T2‐hyperintense lesion volumes, have also been suggested as predictors in previous longitudinal studies 8, 10, showing relatively stronger correlation for T1‐hypointense lesion volumes with disability than T2‐hyperintense lesion volumes 8, although atrophy seems to show stronger correlations 7, 8, 11, 19. However, the results of our long‐term FU did show a predictive role for T1‐hypointense lesion volumes, but not PBVC, suggesting that baseline T1‐hypointense lesion volumes may be valuable for long‐term FU.
Predicting disability versus predicting cognition
Previous longitudinal studies in MS investigating both cognition and disability are scarce. In our study, worsened atrophy and T1‐lesion volumes over the first 2 years were predictive for both cognitive dysfunction and disability, suggesting neurodegeneration as a general marker of poorer prognosis. In addition, some predictors were unique to disability and others to cognition. A more severe increase in EDSS score over the first 2 years was only predictive for disability, suggesting that further study of the underlying mechanism of early increases in disability may provide important future clues. Interestingly, preventing such an early disability progression with an early start of treatment 32 may not be especially relevant for cognition, as the use of DMT in our study was not related to cognition.
In contrast, a lower level of education, which is a known marker of poorer cognitive reserve, was only predictive of cognitive impairment 33. This may indicate that the concept of cognitive reserve may indeed be rather specific for cognition, although more advanced measures of reserve, such as brain reserve, were not included in the present study. Interestingly, a previous study did indicate cognitive reserve as being protective for disability progression 34, which could be due to a different measurement of reserve. Another unique predictor of cognitive decline was male sex, which has previously been shown to be relevant for subcortical atrophy and cognition in this cohort 35. The use of DMT in our cohort was perhaps lower than expected comparing with other cohorts; however, the proportions are in line with DMT use described in the Netherlands 36. Although some predictors were unique to cognition, the explained variance of cognitive decline remained relatively low compared with that of disability. This may indicate that we need more advanced (functional) imaging measures to predict cognitive decline in particular.
The main limitation of our study lies within the low field strength, which was linked with the options available at the time of baseline. Higher field strengths have been shown to increase accuracy, which is promising for future research 37. Additionally, we focused on whole‐brain atrophy and ventricular enlargement, but spinal cord involvement is also highly relevant for disability 20, which was not included given the lack of relevance for cognition. Unfortunately, not all patients were able to return at 12 years. Finally, cognitive performance at baseline and more advanced baseline imaging would also be of great interest in future studies.
Conclusions
In this study, disability as well as cognitive decline were predicted by early signs of neurodegeneration and a progressive disease onset. Male sex and lower educational level only affected cognitive dysfunction. At both FU points, a more severe early change in EDSS score was only predictive for disability, and a lower educational level and male sex were only predictive for cognitive impairment. Where around half of the variance of disability progression could be explained, the prediction of cognitive impairment remains challenging and probably requires more advanced imaging measures. Future research with more advance imaging techniques would be interesting.
Disclosure of conflicts of interest
I.D. received speaking honoraria from Roche. A.J.C.E., V.P. and L.J.B. declare no financial or other conflicts of interest. H.V. reports grants from Novartis, MerckSerono and Teva outside the submitted work, and has a patent brain atrophy standardization method pending. M.P.W. reports personal fees from Biogen, Novartis, Sanofi‐Genzyme, IXICO, Roche, Merck‐Serono and Springer outside the submitted work. B.M.J.U. reports personal fees from Genzyme, Biogen Idec, TEVA, Merck Serono and Roche outside the submitted work. J.K. reports grants and personal fees from Biogen Idec, Novartis, Merck Serono, TEVA and Genzyme, and grants and other from Biogen Idec, Novartis, TEVA, Bayer Schering Pharma, Glaxo Smith Kline and Merck Serono outside the submitted work. J.J.G.G. is an editor of Multiple Sclerosis Journal and serves on the editorial boards of Neurology and Frontiers of Neurology, is president of the Netherlands organization for health research and innovation and has served as a consultant for Merck‐Serono, Biogen, Novartis, Genzyme and Teva Pharmaceuticals. F.B. serves as an editorial board member of Brain, European Radiology, Neurology, Multiple Sclerosis Journal and Radiology, has accepted consulting fees from Bayer‐Schering Pharma, Biogen‐IDEC, TEVA, Merck‐Serono, Novartis, Roche, Jansen Research, Genzyme‐Sanofi, IXICO Ltd, GeNeuro and Apitope Ltd and speaker fees from Biogen‐IDEC and IXICO, has received grants from AMYPAD(IMI), EuroPOND (H2020), UK MS Society, Dutch MS Society, PICTURE (IMDI‐NWO), NIHR UCLH Biomedical Research Centre (BRC), ECTRIMS‐MAGNIMS, and is supported by the NIHR UCLH biomedical research centre. M.M.S. serves on the editorial board of Frontiers of Neurology, receives research support from the Dutch MS Research Foundation (grant number 13‐820) and has received compensation for consulting services or speaker honoraria from ExceMed, Genzyme and Biogen.
Acknowledgments
This research was supported by the Dutch MS Research foundation (grant numbers 08‐650, 09‐358d, 13‐820 and 14‐358e).
[Correction added on 8 February 2019 after first publication: the affiliation of J. J. G. Geurts has been corrected.]
References
- 1. Compston A, Coles A. Multiple sclerosis. Lancet 2008; 372: 1502–1517. [DOI] [PubMed] [Google Scholar]
- 2. Brex PA, Ciccarelli O, O'Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med 2002; 346: 158–164. [DOI] [PubMed] [Google Scholar]
- 3. Barkhof F. The clinico‐radiological paradox in multiple sclerosis revisited. Curr Opin Neurol 2002; 15: 239–245. [DOI] [PubMed] [Google Scholar]
- 4. Perez‐Miralles F, Sastre‐Garriga J, Tintore M, et al Clinical impact of early brain atrophy in clinically isolated syndromes. Mult Scler 2013; 19: 1878–1886. [DOI] [PubMed] [Google Scholar]
- 5. Sastre‐Garriga J, Pareto D, Rovira A. Brain atrophy in multiple sclerosis: clinical relevance and technical aspects. Neuroimaging Clin N Am 2017; 27: 289–300. [DOI] [PubMed] [Google Scholar]
- 6. Horakova D, Dwyer MG, Havrdova E, et al Gray matter atrophy and disability progression in patients with early relapsing‐remitting multiple sclerosis: a 5‐year longitudinal study. J Neurol Sci 2009; 282: 112–119. [DOI] [PubMed] [Google Scholar]
- 7. Lukas C, Minneboo A, de Groot V, et al Early central atrophy rate predicts 5 year clinical outcome in multiple sclerosis. J Neurol Neurosurg Psychiatry 2010; 81: 1351–1356. [DOI] [PubMed] [Google Scholar]
- 8. Jacobsen C, Hagemeier J, Myhr KM, et al Brain atrophy and disability progression in multiple sclerosis patients: a 10‐year follow‐up study. J Neurol Neurosurg Psychiatry 2014; 85: 1109–1115. [DOI] [PubMed] [Google Scholar]
- 9. Rojas JI, Patrucco L, Besada C, Bengolea L, Cristiano E. Brain atrophy at onset and physical disability in multiple sclerosis. Arq Neuropsiquiatr 2012; 70: 765–768. [DOI] [PubMed] [Google Scholar]
- 10. Popescu V, Agosta F, Hulst HE, et al Brain atrophy and lesion load predict long term disability in multiple sclerosis. J Neurol Neurosurg Psychiatry 2013; 84: 1082–1091. [DOI] [PubMed] [Google Scholar]
- 11. Fisher E, Rudick RA, Simon JH, et al Eight‐year follow‐up study of brain atrophy in patients with MS. Neurology 2002; 59: 1412–1420. [DOI] [PubMed] [Google Scholar]
- 12. Chiaravalloti ND, DeLuca J. Cognitive impairment in multiple sclerosis. Lancet Neurol 2008; 7: 1139–1151. [DOI] [PubMed] [Google Scholar]
- 13. Amato MP, Zipoli V, Portaccio E. Multiple sclerosis‐related cognitive changes: a review of cross‐sectional and longitudinal studies. J Neurol Sci 2006; 245: 41–46. [DOI] [PubMed] [Google Scholar]
- 14. Christodoulou C, Krupp LB, Liang Z, et al Cognitive performance and MR markers of cerebral injury in cognitively impaired MS patients. Neurology 2003; 60: 1793–1798. [DOI] [PubMed] [Google Scholar]
- 15. Tekok‐Kilic A, Benedict RH, Weinstock‐Guttman B, et al Independent contributions of cortical gray matter atrophy and ventricle enlargement for predicting neuropsychological impairment in multiple sclerosis. NeuroImage 2007; 36: 1294–1300. [DOI] [PubMed] [Google Scholar]
- 16. Sanchez MP, Nieto A, Barroso J, Martin V, Hernandez MA. Brain atrophy as a marker of cognitive impairment in mildly disabling relapsing‐remitting multiple sclerosis. Eur J Neurol 2008; 15: 1091–1099. [DOI] [PubMed] [Google Scholar]
- 17. Mineev KK, Prakhova LN, Il'ves AG, et al Characteristics of neurological and cognitive status in patients with multiple sclerosis in relation to the location and volumes of demyelination foci and the severity of brain atrophy. Neurosci Behav Physiol 2009; 39: 35–38. [DOI] [PubMed] [Google Scholar]
- 18. Zivadinov R, Sepcic J, Nasuelli D, et al A longitudinal study of brain atrophy and cognitive disturbances in the early phase of relapsing‐remitting multiple sclerosis. J Neurol Neurosurg Psychiatry 2001; 70: 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Filippi M, Preziosa P, Copetti M, et al Gray matter damage predicts the accumulation of disability 13 years later in MS. Neurology 2013; 81: 1759–1767. [DOI] [PubMed] [Google Scholar]
- 20. Sombekke MH, Wattjes MP, Balk LJ, et al Spinal cord lesions in patients with clinically isolated syndrome: a powerful tool in diagnosis and prognosis. Neurology 2013; 80: 69–75. [DOI] [PubMed] [Google Scholar]
- 21. Polman CH, Reingold SC, Edan G, et al Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol 2005; 58: 840–846. [DOI] [PubMed] [Google Scholar]
- 22. Verhage F. Intelligentie en leeftijd: onderzoek bij Nederlanders van twaalf tot zevenenzeventig jaar. Van Gorcum: Assen, Netherlands, 1964. [Google Scholar]
- 23. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33: 1444–1452. [DOI] [PubMed] [Google Scholar]
- 24. Butzkueven H, Kappos L, Pellegrini F, et al Efficacy and safety of natalizumab in multiple sclerosis: interim observational programme results. J Neurol Neurosurg Psychiatry 2014; 85: 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cadavid D, Cohen JA, Freedman MS, et al The EDSS‐Plus, an improved endpoint for disability progression in secondary progressive multiple sclerosis. Mult Scler 2017; 23: 94–105. [DOI] [PubMed] [Google Scholar]
- 26. Eijlers AJ, Meijer KA, Wassenaar TM, et al Increased default‐mode network centrality in cognitively impaired multiple sclerosis patients. Neurology 2017; 88: 952–960. [DOI] [PubMed] [Google Scholar]
- 27. Hickman SI, Barker GJ, Molyneux PD, Miller DH. Technical note: the comparison of hypointense lesions from ‘pseudo‐T1’ and T1‐weighted images in secondary progressive multiple sclerosis. Mult Scler 2002; 8: 433–435. [DOI] [PubMed] [Google Scholar]
- 28. Neacsu V, Jasperse B, Korteweg T, et al Agreement between different input image types in brain atrophy measurement in multiple sclerosis using SIENAX and SIENA. J Magn Reson Imaging 2008; 28: 559–565. [DOI] [PubMed] [Google Scholar]
- 29. Smith SM, Zhang Y, Jenkinson M, et al Accurate, robust, and automated longitudinal and cross‐sectional brain change analysis. NeuroImage 2002; 17: 479–489. [DOI] [PubMed] [Google Scholar]
- 30. Vrenken H, Vos EK, van der Flier WM, et al Validation of the automated method VIENA: an accurate, precise, and robust measure of ventricular enlargement. Hum Brain Mapp 2014; 35: 1101–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benedict RH, Weinstock‐Guttman B, Fishman I, Sharma J, Tjoa CW, Bakshi R. Prediction of neuropsychological impairment in multiple sclerosis: comparison of conventional magnetic resonance imaging measures of atrophy and lesion burden. Arch Neurol 2004; 61: 226–230. [DOI] [PubMed] [Google Scholar]
- 32. Tintore M, Rovira A, Rio J, et al Defining high, medium and low impact prognostic factors for developing multiple sclerosis. Brain 2015; 138(Pt 7): 1863–1874. [DOI] [PubMed] [Google Scholar]
- 33. Rocca MA, Riccitelli GC, Meani A, et al. Cognitive reserve, cognition, and regional brain damage in MS: a 2‐year longitudinal study. Mult Scler 2019; 25: 372–381. [DOI] [PubMed] [Google Scholar]
- 34. D'Hooghe MB, Haentjens P, Van Remoortel A, De Keyser J, Nagels G. Self‐reported levels of education and disability progression in multiple sclerosis. Acta Neurol Scand 2016; 134: 414–419. [DOI] [PubMed] [Google Scholar]
- 35. Schoonheim MM, Popescu V, Rueda Lopes FC, et al Subcortical atrophy and cognition: sex effects in multiple sclerosis. Neurology 2012; 79: 1754–1761. [DOI] [PubMed] [Google Scholar]
- 36. Visser LH, van der Zande A. Reasons patients give to use or not to use immunomodulating agents for multiple sclerosis. Eur J Neurol 2011; 18: 1343–1349. [DOI] [PubMed] [Google Scholar]
- 37. Lysandropoulos AP, Absil J, Metens T, et al Quantifying brain volumes for Multiple Sclerosis patients follow‐up in clinical practice – comparison of 1.5 and 3 Tesla magnetic resonance imaging. Brain Behav 2016; 6: e00422. [DOI] [PMC free article] [PubMed] [Google Scholar]