

Abstract
Background
Diabetes mellitus is caused by a partial or complete lack of insulin production in the body. We have previously shown that a single injection of an adeno‐associated virus serotype 8 (AAV8) vector carrying a modified and codon optimized human insulin gene induced hepatic production of insulin and corrected streptozotocin (STZ)‐induced diabetes in mice for more than 1 year. Insulin production was constitutive, analogous to long‐acting insulin therapy.
Methods
We have developed a single AAV8 vector with a Tet‐Off regulatable system as a safety mechanism to turn off insulin secretion should hypoglycaemia develop in vector‐treated diabetic mice. We first transfected HepG2 cells or freshly isolated rat hepatocytes in vitro with the Tet‐Off system (pAAV‐Tetoffbidir‐Alb‐luc) regulating a luciferase reporter gene. We subsequently incorporated a furin‐cleavable codon‐optimised human proinsulin cDNA into pAAV‐Tetoffbidir backbone to form the doxycycline inducible pAAV‐Tetoffbidir‐Alb‐hINSco.
Results
Using STZ‐induced diabetic mice, we were able to switch off insulin secretion repeatedly with doxycycline administration, and showed full restoration of insulin secretion on withdrawing doxycycline.
Conclusions
The present study provides proof of concept that, under circumstances when inappropriate basal insulin secretion is a safety concern, insulin secretion from AAV8 gene therapy can be turned off reversibly with doxycycline.
Keywords: adeno‐associated virus, diabetes, insulin, inducible, Tet‐off, safety
1. INTRODUCTION
Diabetes mellitus (DM) is caused by a partial or complete lack of insulin production in the body. Patients with type 1 diabetes often require multiple daily insulin injections for glycaemic control. In addition, the progressive nature of type 2 DM means that many patients may require basal insulin at some point in the course of the disease as a result of β‐cell failure. Poor glycaemic control leads to secondary complications, such as cardiovascular disease, amputations of limbs, retinopathy, neuropathy and nephropathy.1 Animal studies and clinical trials have shown that the presence of even a constant low level of basal insulin prevents ketoacidosis, does not cause fasting hypoglycaemia, and may help in decreasing the frequency of chronic complications.2, 3
Many current insulin regimens include a long‐acting basal insulin combined with injections of short‐acting insulin before meals. Basal insulin is designed to provide a steady supply of insulin to meet the body's basal (non‐meal) insulin needs throughout the day and night, supplemented by short‐acting insulin to provide the postprandial requirement for higher insulin secretion.
Pancreas and islet transplantation have been demonstrated to restore long‐term normoglycaemic homeostasis for up to 5–10 years. However, donor scarcity, specialised skills and infrastructure, and strict exclusion criteria have restricted the application of such procedures to only a small number of severely diabetic patients.4, 5 Moreover, substitution of daily insulin injections for lifelong immunosuppression incurs significant risks of other adverse side effects.
Other forms of biological β‐cell replacement therapies using embryonic, induced pluripotent stem cells (iPSCs) and adult stem cells are still experimental. iPSC‐derived insulin producing cells would theoretically be the least immunogenic cell source. However, there is evidence that transplanted autologous iPSC‐derived β‐cells could still be immunogenic, eliciting both auto‐ and allogeneic immune responses.6, 7 Bespoke treatment with autologous cell transplantation would also be extraordinarily expensive (possibly in the region of a million dollars for each patient) to cover the costs of expansion and large scale differentiation of stem cells.
Lentiviral‐based insulin gene therapy has been reported to be efficacious in the long‐term treatment of diabetic rodents.8, 9 However, insertional mutagenesis of the integrated virus is of concern when using this approach and vigorous safety validations are required before it can be considered for feasible clinical application. We and others have recently shown that one‐time injection of an adeno‐associated virus serotype 8 (AAV8) vector carrying the human insulin gene was sufficient to reduce significantly or normalize non‐fasting hyperglycaemia of streptozotocin (STZ)‐induced diabetic mice for more than 1 year.10, 11 The use of AAV8 has already been shown to be efficacious for the long term treatment of human haemophilia B without any significant safety issues for moe than 6 years. There is little concern over the danger of over‐expression of factor IX in these patients because this has no known clinical impact. Restoring constitutive factor IX production to 1–6% of the physiological blood level is sufficent to reduce or abolish frequent bleeding episodes.12 By contrast, gene therapy of diabetes requires that insulin secretion should not cause hypoglycaemia from inappropriate temporary or permanent overexpression. In the present study, we have built in a biosafety mechanism in the AAV8‐insulin vector by incorporating a Tet‐Off switch and tested its ability, in diabetic mice, to turn off insulin expression with doxycycline, as well as reactivate expression upon doxycycline withdrawal. The ability to regulate insulin expression is important in preclinical studies and clinical trials whenever constitutive insulin secretion needs to be reduced to avoid hypoglycaemia.
Vanrell et al.13 developed a compact single Tet‐inducible autoregulated expression system, pTetbidirAlb, that allowed liver‐specific regulated gene expression in AAV8 vectors driven by a hybrid Tet‐on and albumin promoter. They demonstrated rapid induction of the luciferase reporter as well as interleukin (IL)‐12 and interferon‐γ with doxycycline to treat liver cancer. The sustained liver‐secific expression of IL‐12 by the same vector was reported by Gil‐Fariña et al.14 We have modified the AAV8 construct to a TetOffbidir system, by switching the doxycycline‐activated rtTA chimera to tTA, aiming to switch off insulin expression with doxycycline. In the Tet‐Off system, the tetracycline transcriptional activator (tTA) protein is formed by fusing the tetracycline repressor, TetR, from Escherichia coli to the VP16 transactivator from Herpes simplex virus. In the absence of doxycyline, tTA binds to the seven tandem repeats of TetO, which constitute the tetracycline response element (TRE), and activates the adjacent liver‐specific albumin promoter. This results in liver‐specific expression of the gene of interest. In the presence of doxycycline, tTA detaches from TetO and inactivates transgene expression.15
We employed a non‐diabetic mouse model with the luciferase reporter gene to confirm the liver‐ specificity and also the kinetics of the pAAV‐TetOffbidir‐Alb‐luc vector using a live imaging system. These experiments provide information for researchers who may be interested in using this vector to switch off their gene of interest under non‐diabetic conditions. Using the pAAV‐TetOffbidir‐hInsco vector system in STZ‐induced diabetic mice, we showed that insulin gene expression could be reversibly turned off and on, multiple times, without loss of signal.
2. MATERIALS AND METHODS
2.1. Assembly of AAV vectors
Standard cloning methods were used for the cloning of recombinant AAV vectors. The AAV vectors in the present study were modified from the Tet‐on AAV vector pAAV‐Tetbidir‐Alb‐luc.13 pAAV‐Tetbidir‐Alb‐luc was converted to pAAV‐Tetoffbidir‐Alb‐luc by substituting the Tet‐On rtTA transactivator sequence flanked by EcoRI‐BamHI restriction sites with the Tet‐off tTA‐Advanced (tTA‐2S) transactivator sequence (Takara Bio Inc., Otsu, Japan) with the same restriction sites. pAAV‐Tetoffbidir‐Alb‐hINSco was modified from pAAV‐Tetoffbidir‐Alb‐luc by applying restriction enzymes SalI and NotI and replacing the luciferase gene with the furin‐cleavable codon‐optimized human proinsulin gene, hINSco,10 flanked by the same restriction enzymes.
2.2. Cell transfection and Luc measurement
3 × 104 HepG2 cells or 4 × 104 freshly isolated rat hepatocytes were seeded in each well of 96‐well plates. Cells were transfected with 100 ng of the constitutive pAAV‐HLP‐Luc or pAAV‐Tetoffbidir‐Alb‐luc and 25 ng of pSV40‐NlucP (ratio 3:1) using Lipofectamine 3000 reagent (Thermo‐Fisher Scientific, Waltham, MA, USA). Various concentrations of doxycycline were added at the indicated times and luciferase assays using the Nano‐Glo® Dual‐Luciferase® Reporter (NanoDLR™) Assay System (Promega, Madison, WI, USA) were performed at the indicated time points, in accordance with the manufacturer's instructions. Cell culture experiments were performed once in triplicate.
2.3. Packaging, purification and titration of viruses
The AAV8 pseudotyped vector particles used in the present study were packaged with the polyethylenimine triple plasmid transfection into 293 T cells using the transgene plasmid, an adenoviral helper plasmid (HGT1) and a chimeric AAV2 Rep‐8Cap packaging plasmid (pAAV2–8) at a ratio of 3:10:3, as described in a previous study.10 The vectors from transfected cells were harvested and purified using discontinuous iodixanol gradient ultracentrifugation.16 Vector particles were titrated by a quantitative polymerase chain reaction, as described previously,17 and the results were used to determine the vector dose for the mice.
2.4. Animal studies
NOD.cg‐PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were derived from NOD‐SCID parental line and STZ‐induced diabetic NSG mice have been used as a model for type 1 diabetes in previous studies.10, 18, 19, 20 The NSG breeders were purchased from The Jackson Laboratory (Bar Harbor, ME, USA), bred and maintained in the specific pathogen‐free facility within the University in accordance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the National University of Singapore. In addition, the Guide for the care and use of laboratory animals, eighth edition (2011) (http://grants.nih.gov/grants/olaw/guide‐for‐the‐care‐and‐use‐of‐laboratory‐animals.pdf) was followed. The mice were subjected to regular 12:12 hour dark/light photocycles and provided with normal feed and water ad libitum unless otherwise indicated. No randomization was performed in the allocation of animals to each group. The in vivo experiments with pAAV‐Tetoffbidir‐Alb‐luc were done in non‐STZ‐induced male NSG mice (8–12 weeks old). For experiments involving pAAV‐Tetoffbidir‐Alb‐hINSco, the mice received a single intraperitoneal (i.p.) injection of 120 mg/kg STZ (Sigma‐Aldrich, St Louis, MO, USA) to induce diabetes. Body weight was measured and a drop of blood was obtained via the tail vein for non‐fasting blood glucose measurement with an Accu‐Chek Active blood glucose monitor (Roche, Basel, Switzerland). Mice that maintained their non‐fasting blood glucose levels at > 20 mM for 2 consecutive days were considered diabetic and included in the study. The AAV8‐Tetoffbidir‐Alb‐luc or AAV8‐Tetoffbidir‐Alb‐hINSco vectors were injected into the diabetic mice via the tail vein with specified numbers of viral genome copies (vg) per mouse. Mice that became moribund or severely underweight were euthanized using carbon dioxide. All efforts were made to minimize suffering. Adminstration of doxycycline (Sigma‐Aldrich) was by i.p. injection, gavage or in the animal feed (Envigo, Huntingdon, UK). Blood was drawn from the facial vein and processed to obtain serum for a human C‐peptide enzyme‐linked immunosorbent assay (ELISA).
2.5. Bioluminescence imaging
Mice were anesthetized with 1–2.5% isoflurane, supplied with 2 L/min oxygen and placed in a dark chamber for light acquisition in an IVIS Spectrum In Vivo Imaging System (Perkin Elmer, Waltham, MA, USA). No blinding was performed. The data were analyzed using Living Image 4 (Perkin Elmer). The substrate, Xenolight d‐luciferin (Perkin Elmer), was dissolved in saline at 30 mg/ml and injected i.p. at 160 mg/kg. The region of interest (ROI) covering the whole animal was defined, and quantification of light emission was performed in photons/second before and after luciferin (substrate) injection. Images were obtained at 1‐minute intervals (10‐second exposures) for the first 12 to 16 minutes after luciferin administration. The peak signal for each mouse was used as the data point for the experiment.
2.6. C‐peptide ELISA
Serum C‐peptide was measured using an ELISA kit (EZHCP‐20 K) for human C‐peptide (EMD Millipore, Billerica, MA, USA) at the indicated times in the respective experiments. The antibody was specific for human C‐peptide and did not cross‐react with mouse C‐peptide.
2.7. Statistical analysis
Statistical analysis were performed using the Prism (GraphPad Software Inc., San Diego, CA, USA). The results were expressed as the mean ± SEM. Comparisons of human C‐peptide levels in the serum of treated mice in the absence of doxycycline, induced with doxycyline and after withdrawal of doxycycline, were analyzed. Statistical significance was determined using the Holm–Sidak method, with alpha = 5.000%, without assuming a consistent SD.
3. RESULTS
3.1. In vitro characterization of the pAAV‐Tetoffbidir‐Alb‐luc vector
The Tet‐Off vector constructs are represented schematically in Figure 1A. Control of the pAAV‐Tetoffbidir‐Alb‐luc vector by doxycycline was tested in plasmid‐transfected cells in vitro before packaging into virus for the in vivo experiments. Luciferase activity of pAAV‐Tetoffbidir‐Alb‐luc was compared with the constitutive liver‐specific pAAV‐HLP‐Luc in HepG2, a human liver carcinoma cell line. Transfected pAAV‐HLP‐Luc was not regulated by doxycycline, whereas luciferase activity of cells transfected with pAAV‐Tetoffbidir‐Alb‐luc was attenuated by 97% with the addition of doxycycline at both 0.5 μg/ml and 1 μg/ml (Figure 1B). In a subsequent experiment shown in Figure 2A, pAAV‐Tetoffbidir‐Alb‐luc transfected HepG2 cells were subjected to different concentrations of doxycycline 24 hours post‐transfection, and luciferase acitivities were determined 24 hours after the addition of doxycycline. Significant reductions in activities were observed in cells exposed to 1, 10, 100 and 1000 ng/ml of doxycycline (Figure 2B; see also Supporting information, Table S1). We investigated the reversibility of doxycycline‐induced luciferase repression in transfected cells. Twenty‐four hours after transfection, cells were exposed to doxycycline for 24 hours (48 hours post transfection) and subsequently withdrawn for 24 hours (72 hours post transfection) and then compared with parallel cultures in the absence of doxycycline. It was found that 0.1 ng/ml doxycycline did not attenuate the luciferase activity at 24 hours post doxycycline induction. Luciferase activities of cells exposed to 1 ng/ml decreased to 24.1% of the non‐induced signal after induction for 24 hours, and returned to 96.7% of the non‐doxycycline‐induced control at 24 hours upon doxycycline withdrawal. Cells induced with 10 ng/ml of doxycycline decreased to 4.3% of the non‐induced level, and returned to 75.2% of the level of control cells that were not exposed to doxycycline. The luciferase activity of cells that were exposed to 100 and 1000 ng/ml remained low, with no signicant recovery of activity 24 hours after doxycycline withdrawal. A similar trend of reduction in luciferase activity by different concentrations of doxycycline was observed in transfected rat hepatocytes (see Supporting information, Figure S1). The kinetics of the decrease and increase in luciferase activity by the addition and withdrawal of doxycycline, respectively, were examined over 24 hours. Doxycycline at 1 ng/ml reduced luciferase activity by 50% within approximately 10 hours, and to the basal level within 24 hours. Luciferase activity returned to near maximum activity within 24 hours after the withdrawal of doxycycline (Figure 3A). Transfected cells that were exposed to 10 ng/ml of doxycycline took approximately 5 hours to reduce luciferase activity by 50% (Figure 3B). However, more than 24 hours was required to regain the maximum signal after doxycycline withdrawal.
Figure 1.
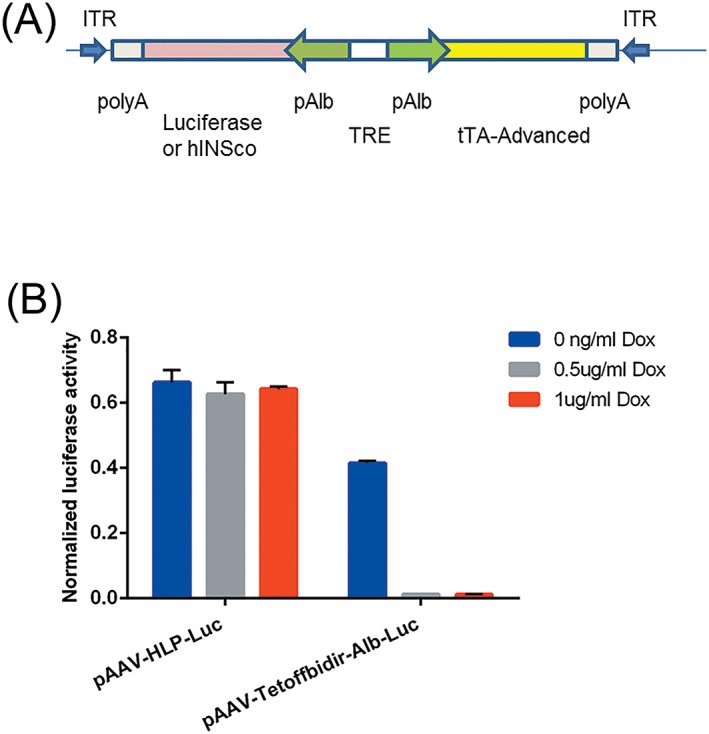
(A) Schematic diagram of a single Tet‐off autoregulated AAV expression system: pAAV‐Tetoffbidir‐Alb‐luc vector. Vector encodes luciferase or furin‐cleavable codon optimized modified human proinsulin gene under the control of Alb, the albumin promoter. ITR, inverted terminal repeat; luc, luciferase gene; polyA, SV40 fragment containing the early and late polyadenylation signals; tTA‐advanced, mutated tetracycline transactivator; TRE‐tetracycline response element that contains seven copies of tetracycline operator (TetO). (B) Constitutive versus regulatable luciferase expression by doxycycline. HepG2 cells were transfected with either a constitutive liver specific pAAV‐HLP‐Luc vector or pAAV‐Tetoffbidir‐Alb‐luc vector. The transfected cells were induced with 0, 0.5 and 1 μg/ml doxycycline for 24 hours. Luciferase activities were assayed using Nano‐Glo® dual‐luciferase® reporter (NanoDLR™) assay system (Promega). Readings from each well were normalized to signals from co‐transfected controls (pSV40‐NlucP). Data are reported as the mean ± SEM from triplicate wells in a single experiment
Figure 2.
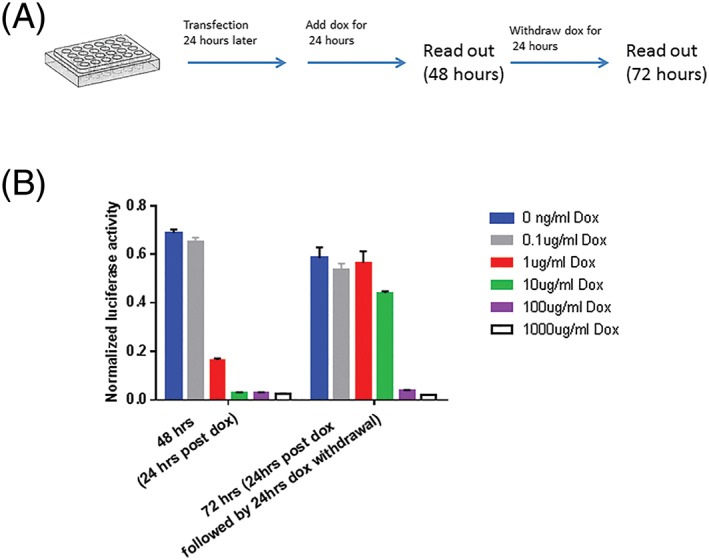
(A) Schematic diagram of doxycycline addition and withdrawal after transfection of HepG2 cells with pAAV‐Tetoffbidir‐Alb‐luc vector. (B) Transfected cells were induced with 0, 0.1, 1, 10, 100 and 1000 μg/ml doxycycline for 24 hours, 1 day after transfection, and activities were read at 48 hours after transfection. The same number of wells and conditions that were continued with doxycycline exposure had doxycycline withdrawn for a further 24 hour, and luciferase activities were assayed at 72 hours after transfection, using Nano‐Glo® dual‐luciferase® reporter (NanoDLR™) assay system (Promega). Readings from each well were normalized to signals from co‐transfected controls (pSV40‐NlucP). Data are reported as the mean ± SEM from triplicate wells in a single experiment
Figure 3.
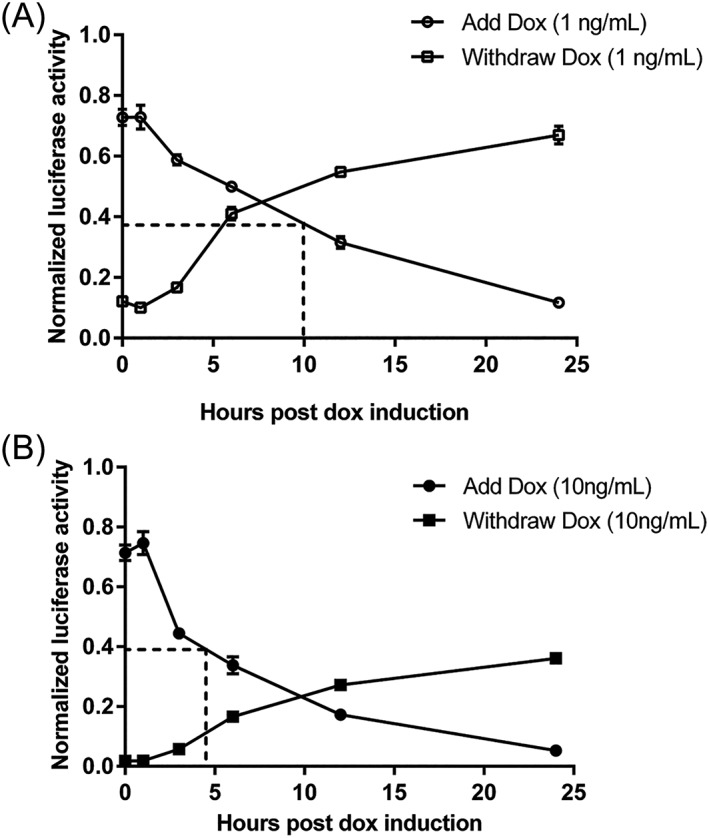
Kinetics of the switching off and on of Tet‐off system in HepG2 (A) doxycycline at 1 ng/ml. (B) Doxycycline at 10 ng/ml. Cells were exposed to doxycycline for 24 hours, 1 day after transfection. Doxycycline was removed for the subsequent 24 hours, and activities were assayed at 0, 1, 3, 6, 12 and 24 hours. Luciferase activities were assayed using Nano‐Glo® dual‐luciferase® reporter (NanoDLR™) assay system (Promega). Readings from each well were normalized to signals from co‐transfected controls (pSV40‐NlucP). Dotted lines depicts the time taken for the luciferase signal to reduce by 50%. Data are reported as the mean ± SEM from triplicate wells in a single experiment.
3.2. In vivo imaging of luciferase expression of the AAV8‐Tetoffbidir‐Alb‐luc vector
The liver specificity of the vector was confirmed by real time in vivo imaging. AAV8‐Tetoffbidir‐Alb‐luc vector (1.6 × 1010 vg/mouse) was injected into the tail vein of mice. The background luciferase activity of non‐vector‐injected control mice before and after luciferin (substrate) injection is shown in the Supporting information (Figure S2). The signals are very similar between the two and also to that found in the vector‐injected mice before luciferin was injected. The background relative luciferase activity is in the range of 1–1.5 × 105. The pAAV‐Tetoffbidir‐Alb‐luc vector‐injected mice, when injected with luciferin, had relative luciferase activities up to 1 × 108, which is 1000‐fold above the background signal.
Luciferase expression was monitored using a live imaging system and found to be confined to the liver (Figure 4A). Upon a single gavage with 0.4 mg of doxycycline, the signal started to decrease when measured 6 hours later, and continued to fall to the basal level at 24 hours post‐gavage. The signal was maintained at basal level for approximately 24 hours and subsequently increased to near original level within 72 hours post‐gavage. Luciferase activity of controls injected with AAV8‐Tetoffbidir‐Alb‐luc vector without doxycycline remained stable throughout the course of study (Figure 4B). The doxycycline dose was decreased further aiming to determine the lowest possible dose to attenuate luciferase gene expression and enable a return to uninhibited expression with doxycycline removal. A reduction of the dose by half (0.2 mg) was not sufficient to significantly shorten the recovery time (see Supporting information, Figure S3). Further reduction of doses to 2, 4 or 6ug per mouse resulted in the inhibition of luciferase acitivities of 84%, 87% and 99%, respectively, and a recovery time of approximately 40 hours post‐gavage (see Supporting information, Figure S3). Doxycycline doses ≤ 0.8 μg per mouse had no effect with respect to inhibiting the expression of luciferase activity (see Supporting information, Figure S3). The same mice (identified by specific Experiment number followed by mouse ID; e.g. AA6_1, AA11_1) could be repeatedly exposed to doxycycline and showed dose‐dependent reductions in luciferase expression (Figure 4; see also Supporting information, Figure S3). Higher doxycycline doses lowered luciferase expression more rapidly and to a greater degree, although more time was required to return to the original expression level.
Figure 4.
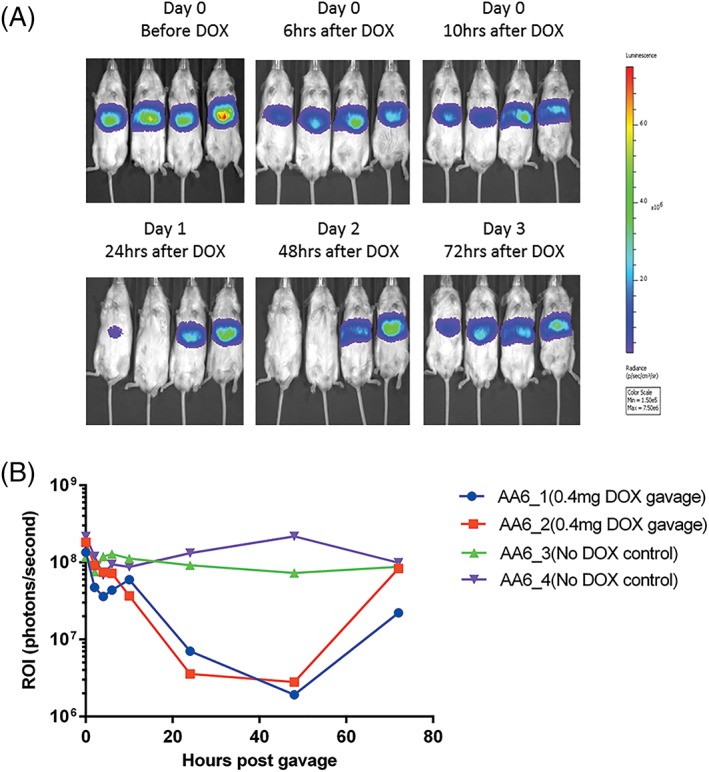
In vivo imaging of mice injected with 1.6 × 1010 vg/mouse AAV8‐Tetoffbidir‐Alb‐luc vector. (A) Optical CCD images for luciferase expression of mice before and after doxcycline induction. Left to right: AA6_ 1 and AA6_2: Gavaged with 0.4 mg of doxycycline. AA6_3 and AA6_4: Controls without doxycycline. (B) Luciferase expression was quantified and represented as photons/second of the ROI
We have carried out the luciferase experiments with repeated dox on and off induction experiments on each mouse, over a period of 8 months, without a loss in the magnitude of the signal of luciferase activity at the begining of each dox induction time point. The relative luciferase activity of mice at day 34 is not significantly different from that readings taken from the same mice 8 months (day 236) later (see Supporting information, Figure S4).
3.3. Comparison of gavage with i.p. injections of doxycycline
Doxycycline administrations by gavage or i.p. injection using 0.2 mg or 0.02 mg per mouse was carried out to determine the time taken for luciferase gene expression to decrease to the basal level, as well as the time taken to return to the maximum signal (Figure 5). The time taken for luciferase activity to be inhibited by more than 98% was 30 hours for both i.p. injected and gavaged mice at 0.2 mg doxycycline per mouse. The time taken for luciferase expression to return to the original uninhibited expression was shorter using gavage (approximately 3 days) compared to i.p. injection (6–8 days). Also, the kinetics of the doxycycline effect appeared to be less variable when administered by gavage (Figure 5A). There was no significant difference between the two routes of administration when the doxycycline dose was reduced to 0.02 mg per mouse (Figure 5B). The time taken for 90–95% inhibition was approximately 24 hours for both i.p. and gavage routes and the time taken to return to > 98% of the original signal was within 30 hours.
Figure 5.
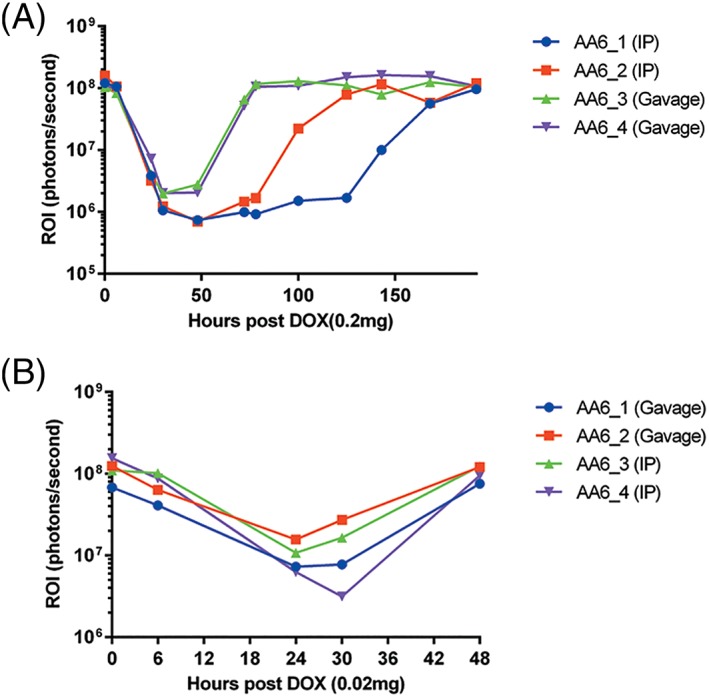
Intraperitoneal injection versus gavage of doxycycline administration. In vivo imaging of mice injected with 1.6 × 1010 vg/mouse AAV8‐Tetoffbidir‐Alb‐luc vector. Luciferase expression of mice before and after (A) 0.2 mg/mouse (B) 0.02 mg/mouse doxycycline induction
3.4. In vivo switching off of insulin expression in vector‐treated diabetic mice
Diabetic mice were injected with 3–5 × 1010 vg/mouse of AAV8‐Tetoffbidir‐Alb‐hINSco vector and blood glucose and body weight were monitored at least twice a week. Blood glucose levels decreased gradually over a month. Commercially available formulated doxycycline feed (200 ppm or 200 mg/kg diet) was introduced on day 28 and withdrawn on day 31 post‐virus injection. Doxycycline feed was equivalent to a daily dose of 0.5 mg to 1 mg doxycycline when the mice were fed ad libitum, 3–5 grams of feed per day. The average blood glucose levels and body weight profile are shown in Figure 6A, and the individual profiles are shown in the Supporting information (Figure S5). The body weights of the mice correlated inversely with blood glucose levels. We further reduced doxycycline concentration of the feed to 20 ppm (20 mg doxycycline/kg diet equivalent to a daily dose of 0.05 mg to 0.1 mg doxycycline) to the same animals and were still able to inhibit insulin secretion (Figure 6B; see also Supporting information, Figure S6).
Figure 6.
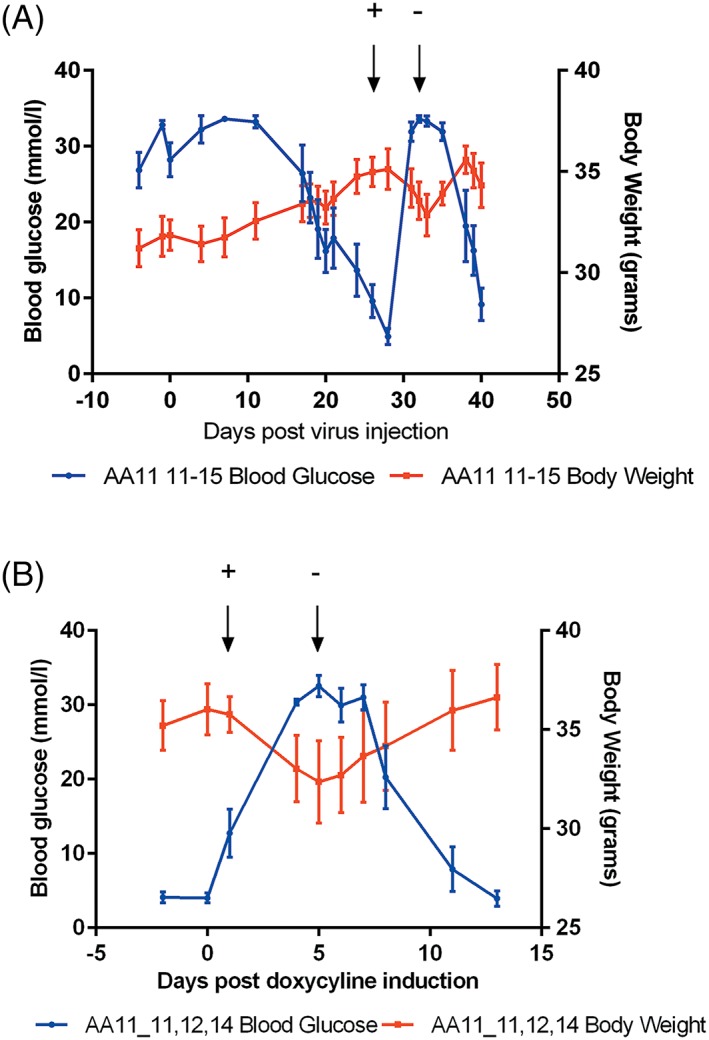
Effects of doxycycline on blood glucose and body weight of AAV8‐Tetoffbidir‐Alb‐hINSco treated diabetic mice. Mice were rendered diabetic with STZ and 5 × 1010 vg/mouse AAV8‐Tetoffbidir‐Alb‐hINSco was injected via tail vein. (A) Feed containing doxycycline (200 ppm equivalent to 200 mg doxycyline/kg feed) was given on day 28 post virus injection and withdrawn on day 31 as indicated by arrows with + and – respectively. Weight decrease with increase of blood glucose and vice versa (n = 5) (individual mouse data are provided in Supporting information Figure S5). (B) Mice were subjected to a 20 ppm doxycycline feed (20 mg/kg feed) at time point day 1 and this was withdrawn on day 5 post induction. Blood glucose and weight were monitored for a further 8 days after withdrawal (n = 3). Data are reported as the mean ± SEM (individual mouse data are provided in Supporting information, Figure S6)
The mice were injected with different doses of doxycycline and the time taken to inhibit insulin secretion and for blood glucose to increase to the peak was similar with the adminstrations of 0.2, 0.1 and 0.05 mg per mouse (approximately 40 hours). However, the time to return to pre‐doxycycline blood glucose levels was inversely proportional to the administered dose of doxycycline, approximately 400 hours for 0.2 mg, 200 hours for 0.1 mg and 100 hours for 0.05 mg doxycycline per mouse (Figure 7; see also Supporting information, Figure S7).
Figure 7.
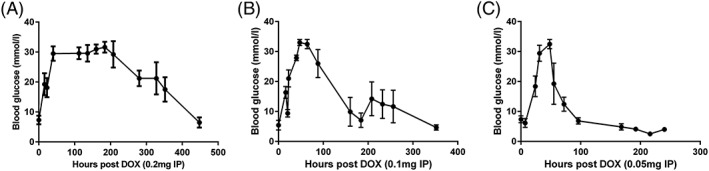
Time taken for blood glucose to peak and attenuate with the i.p. injections of different doses: (A) 0.2 mg on day 30 post vector injection; (B) 0.1 mg on day 50 post vector injection; and (C) 0.05 mg of doxycycline on day 76 post vector injection (n = 3). Data are reported as the mean ± SEM. (individual mouse data are provided in Supporting information, Figure S7)
To demonstrate the direct inhibitory effect of doxycycline on the insulin expression, we measured serum human C‐peptide levels before and after the mice were fed with doxycycline chow. Human C‐peptide was used as a measure of the human insulin secreted in the serum because it was produced in equimolar concentrations compared to insulin and it is species specific. Table 1 shows that doxycycline switched off insulin expression as reflected by undetectable human C‐peptide in serum.
Table 1.
Blood glucose and corresponding serum human C‐peptide levels before and after doxycycline feed
| Animal ID | Blood glucose (mmol/l) after AAV8‐Tetoffbidir‐Alb‐hInsco treatment. | Corresponding human C‐peptide (ng/ml) | Blood glucose (mmol/l) after doxycycline feed | Corresponding human C‐peptide (ng/ml) |
|---|---|---|---|---|
| AA11_12 | 5.8 | 4.9 | ≥ 34 | Not detected |
| AA11_14 | 4.4 | 5.6 | ≥ 34 | Not detected |
| AA12_2 | 8.9 | 4.5 | ≥ 34 | Not detected |
| AA12_13 | 2.9 | 12.3 | ≥ 34 | Not detected |
Blood glucose levels of mice were measured and blood was drawn at the same time for serum human C‐peptide ELISA. Doxycycline feed (20 ppm) was given for 8 days, after which blood glucose and serum human C‐peptide levels were determined (the lowest threshold detection level of the C‐peptide ELISA was 0.05 ng/ml).
To confirm that the insulin could be restored upon withdrawal of doxycycline, serum samples were obtained from the another group of vector‐treated diabetic mice at different stages: (i) when the blood glucose had reverted to near normoglycemia after the injection of vector and, in the absence of doxycycline, (ii) when the mice has become hyperglycemia as a result of doxycycline induction and (iii) when the blood glucose has once again returned to near nomoglycemia upon withdrawal of doxycycline. The human C‐peptide measurements were obtained, showing that the levels decreased with doxycycline induction and were restored upon doxycycline withdrawal. The results are shown in Figure 8.
Figure 8.
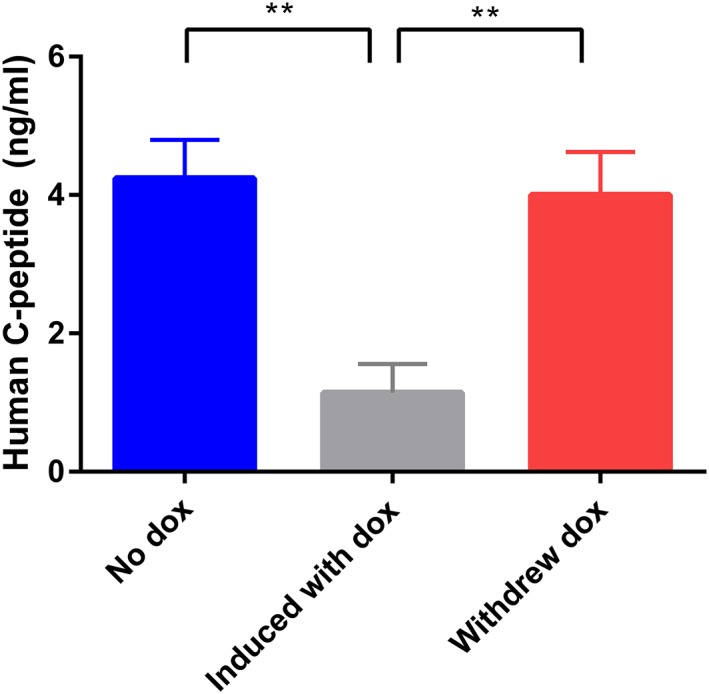
Effect of doxycycline on human insulin expression, as measured by human C‐peptide. Mice were rendered diabetic with STZ and 2.5 × 1010 vg/mouse AAV8‐Tetoffbidir‐Alb‐hINSco was injected via the tail vein. When the blood glucose had reverted to near normoglycemia over 1 month post injection, blood was drawn from the mice and serum kept frozen for the human C‐peptide asssay. Doxycycline was administered until the mice became hyperglycemia, and blood was drawn and the serum kept frozen. The doxycycline was withdrawn and blood was drawn again when the blood glucose of mice returned to near‐normoglycemia. Human C‐peptide ELISA was performed. Data are reported as the mean ± SEM (n = 7). **p < 0.005
Upon euthanasia of mice at the end point, that there was no insulin expression in the pancreas of the treated mice, indicating that the restoration of the blood glucose level was a result of the insulin vector and not spontaneous pancreas regeneration (see Supporting information, Figure S8).
4. DISCUSSION
Viral insulin gene therapy has been shown to be effective for treating animals with model type 1 diabetes.8, 9, 10, 21 Although near euglycaemia could be achieved and maintained for a long period of time with a single optimal dose injection without loss of insulin expression or hypoglycaemia in the diabetic mice, the inadvertent administration of a more‐than‐optimal dose may result in hypoglycaemia, even when the insulin transgene is driven by glucose responsive promoters.10, 11 In such instances, it is absolutely necessary to have a mechanism whereby the insulin gene can be attenuated for a controllable duration or turned off for a prolonged period. We would therefore favour the use of the insulin gene therapy approach to provide a sustained baseline insulin secretion, in a similar fashion to the current long‐acting insulin injection treatments by degludec, with a half life of 40 hours, or glargine, with a half life of 20 hours. These insulin analogues are used in many patients with type 1 or type 2 diabetes. A similar provision of fixed low‐dose basal insulin by low‐dose liver transduction could stabilize glycaemic control, improve HbA1C, and potentially lessen the risk of secondary diabetic complications. For this to be translated to a preclinical or clinical setting, it is important to minimize the possibility of hypoglycaemia occuring when the patients treated with insulin gene therapy are unable to receive normal food intake or require fasting for a period (e.g. before a surgical procedure).
The insulin secretion can be attenuated significantly within 24 hours with the range of doxycycline tested. The time taken to return to the original expression level was dose‐dependent. This could be because of the long half life of the doxycycline,22 as well as the accumulation of doxycycline in some tissues or organs.
We have shown in the present study that luciferase and insulin gene expression can be turned off using sub‐antimicrobial concentrations, which have previously been shown not to cause any changes in the gut microbiome, nor result in drug resistance with long term usage.23 Patients with acne take similar antibiotics for long periods, and the risk of antibiotic resistance does not appear to be a clinical problem.24 There are also doxycycline analogues such as minocycline and 4‐epidoxycycline that have similar efficacies on the Tet‐On/Off systems and are less toxic25 or without antibiotic effects.26 For longer‐term switching off of insulin gene expression, doxycycline was added to the feed instead of water because previous studies have reported adverse effects resulting in dehydration and weight loss.27, 28 The addition of sucrose to the drinking water to lessen the bitterness of doxycycline was not an option in the present study because it would affect the blood glucose readouts of the treated mice. Intraperitoneal injections could also be administered independent of the food or fluid intake to switch off the insulin gene. Body weights were inversely correlated with blood glucose levels, which were regulated by insulin expression from the AAV vector. The increase of body weight was an indication of recovery from diabetes because the mice were no longer losing weight from glucosuria. When insulin expression was stalled by doxycycline, the diabetes condition returned with a corresponding weight loss.
The present study addressed the need for a built‐in safety mechanism as an important component of insulin gene therapy of diabetes using developing a Tet‐Off system. We have also shown this proof of concept strategy to be effective and robust during multiple cycles of switching off and on without the loss of signal over time. The response time for turning off, although not instantaneous, was complete. Oral doxycycline attains peak plasma concentration in 2–3 hours in humans.29, 30 In the context of switching off insulin transcription, subpeak levels will begin to correct hypoglycemia or may already be sufficient to restore normoglycemia within a short period of time. Oral minocycline appears to peak in serum slightly earlier than doxycycline (2 h versus 2–3 h). This further suggests that more rapidly absorbed analogues could be used in place of doxycycline. If the occurrence of hypoglycaemia needs to be rectified immediately, sweet drinks or boluses of glucagon injections could be given as emergency measures to increase the blood glucose rapidly.
This system would also be applicable if the vector dose has been miscalculated inadvertently or if, for any other reason, there is excessive and dangerous insulin hypersecretion, and the vector could not be retrieved and removed. We could maintain the prolonged switching off of insulin by continuous oral intake of a submicrobial dose of a conventional or slow release form of doxycycline or analogues. The patient would have to resume the insulin injections prior to the gene therapy treatment. In addition to the Tet‐Off gene casette, the insulin or Tet‐Off insulin cassette could be flanked by loxP sites to enable excision of the insulin transgene by cre‐recombinase. This will permanently abolish insulin expression by the AAV vector.
There may be concern about the immune response directed against the Tet regulated system. Vanrell et al.,13 as well as previous studies31, 32 did not encounter immune rejection in their immune competent rodent models. The immune responses against Tet regulatory elements observed by some in nonhuman primates after intramuscular injection of Tet‐on encoding vectors raise concerns about the clinical value of Tet‐regulated vectors.33, 34 However, previous studies have not examined immune responses following injection of hepatotropic AAV vectors into large animal models. Other studies are looking into developing immunologically inert tetracycline regulatable systems35 and also generating new chimeric protein switches that do not trigger the host immune system in large animals. The immunogenicity of the Tet‐Off system in the liver of large animal models consitutes an area worthy of further study.
AUTHOR CONTRIBUTIONS
GSU was responsible for writing the paper and the original draft. RC, LKO, KOL and SKC were responsible for reviewing and editing the paper. GSU, RC and LKO were responsible for the study conception. GSU, FZY and SKC were responsible for the investigation and formal analyses. FZY, SKC and GSU were responsible for the methodology. RC, LKO and GSU were responsible for the acquisition of funding.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflicts of interest.
Supporting information
Figure S1. Percentage reduction in the mean signal compared to the control (no doxycycline) as shown in Figure 2B.
Figure S1 (a) Schematic diagram of doxycycline addition and withdrawal after transfection of rat hepatocytes cells with pAAV‐Tetoffbidir‐Alb‐luc vector. (b) The transfected cells were induced with 0, 0.1,1,10,100 and 1000 μg/ml doxycycline for 24 hours and activities were read. The same number of wells and conditions were continued with doxycycline exposure and another set had doxycycline withdrawn for a further 24 hours. Luciferase activities were assayed using the Nano‐Glo® Dual‐Luciferase® Reporter (NanoDLR™) Assay System (Promega). Readings from each well were normalized to signals from co‐transfected controls (pSV40‐NlucP). Data are reported as the mean ± SEM for each condition. (c) Percentage reduction in the mean signal compared to the control (no doxycycline).
Figure S2 (a) Imaging of control mice not injected with vector, before and after administration of luciferin. (b) Mean ± SEM ROI of four mice before and after luciferin administration. Paired t‐test (two tailed) (p = 0.298).
Figure S3 The effect of doxycycline concentration on the inhibition of luciferase activities in vivo. AA6–1, 2, 3 and 4 were injected with 1.6 × 1010 vg/mouse of AAV8‐Tetoffbidir‐Alb‐luc vector. (a) Luciferase activities of 2 mice (AA6_1 and AA6_2) gavaged with 0.2 mg of doxycycline in a volume of 100 μl. AA6_3 and AA6_4 served as negative controls with stable luciferase activity without doxycycline. (b) Relative luciferase activities of three mice (AA6_1, AA6_2 and AA6_3) gavaged with 2, 4 and 6 μg of doxycycline in a volume of 100 μl, respectively. AA6_4 served as negative control with stable luciferase activity without doxycycline. (c) Relative luciferase activities of three mice (AA6_1, AA6_2 and AA6_4) gavaged with 0.08, 0.8 and 4 μg of doxycycline in a volume of 100 μl, respectively. AA6_3 served as negative control with stable luciferase activity without doxycycline.
Figure S4The luciferase activity (in the absence of doxycycline) in mice injected with AAV8‐Tetoffbidir‐Alb‐luc over time. Luciferase activities of the mice were measured on day 32 (approximately 1 month) and day 236 (approximately 8 months). Data are reported as the mean ± SEM (n = 4). Wilcoxon matched‐pairs signed rank test was performed and found not to be significant.
Figure S5. Profile of individual (a) blood glucose and (b) body weight before and after doxycycline induction via feed and withdrawal. Mean blood glucose and body weight depicted in Figure 6A.
Figure S6 Profile of individual (a) blood glucose and (b) body weight before and after doxycycline induction via feed and withdrawal. Mean blood glucose and body weight depicted in Figure 6B.
Figure S7Blood glucose profiles in response to different doses (0.2 mg, 0.1 mg and 0.05 mg per mouse) of doxycycline via i.p. injections. (a) Mouse AA11_11. (b) Mouse AA11_12. (c) Mouse AA11_14.
Figure S8 Immunohistochemical staining of pancreas of treated diabetic mice and healthy control mice using anti‐insulin antibody. (a) Mouse AA11_14. (b) Mouse AA12_13. (c) AA12_15. (d) Healthy control mouse. Magnification 40×.
ACKNOWLEDGEMENTS
The Kidney Dialysis Foundation, Singapore, is thanked for funding our project. Professor Hanry Yu and Ms Phoebe Koh Kang Sheing, NUS, Department of Physiology, provided us with rat hepatocytes. Dr Gloria Gonzalez‐Aseguinolaza is thanked for the kind gift of pAAV‐Tetbidir‐Alb‐luc vector. The authors would like to acknowledge the other members of our diabetes workgroup who provided critical review and suggestions, although they are not responsible for the content of this article: Dr Anne Cooke, Dr Andrew Lever, Dr Amit Nathwani, Dr Maja Wallberg, Dr Rob Foale and Dr Asha Recino.
Gan SU, Fu Z, Sia KC, Kon OL, Calne R, Lee KO. Development of a liver‐specific Tet‐off AAV8 vector for improved safety of insulin gene therapy for diabetes. J Gene Med. 2019;21:e3067 10.1002/jgm.3067
REFERENCES
- 1. Wu T, Qiao S, Shi C, Wang S, Ji G. The metabolomics window into diabetic complications. J Diabetes Investig. 2017;9:244‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dong H, Altomonte J, Morral N, Meseck M, Thung SN, Woo SLC. Basal insulin gene expression significantly improves conventional insulin therapy in type 1 diabetic rats. Diabetes. 2002;51:130‐138. [DOI] [PubMed] [Google Scholar]
- 3. Marso SP, McGuire DK, Zinman B, et al. Efficacy and safety of degludec versus glargine in type 2 diabetes. N Engl J Med. 2017;377:723‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shapiro AM, Pokrywczynska M, Ricordi C. Clinical pancreatic islet transplantation. Nat Rev Endocrinol. 2017;13:268‐277. [DOI] [PubMed] [Google Scholar]
- 5. Dean PG, Kukla A, Stegall MD, Kudva YC. Pancreas transplantation. BMJ. 2017;357:j1321. [DOI] [PubMed] [Google Scholar]
- 6. Liu X, Li W, Fu X, Xu Y. The immunogenicity and immune tolerance of pluripotent stem cell derivatives. Front Immunol. 2017;8:645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sackett SD, Brown ME, Tremmel DM, Ellis T, Burlingham WJ, Odorico JS. Modulation of human allogeneic and syngeneic pluripotent stem cells and immunological implications for transplantation. Transplant Rev (Orlando). 2016;30:61‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elsner M, Terbish T, Jorns A, et al. Reversal of diabetes through gene therapy of diabetic rats by hepatic insulin expression via lentiviral transduction. Mol Ther. 2012;20:918‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ren B, O'Brien BA, Swan MA, et al. Long‐term correction of diabetes in rats after lentiviral hepatic insulin gene therapy. Diabetologia. 2007;50:1910‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gan SU, Notaridou M, Fu ZY, et al. Correction of murine diabetic hyperglycaemia with a single systemic administration of an AAV2/8 vector containing a novel codon optimized human insulin gene. Curr Gene Ther. 2016;16:65‐72. [DOI] [PubMed] [Google Scholar]
- 11. Thule PM, Campbell AG, Jia D, et al. Long‐term glycemic control with hepatic insulin gene therapy in streptozotocin‐diabetic mice. J Gene Med. 2015;17:141‐152. [DOI] [PubMed] [Google Scholar]
- 12. Nathwani AC, Reiss UM, Tuddenham EG, et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371:1994‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vanrell L, Di Scala M, Blanco L, et al. Development of a liver‐specific Tet‐on inducible system for AAV vectors and its application in the treatment of liver cancer. Mol Ther. 2011;19:1245‐1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gil‐Farina I, Di Scala M, Vanrell L, et al. IL12‐mediated liver inflammation reduces the formation of AAV transcriptionally active forms but has no effect over preexisting AAV transgene expression. PLoS ONE. 2013;8:e67748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Furth PA, St Onge L, Boger H, et al. Temporal control of gene expression in transgenic mice by a tetracycline‐responsive promoter. Proc Natl Acad Sci U S A. 1994;91:9302‐9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno‐associated viral vectors. Nat Protoc. 2006;1:1412‐1428. [DOI] [PubMed] [Google Scholar]
- 17. Nathwani AC, Rosales C, McIntosh J, et al. Long‐term safety and efficacy following systemic administration of a self‐complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther. 2011;19:876‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jurczyk A, Diiorio P, Brostowin D, et al. Improved function and proliferation of adult human beta cells engrafted in diabetic immunodeficient NOD‐scid IL2rgamma(null) mice treated with alogliptin. Diabetes Metab Syndr Obes. 2013;6:493‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li K, Zhu S, Russ HA, et al. Small molecules facilitate the reprogramming of mouse fibroblasts into pancreatic lineages. Cell Stem Cell. 2014;14:228‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Purwana I, Zheng J, Li X, et al. GABA promotes human beta‐cell proliferation and modulates glucose homeostasis. Diabetes. 2014;63:4197‐4205. [DOI] [PubMed] [Google Scholar]
- 21. Kozlowski M, Olson DE, Rubin J, Lyszkowicz D, Campbell A, Thulé PM. Adeno‐associated viral delivery of a metabolically regulated insulin transgene to hepatocytes. Mol Cell Endocrinol. 2007;273:6‐15. [DOI] [PubMed] [Google Scholar]
- 22. Redelsperger IM, Taldone T, Riedel ER, Lepherd ML, Lipman NS, Wolf FR. Stability of doxycycline in feed and water and minimal effective doses in tetracycline‐inducible systems. J Am Assoc Lab Anim Sci. 2016;55:467‐474. [PMC free article] [PubMed] [Google Scholar]
- 23. Thomas J, Walker C, Bradshaw M. Long‐term use of subantimicrobial dose doxycycline does not lead to changes in antimicrobial susceptibility. J Periodontol. 2000;71:1472‐1483. [DOI] [PubMed] [Google Scholar]
- 24. Skidmore R, Kovach R, Walker C, et al. Effects of subantimicrobial‐dose doxycycline in the treatment of moderate acne. Arch Dermatol. 2003;139:459‐464. [DOI] [PubMed] [Google Scholar]
- 25. Chtarto A, Tenenbaum L, Velu T, Brotchi J, Levivier M, Blum D. Minocycline‐induced activation of tetracycline‐responsive promoter. Neurosci Lett. 2003;352:155‐158. [DOI] [PubMed] [Google Scholar]
- 26. Eger K, Hermes M, Uhlemann K, et al. 4‐Epidoxycycline: an alternative to doxycycline to control gene expression in conditional mouse models. Biochem Biophys Res Commun. 2004;323:979‐986. [DOI] [PubMed] [Google Scholar]
- 27. Cawthorne C, Swindell R, Stratford IJ, Dive C, Welman A. Comparison of doxycycline delivery methods for Tet‐inducible gene expression in a subcutaneous xenograft model. J Biomol Tech. 2007;18:120‐123. [PMC free article] [PubMed] [Google Scholar]
- 28. Hojman P, Eriksen J, Gehl J. Tet‐on induction with doxycycline after gene transfer in mice: sweetening of drinking water is not a good idea. Anim Biotechnol. 2007;18:183‐188. [DOI] [PubMed] [Google Scholar]
- 29. Welling PG, Koch PA, Lau CC, Craig WA. Bioavailability of tetracycline and doxycycline in fasted and nonfasted subjects. Antimicrob Agents Chemother. 1977;11:462‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Newton PN, Chaulet JF, Brockman A, et al. Pharmacokinetics of oral doxycycline during combination treatment of severe falciparum malaria. Antimicrob Agents Chemother. 2005;49:1622‐1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bohl D, Salvetti A, Moullier P, Heard JM. Control of erythropoietin delivery by doxycycline in mice after intramuscular injection of adeno‐associated vector. Blood. 1998;92:1512‐1517. [PubMed] [Google Scholar]
- 32. Rendahl KG, Leff SE, Otten GR, et al. Regulation of gene expression in vivo following transduction by two separate rAAV vectors. Nat Biotechnol. 1998;16:757‐761. [DOI] [PubMed] [Google Scholar]
- 33. Le Guiner C, Stieger K, Toromanoff A, et al. Transgene regulation using the tetracycline‐inducible TetR‐KRAB system after AAV‐mediated gene transfer in rodents and nonhuman primates. PLoS ONE. 2014;9:e102538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Favre D, Blouin V, Provost N, et al. Lack of an immune response against the tetracycline‐dependent transactivator correlates with long‐term doxycycline‐regulated transgene expression in nonhuman primates after intramuscular injection of recombinant adeno‐associated virus. J Virol. 2002;76:11605‐11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoyng SA, Gnavi S, de Winter F, et al. Developing a potentially immunologically inert tetracycline‐regulatable viral vector for gene therapy in the peripheral nerve. Gene Ther. 2014;21:549‐557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Percentage reduction in the mean signal compared to the control (no doxycycline) as shown in Figure 2B.
Figure S1 (a) Schematic diagram of doxycycline addition and withdrawal after transfection of rat hepatocytes cells with pAAV‐Tetoffbidir‐Alb‐luc vector. (b) The transfected cells were induced with 0, 0.1,1,10,100 and 1000 μg/ml doxycycline for 24 hours and activities were read. The same number of wells and conditions were continued with doxycycline exposure and another set had doxycycline withdrawn for a further 24 hours. Luciferase activities were assayed using the Nano‐Glo® Dual‐Luciferase® Reporter (NanoDLR™) Assay System (Promega). Readings from each well were normalized to signals from co‐transfected controls (pSV40‐NlucP). Data are reported as the mean ± SEM for each condition. (c) Percentage reduction in the mean signal compared to the control (no doxycycline).
Figure S2 (a) Imaging of control mice not injected with vector, before and after administration of luciferin. (b) Mean ± SEM ROI of four mice before and after luciferin administration. Paired t‐test (two tailed) (p = 0.298).
Figure S3 The effect of doxycycline concentration on the inhibition of luciferase activities in vivo. AA6–1, 2, 3 and 4 were injected with 1.6 × 1010 vg/mouse of AAV8‐Tetoffbidir‐Alb‐luc vector. (a) Luciferase activities of 2 mice (AA6_1 and AA6_2) gavaged with 0.2 mg of doxycycline in a volume of 100 μl. AA6_3 and AA6_4 served as negative controls with stable luciferase activity without doxycycline. (b) Relative luciferase activities of three mice (AA6_1, AA6_2 and AA6_3) gavaged with 2, 4 and 6 μg of doxycycline in a volume of 100 μl, respectively. AA6_4 served as negative control with stable luciferase activity without doxycycline. (c) Relative luciferase activities of three mice (AA6_1, AA6_2 and AA6_4) gavaged with 0.08, 0.8 and 4 μg of doxycycline in a volume of 100 μl, respectively. AA6_3 served as negative control with stable luciferase activity without doxycycline.
Figure S4The luciferase activity (in the absence of doxycycline) in mice injected with AAV8‐Tetoffbidir‐Alb‐luc over time. Luciferase activities of the mice were measured on day 32 (approximately 1 month) and day 236 (approximately 8 months). Data are reported as the mean ± SEM (n = 4). Wilcoxon matched‐pairs signed rank test was performed and found not to be significant.
Figure S5. Profile of individual (a) blood glucose and (b) body weight before and after doxycycline induction via feed and withdrawal. Mean blood glucose and body weight depicted in Figure 6A.
Figure S6 Profile of individual (a) blood glucose and (b) body weight before and after doxycycline induction via feed and withdrawal. Mean blood glucose and body weight depicted in Figure 6B.
Figure S7Blood glucose profiles in response to different doses (0.2 mg, 0.1 mg and 0.05 mg per mouse) of doxycycline via i.p. injections. (a) Mouse AA11_11. (b) Mouse AA11_12. (c) Mouse AA11_14.
Figure S8 Immunohistochemical staining of pancreas of treated diabetic mice and healthy control mice using anti‐insulin antibody. (a) Mouse AA11_14. (b) Mouse AA12_13. (c) AA12_15. (d) Healthy control mouse. Magnification 40×.