

Abstract
Pediatric drug development is a challenging process due to the rarity of the population, the need to meet regulatory requirements across the globe, the associated uncertainty in extrapolating data from adults, the paucity of validated biomarkers, and the lack of systematic testing of drugs in pediatric patients. In oncology, pediatric drug development has additional challenges that have historically delayed availability of safe and effective medicines for children. In particular, the traditional approach to pediatric oncology drug development involves conducting phase 1 studies in children once the drug has been characterized and in some cases approved for use in adults. The objective of this article is to describe clinical pharmacology factors that influence pediatric oncology trial design and execution and to highlight efficient approaches for designing and expediting oncology drug development in children. The topics highlighted in this article include (1) study design considerations, (2) updated dosing approaches, (3) ways to overcome the significant biopharmaceutical challenges unique to the oncology pediatric population, and (4) use of data analysis strategies for extrapolating data from adults, with case studies. Finally, suggestions for ways to use clinical pharmacology approaches to accelerate pediatric oncology drug development are provided.
Keywords: clinical pharmacology, drug development, modeling and simulation, oncology, pediatric, PK/PD
Although pharmaceutical companies strive to be first‐in‐class for adult oncology indications, they almost never do so in pediatrics due to the rarity of these cancers and the difficulty in enrolling meaningful numbers of pediatric patients in the clinical trials. Worldwide, the number of drugs that have been studied in children and young adults has grown due to regulatory legislation increasing the transparency and accountability of pediatric drug development.1 There are available mechanisms for seeking formal or informal advice from the regulatory agencies such as at the pre–investigational new drug stage through discussions with the US Food and Drug Administration's (FDA) Office of Clinical Pharmacology, Office of New Drugs for pediatrics, pediatric Oncology Drug Advisory committee, international collaboration initiative by the Office of Pediatric Therapeutics, and the European Medicine Agency's (EMA) Paediatric Committee. However, the pathways by which regulatory approval of pediatric studies is sought differ with respect to scope and timing. This lack of alignment may hamper and/or delay global approaches to pediatric drug development. Since 2002, results of pediatric studies have been added to the labels of 34 cancer drugs,2, 3 of which 15 have approved pediatric indications (Table 1). Despite this progress, there are still more opportunities to increase the number of drugs available to children and to speed their availability. A recent review of molecularly targeted oncology drugs suggested that studies in pediatrics based on molecular mechanism of action, rather than indication, may accelerate drug development in pediatrics.4 For prescribers and patients, information on the use of drugs in children may not be available until several years after the initial approval in adults,5 leading to substantial off‐label drug use in the pediatric population. New strategies for designing and conducting clinical trials have emerged to overcome challenges associated with conducting studies in children. Perhaps one of the greatest challenges for drug developers is the lack of empirical data in the target population that can be used to inform the initial dose, particularly for oncology drugs. Problems with dose selection and trial design in pediatric oncology drug development, where new drugs are often added to existing treatment regimens, complicates data interpretation6 and can then lead to high rates of clinical trial failure,5 delaying patients’ access to potentially effective treatments.
Table 1.
Summary of Oncology Drugs That Have Received US FDA Approval for Use in Pediatrics Since 2002 and Comparison of Dosing Regimens Between Pediatric and Adult Patients for Specific Indications
| Trade Name (Generic Name) | Pediatric Labeling Datea | Pediatric Indication(s) | Pediatric Dosing Regimen and Supporting Evidence | Adult Indication(s) | Adult Dosing Regimen | S/H | Was Efficacy in Children Similar to Adults? (Per Approved Label) |
|---|---|---|---|---|---|---|---|
| Arranon (nelarabine) | 10/2005 | T‐cell ALL; T‐cell lymphoblastic lymphoma (age range studied: 2.5‐21.7 years) | 650 mg/m² IV over 1 hour daily for 5 consecutive days repeated every 21 days; based on 1 clinical trial in pediatric patients | T‐cell ALL and T‐cell lymphoblastic lymphoma | 1500 mg/m² IV over 2 hours on days 1, 3, and 5 repeated every 21 days | H | Yes; based on complete response data |
| Afinitor (everolimus) | 10/2010 and 8/2012 | Subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis in patients ≥3 years of age | 4.5 mg/m2 orally QD; adjust dose to attain trough concentrations of 5‐15 ng/mL; based on 1 clinical trial in pediatric | Advanced hormone receptor‐positive, HER2‐negative breast cancer; progressive neuroendocrine tumors; advanced renal cell carcinoma; renal angiomyolipoma and tuberous sclerosis complex; SEGA | 10 mg orally QD | S | Yes; for SEGA based on 1 trial with pediatrics and young adults patients |
| Bavencio (avelumab) | 3/2017 | Metastatic MCC in patients aged ≥12 years | 10 mg/kg IV over 60 minutes every 2 weeks; based on several clinical trials in adult patients and population pharmacokinetics data to support use in pediatric patients aged ≥12 years | Metastatic MCC | Same as pediatric | S | Yes; based on full extrapolation from adults using population pharmacokinetics data and assuming the course of MCC is sufficiently similar in adults and pediatric patients |
| Blincyto (blinatumomab) | 8/2016 | B‐cell precursor ALL in first or second complete remission with MRD ≥0.1%; R/R B‐cell precursor ALL (age range studied: <1‐17 years) |
|
B‐cell precursor ALL in first or second complete remission with MRD ≥0.1% or R/R B‐cell precursor ALL | Based on weight; same as pediatric | H | Yes; based on partial extrapolation from adults with MRD‐positive B‐cell precursor ALL |
| Busulfex (busulfan) | 1/2003 | Part of a conditioning regimen in hematopoietic stem cell transplant (age range studied: 5 months to 16 years) |
|
Part of a conditioning regimen prior to allogeneic hematopoietic progenitor cell transplant for CML | 0.8 mg/kg of ideal body weight or actual body weight as a 2‐hour IV infusion every 6 hours for 4 consecutive days for a total of 16 doses | H | Yes; based on full extrapolation from adult CML |
| Clolar (clofarabine) | 12/2004 | R/R ALL in patients aged 1‐21 years | 52 mg/m2 IV over 2 hours daily for 5 consecutive days of a 28‐day cycle; based on one clinical trial in R/R ALL in pediatrics and young adults | NA | NA | H | Yes; based on data in pediatrics and young adults in R/R ALL |
| Erwinaze (asparaginase Erwinia chryanthemi) | 11/2011 | ALL in patients with hypersensitivity to E coli‐derived asparaginase (age range studied: 2–18, 1‐17, and 0‐76 years) | 25,000 IU/m2 IM or IV 3 times a week; based on 2 clinical trials in pediatric patients | NA | NA | H | Efficacy not established in adults |
| Gleevec (imatinib mesylate) | 9/2006 | Newly diagnosed Ph+ CML in chronic phase; Ph+ acute ALL (age range studied: ≥1 year) | 340 mg/m2/day orally QD (not to exceed 600 mg) for Ph+ CML or Ph+ ALL or as a split dose in the morning and evening for Ph+ CML; based on several clinical trials in pediatric patients | Ph+ CML; Ph+ ALL; myelodysplastic/myeloproliferative diseases; aggressive systemic mastocytosis; hypereosinophilic syndrome and/or chronic eosinophilic leukemia; dermatofibrosarcoma protuberans; malignant gastrointestinal stromal tumors | 400‐800 mg orally QD depending upon the indication | H | Yes; based on complete cytogenic response rate for Ph+ CML in chronic phase. Ph+ acute ALL used a different end point relative to adults |
| Keytruda (pembrolizumab) | 11/2017 | Refractory cHL; refractory PMBCL; unresectable or metastatic MSI‐H or mismatch repair deficient solid tumors; colorectal cancer (age range studied: 2‐18 years) | 2 mg/kg (up to 200 mg) IV every 3 weeks; based on limited clinical studies in pediatric patients and extrapolation from several studies in adults | Melanoma; NSCLC; HNSCC; classical Hodgkin lymphoma, PMBCL; urothelial carcinoma; microsatellite instability‐high cancer; gastric cancer; cervical cancer | 200 mg IV over 30 minutes every 3 weeks | S/H | Yes; based on full extrapolation from adults data in cHL, PMBCL and MSI‐H |
| Kymriah (tisagenlecleucel) | 8/2017 | B‐cell precursor ALL refractory or in second or later relapse in patients aged ≤25 years |
|
B‐cell ALL (young adults); R/R DLBCL after 2+ lines of systemic therapy | 0.6 to 6.0 × 108 CAR‐positive viable T cells administered IV (R/R DLBCL) | H | Yes; based on CR rate in r/r B‐cell ALL |
| Mylotarg (gemtuzumab ozogamicin) | 9/2017 | R/R CD33+ AML in patients aged ≥2 years | 3 mg/m2 (up to one 4.5 mg vial) IV on days 1, 4, and 7; based on 1 clinical trial in pediatric r/r AML patients | Newly diagnosed or R/R CD33+ AML | 3 mg/m2 (up to one 4.5 mg vial) IV on days 1, 4, and 7 in combination with daunorubicin and cytarabine; 6 mg/m2 IV on day 1 and 3 mg/m2 on day 8 as a single agent | H | Yes; based on CR rates in r/r AML pediatric and young adults |
| Sprycel (dasatinib) | 11/2017 | Ph+ CML in chronic phase (age range studied: ≥1 year) | Starting dose of 40‐100 mg orally QD based on body weight; tablet dosing not recommended in patients weighing <10 kg; based on 2 clinical trials in pediatric patients with chronic‐phase CML | Newly diagnosed Ph+ CML in chronic phase; chronic, accelerated, or myeloid or lymphoid blast phase Ph+ CML resistant or intolerant to prior therapy including imatinib; Ph+ chromosome‐positive ALL with resistance or intolerance to priory therapy | Starting dose of 100‐140 mg orally QD | H | Higher efficacy in pediatrics based on comparison of complete cytogenetic response rates within 12 months |
| Tasigna (nilotinib) | 3/2018 | Newly diagnosed Ph+ CML in chronic phase or newly diagnosed Ph+ CML in chronic phase resistant or intolerant to prior TKI therapy in patients aged ≥1 year | 230 mg/m2 orally BID, rounded to the nearest 50‐mg dose (to a maximum single dose of 400 mg); based on two clinical trials in pediatric patients | Newly diagnosed Ph+ CML in chronic phase; chronic phase and accelerated phase Ph+ CML resistant/intolerant to prior therapy including imatinib | 300‐400 mg orally BID | H | Yes; for newly diagnosed Ph+ CML |
| Unituxin (dinutuximab) | 3/2015 | High‐risk neuroblastoma (in combination with with GM‐CSF, IL‐2, and 13‐cis‐retinoic acid) (age range studied: 11 months to 15 years) | 17.5 mg/m2/day IV over 10‐20 hours for 4 consecutive days for up to 5 cycles; based on 1 clinical trial in pediatric patients | NA | NA | S | Efficacy not established in adults; neuroblastoma is a pediatric‐specific cancer |
| Xgeva (denosumab) | 6/2013 | Giant cell tumor of bone in skeletally mature adolescents (age range studied: 13‐17 years) | 120 mg SC every 4 weeks with additional 120 mg doses on days 8 and 15 of the first month of therapy; based on one clinical trial in adolescent patients. | Prevention of skeletal‐related events in patients with MM and patients with bone metastases from solid tumors; giant cell tumor of bone | 120 mg SC every 4 weeks; 120 mg SC every 4 weeks with additional 120 mg doses on days 8 and 15 of the first month of therapy for patients with giant cell tumor of bone | S | Yes; based on efficacy in skeletally mature adolescents and adults |
ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; BID, twice daily; CAR, chimeric antigen receptor; cHL, classical Hodgkin lymphoma; CML, chronic myelogenous leukemia; DLBCL, diffuse large B‐cell lymphoma; GM‐CSF, granulocyte‐macrophage colony stimulating factor; HER2, human epidermal growth factor receptor 2; HNSCC; head and neck squamous cell carcinoma; IL‐2, interleukin‐2; IM, intramuscular; IV, intravenous; MCC, Merkel cell carcinoma; MM, multiple myeloma; MRD, minimal residual disease; MSI‐H, microsatellite instability‐high; NA, not applicable; NSCLC, non–small cell lung cancer; Ph +, Philadelphia chromosome positive; PMBCL, primary mediastinal B‐cell lymphoma; QD, once daily; R/R, relapsed or refractory; SC, subcutaneous; S/H, solid tumor or hematological malignancy; TKI, tyrosine kinase inhibitor.
Based on US FDA approvals obtained since 2002 (adapted from FDA2,3). The indications and dosing regimens have been summarized for brevity; consult approved labeling for complete descriptions of indications and dosing regimens.
In a concept paper issued in 2017, the EMA recognized that a revision of the guideline on the role of pharmacokinetics (PK) in pediatric drug development is needed to address recent advances in the field and to take into account new knowledge that has been gained as a result of the pediatric regulation coming into force.7 The FDA and the EMA have also issued guidance documents supporting the use of quantitative clinical pharmacology modeling and simulation analyses in pediatric drug development.8, 9, 10 These documents plus several subsequent concept papers and addenda1, 11, 12 outline considerations for the use of quantitative approaches in pediatric drug development, which have aided drug developers in applying these approaches. The aim of the current work is to summarize the role of clinical pharmacology approaches in pediatric drug development with a specific focus on the challenges encountered in pediatric oncology drug development (Figure 1). The examples described herein are intended to highlight practical considerations for implementing the clinical pharmacology approaches described in the regulatory guidances.
Figure 1.
Role of clinical pharmacology in pediatric oncology drug development. Strategically designed PK/PD and biomarker sampling in pediatric oncology trials enables collection of the right data, which feed into advanced and prespecified modeling and simulation approaches, thus enabling rational dose selection for modern molecularly targeted cancer therapies in children. Extrapolation of relative bioavailability information to inform dosing using physiologically based PK modeling enables acceleration of pediatric oncology drug development when studies in adults are limited or absent. Clinical pharmacology tools represent an essential piece of the challenging pediatric oncology drug development puzzle. PD, pharmacodynamics; PK, pharmacokinetics.
Clinical Pharmacology Considerations
Study Plan
Overall Timeline
Pediatric studies lag behind adult studies primarily because of the rarity in the population. Assuming that a compound is expected to be effective in a common cancer and a rare cancer, sponsors would typically choose to conduct clinical trials for the common cancer because of enrollment efficiencies as well as economic advantages. In Europe, the regulatory requirement is to have a pediatric investigational program in place at the end of the phase 1 trial. In the United States, the requirement for a pediatric study plan is at the end of phase 2. Even more problematic is the fact that adults, adolescents, and younger children often do not have the same cancer disease types. Of the 10 most common cancer types, only non‐Hodgkin lymphoma occurs in all 3 age groups, and even then, the prevalent histologic subtypes in children differ greatly from those in adults.13 Because all pediatric solid tumors and lymphoid malignances are rare, dedicated development of drugs for a pediatric indication are limited, with the exception of drugs for neuroblastoma.
Concerns about lack of long‐term safety data before enrolling children in clinical trials early are warranted. Preclinical toxicology studies are not able to address these concerns. However, utilizing the argument that children should not be treated until the long‐term effects are known would delay pediatric development and access to therapies in a timely fashion. In a recent analysis of 25 molecularly targeted agents approved by the FDA or EMA for adult cancers and evaluated in pediatric cancers, the toxicities in children associated with 24 drugs were usually similar as those observed in adult patients, with the main PK parameters being comparable as well, except for drugs with a narrow therapeutic index.14, 15 Another review of nononcology drugs for pediatrics concluded that the observed toxicities may not be predicted in children based exclusively on adults.16 In the context of unknown long‐term side effects, the patient and the treating physician must make a decision based on potential benefits associated with response, compared to unknown or suspected long‐term consequences. For these reasons, accelerating drug development in children could be achieved by allowing adolescents to enroll into adult clinical trials and first‐in‐human phase 1 studies.17 Subsequently, children can be enrolled after the initial dose escalation phase in adults is completed, once a maximum tolerated dose or recommended phase 2 dose has been identified, and the adult equivalent doses for children are estimated.
Study Design
From a clinical pharmacology point of view, characterization of PK and the PK/pharmacodynamic (PK/PD) relationship to determine the optimal dose are primary objectives. The PK and PK/PD relationships are typically characterized in early studies and then confirmed in late‐stage studies.
Oncology phase 1 trials typically include a dose‐escalation phase and dose‐expansion phase. The objective of the dose‐escalation phase is to characterize the PK and safety in adult subjects. Studies in pediatric subjects are typically conducted after a therapeutic dose has been determined. If the disease progression and PK/PD relationship can be expected to be similar between pediatric subjects and adults, then an initial cohort of subjects for PK characterization to select a dose that provides similar exposure to the adult therapeutic dose can be followed by a larger expansion cohort, without evaluation of multiple doses. This is especially true for targeted therapies that do not have a narrow therapeutic index and tend to have better safety profiles compared to cytotoxic drugs. An example of this approach is the proposed design for a nivolumab pediatric study in which the target adult dose of 3 mg/kg is the only dose that is being proposed with an option to go to a lower dose if needed.18 For combination with ipilumimab, a single dose of nivolumab with 2 doses of ipilumimab is being proposed.18 The venetoclax pediatric study is designed similarly, with the initial pediatric subjects receiving an age‐ and weight‐adjusted dose to match the adult plasma exposure at the target dose of 800 mg daily for adult patients with acute myeloid leukemia and multiple myeloma. The lack of serious toxicities related to venetoclax and the relatively wide therapeutic window allowed for the use of this starting dose strategy in the dose determination portion of the pediatric study.19
Another challenge in pediatric phase 1 trials in which PK characterization is the primary objective is the inclusion of a wide variety of tumor types and age ranges (neonates to <18 years), due to the rarity of pediatric cancers in general, making up less than 1% of all cancers diagnosed each year. Because these trials typically enroll a small number of subjects for each dose group (eg, 3‐6) due to typical statistical designs, characterizing the PK across the age range and tumor spectrum is difficult. The above‐mentioned approach of limiting the initial or dose‐escalation cohort to 1 or 2 doses allows enrollment of sufficient numbers of subjects across age ranges to provide a better understanding of PK comparability to the adult population.
If the assumptions of disease similarity cannot be reasonably made, which is often the case in pediatric cancers in which the disease is not attributed to factors that play a role in disease progression or initiation in adults (ie, environmental or lifestyle factors), a more extensive program for PK characterization and dose finding may be required prior to conducting the confirmatory safety and efficacy studies. While typical study designs like the 3+320 or rolling 621 are commonly used for dose escalation, Bayesian designs like the continual reassessment method21, 22 or adaptive logistic regression23 design may be preferable and should be considered in pediatric studies. In the case where the toxicity profile of the drug has been characterized in adult subjects, the continual reassessment method design might be more appropriate than the 3+3 design in pediatric studies. An extensive review of pediatric dose‐finding studies showed that for approximately 75% of the molecularly targeted agents studied, the pediatric recommended phase 2 dose was very similar (90%‐130% of the adult recommended phase 2 dose) to the FDA‐approved dose in adults.15 For the dose‐expansion phase, an adaptive and Bayesian design can be employed to ensure that an unnecessarily large number of subjects are not exposed to a drug that does not show therapeutic benefit. The Bayesian optimal interval design is one such method that is being proposed in the dose‐expansion phase of venetoclax pediatric trials.19, 24
Another design for including younger children in first‐in‐human phase 1 oncology trials is to begin with dose escalation in adults. If a maximum tolerated dose has not been reached, then it would be feasible to open an expansion cohort of children to be dosed at the same recommended phase 2 dose as adults. If a maximum tolerated dose was reached, then dosing in children could begin at approximately 80% of the adult dose, or at the next lower tested dose level, followed by dose escalation if tolerated and assuming the administered dose will provide exposure equivalent to target exposure in adult patients. Starting at 80% increases the likelihood that the patient will benefit, as it is close to the adult dose, but it does presume that children will have a similar threshold for toxicity as adults.25 A dose lower than 80% or dose similar to adults should also be considered based on the available exposure‐response data and clinical profile of the drug. The ability to eliminate lower dose levels also minimizes the number of children required for the study. Exceptions to this paradigm may include drugs with a narrow therapeutic index, drugs with suspected differences in the volume of distribution between adults and children, and drugs for infants (ages 28 days to 2 years), where a more traditional escalation schedule with an emphasis on safety and PK assessments should be considered. These recommendations are similar to those endorsed by the Innovative Therapies for Children with Cancer Consortium in the European Union.14
Age Groups
Many physiologic characteristics are shared between older children and adults; for example, children 12 years of age and older have the same percentages of total body water, extracellular fluid, and intracellular fluid and the same creatinine clearance as adults.26 This suggests that the volume of distribution of drugs will be similar for children 12 years of age and older compared with adults. The drug‐metabolizing isoenzymes in the liver and intestine are immature at birth and mature over time, ranging from 2 years for cytochrome P450 (CYP) 3A to 12 years for CYP2D627 to reach full maturation. Therefore, by the age of 12, all isoenzymes have reached the adult levels, leading to similar drug clearance. Thus, from a PK perspective, no difference would be expected between adolescents 12 to 17 years of age and adults. A cross‐functional analysis by multiple departments at AbbVie found that there were no absolute contraindications from a scientific or medical perspective to enrolling adolescents in oncology adult clinical trials, for the same indication. AbbVie Oncology has thus implemented a new strategy whereby oncology protocols, including first‐in‐human phase 1 adult trials that may be relevant to the adolescent population, will have age inclusion criteria of ≥12 years of age. This strategy has also been endorsed by the American Society of Clinical Oncology/Friends of Cancer Research,28 the Innovative Therapies for Children with Cancer,14 ACCELERATE,29 and more recently by the FDA.17
The paradigm for younger children is different. Additional concerns for this age group include developmental factors that lead to different volumes of distribution, organ maturation, drug‐metabolizing enzyme and transporter ontogeny for small molecules, maturity of catabolizing enzymes and receptor‐mediated endocytosis for large molecules, inability to swallow pills and need for a suitable age‐appropriate formulation, and the desire to avoid exposure to subtherapeutic doses. Study design considerations that were highlighted in the previous section are suggested to account for some of these factors.
Pharmacokinetic/Pharmacodynamic Sample Acquisition
There are presently no clear global guidelines on the limits of blood volume and frequency of blood draws for children of various ages. A typical intensive blood‐sampling schema to establish a PK/PD profile in adults may not be achievable in children, due to limits on maximum of blood volume drawn in a given time period (and equally the lack of universal guidelines for blood collection), ability to draw blood samples during hospitalization vs a clinical visit, and compliance (especially for very young children or infants) where multiple needle pricks are applied per visit to collect the required multiple samples. In pediatric oncology patients, who may be suffering from hematologic malignancies or toxicities due to chemotherapy, the bone marrow dysfunction can inhibit erythropoiesis and shorten red cell survival, thereby reducing the body's ability to replenish the blood taken through sampling. These disease factors impose limitations on the amount and number of blood samples to be taken from pediatric oncology patients.30 Therefore, it is recommended to minimize the number of blood draws for PK/PD sampling, without compromising the quality of the collected data, in order to avoid additional burden on pediatric cancer patients.
A useful survey by Altamimi et al31 found that across age groups there was no difference in the collected volume per sample or per child, while a significant difference was found in the frequency of sampling between sparse PK sampling (also known as population PK) and traditional intensive PK sampling studies in children. This suggests that clinical pharmacologists should emphasize the use of population‐based approaches to optimize the blood collection time‐points to avoid the limitations described above. It can be further argued that it is an ethical responsibility for sponsors and in particular clinical pharmacologists to use modern population PK approaches that optimize the PK/PD sampling schema by minimizing the number of samples needed while maintaining quality of the collected PK/PD information, with the objective of minimizing the patients’ burden in this special population. As shown by Wade et al,32 population PK modeling can allow sparse PK sampling in the pediatric population. Suyagh et al33 employed dried blood spotting, a microvolume sampling technique to analyze PK of metronidazole in neonates. Collectively, these clinical pharmacology approaches should be utilized to minimize collection of frequent samples while ensuring optimal design for collecting necessary information and data in the pediatric oncology population.
Dosing
Starting Dose Projections
Historically, the starting dose for pediatric oncology phase 1 studies was generally based on the adult dose adjusted for body size, and mostly based on body surface area.34 This approach was implemented during the era of cytotoxic drug development, where the aim was to minimize toxicity among pediatric patients by correcting for body surface area. Although this approach is still used in clinical practice (ie, chemotherapy dosing) and may still be appropriate for large‐molecule drugs (ie, monoclonal antibody therapeutics), it is almost abandoned for newer targeted oral therapies due to the lack of scientific evidence and justification for body size–adjusted dosing.35 Other scaling and dose projection approaches are now being adopted for oral targeted therapies based on an understanding of the drug PK and PD properties in adults. These approaches include allometric scaling of adult PK parameters (ie, clearance and volume of distribution) based on body weight in children, and accounting for developmental factors such as organ and enzyme maturation where ontogeny functions are added to the dose calculation algorithms.19, 24 Although general PK principles apply to both small‐molecule and large‐molecule biologics, the underlying mechanisms that affect absorption, distribution, metabolism, and excretion are quite different between these 2 modalities. Therefore, developmental changes in pediatrics (ie, total body water, catabolic enzymes, receptor‐mediated endocytosis, and fat content) must also be accounted for when large‐molecule doses are evaluated for pediatric patients, especially when simple body size adjustment relative to adults may not offer optimal dosing in children.
From a practical point of view, fixed flat dosing is recommended for oral, targeted drugs to preclude the need for body size normalization and dosing errors, especially in the infant group where accurate determination of body surface area may not be possible. For large molecules, an attempt to develop flat fixed dosing per age or body size group would be advantageous if warranted, to avoid dosing errors in small patients.
The disease similarity or difference between adult and pediatric patients dictates the ability for extrapolation of efficacy and hence the selection of an efficacious starting dose. For pediatric oncology studies, it is not feasible to study a range of doses where a dose may be subtherapeutic due to ethical considerations. This presents an opportunity for using modeling and simulation to select a starting pediatric dose, for example, based on an understanding of exposure‐response relationships in adults and matching target exposures in children after accounting for factors that influence the PK, assuming the relationship in adults and children is sufficiently similar. For example, in case of venetoclax, the initial target dose was selected to be an adult exposure equivalent of 800 mg daily. The 800 mg daily adult dose was selected based on safety and efficacy data of venetoclax available in adults with various hematologic malignancies. The pediatric target dose was weight or age adjusted, taking into consideration the maturation of CYP3A4, which metabolizes venetoclax.19 Another commonly used approach for pediatric starting dose determination is physiologically based pharmacokinetics (PBPK) modeling and simulation. Rioux and Waters36 have discussed several examples where PBPK modeling is used to predict exposures in pediatric subjects. The FDA guidance for General Clinical Pharmacology Considerations for Pediatric Studies recommends several approaches for conducting pediatric clinical studies and extrapolation algorithms.8 Because pediatric oncology indications are mostly unique and dissimilar to the disease in adults (ie, disease progression is not similar to adults), full or partial extrapolation approaches based on PK and PK/PD are generally not possible and the pediatric study objectives would focus on providing evidence of effectiveness and safety and to characterize the PK and exposure‐response relationships in pediatric oncology patients to enable optimal dosing. Understanding of tumor biology and markers of disease in pediatric patients along with bridging of knowledge from trials in adult patients may offer more opportunity to inform pediatric clinical trials.37 Despite these challenges, an analysis of the labels of 34 cancer drugs in Table 1 where the pediatric cancer was evaluated in adults (except for conditions that are exclusively in pediatrics only) revealed that after correcting for body size (mg/kg or mg/m2), for approximately 50% of these drugs the pediatric and adult doses were the same, 13% were within 70% to 130% of the adult dose, 20% had a higher adult dose (≥50%), and 20% did not have an adult dose (due to condition existing only in pediatrics, such as neuroblastoma) for comparison (Figure 2). Overall, the majority of cancer drugs had similar body size–adjusted pediatric and adult doses on their labels.
Figure 2.
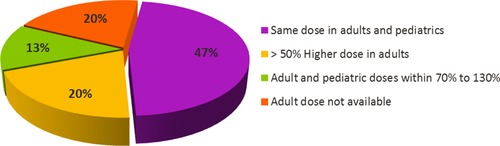
Comparison of adult and pediatric doses for 15 oncology drugs that obtained approval of a pediatric indication since 2002.
Biopharmaceutical Considerations
Pediatric Formulations
When the pediatric requirements went into effect, not enough attention and effort was devoted to developing age‐appropriate formulations. In many instances, extemporaneous formulations were prepared at home by grinding the adult dosage form and suspending it in an appropriate liquid vehicle. This created dosing and administration problems as well as stability issues. Since then, most regulatory authorities require age‐appropriate formulations that do not restrict dosing flexibility or present palatability or dosing issues and that would be stable over the duration of treatment.38 As a consequence, most drug companies embarked on extensive pediatric formulation development efforts that required the conduct of in vivo studies, usually in healthy adult volunteers. It was assumed that if the formulations were found to be bioequivalent in adults, then they were assumed to be bioequivalent in the target pediatric population. Close attention should be paid to the choice of excipients used in the pediatric formulations, particularly liquid formulations. For example, it is well known that sorbitol and most other sugar alcohols can have a profound effect on absorption and bioavailability.39 Sorbitol and alcoholic sugars can increase gastrointestinal fluid volume, leading to reduced small intestinal transit time. Consequently, the decreased bioavailability could result in decreased efficacy in certain pediatric age groups, necessitating an increase in dose to match the adult doses, as was the case with the lamuvidine oral solution dosage form, where the dose was increased by 25%.40 Moreover, development of formulations for oncology indications can present unique challenges in that in many instances the drugs cannot be tested in healthy volunteers due to toxicity issues. Despite these issues, the assessment of relative bioavailability and palatability of the pediatric formulations is typically performed in adults to have a general idea on the exposure achieved with these formulations and, if needed, adjust the dose to be given to the pediatric population.
Effect of Food on Bioavailability in Children
Lipophilic drugs with poor bioavailability formulated in such a way that they are always given with food pose a particular challenge. Because the adult diet could be somewhat different and the fact that pediatric oncology patients may not eat full meals or might be on a specific diet due to their illness, it is not possible to achieve the full exposure levels that are achieved with adults in the fed state. For all the above considerations, formulations that need to always be given with food should be avoided. However, it might be possible to overcome this challenge by formulating the drug as sprinkles or mini‐tablets that can be dispersed in a vehicle such as apple sauce, yogurt, or a vehicle that is suitable for children. The bioequivalence or at least the establishment that the exposure levels obtained from such a mode of administration are sufficiently similar to those achieved by the formulation used in the clinical trials is a prerequisite to the acceptability of such an approach.
Extrapolation of Drug‐Drug Interactions From Adults
Because it may be unethical and infeasible to conduct drug‐drug interaction (DDI) trials in pediatrics in general and in oncology patients in particular, drug interaction results in adults are typically extrapolated to pediatrics based on the dosing considerations described above (ie, intrinsic factors that influence drug clearance). However, due to known physiologic and developmental differences between adults and children (ie, drug‐metabolizing enzyme ontogeny), DDIs are often extrapolated using modeling and simulation approaches such as PBPK, when the ontogeny of drug elimination pathways is well understood such as in the case of CYP3A‐mediated metabolism. Not only does this provide a more scientifically robust approach to provide DDI recommendations and dose adjustment in children, but it also provides a platform to simulate scenarios that are otherwise impossible to test clinically and ethically. Furthermore, some chemotherapy regimens differ even when the disease type is the same in children and adults, often due to the inability of older adults to tolerate a more intensive regimen (ie, PEG‐L‐asparaginase is always used in pediatric acute lymphocytic leukemia protocols but rarely in adult protocols). Therefore, DDI information on such combinations would be completely absent. It also may be impossible to adjust the chemotherapy dose (ie, lower dose/intensity) when introducing the new drug if DDI is suspected, since the new drug may not provide efficacy and the loss of benefit to the child from adjusting backbone therapy would leave the patients in a subtherapeutic window. This again highlights the need for a model‐informed drug development approach to provide the appropriate dosing instructions for concomitant drug administration in children. Kersting et al41 developed a PBPK model for etoposide using data from adult patients and successfully scaled it down to predict plasma concentrations in children. Comedications impacting the metabolism and excretion of etoposide were also taken into consideration in this model to inform dosing in children. Overall, simulated and observed plasma concentration‐time profiles were in agreement for different age groups.41 This PBPK approach presents an opportunity for quantitative pharmacology to improve study designs and dosing, where DDI data from adults are absent and thus extrapolation of DDI to children would not be possible, considering that the ontogeny of drug elimination pathways are well understood.
Data Analysis and Modeling
Data analysis and modeling strategies need to be aligned with the major objectives of the study, which are informed by the pediatric study decision tree.6 Generally, there appear to be 3 major objectives for which the data analysis and modeling approaches are considered, as described in more detail below.
PK Bridging
For this type of study, modeling and simulation can help inform the starting dose, dose regimen optimization, and optimization of the study design. PBPK and allometry represent the most common scaling approaches to leverage available PK data from adult oncology patients to help define pediatric doses and regimens. The selection of the appropriate methodology is a function of the age range to be studied in the trial. Allometric approaches may not work well for very young children where age‐dependent enzyme and transporter function may alter drug elimination42; however, when enough data are available to empirically determine the allometric age‐dependent exponents in the pediatric population for the relevant PK parameters, allometry may provide similar results in the young children group compared to PBPK.43 Summaries of the pediatric dosing regimens for cancer drugs with an FDA‐approved pediatric indication obtained since 2002 and comparisons to the dosing regimens in adults are shown in Table 1.
Key applications of PBPK modeling include support for the first pediatric trial, starting dose selection, prediction of drug exposures across the age continuum, optimization of blood sample collection, prediction of target organ exposure, and evaluation of the drug interaction potential.36, 42 There are a number of retrospective examples to show the utility of the approach, and prospective examples in pediatric oncology are beginning to emerge.44 Thai et al45 developed a PBPK model for docetaxel, incorporating in vitro absorption, distribution, metabolism, and excretion and intravenous PK data from over 500 cancer patients after either single or multiple dosing to optimize dose and sampling times for a pediatric PK bridging study. The model appropriately predicted docetaxel plasma concentration‐time profiles in neonates to 18‐year‐old patients. Challenges and limitations associated with the application of PBPK modeling in pediatrics include the lack of complete information on drug absorption processes, drug‐metabolizing enzymes, and transporters. In addition, as highlighted by the example for docetaxel,45 the PBPK approach tended to underpredict the intersubject variability in drug exposure, highlighting an additional challenge in defining variability in ontogeny functions in pediatrics. It will be important to continue to accrue examples of prospective DDI predictions in pediatrics given the role of combination drug therapies in attaining successful patient outcomes.
For study design optimization, it is necessary to use population PK approaches to define sparse sampling schedules to minimize blood volume collection and maximize the value of PK data collection in pediatric oncology patients due to the challenges described in the previous section on dosing. In addition, these approaches can help inform sample size for sufficient precision of PK parameters.8
Pharmacokinetic/Pharmacodynamic Studies
The main objective of PK/PD studies is to characterize the relationship between exposure and pharmacodynamic end points in cases in which a difference between adult and pediatric patients is suspected. A key challenge for oncology in general revolves around the development and validation of PD end points to characterize target engagement and/or pathway modulation required to provide support for optimal dose and regimen selection.46 One general paradigm involves the use of longitudinal tumor volume measurements and their ability to predict outcomes such as progression‐free survival or overall survival. For tumors that arise predominantly in a pediatric population (ie, Wilms or neuroblastoma), there is limited opportunity to validate the translational value of a PD end point to inform drug development and further emphasizes the need for dedicated pediatric studies in cancers that affect only children to further inform translational value of PD end points.
In oncology, biomarkers are frequently used to identify subsets of patients with a greater chance of responding to a particular therapy. The phase 1 study of crizotinib in pediatric patients21 highlights the importance of exploiting a known biomarker across a variety of adult and pediatric cancers. Crizotinib, an ALK inhibitor, has shown excellent response rates in chemotherapy‐refractory non‐small cell lung cancer (NSCLC) patients with anaplastic lymphoma kinase (ALK) translocations. The ALK oncogene is frequently mutated in patients with advanced neuroblastoma, and it is also commonly overexpressed in lung cancer, thyroid cancer, anaplastic large‐cell lymphoma, rhabdomyosarcoma, and glioblastoma multiforme. In the phase 1 study in pediatric patients, for the dose‐escalation part of the study, patients between the ages of 12 months and 22 years with solid or central nervous system tumors, anaplastic large‐cell lymphoma, or any other cancers for which there is no known curative treatment were eligible to enroll. For the other 2 parts of the study, only patients with confirmed ALK translocations, mutations, or amplifications were enrolled. Currently, further clinical studies are under way in children with neuroblastoma and anaplastic large‐cell lymphoma. Continuing to define the quantitative PK/PD relationships for biomarkers will aid in the ability to efficiently develop new treatments in pediatric patients by leveraging prior experience in adult populations.
Full Development Studies Including PK, Safety, and Efficacy
Traditional exposure‐efficacy and exposure‐safety analyses can be used in conjunction with biomarker and safety data to support the proposed dosing regimen in pediatric oncology. Frequently, a dosing paradigm based on the maximum tolerated dose is selected for oncology therapeutics, necessitating a single dose level in safety and efficacy studies. Given that the range of drug exposures associated with a single dosing intensity is much smaller than the range achieved when an array of doses is selected, it may be challenging to extrapolate the results from a single dose level to other cases with different doses or regimens. Therefore, the goal of exposure‐response analyses is to identify patients who are suboptimally exposed to the drug due to covariate factors. For example, the population PK of imatinib, a small molecule that inhibits the constitutive abnormal bcr‐abl tyrosine kinase created by the Philadelphia chromosome abnormality in chronic myeloid leukemia, was characterized in a combined analysis including children and adults.47 Adult patients (n = 34; age ≥18 years) received oral imatinib 400 mg/day while children, adolescents, and young adults (n = 33; age <21 years) received oral imatinib 340 mg/m2/day with PK samples collected after the first dose and on days 30 and 60. A base population PK model adequately described the concentration‐time profile of imatinib and its primary metabolite CGP74588 in children and adults. Body weight, alpha acid glycoprotein plasma level, and albuminemia were identified as covariates that influenced imatinib clearance, and inclusion of these covariates reduced the intersubject variability in clearance from 47% to 19%. Children had a slightly higher imatinib exposure compared to adults due to a higher dose (417 mg/day vs 400 mg/day) and lower clearance (6.2 L/h vs 8.22 L/h) in children and adults, respectively. This study illustrates the value of characterizing the PK properties in children and adults, and the significant impact that population PK/PD analyses provide in selecting the optimal dosages for pediatric oncology patients.
Conclusions and Recommendations
This work has highlighted the opportunities in which clinical pharmacology–based approaches can be (and arguably must be) used to overcome the challenges encountered in pediatric oncology drug development. In the past, pediatric studies have lagged behind adult studies due to the difficulty in enrolling sufficient numbers of pediatric patients and the lack of long‐term safety data from adults. Moving forward, on the basis of scientific evidence where the disease, safety, PK, and dosages in adolescents were comparable to those in adults,14, 15 it is recommended that adolescents be included in adult oncology clinical trials and/or first‐in‐patient studies to avoid the lag time that currently hinders pediatric drug development and access to useful therapies.17 Similarly, study designs can be modified and Bayesian methodologies and pediatric expansion cohorts can be employed to establish optimal doses in children sooner and with greater confidence, especially for targeted therapies. Because sample acquisition remains a concern and there are no clear guidelines on the frequency or volumes of sample collection in children of various ages, population‐based PK analyses that use sparse PK sampling and optimized PK/PD sampling designs such as simulations48 or D‐optimality49 approaches can significantly minimize excessive sampling in children, especially in cancer patients who suffer from hematologic deficiencies due to disease or chemotherapy. Establishing an optimal starting dose of a convenient, age‐appropriate formulation remains important for successful pediatric oncology drug development. Use of modeling and simulation approaches must continue to advance to inform selection of safe and efficacious doses as early in the drug development process as possible and to avoid overexposing children to subtherapeutic doses. It is imperative that modeling and simulation approaches, together with identification and validation of additional PD end points, continue to evolve to enable development of the most effective therapies in the shortest amount of time. Additional research in both of these areas is warranted.
Declaration of Conflicting Interests
All authors are employees and shareholders of AbbVie Inc. Medical writing support was provided by Allison Kitten, an employee of AbbVie.
References
- 1. Zisowsky J, Krause A, Dingemanse J. Drug development for pediatric populations: regulatory aspects. Pharmaceutics. 2010;2(4):364–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. FDA . United States Food and Drug Administration. New pediatric labeling information database. https://www.accessdata.fda.gov/scripts/sda/sdNavigation.cfm?sd=labelingdatabase&displayAll=false&page=2. Accessed June 29, 2018.
- 3. FDA . United States Food and Drug Administration. Hematology/Oncology (cancer) approvals & safety notifications. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm279174.htm. Accessed June 29, 2018.
- 4. Patel KS, Leong R, Zhao H, et al. Pediatric development of molecularly targeted oncology drugs. Clin Pharmacol Ther. 2018;104(10):384–389. [DOI] [PubMed] [Google Scholar]
- 5. Mehrotra N, Bhattaram A, Earp JC, et al. Role of quantitative clinical pharmacology in pediatric approval and labeling. Drug Metab Dispos. 2016;44(7):924–933. [DOI] [PubMed] [Google Scholar]
- 6. van Hasselt JG, van Eijkelenburg NK, Beijnen JH, Schellens JH, Huitema AD. Optimizing drug development of anti‐cancer drugs in children using modelling and simulation. Br J Clin Pharmacol. 2013;76(1):30–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. EMA. European Medicines Agency . Concept paper on the revision of the guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/05/WC500226652.pdf. Published April 21, 2017. Accessed May 2018.
- 8. FDA. United States Food and Drug Administration . General clinical pharmacology considerations for pediatric studies for drugs and biological products. Guidance for Industry. https://www.fda.gov/downloads/drugs/guidances/ucm425885.pdf. Published December 2014. Accessed May 2017.
- 9. EMA. European Medicines Agency . Guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003066.pdf. Published June 28, 2006. Accessed May 2017.
- 10. EMA. European Medicines Agency . Reflection paper on the use of extrapolation in the development of medicines for paediatrics. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/10/WC500236640.pdf. Published October 9, 2017. Accessed May 2018.
- 11. Green DJ, Zineh I, Burckart GJ. Pediatric drug development: outlook for science‐based innovation. Clin Pharmacol Ther. 2018;103(3):376–378. [DOI] [PubMed] [Google Scholar]
- 12. Barrett JS, Bishai R, Bucci‐Rechtweg C, et al. Challenges and opportunities in the development of medical therapies for pediatric populations and the role of extrapolation. Clin Pharmacol Ther. 2018;103(3):419–433. [DOI] [PubMed] [Google Scholar]
- 13. Sandlund JT, Martin MG. Non‐Hodgkin lymphoma across the pediatric and adolescent and young adult age spectrum. Hematology Am Soc Hematol Educ Program. 2016;2016(1):589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moreno L, Pearson ADJ, Paoletti X, et al. Early phase clinical trials of anticancer agents in children and adolescents—an ITCC perspective. Nat Rev Clin Oncol. 2017;14(8):497–507. [DOI] [PubMed] [Google Scholar]
- 15. Paoletti X, Geoerger B, Doz F, Baruchel A, Lokiec F, Le Tourneau C. A comparative analysis of paediatric dose‐finding trials of molecularly targeted agent with adults’ trials. Eur J Cancer. 2013;49(10):2392–2402. [DOI] [PubMed] [Google Scholar]
- 16. Momper JD, Chang Y, Jackson M, et al. Adverse event detection and labeling in pediatric drug development: antiretroviral drugs. Therap Innov Regul Sci. 2015;49(2):302–309. [DOI] [PubMed] [Google Scholar]
- 17. FDA. United States Food and Drug Administration . Considerations for the inclusion of adolescent patients in adult oncology clinical trials. Guidance for Industry. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM609513.pdf. Published June 2018. Accessed June 2018.
- 18. Davis KL, Fox E, Reid JM, et al. ADVL1412: initial results of a phase I/II study of nivolumab and ipilimumab in pediatric patients with relapsed/refractory solid tumors—a COG study. J Clin Oncol. 2017;35(15 suppl):10526–10526. [Google Scholar]
- 19. Place AE, Goldsmith K, Bourquin JP, et al. Accelerating drug development in pediatric cancer: a novel phase I study design of venetoclax in relapsed/refractory malignancies. Future Oncol. 2018;14(1):2115–2129. [DOI] [PubMed] [Google Scholar]
- 20. Fouladi M, Laningham F, Wu J, et al. Phase I study of everolimus in pediatric patients with refractory solid tumors. J Clin Oncol. 2007;25(30):4806–4812. [DOI] [PubMed] [Google Scholar]
- 21. Mosse YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large‐cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14(6):472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Banerjee A, Jakacki RI, Onar‐Thomas A, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low‐grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol. 2017;19(8):1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kieran MW, Chisholm J, Casanova M, et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017;19(11):1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. AbbVie. Advisory Committee Briefing Document . Venetoclax for the treatment of pediatric patients with relapsed/refractory cancers. Oncologic Drugs Advisory Committee Pediatric Subcommittee. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM508665.pdf. Published June 28, 2016. Accessed May 2017.
- 25. Smith M, Bernstein M, Bleyer WA, et al. Conduct of phase I trials in children with cancer. J Clin Oncol. 1998;16(3):966–978. [DOI] [PubMed] [Google Scholar]
- 26. Shi R, Derendorf H. Pediatric dosing and body size in biotherapeutics. Pharmaceutics. 2010;2(4):389–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fernandez E, Perez R, Hernandez A, Tejada P, Arteta M, Ramos JT. Factors and mechanisms for pharmacokinetic differences between pediatric population and adults. Pharmaceutics. 2011;3(1):53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gore L, Ivy SP, Balis FM, et al. Modernizing clinical trial eligibility: recommendations of the American Society of Clinical Oncology–Friends of Cancer Research Minimum Age Working Group. J Clin Oncol. 2017;35(33):3781–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaspar N, Marshall LV, Binner D, et al. Joint adolescent‐adult early phase clinical trials to improve access to new drugs for adolescents with cancer proposals from the multi‐stakeholder platform—ACCELERATE. Ann Oncol. 2018;29(3):766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Howie SR. Blood sample volumes in child health research: review of safe limits. Bull World Health Organ. 2011;89(1):46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Altamimi MI, Choonara I, Sammons H. Invasiveness of pharmacokinetic studies in children: a systematic review. BMJ Open. 2016;6(7):e010484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wade KC, Wu D, Kaufman DA, et al. Population pharmacokinetics of fluconazole in young infants. Antimicrob Agents Chemother. 2008;52(11):4043–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suyagh M, Collier PS, Millership JS, et al. Metronidazole population pharmacokinetics in preterm neonates using dried blood‐spot sampling. Pediatrics. 2011;127(2):e367–e374. [DOI] [PubMed] [Google Scholar]
- 34. Sawyer M, Ratain MJ. Body surface area as a determinant of pharmacokinetics and drug dosing. Invest New Drugs. 2001;19(2):171–177. [DOI] [PubMed] [Google Scholar]
- 35. Bins S, Ratain MJ, Mathijssen RH. Conventional dosing of anticancer agents: precisely wrong or just inaccurate? Clin Pharmacol Ther. 2014;95(4):361–364. [DOI] [PubMed] [Google Scholar]
- 36. Rioux N, Waters NJ. Physiologically based pharmacokinetic modeling in pediatric oncology drug development. Drug Metab Dispos. 2016;44(7):934–943. [DOI] [PubMed] [Google Scholar]
- 37. Leong R, Zhao H, Reaman G, et al. Bridging adult experience to pediatrics in oncology drug development. J Clin Pharmacol. 2017;57(S10):S129–S135. [DOI] [PubMed] [Google Scholar]
- 38. Ivanovska V, Rademaker CM, van Dijk L, Mantel‐Teeuwisse AK. Pediatric drug formulations: a review of challenges and progress. Pediatrics. 2014;134(2):361–372. [DOI] [PubMed] [Google Scholar]
- 39. Chen ML, Sadrieh N, Yu L. Impact of osmotically active excipients on bioavailability and bioequivalence of BCS class III drugs. AAPS J. 2013;15(4):1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adkison K, Wolstenholme A, Lou Y, et al. Effect of sorbitol on the pharmacokinetic profile of lamivudine oral solution in adults: an open‐label, randomized study. Clin Pharmacol Ther. 2018;103(3):402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kersting G, Willmann S, Wurthwein G, Lippert J, Boos J, Hempel G. Physiologically based pharmacokinetic modelling of high‐ and low‐dose etoposide: from adults to children. Cancer Chemother Pharmacol. 2012;69(2):397–405. [DOI] [PubMed] [Google Scholar]
- 42. Barrett JS, Della Casa Alberighi O, Laer S, Meibohm B. Physiologically based pharmacokinetic (PBPK) modeling in children. Clin Pharmacol Ther. 2012;92(1):40–49. [DOI] [PubMed] [Google Scholar]
- 43. Mahmood I, Tegenge MA. A comparative study between allometric scaling and physiologically based pharmacokinetic modeling for the prediction of drug clearance from neonates to adolescents. J Clin Pharmacol. 2019;59(2):189–197. [DOI] [PubMed] [Google Scholar]
- 44. Shebley M, Sandhu P, Emami Riedmaier A, et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin Pharmacol Ther. 2018;104(1):88–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thai HT, Mazuir F, Cartot‐Cotton S, Veyrat‐Follet C. Optimizing pharmacokinetic bridging studies in paediatric oncology using physiologically‐based pharmacokinetic modelling: application to docetaxel. Br J Clin Pharmacol. 2015;80(3):534–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Venkatakrishnan K, Ecsedy JA. Enhancing value of clinical pharmacodynamics in oncology drug development: an alliance between quantitative pharmacology and translational science. Clin Pharmacol Ther. 2017;101(1):99–113. [DOI] [PubMed] [Google Scholar]
- 47. Petain A, Kattygnarath D, Azard J, et al. Population pharmacokinetics and pharmacogenetics of imatinib in children and adults. Clin Cancer Res. 2008;14(21):7102–7109. [DOI] [PubMed] [Google Scholar]
- 48. Barrett J. Pharmacometrics in pediatrics In: Schmidt S., Derendorf H., eds. Applied Pharmacometrics. New York: Springer; 2014:83–128. [Google Scholar]
- 49. Davda JP, Dodds MG, Gibbs MA, et al. A model‐based meta‐analysis of monoclonal antibody pharmacokinetics to guide optimal first‐in‐human study design. MAbs. 2014;6(4):1094–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]