

Abstract
Endothelial–mesenchymal transition occurs during intimal hyperplasia and neointima formation via mechanisms that are incompletely understood. Endothelial MAPK7 signaling is a key mechanosensitive factor that protects against endothelial–mesenchymal transition, but its signaling activity is lost in vessel areas that are undergoing pathological remodeling. At sites of vascular remodeling in mice and pigs, endothelial MAPK7 signaling was lost. The TGFβ‐induced microRNA‐374b targets MAPK7 and its downstream effectors in endothelial cells, and its expression induces endothelial–mesenchymal transition. Gain‐of‐function experiments, where endothelial MAPK7 signaling was restored, precluded endothelial–mesenchymal transition. In human coronary artery disease, disease severity is associated with decreased MAPK7 expression levels and increased miR‐374b expression levels. Endothelial–mesenchymal transition occurs in intimal hyperplasia and early lesion formation and is governed in part by microRNA‐374b‐induced silencing of MAPK7 signaling. Restoration of MAPK7 signaling abrogated these pathological effects in endothelial cells expressing miR‐374b. Thus, our data suggest that the TGFβ‐miR‐374b‐MAPK7 axis plays a key role in the induction of endothelial–mesenchymal transition during intimal hyperplasia and early lesion formation and might pose an interesting target for antiatherosclerosis therapy. © 2018 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: microRNA, MAPK7, shear stress, endothelial–mesenchymal transition (EndMT), atherosclerosis
Introduction
The development of atherosclerosis is preceded by intimal hyperplasia and neointima formation 1. Although the commonly recognized risk factors for the development of lesions and atherosclerosis are present at the systemic level 2, neointima formation and atherosclerosis manifest focally in so‐called atheroprone regions 3. These atheroprone regions are characterized by low levels of nonuniform shear stress, typically encountered at the outer walls of vascular bifurcations and at the inner wall of vascular curvatures, whereas atheroprotected regions are characterized by high levels of uniform laminar shear stress (LSS) 4.
We and others have recently described a major contribution of endothelial cells to intimal hyperplasia and atherosclerosis development 5, 6, 7. Upon exposure to inflammatory and profibrotic growth factors (i.e. TGFβ) and cytokines, endothelial cells lose their endothelial cell markers and functionality, start to express markers of the mesenchymal lineage, and gain contractile behavior 8, 9, 10. During early lesion formation, endothelial cells that undergo this endothelial–mesenchymal transition (EndMT) acquire a fibroproliferative mesenchymal phenotype and migrate from the endothelial monolayer to the underlying lesion 5, 6.
TGFβ is a major inducer of EndMT and is highly expressed in neointimal lesions 11, which may cause EndMT. We discovered that high levels of uniform LSS – observed at atheroprotected regions of the arteries – activates a specific mitogen‐activated kinase (MAPK), namely, MAPK7 [also known as Erk5 or Big MAPK (BMK1)], which inhibits the induction of EndMT by TGFβ1 5. Corroboratively, knockdown of MAPK7 in endothelial cells causes EndMT, even in the absence of exogenous TGFβ1 5, suggesting a pivotal balance between TGFβ and MAPK7 signaling in the induction of EndMT and the formation of early lesions. Indeed, TGFβ1 represses endothelial MAPK7 expression through unidentified mechanisms (unpublished data), and the loss of endothelial MAPK7 signaling aggravates atherosclerosis development 12.
MicroRNAs (miRNAs) are small, noncoding RNAs that cause post‐translational repression of their target genes. MicroRNAs elicit translational repression by imperfect base‐pairing to the 3′‐UTR of their gene targets 13, which allows any specific microRNA to target multiple genes simultaneously. Moreover, multiplicity of microRNA targets might also allow one microRNA to specifically target multiple genes within one signal transduction cascade 14, efficiently abolishing its activity.
TGFβ induces a shift in endothelial miRNA expression levels 15, 16, 17, 18 that may reduce MAPK7 signaling and thus facilitate EndMT induction. Here, we hypothesized that TGFβ1 induces the expression of a specific miRNA that targets MAPK7 and its signaling intermediates, resulting in the induction of EndMT.
Materials and methods
Additional details are provided in supplementary materials, Supplementary materials and methods.
Clinical samples
Human coronary arteries were obtained from autopsy specimens from 10 patients who died from an acute coronary episode at the Heart Institute (InCor), Sao Paulo, Brazil. During necropsy, each dissected coronary artery was fixed in neutral‐buffered formalin with constant perfusion at a quasinormal perfusion pressure. The mean age of subjects contributing pathology specimens was 65 years. Hypertension was present in nine subjects and diabetes in six. Five individuals were active smokers. The patients' next of kin gave informed consent, and the investigation was performed according to institutional guidelines (InCor, Sao Paulo; #SDC 3723/11/141 and #CAPPesq 482/11) and the Declaration of Helsinki.
Animals and surgical procedures
Porcine abdominal trifurcations were obtained from healthy male slaughterhouse Yorkshire pigs (12–13 weeks of age; body weight 30–35 kg, n = 3, V.O.F. van Beek, Lelystad, The Netherlands). Animals were fed a normal diet and were euthanized under anesthesia [ketamine (Nimatek) and midazolam with a bolus of pentobarbital and heparin (Actrapid)]. No ethical approval is needed for the use of slaughterhouse materials according to Dutch law. Male C57Bl/6j wild‐type mice (8–12 weeks of age, n = 8, Harlan, Horst, The Netherlands) were subjected to transverse aortic constriction (TAC) under anesthesia [2% Isofluorane (Forene; Abbott, Zwolle, The Netherlands) and oxygen] and analgesia (Carprofen, 5 mg/kg). In brief, an incision was made in the second intercostal space, and a small incision was made in the parietal pleura to expose the ascending loop of the aorta. The aorta was supported with a 27G needle, and a suture was placed around the aorta and drawn tight, after which the needle was removed. Next, the pleura, muscle layers, and skin were closed by sutures. Animals received postoperative analgesia (Carprofen, 5 mg/kg/day for 48 h). Animals were kept on a 12 h light:dark cycle with access ad libitum to standard laboratory chow and water. Eight weeks after aortic constriction, animals were sacrificed under deep anesthesia (3% Isofluorane by exsanguination), after which the thoracic aorta was explanted. Experiments on mice were approved by the local Institutional Animal Care and Use Committee (University of Groningen, #DEC‐5910).
Human umbilical vein endothelial cell culture
Human umbilical vein endothelial cells (HUVEC, Lonza, Walkersville, MD, USA) were cultured in endothelial cell medium up to passage 5 as described previously 8. EndMT was initiated by replating the HUVEC in RPMI1640, supplemented with 20% FCS, 1% penicillin–streptomycin, 2 mm l‐glutamine, 5 U/ml heparin, and 10 ng/ml TGFβ1 (Peprotech, NJ, USA). For shear stress experiments, HUVEC were plated on 1% gelatin‐coated μ‐Slides (Ibidi, Martinsried, Germany) and grown to confluence prior to exposure to 20 dynes per·cm of unidirectional uniform LSS. LSS was generated using the Ibidi Pump System (Ibidi).
3′‐UTR reporter analysis
Gene‐specific 3′‐UTR fragments were isolated from a cDNA pool derived from various human tissues using oligonucleotides extended with SgfI (GCGATCGC) and NotI (GCGGCCGC) restriction sequences in the sense and antisense primer, respectively (see supplementary material, Table S1). DNA amplification was performed using the DyNAzyme EXT PCR kit (Finnzymes, Vantaa, Finland) according to the manufacturer's instructions. Amplicon size was validated by gel electrophoresis on 1% agarose gels. 3′‐UTR fragments were cloned into the SgfI/NotI‐sites of the psiCHECK‐2 dual luciferase reporter vector (Promega, Madison, WI, USA). COS‐7 cells were transfected with 100 ng/ml 3′‐UTR reporter plasmid and 50 nm miR‐374b mimic or scrambled control (Life Technologies, Carlsbad, CA, USA) using Lipofectamine2000 (ThermoFisher, Waltham, MA, USA). Luciferase activity was assayed 48 h post‐transfection using the DualGlo Luciferase assay system (Promega) and recorded for 1 s on a Luminoskan ASCENT photometer (ThermoFisher).
Plasmids and lentiviral expression of miR‐374b, shRNA, and MAPK7 signaling members
For lentiviral expression of miR‐374b and small hairpin RNA (shRNA) against MAPK7 signaling members, DNA oligonucleotides containing the endogenous miR‐374b hairpin or specific 21‐mer targeting sequences for human MAP3K3, MAPK7, MEF2D, or KLF4 (all Sigma‐Aldrich, St. Louis, MO, USA, see supplementary material, Table S2) were cloned into the BamHI/EcoRI sites of the pGreenPuro shRNA expression vector (System Biosciences, Palo Alto, CA, USA), Scrambled sequences were used as control.
Gene‐CDS fragments were isolated from a cDNA pool derived from various human tissues using oligonucleotides extended with EcoRI (GAATTC) and BamHI (GGATCC) restriction sequences in the sense and antisense primer, respectively (see supplementary material, Table S3). Amplification was performed using the DyNAzyme EXT PCR kit (Finnzymes, Vantaa, Finland) according to the manufacturer's instructions. Amplicon size was validated by gel electrophoresis on 1% agarose gels. Gene‐CDS fragments were cloned into the EcoRI/BamHI sites of the pCDH‐CMV‐MCS‐EF1‐Puro lentiviral expression vector (System Biosciences). Empty vectors served as control.
For lentiviral transduction, HEK293 cells were transfected with pGreenPuro or pCDH‐ CMV‐MCS‐EF1‐Puro shuttle vectors and second‐generation lentiviral helper plasmids using Endofectin (GeneCopoeia, MD, USA). Viral supernatants were collected every 24 h, supplemented with 6 μg/ml polybrene and directly transferred to HUVEC cultures for two consecutive rounds. Transduced cells were selected for puromycin resistance 72 h post‐transduction (4 μg/ml puromycin) for 24 h and reseeded into a puromycin‐free medium for the experiments. Transduced cells did not differ in cell viability or proliferative capacity from nontransduced cells or cells transduced with control vectors.
MicroRNA and gene transcript analysis
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer's instructions. For microRNA transcript analyses, 20 ng of total RNA was reverse transcribed using a Taqman MicroRNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA) using microRNA‐specific stemloop primers (see supplementary material, Table S4). For gene transcript analysis, 1 μg of total RNA was reverse transcribed using the RevertAid First Strand cDNA Synthesis Kit (Applied Biosystems) according to manufacturer's protocol. Quantitative PCR expression analysis was performed on a reaction mixture containing 10 ng cDNA equivalent, 0.5 μm sense primers, and 0.5 μm antisense primers (for microRNA primers see supplementary material, Table S4 and gene transcript primers see supplementary material, Table S5, all Sigma‐Aldrich, St. Louis, MO, USA) and FastStart SYBR Green (Roche, Almere, The Netherlands). Analyses were run on a Viia7 real‐time PCR system (Applied Biosystems).
Immnunofluorescence
Heat‐induced antigen retrieval was performed on formalin‐fixed paraffin‐embedded sections using 0.1 M Tris–HCl (pH 9.0, 80 °C, 20 min) prior to immunohistochemistry. Sections were incubated with primary antibodies at room temperature for 2 h, followed by incubation with secondary antibodies at room temperature for 1 h (Table S6). Detailed descriptions of the imaging procedures are provided in supplementary material, Supplementary materials and methods.
microRNA in situ hybridization
Proteinase K was used to demask microRNAs in the formalin‐fixed paraffin‐embedded sections prior to in situ hybridization. Sections were incubated with double DIG‐labelled probes against miR‐374b (Exiqon/Qiagen, Vedbaek, Denmark) at 44 °C according to the manufacturer's protocol. Detection of miR‐374b was performed using 1 μg/ml anti‐DIG‐fluorescein antibodies (Roche).
Immunoblotting
Whole‐cell lysates were prepared in RIPA buffer (ThermoFisher) supplemented with 1% protease inhibitor cocktail (Sigma‐Aldrich) and 1% HALT‐phosphatase inhibitor cocktail (ThermoFisher). Protein concentrations were determined using the detergent‐compatible protein assay (Bio‐Rad, Hercules, CA, USA) according to manufacturer's protocol. Equal amounts of protein were loaded on a 10% denaturing SDS–polyacrylamide gel and separated by gel electrophoresis (110 V). Next, proteins were blotted onto nitrocellulose membranes using the Trans‐Blot Turbo System (Bio‐Rad) according to manufacturer's instructions. Blots were blocked in Odyssey Blocking Buffer (Li‐COR Biosciences, Lincoln, NE, USA) at room temperature for 30 min and incubated at 4 °C with primary antibodies (see supplementary material, Table S7) in Odyssey Blocking Buffer overnight, after which membranes were incubated with secondary antibodies (see supplementary material, Table S7) in Odyssey Blocking Buffer at room temperature for 1 h. Protein was detected using the Odyssey Infrared Imaging System (Li‐COR Biosciences). Densitometric analysis was performed using Totallab 120 (Nonlinear Dynamics, Newcastle upon Tyne, UK).
Angiogenic sprouting capacity
A total of 10 μl of MatriGel (BD Biosciences, San Jose, CA, USA) was solidified in μ‐Slide Angiogenesis (Ibidi); 10 000 cells per well were cultured on the solidified MatriGel in endothelial growth medium, and the formation of sprouts was analyzed by conventional light microscopic analysis after overnight incubation.
Collagen contraction assay
Cells were suspended in a solution of rat tail Collagen type I (Corning, Corning, NY, USA) containing 3 mg/ml NaHCO3 and 0.1 m Na2HPO4. Aliquots of 50 μl (containing 100 000 cells and 0.5 mg Collagen type I) were solidified at 37 °C in a humidified incubator with 5% CO2 for 30 min. The collagen gels were released from the culture dishes by the addition of 2 ml endothelial growth medium and were imaged using a common Flatbed scanner and allowed to contract for an additional 24 h. The degree of gel contraction was determined by measuring the total gel area and dividing the areas of the contracted gels by the areas of the gels before contraction.
Statistical analysis
Data are presented as means ± SEM. n values relate to independent experiments/samples. P values were calculated using one‐way ANOVA followed by Bonferroni's post hoc comparisons tests using GraphPad Prism (GraphPad Software, San Diego, CA, USA). p < 0.05 was considered statistically significant.
Results
MicroRNA‐374b targets MAPK7 signaling
Database analysis (miRanda 19, 20) identified 12 microRNAs that putatively target MAPK7 (see supplementary material, Figure S1). We crosschecked these microRNAs against other genes in the MAPK7 signaling cascade (see supplementary material, Figure S1) and found that the miR‐374 family (miR‐374a and miR‐374b) target not only MAPK7 but also its upstream activators Rac1, MAP3K3, and MAP3K7. Moreover, miR‐374 also putatively targets the downstream transcription factors of the myocyte enhancer factor (MEF)‐2 family as well as Krüppel‐like factor (KLF)‐4 (see supplementary material, Figure S2). TGFβ1 induced the expression of miR‐374a, miR‐374b, miR‐143, miR‐24, and miR‐410 in endothelial cells (p < 0.05 versus unstimulated control cells), whereas it decreased the expression of miR‐488 (p < 0.05 versus unstimulated control cells). The expression of miR‐429, miR‐200b, miR‐200c, miR‐183, miR‐124, and miR‐506 were unchanged (see supplementary material, Figure S3) upon TGFβ1 treatment.
MicroRNA‐374a and miR‐374b collectively have 1305 putative gene targets, of which 527 and 434 are unique to miR‐374a and miR‐374b, respectively. A total of 344 putative gene targets are shared between miR‐374a and miR‐374b (see supplementary material, Figure S2). Analysis of genes within the MAPK7 signaling cascade indicates that MAP3K7 (TAK1), MAPK7 (ERK5), MEF2A, and MEF2C are putative targets of miR‐374a and miR‐374b, whereas Rac1, MAP3K3 (MEKK3), MEF2D, and KLF4 are targets of miR‐374b only. None of the genes within the MAPK7 signaling cascade where unique targets to miR‐374a (see supplementary material, Figure S2).
MicroRNA‐374b and MAPK7 are differentially expressed at atheroprone regions
EndMT contributes to the formation of intimal hyperplasia 5, which is aggravated by the loss of protective MAPK7 signaling 12. Cells coexpressing an endothelial‐specific molecule (ESM)‐1 and the mesenchymal protein SM22α were abundantly present in the hyperplastic intima of the porcine trifurcation – a well‐documented model of early lesion formation 21, 22, 23 – compared to the nonhyperplastic intima (11.5 versus 1.0%; p < 0.001, Figure 1A). In the atheroprone hyperplastic regions, endothelial MAPK7 expression was decreased two‐fold (p = 0.01) compared to endothelial cells in the atheroprotected regions within the same trifurcation (Figure 1B). We dissected the atheroprone and atheroprotected areas of the porcine trifurcation and found increased expression levels of miR‐374b in the atheroprone regions (∼three‐fold, p = 0.04, Figure 1C). Concurrently, in mice with transverse aortic banding, atheroprone regions characterized by disturbed fluid shear stress were characterized by the accumulation of cells expressing both the endothelial marker CD31 and the mesenchymal protein SM22α (12.3%, Figure 1D), indicative of EndMT. At these atheroprone sites, endothelial MAPK7 expression was decreased (1.6‐fold, p = 0.04, Figure 1E), whereas the expression of miR‐374b was elevated (four‐fold, p = 0.01, Figure 1F). We assessed the expression pattern and level of miR‐374b by fluorescence in situ hybridization in mice with aortic banding. At atheroprotected sites, miR374b levels were low and primarily present in endothelial cells, whereas miR‐374b was evident in the endothelium (CD31‐positive cells) at atheroprone regions where its abundance was increased ∼three‐fold (p < 0.001; Figure 1G).
Figure 1.
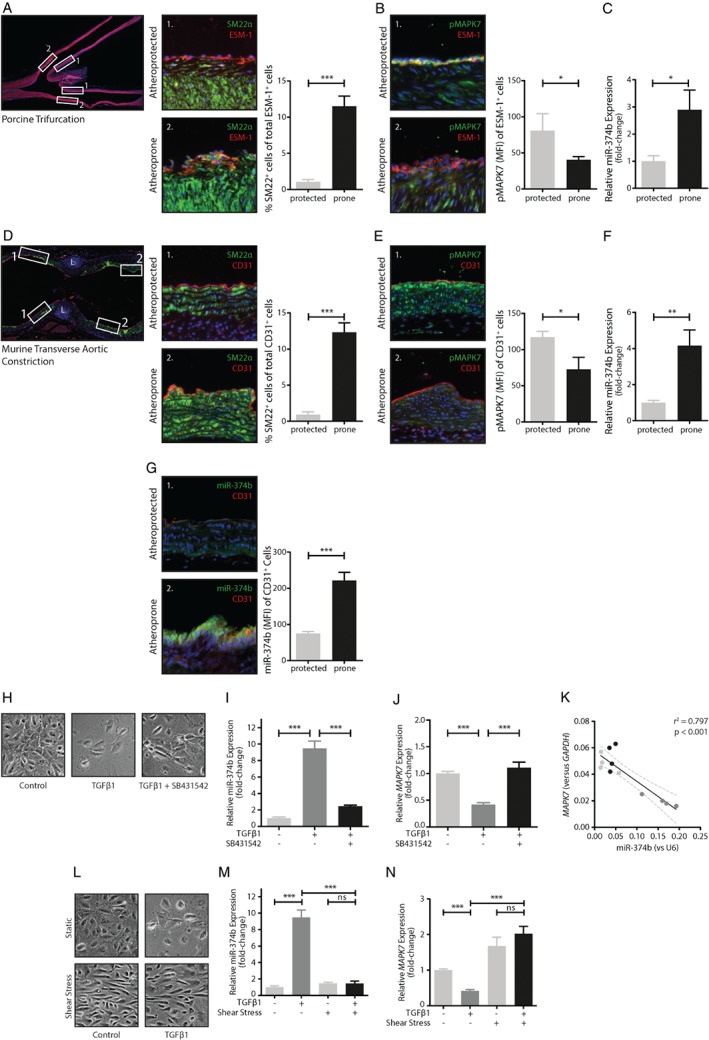
MicroRNA‐374b and MAPK7 are differentially expressed in early lesions. (A) Porcine intimal hyperplastic lesions contain myoendothelial cells (myoEC) that coexpress the endothelial cell marker ESM‐1 (red) and mesenchymal cell marker SM22α (green). Nuclei are stained with DAPI (blue). The atheroprotected regions are indicated by 1, and atheroprone regions are indicated by 2. (B) The expression of MAPK7 (pMAPK7, green) is reduced in endothelial cells (ESM‐1, red) in the atheroprone regions of the porcine trifurcation (n = 5), whereas in (C), the expression of miR‐374b was increased in the atheroprone areas compared to atheroprotected areas of the same porcine trifurcation (n = 5). Mice were subjected to TAC. (D) Eight weeks after TAC, myoEC that coexpress the endothelial cell marker CD31 (red) and the mesenchymal cell marker SM22α (green) were detected at the areas exposed to disturbed flow (annotated 2, n = 5, *=ligation). (E) The expression of MAPK7 (pMAPK7, green) was reduced in endothelial cells (CD31, red) in the areas exposed to disturbed flow compared to areas exposed to laminar flow (n = 5), which coincides with (F) an increase in the expression of miR‐374b (n = 5). (G) The expression of miR‐374b was confirmed by fluorescence in situ hybridization, which indicates a strong endothelial signal, especially at aortic regions exposed to disturbed shear stress (n = 4). H) In vitro, endothelial cells treated with TGFβ1 were hyperplastic (which was inhibited by the ALK5‐inhibitor SB431542 at 10 μm) and (I) increased their expression of miR‐374b (n = 6). (J) The expression of MAPK7 was reduced in TGFβ1‐stimulated endothelial cells, compared to untreated control cells or endothelial cells treated with TGFβ1 and the ALK5‐inhibitor SB431542 (n = 6). (K) The expression levels of miR‐374b were inversely associated with MAPK7 expression levels (n = 6). (L) Laminar fluid shear stress (20 dyne·cm2) antagonized the cellular hypertrophy induced by TGFβ1 in endothelial cells and (M) inhibited the increase in miR‐374b expression (n = 5). (N) Concurrently, when exposed to fluid shear stress, MAPK7 expression was unaltered in TGFβ1‐treated endothelial cells compared to unstimulated control cells (n = 5). T‐test for comparison between two groups; one‐way ANOVA for analyses between multiple groups. *p < 0.05, **p < 0.01, ***p < 0.001.
In vitro, endothelial cells treated with TGFβ1 displayed cell death and hypertrophy (Figure 1H) and had increased levels of miR‐374b (∼nine‐fold compared to nonstimulated cells, p < 0.001), which was completely abolished by the addition of a small molecule inhibitor of ALK5 (SB431542) (Figure 1H,I). The hypertrophic endothelial cells are reminiscent of senescent endothelial cells; however, no change in expression of senescence‐associated (SA) expression of p16INK4 or p21 was detected, nor was a change in SA‐β‐Galactosidase expression found (see supplementary material, Figure S4). The increase in miR‐374b expression associated with decreased expression of MAPK7 (r 2 = 0.797, p < 0.001, Figure 1J,K) and intermediates of MAPK7 signaling, that is, Rac1 (r 2 = 0.595, p < 0.01), MAP3K7 (r 2 = 0.688, p < 0.01), MAPK7 (r 2 = 0.797, p < 0.01), MEF2D (r 2 = 0.682, p < 0.001), and KLF4 (r 2 = 0.555, p < 0.01, see supplementary material, Figure S5A), as well as a decreased expression of endothelial markers, CDH5 (VE‐cadherin; r 2 = 0.678, p < 0.001) and NOS3 (eNOS; r 2 = 0.546, p < 0.01, see supplementary material, Figure S5B) and increased expression of mesenchymal markers TAGLN (SM22α; r 2 = 0.872, p < 0.001) and CNN1 (r 2 = 0.814, p < 0.001, see supplementary material, Figure S5C).
Under LSS, endothelial cells aligned in the direction of flow, indicating mechanosensory behavior (Figure 1L). In contrast to the static cell cultures, endothelial cells exposed to LSS did not became hypertrophic and did not increase their expression of miR‐374b, even when exogenous TGFβ1 was applied (Figure 1M). Corroboratively, MAPK7 expression, which is diminished by TGFβ1 in static cultures, remained high under LSS even in the presence of TGFβ1 (Figure 1N).
MiR‐374b targets multiple members of the MAPK7 signaling cascade
Computational analysis of putative miR‐374b targets identified multiple members of the MAPK7 signaling cascade (Figure 2A). Reporter assays, wherein the 3′‐UTR regions of miR‐374b gene targets are coupled to a luciferase gene, demonstrated that the MAPK7 signaling members Rac1, MAP3K3, MAP3K7, MAPK7, MEF2d, and KLF4 are genuine miR‐374b target genes (Figure 2B) because cotransfection of these reporter plasmids with miR‐374b mimics reduced luciferase activity (all p < 0.01). In contrast, cotransfection of reporter plasmids with a scrambled miR‐374b sequence did not alter luciferase activity (Figure 2B). MEF2a and MEF2c were identified as putative gene targets of miR‐374b by in silico analyses; however, cotransfection of their respective reporter constructs with miR‐374b mimics did not result in decreased luciferase activity (Figure 2B), implying that MEF2a and MEF2c are not genuine targets of miR‐374b.
Figure 2.
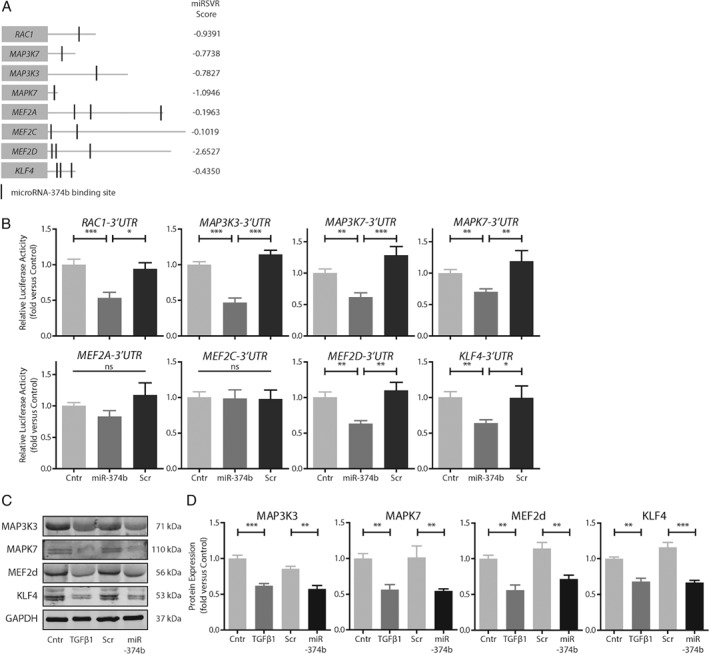
MicroRNA‐374b interferes with MAPK7 signaling at multiple levels. (A) In silico analysis (TarBase 24) demonstrates that miR‐374b putatively targets multiple genes within the MAPK7 signaling cascade with different efficacies (miRSVR‐scores indicate the predicted target site efficacy 20). (B) Cotransfection of 3′‐UTR reporter constructs with miR‐374b mimics or scrambled control sequences in COS7 cells suggests that Rac1, MAP3K3, MAP3K7, MAPK7, MEF2D, and KLF4 are genuine miR‐374b target genes (n = 5). (C, D) Transfection of miR‐374b mimics in endothelial cells mimics the TGFβ1‐induced decrease in expression of MAPK7 signaling members (n = 3). One‐way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001.
To establish if MAPK7 signaling members are in fact endogenous miR‐374b targets in mature endothelial cells, we used lentiviral overexpression of miR‐374b or its scrambled sequence. Unstimulated control cells readily expressed MAPK7 and its signaling members (Figure 2C). Stimulation of mature endothelial cells with TGFβ1 drastically reduced the expression of these molecules (Figure 2C) to approximately 50% of control endothelial cell levels (all p < 0.01). Endothelial cells that expressed miR‐374b had reduced expression of MAP3K3 (∼33%, p < 0.01), MAPK7 (∼47%, p < 0.01), MEF2d (∼37%, p < 0.01), and KLF4 (∼42%, p < 0.001) compared to endothelial cells treated with scrambled controls (Figure 2C). We questioned if the loss of MAPK7 expression would also augment MAPK7 signaling activity and therefore assessed the expression of MAPK7 gene targets KLF2, KLF4 and endothelial nitric oxide synthase (NOS3). Endothelial cells that express miR‐374b had decreased gene expression of KLF2 (∼44%, p < 0.001), KLF4 (∼45%, p < 0.01) and NOS3 (∼41%, p < 0.01). As KLF2 and NOS3 do not have a binding site for miR‐374b in their 3′‐UTR, these data indicate that miR‐374b decreases endogenous MAPK7 signaling beyond direct targets of miR‐374b (see supplementary material, Figure S6).
TGF‐β1 suppresses MAPK7 expression through induction of miR‐374b and causes endothelial–mesenchymal transition
We questioned whether ectopic expression of miR‐374b in endothelial cells would facilitate EndMT. Lentiviral expression caused a ∼four‐fold increase of miR‐374b in endothelial cells (Figure 3A). Consequently, endothelial cells lost their typical cobblestone morphology and started to show signs of hypertrophy (Figure 3B). Consistent with EndMT, the expression of typical endothelial cell markers, that is, VE‐cadherin and eNOS, was lost (Figure 3B,C), whereas expression of mesenchymal cell markers, that is, SM22α and Calponin 1 (CNN1), was induced (Figure 3B,C). In addition, miR‐374b gain of function in endothelial cells reduced the ability to form capillary‐like sprouts on Matrigel (Figure 3D) and fostered the gain of contractile behavior (Figure 3E), all consistent with mesenchymal transition.
Figure 3.
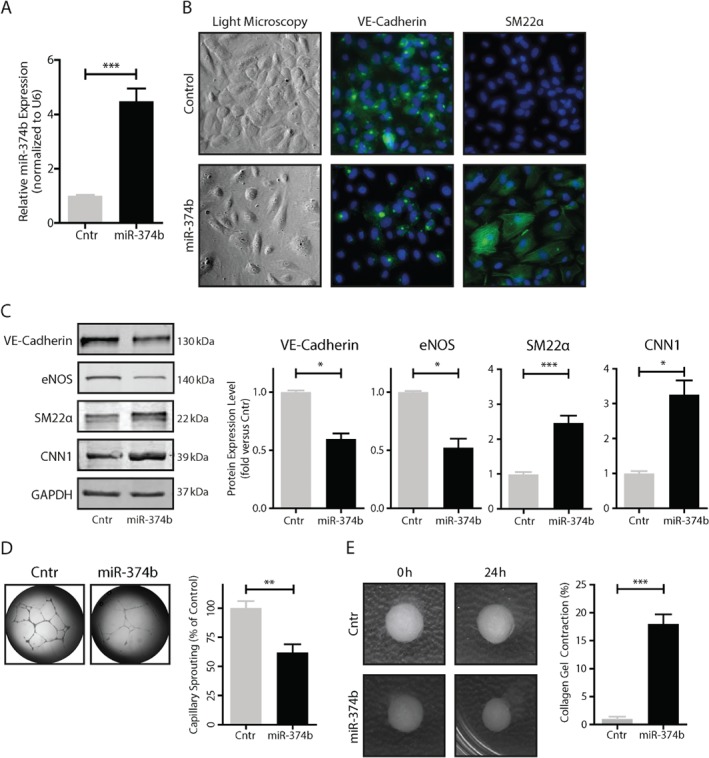
MicroRNA‐374b gain‐of‐function induces EndMT. (A) Transformation of endothelial cells with a lentivirus encoding the stemloop sequence of miR‐374b increased its expression ∼four‐fold. (B) MiR‐374b‐expressing endothelial cells were hypertrophic and decreased their expression of the endothelial cell marker VE‐cadherin and increased expression of the mesenchymal cell marker SM22α, compared to endothelial cells that expressed a scrambled control sequence. (C) VE‐cadherin and eNOS expression were decreased in endothelial cells expressing miR‐374b, whereas the expression of SM22α and CNN1 was increased. (D) Endothelial cells expressing miR‐374b had a reduced angiogenic sprouting capacity and (E) increased contractile capacity compared to endothelial cells that expressed a scrambled control sequence. All n = 5, t‐test for comparison between two groups, *p < 0.05, **p < 0.01, ***p < 0.001.
Loss of specific miR‐374b targets induces endothelial–mesenchymal transition
We questioned whether the loss of function of specific miR‐374b targets would suffice for EndMT induction and used a shRNA approach to specifically decrease MAP3K3, MAPK7, MEF2d, or KLF4 expression in endothelial cells (see supplementary material, Figure S7). Decreased expression of MAPK7 signaling members caused the dissociation of endothelial cell–cell contacts and decreased the expression of VE‐cadherin. Concurrently, the expression of SM22α was increased upon loss of MAPK7 signaling (Figure 4A). The expression of endothelial markers VE‐cadherin and eNOS was abrogated, and the expression of mesenchymal markers SM22α and Calponin was induced (Figure 4B,C). Moreover, angiogenic sprouting ability decreased (Figure 4D). In contrast to the EndMT induction by the loss of downstream MAPK7 signaling members, MAP3K3 deficiency did not alter the expression of endothelial or mesenchymal marker proteins (Figure 4B,C). Yet, angiogenic sprouting ability was lost (Figure 4D), and contractile behavior acquired (Figure 4E) by endothelial cells was deficient in MAP3K3.
Figure 4.

Knockdown of specific microRNA‐374b gene targets induces EndMT. (A) Transformation of endothelial cells with lentiviruses encoding shRNA sequences to MAP3K3, MAPK7, MEF2D, and KLF4 induced cellular hypertrophy, decreased the expression of the endothelial cell marker VE‐cadherin, and increased expression of the mesenchymal cell marker SM22α, compared to endothelial cells that expressed scrambled control sequences. (B, C) VE‐cadherin and eNOS expression were decreased in endothelial cells expressing shMAPK7, shMEF2D, and shKLF4, whereas their expression was unaltered in endothelial cells expressing shMAP3K3. The expression of SM22α and CNN1 was increased in endothelial cells expressing shMAPK7, shMEF2D, and shKLF4. (D) Endothelial cells expressing shRNA sequences to MAP3K3, MAPK7, MEF2D, and KLF4 had a reduced angiogenic sprouting capacity and (E) increased contractile capacity compared to endothelial cells that expressed a scrambled control sequence. All n = 6, one‐way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001.
Maintenance of endothelial MAPK7 signaling abrogates miR‐374b‐induced endothelial–mesenchymal transition
To investigate whether restoration of MAPK7 signaling members could block miR‐374b‐induced EndMT, we used lentiviral expression of miR‐374b together with the protein‐coding sequences of MAP3K3, MAPK7, MEF2D, and KLF4 (Figure 5A). VE‐cadherin expression was reduced in endothelial cells that express miR‐374b, whereas the expression of SM22α was induced. In contrast, cells that coexpressed MAP3K3 or MAPK7 together with miR‐374b maintained their expression of VE‐cadherin and eNOS and were refractory to the induction of SM22a and Calponin by miR‐374b (Figure 3B–D), whereas cells coexpressing KLF4 and miR‐374b maintained only the expression of eNOS (Figure 3C,D). Restoration of MEF2D in endothelial cells expressing miR‐374b inhibited the expression of the mesenchymal proteins SM22α and Calponin but failed to maintain the expression of endothelial marker proteins VE‐cadherin and eNOS (Figure 3B–D). Restoration of all MAPK7 signaling members in endothelial cells expressing miR‐374b maintained the angiogenic ability (Figure 3E) and precluded the contractile behavior induced by miR‐374b expression (Figure 3F).
Figure 5.
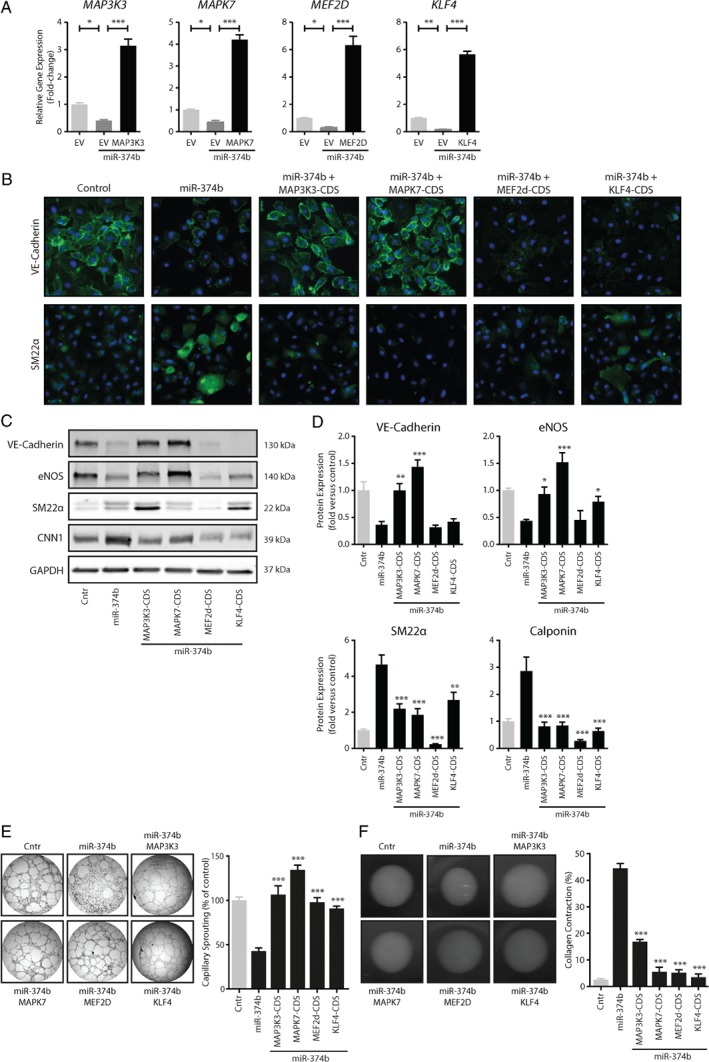
Re‐expression of microRNA‐374b target genes inhibits EndMT. (A) The expression of miR‐374b target genes was restored by lentiviral transformation using plasmids encoding the coding sequences (CDS) of MAP3K3, MAPK7, MEF2D, and KLF4 in endothelial cells that overexpressed miR‐374b. The (re‐)expression of these genes was confirmed by RT‐qPCR. (B–D) Endothelial cells that express miR‐374b decreased their expression of VE‐cadherin and increased their expression of SM22α. These effects were counteracted by the expression of MAPK7 signaling members. (E) Endothelial cells that expressed MAP3K3, MAPK7, MEF2D, or KLF4 have an enhanced angiogenic sprouting capacity and (F) decreased contractile capacity compared to endothelial cell that express miR‐374b. The levels of angiogenic sprouting and contractile behavior were similar to that of endothelial cells transformed using scrambled control sequences. All n = 5, one‐way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001.
MicroRNA‐374b levels are increased in human coronary artery stenosis
We assessed the expression levels of miR‐374b and MAPK7 in samples from progressive human coronary artery stenosis. Increasing intima media thickness (IMT) is associated with the severity of coronary stenosis (Figure 5A,B) and increased miR‐374b expression (Figure 6C). Moreover, miR‐374b levels were associated with IMT (r 2 = 0.5874, p < 0.01, Figure 6D). Conversely, MAPK7 expression levels progressively decrease with increasing stenosis (Figure 6E). In human coronary artery stenosis, the levels of miR‐374b are associated with the expression levels of MAPK7 (r 2 = 0.3341, p < 0.01, Figure 6F), suggesting that the miR‐374b‐induced loss of MAPK7 signaling might contribute to stenosis development and progression.
Figure 6.
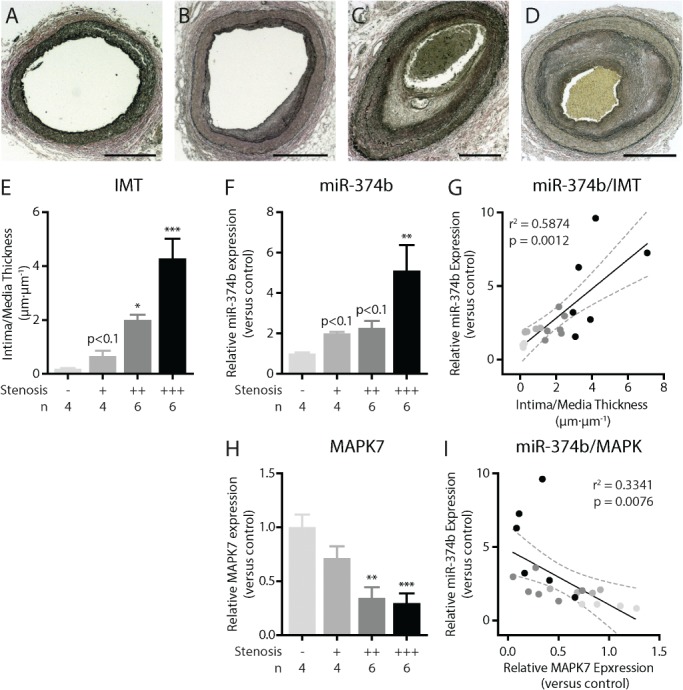
MicroRNA‐374b is increased in human coronary artery stenosis. (A–D) Verhoeff stain of progressive coronary artery stenosis characterized by (E) an increasing intima/media ratio. (F) miR‐374b expression is elevated in human coronary artery stenosis and (G) correlated to the degree of stenosis. (H) Conversely, the expression levels of MAPK7 decrease with progressive stenosis and (I) are inversely associated to the expression level of miR‐374b. One‐way ANOVA for comparison between groups, Pearson correlations, two‐tailed. **p < 0.01, ***p < 0.001.
Discussion
Here, we have shown that EndMT occurs in intimal hyperplasia and early lesion formation and is governed in part by microRNA‐374b. We have previously identified the inhibitory effects of MAPK7 signaling on EndMT 5 and questioned whether MAPK7 signaling is silenced at atheroprone areas in the vasculature. We uncovered that the TGFβ‐induced microRNA‐374b silences MAPK7 signaling and induces EndMT in the absence of exogenous TGFβ (see supplementary material, Figure S8). Moreover, restoration of MAPK7 signaling abrogated these pathological effects in endothelial cells expressing miR‐374b. Interestingly, we uncovered that miR‐374b levels are elevated in human coronary artery disease and inversely related with MAPK7 expression. These data suggest that the TGFβ‐miR‐374b‐MAPK7 axis plays a detrimental role in the induction of EndMT during intimal hyperplasia and early lesion formation and might pose an interesting target for antiatherosclerosis therapy.
Atherosclerosis is characterized by systemic risk factors, and antiatherosclerosis therapies are focused on maintaining these systemic contributors within clinically acceptable ranges (e.g. antihypertensives, anti‐inflammatory agents, and lipid‐lowering drugs) 25, 26, 27. Yet, it is becoming increasingly clear that focal risk factors, such as fluid shear stress levels, play a major role in the pathogenesis of atherosclerosis. Indeed, endothelial MAPK7 signaling has been identified as a major contributor to the initiation and severity of atherosclerosis 5, 12. The atheroprotective effects of MAPK7 include anti‐inflammatory effects, the reduction of oxidative stress, and the increased biosynthesis of nitric oxide 28, 29, 30, which decrease smooth muscle cell proliferation and inflammatory cell infiltration into the atherosclerotic neointima. We recently uncovered that endothelial MAPK7 signaling additionally confers atheroprotective effects through the inhibition of EndMT 5, a process increasingly recognized in the initial phases of intimal hyperplasia and early lesion formation 5, 6, 7, 31. Indeed, the inhibition of MAPK7 activity by SUMOylation increases atherosclerosis 32, 33, and endothelial‐specific deletion of MAPK7 aggravates atherosclerosis development and progression 12. MAPK7 activity culminates in the increased expression of SMAD7 34, 35, a repressor of canonical TGFβ signaling, which might explain the inhibitory effects on EndMT by MAPK7 expression (see supplementary material, Figure S8).
Here, we have used two models of early lesion formation, where biomechanics play a major role. First, the porcine trifurcation model of early lesion formation is a well‐established preclinical model where pigs on a normal diet develop naturally occurring early lesions that closely resemble early lesions in humans 21. In these lesions, there is abundant lipid 22, and smooth muscle cell accumulation is evident at an early age. If the cholesterol diet of the animals is raised for a number of months, these early lesions progress into elastic–hyperplastic lesions and then to lesions that contain a necrotic core 22, 23. We recently identified a high number of EndMT‐derived fibroproliferative cells in the early lesions of porcine trifurcations coexpressing markers of the endothelium and mesenchymal lineage 5. Second, the murine model of thoracic aortic constriction for the induction of cardiac failure was previously described by us to induce robust EndMT just adjacent to the aortic constriction 5. In the current study, we corroborate these findings by the identification of cells coexpressing the endothelial cell marker ESM‐1 (porcine model) or CD31 (murine model) and the mesenchymal cell marker SM22α. We extend these observations by showing a loss of MAPK7 expression at these atheroprone sites in both models.
Although EndMT is evident in both the porcine and murine model of early lesion formation, we did not investigate whether TGFβ signaling is increased in these lesions. EndMT can be induced by a variety of stimuli, such as TGFβ signaling, hypoxia, inflammatory signaling, changes in endothelial cell metabolism, or the loss of FGF2 signaling 36, 37. Here, we show that altered biomechanical signaling, potentially through MAPK7 signaling, is associated with the induction of EndMT. Interestingly, disturbed endothelial shear stress has been implicated in the activation of TGFβ and inflammatory signaling 38, 39, the reduction of FGF2 signaling 6, and the repression of cell metabolism 40 in endothelial cells.
MicroRNAs are involved in atherosclerosis development and progression 41, and microRNA‐based therapies that target endothelial dysfunction reduce atherosclerosis development in mice 42, 43, 44. MicroRNAs regulate gene expression by imperfect base‐pairing with the 3′‐UTR region of their gene target, causing translational repression 45. This imperfect base‐pairing allows for gene target multiplicity, where one microRNA targets multiple genes with a specific signaling cascade 14. Hence, we questioned if MAPK7 signaling would be regulated by a specific microRNAs and if such microRNA would be differentially expressed at atheroprone and atheroprotected sites. We found that miR‐374b expression is elevated at atheroprone areas in the vessel wall and is associated with decreased MAPK7 expression.
A role for miR‐374b in atherosclerosis has not been described before, yet elevated levels of miR‐374b have been reported in the plasma of acute coronary syndrome patients 46 and in stenosis of the arteriovenous fistulae of dialysis patients 47; however, its relevance in these pathologies remains elusive. Here, we show that miR‐374b is a shear stress‐sensitive microRNA that targets MAPK7 signaling at multiple levels ranging from its upstream activating kinase (i.e. MAP3K3) to its downstream transcription factor (i.e. KLF4). The loss of MAPK7 signaling culminates in the induction of EndMT (see supplementary material, Figure S8) in the absence of exogenous triggers, which contributes to intimal hyperplasia and early lesion formation. Moreover, we show that restoration of MAPK7 signaling in endothelial cells that express miR‐374b abolishes EndMT. From a clinical perspective, our data imply that targeting miR‐374b in atherosclerosis might restore endothelial MAPK7 expression and limit lesion formation through the inhibition of EndMT.
Interestingly, it has been suggested recently that cardiac fibroblasts might transition into endothelial cells upon ischemic stress 48. This mesenchymal–endothelial transition (MEndT) yields cells coexpressing endothelial and mesenchymal cell markers, similar to the cells we describe here. Hence, it is conceivable that a decreased expression of miR‐374b in fibroblasts might facilitate MEndT, culminating in cell expressing both lineage markers. Although we cannot exclude the presence of MEndT by the data presented here, EndMT‐like cells were only found in the subendothelial layer and not in the adventitial layer where the vascular fibroblasts reside, nor did we find a difference in miR‐374b expression in the medial or adventitial cell layers. Moreover, lineage‐tracing studies in atherosclerosis favor the presence of EndMT over MEndT 6. Although these observations would argue against MEndT in atherosclerosis development, further lineage‐tracing experiments should be performed to make a definite conclusion.
In conclusion, here, we have shown that miR‐374b expression is elevated in coronary artery stenosis and early lesion formation and abolishes endothelial MAPK7 expression, culminating in EndMT. The restoration of endothelial MAPK7 expression surmounts the induction of EndMT by miR‐374b.
Author contributions statement
JRM and GK designed the study. BV, EO, JF, MGB, and GK conceived and carried out experiments. BK and AP obtained the human samples and carried out data acquisition. BV, EO, JF, JRM, and GK analyzed and interpreted the data. All authors were involved in writing the paper and had final approval of the submitted and published versions.
SUPPLEMENTARY MATERIAL ONLINE.
Supplementary materials and methods
Figure S1. In silico analysis identifies miR‐374 as putative regulator of MAPK7 signaling
Figure S2. MicroRNA‐374b targets MAPK7 signaling
Figure S3. TGFβ1‐induced microRNA expression
Figure S4. EndMT induction is not associated with senescence
Figure S5. miR‐374b expression levels associated with decreased expression of MAPK7 signaling members and EndMT
Figure S6. miR‐374b induces a loss in MAPK7 signaling activity
Figure S7. RNAi‐mediated repression of MAPK7 signaling members
Figure S8. Schematic representation of miR‐374/MAPK7 signaling in atheroprone and atheroprotected areas
Table S1. Primers used for preparation of 3′‐UTR‐reporter constructs
Table S2. Oligonucleotides used for lentiviral miR‐374b and shRNA expression
Table S3. Primers used for the cloning of MAPK7 signaling members
Table S4. Primers used for microRNA transcript analysis
Table S5. Primers used for mRNA transcript analysis
Table S6. Antibodies used for immunofluorescence
Table S7. Antibodies used for immunoblotting
Supporting information
Supplementary materials and methods
Figure S1. In silico analysis identifies miR‐374 as putative regulator of MAPK7 signaling
Figure S2. MicroRNA‐374b targets MAPK7 signaling
Figure S3. TGFβ1‐induced microRNA expression
Figure S4. EndMT induction is not associated with senescence
Figure S5. miR‐374b expression levels associate with decreased expression of MAPK7 signaling members and EndMT
Figure S6. miR‐374b induces a loss in MAPK7 signaling activity
Figure S7. RNAi‐mediated repression of MAPK7 signaling members
Figure S8. Schematic representation of miR‐374/MAPK7 signaling in atheroprone and atheroprotected areas
Table S1. Primers used for preparation of 3′‐UTR‐reporter constructs
Table S2. Oligonucleotides used for lentiviral miR‐374b and shRNA expression
Table S3. Primers used for the cloning of MAPK7 signaling members
Table S4. Primers used for microRNA transcript analysis
Table S5. Primers used for mRNA transcript analysis
Table S6. Antibodies used for immunofluorescence
Table S7. Antibodies used for immunoblotting
Acknowledgements
BV is supported by the State Training Foundation Scholarship from the Ministry of Education and Science of Mongolia (grant #621). BK was supported by a FAPESP Scholarship from the São Paulo Research Foundation, Brazil (grant #2012/11871‐0). JF is supported by the International Research Training Network on Diabetic Microvascular Complications (GRK/1874 DIAMICOM). MCH is supported by a “Science without Borders”‐grant from the Brazilian Federal Government (grant #401749/2012‐6). JRM is supported by the Dutch Heart Foundation (grant #2013T116) and the Netherlands CardioVascular Research Initiative (CVON): the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Sciences (CVON Phaedra 2012‐08). GK is supported by innovational research incentive grants from the Netherlands Organization for Scientific Research (NWO) and the Netherlands Organization for Health Research and Development (ZonMW, grant #916.11.022 and #917.16.446). Imaging was performed at the UMCG Imaging Center (UMIC), supported by the Netherlands Organization for Health Research and Development (ZonMW grant #40‐00506‐98‐9021).
No conflicts of interest were declared.
References
*Cited only in supplementary material.
- 1. Ikari Y, McManus BM, Kenyon J, et al Neonatal intima formation in the human coronary artery. Arterioscler Thromb Vasc Biol 1999; 19: 2036–2040. [DOI] [PubMed] [Google Scholar]
- 2. Vogel RA. Coronary risk factors, endothelial function, and atherosclerosis: a review. Clin Cardiol 1997; 20: 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol 2009; 10: 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Traub O, Berk BC. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler Thromb Vasc Biol 1998; 18: 677–685. [DOI] [PubMed] [Google Scholar]
- 5. Moonen JA, Lee ES, Schmidt M, et al Endothelial‐to‐mesenchymal transition contributes to fibro‐proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc Res 2015; 108: 377–386. [DOI] [PubMed] [Google Scholar]
- 6. Chen P‐Y, Qin L, Baeyens N, et al Endothelial‐to‐mesenchymal transition drives atherosclerosis progression. J Clin Invest 2015; 125: 4514–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Evrard SM, Lecce L, Michelis KC, et al Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun 2016; 7: 11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krenning G, Moonen JR, van Luyn MJ, et al Vascular smooth muscle cells for use in vascular tissue engineering obtained by endothelial‐to‐mesenchymal transdifferentiation (EnMT) on collagen matrices. Biomaterials 2008; 29: 3703–3711. [DOI] [PubMed] [Google Scholar]
- 9. Moonen JR, Krenning G, Brinker MG, et al Endothelial progenitor cells give rise to pro‐angiogenic smooth muscle‐like progeny. Cardiovasc Res 2010; 86: 506–515. [DOI] [PubMed] [Google Scholar]
- 10. Maleszewska M, Moonen JR, Huijkman N, et al IL‐1beta and TGFbeta2 synergistically induce endothelial to mesenchymal transition in an NFkappaB‐dependent manner. Immunobiology 2013; 218: 443–454. [DOI] [PubMed] [Google Scholar]
- 11. Sinha S, Heagerty AM, Shuttleworth CA, et al Expression of latent TGF‐beta binding proteins and association with TGF‐beta 1 and fibrillin‐1 following arterial injury. Cardiovasc Res 2002; 53: 971–983. [DOI] [PubMed] [Google Scholar]
- 12. Le NT, Heo KS, Takei Y, et al A crucial role for p90RSK‐mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation 2013; 127: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brennecke J, Stark A, Russell RB, et al Principles of microRNA‐target recognition. PLoS Biol 2005; 3: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011; 469: 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghosh AK, Nagpal V, Covington JW, et al Molecular basis of cardiac endothelial‐to‐mesenchymal transition (EndMT): differential expression of microRNAs during EndMT. Cell Signal 2012; 24: 1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumarswamy R, Volkmann I, Jazbutyte V, et al Transforming growth factor‐β–induced endothelial‐to‐mesenchymal transition is partly mediated by microRNA‐21. Arterioscler Thromb Vasc Biol 2012; 32: 361–369. [DOI] [PubMed] [Google Scholar]
- 17. Correia AC, Moonen J‐RA, Brinker MG, et al FGF‐2 inhibits endothelial‐mesenchymal transition through microRNA‐20a‐mediated repression of canonical TGF‐β signaling. J Cell Sci 2016; 129: 569–579. [DOI] [PubMed] [Google Scholar]
- 18. Chen P‐Y, Qin L, Barnes C, et al FGF regulates TGF‐β signaling and endothelial‐to‐mesenchymal transition via control of let‐7 miRNA expression. Cell Rep 2012; 2: 1684–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Betel D, Wilson M, Gabow A, et al The microRNA.org resource: targets and expression. Nucl Acids Res 2008; 36: D149–D153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Betel D, Koppal A, Agius P, et al Comprehensive modeling of microRNA targets predicts functional non‐conserved and non‐canonical sites. Genome Biol 2010; 11: R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murphy EA, Rowsell HC, Downie HG, et al Encrustation and atherosclerosis: the analogy between early in vivo lesions and deposits which occur in extracorporeal circulations. Can Med Assoc J 1962; 87: 259–274. [PMC free article] [PubMed] [Google Scholar]
- 22. Fuster V, Bowie EJW, Lewis JC, et al Resistance to arteriosclerosis in pigs with von Willebrand's disease: spontaneous and high cholesterol diet‐induced arteriosclerosis. J Clin Invest 1978; 61: 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Griggs TR, Reddick RL, Sultzer D, et al Susceptibility to atherosclerosis in aortas and coronary arteries of swine with von Willebrand's disease. Am J Pathol 1981; 102: 137–145. [PMC free article] [PubMed] [Google Scholar]
- 24. Vlachos IS, Paraskevopoulou MD, Karagkouni D, et al DIANA‐TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucl Acids Res 2015; 43: D153–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Back M, Hansson GK. Anti‐inflammatory therapies for atherosclerosis. Nat Rev Cardiol 2015; 12: 199–211. [DOI] [PubMed] [Google Scholar]
- 26. Stone NJ, Robinson JG, Lichtenstein AH, et al Treatment of blood cholesterol to reduce atherosclerotic cardiovascular disease risk in adults: synopsis of the 2013 American College of Cardiology/American Heart Association cholesterol guideline. Ann Intern Med 2014; 160: 339–343. [DOI] [PubMed] [Google Scholar]
- 27. Rosendorff C, Lackland DT, Allison M, et al Treatment of hypertension in patients with coronary artery disease. Hypertension 2015; 65: 1372–1407. [DOI] [PubMed] [Google Scholar]
- 28. Kim M, Kim S, Lim JH, et al Laminar flow activation of ERK5 protein in vascular endothelium leads to atheroprotective effect via NF‐E2‐related factor 2 (Nrf2) activation. J Biol Chem 2012; 287: 40722–40731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clark PR, Jensen TJ, Kluger MS, et al MEK5 is activated by shear stress, activates ERK5 and induces KLF4 to modulate TNF responses in human dermal microvascular endothelial cells. Microcirculation 2011; 18: 102–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ohnesorge N, Viemann D, Schmidt N, et al Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel‐like factor 4 (KLF4). J Biol Chem 2010; 285: 26199–26210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mahmoud MM, Serbanovic‐Canic J, Feng S, et al Shear stress induces endothelial‐to‐mesenchymal transition via the transcription factor snail. Sci Rep 2017; 7: 3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Woo CH, Shishido T, McClain C, et al Extracellular signal‐regulated kinase 5 SUMOylation antagonizes shear stress‐induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res 2008; 102: 538–545. [DOI] [PubMed] [Google Scholar]
- 33. Heo KS, Chang E, Le NT, et al De‐SUMOylation enzyme of sentrin/SUMO‐specific protease 2 regulates disturbed flow‐induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res 2013; 112: 911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boon RA, Fledderus JO, Volger OL, et al KLF2 suppresses TGF‐beta signaling in endothelium through induction of Smad7 and inhibition of AP‐1. Arterioscler Thromb Vasc Biol 2007; 27: 532–539. [DOI] [PubMed] [Google Scholar]
- 35. Lee ES, Boldo LS, Fernandez BO, et al Suppression of TAK1 pathway by shear stress counteracts the inflammatory endothelial cell phenotype induced by oxidative stress and TGF‐beta1. Sci Rep 2017; 7: 42487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Souilhol C, Harmsen MC, Evans PC, et al Endothelial‐mesenchymal transition in atherosclerosis. Cardiovasc Res 2018; 114: 565–577. [DOI] [PubMed] [Google Scholar]
- 37. Xiong J, Kawagishi H, Yan Y, et al A metabolic basis for endothelial‐to‐mesenchymal transition. Mol Cell 2018; 69: 689–698 e687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lum RM, Wiley LM, Barakat AI. Influence of different forms of fluid shear stress on vascular endothelial TGF‐beta1 mRNA expression. Int J Mol Med 2000; 5: 635–641. [DOI] [PubMed] [Google Scholar]
- 39. Punchard MA, Stenson‐Cox C, O'Cearbhaill ED, et al Endothelial cell response to biomechanical forces under simulated vascular loading conditions. J Biomech 2007; 40: 3146–3154. [DOI] [PubMed] [Google Scholar]
- 40. Doddaballapur A, Michalik KM, Manavski Y, et al Laminar shear stress inhibits endothelial cell metabolism via KLF2‐mediated repression of PFKFB3. Arterioscler Thromb Vasc Biol 2015; 35: 137–145. [DOI] [PubMed] [Google Scholar]
- 41. Feinberg MW, Moore KJ. MicroRNA regulation of atherosclerosis. Circ Res 2016; 118: 703–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun X, He S, Wara AK, et al Systemic delivery of microRNA‐181b inhibits NF‐κB activation, vascular inflammation, and atherosclerosis in apolipoprotein E‐deficient mice. Circ Res 2014; 114: 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Loyer X, Potteaux S, Vion AC, et al Inhibition of microRNA‐92a prevents endothelial dysfunction and atherosclerosis in mice. Circ Res 2014; 114: 434–443. [DOI] [PubMed] [Google Scholar]
- 44. Schober A, Nazari‐Jahantigh M, Wei Y, et al MicroRNA‐126‐5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med 2014; 20: 368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ward JA, Esa N, Pidikiti R, et al Circulating cell and plasma microRNA profiles differ between non‐ST‐segment and ST‐segment‐elevation myocardial infarction. Fam Med Med Sci Res 2013; 2: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lv L, Huang W, Zhang J, et al Altered microRNA expression in stenoses of native arteriovenous fistulas in hemodialysis patients. J Vasc Surg 2016; 63: 1034–1043.e3. [DOI] [PubMed] [Google Scholar]
- 48. Ubil E, Duan J, Pillai ICL, et al Mesenchymal‐endothelial‐transition contributes to cardiac neovascularization. Nature 2014; 514: 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burgess WH, Mehlman T, Friesel R, et al Multiple forms of endothelial cell growth factor. Rapid isolation and biological and chemical characterization. J Biol Chem 1985; 260: 11389–11392. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. In silico analysis identifies miR‐374 as putative regulator of MAPK7 signaling
Figure S2. MicroRNA‐374b targets MAPK7 signaling
Figure S3. TGFβ1‐induced microRNA expression
Figure S4. EndMT induction is not associated with senescence
Figure S5. miR‐374b expression levels associate with decreased expression of MAPK7 signaling members and EndMT
Figure S6. miR‐374b induces a loss in MAPK7 signaling activity
Figure S7. RNAi‐mediated repression of MAPK7 signaling members
Figure S8. Schematic representation of miR‐374/MAPK7 signaling in atheroprone and atheroprotected areas
Table S1. Primers used for preparation of 3′‐UTR‐reporter constructs
Table S2. Oligonucleotides used for lentiviral miR‐374b and shRNA expression
Table S3. Primers used for the cloning of MAPK7 signaling members
Table S4. Primers used for microRNA transcript analysis
Table S5. Primers used for mRNA transcript analysis
Table S6. Antibodies used for immunofluorescence
Table S7. Antibodies used for immunoblotting