

Abstract
miRNAs play a central role in the complex signaling network of cancer cells with the tumor microenvironment. Little is known on the origin of circulating miRNAs and their relationship with the tumor microenvironment in lung cancer. Here, we focused on the cellular source and relative contribution of different cell types to circulating miRNAs composing our risk classifier of lung cancer using in vitro/in vivo models and clinical samples. A cell‐type specific expression pattern and topography of several miRNAs such as mir‐145 in fibroblasts, mir‐126 in endothelial cells, mir‐133a in skeletal muscle cells was observed in normal and lung cancer tissues. Granulocytes and platelets are the major contributors of miRNAs release in blood. miRNAs modulation observed in plasma of lung cancer subjects was consistent with de‐regulation of the same miRNAs observed during immunosuppressive conversion of immune cells. In particular, activated neutrophils showed a miRNA profile mirroring that observed in plasma of lung cancer subjects. Interestingly mir‐320a secreted by neutrophils of high‐risk heavy‐smokers promoted an M2‐like protumorigenic phenotype through downregulation of STAT4 when shuttled into macrophages. These findings suggest a multifactorial and nonepithelial cell‐autonomous origin of circulating miRNAs associated with risk of lung cancer and that circulating miRNAs may act in paracrine signaling with causative role in lung carcinogenesis and immunosuppression.
Keywords: microRNA, lung cancer, microenvironment
Short abstract
What's new?
microRNAs play a central role in the complex signaling network of cancer cells with the tumor microenvironment. However, little is known on the origin of circulating miRNAs and their mechanisms of action. This study found a multifactorial and non‐epithelial cell‐autonomous origin of circulating miRNAs associated with lung cancer risk. The findings also suggest a link between an immunosuppressive and pro‐tumorigenic microenvironment and modulation of circulating miRNAs associated with lung cancer risk. The authors propose a novel mechanism whereby miRNA released by neutrophils induce macrophage polarization to support lung cancer growth, highlighting the potential for reprogramming macrophages toward an anti‐tumor polarization.
Introduction
Lung cancer takes more lives annually than breast, pancreas and prostate cancer combined in Europe.1
The importance of identifying noninvasive biomarkers for lung cancer risk and diagnosis to reduce patient's mortality has been investigated by several studies.2, 3 Increasing attention is being paid to the role of microRNAs (miRNAs), small noncoding RNA molecules with regulatory function on gene expression, in lung carcinogenesis.4
Numerous studies have reported the diagnostic and prognostic potential of the detection of miRNAs circulating in plasma or serum in retrospective clinical series,5 but their biological significance and functional role are currently unknown.
Recently, miRNA‐based biomarkers have been described with prognostic or predictive potential for response to immunocheckpoint inhibitors.6
The use of circulating miRNAs for early detection of lung cancer has been assessed in the context of LDCT screening in large retrospective studies.7, 8 Notably, we have described a lung cancer risk classifier based on reciprocal ratios of 24 plasma miRNAs (miRNA‐signature classifier, MSC) with predictive, diagnostic, and prognostic potential in heavy smokers enrolled in two independent screening trials for lung cancer with low dose computed tomography (LDCT).7, 9 The diagnostic performance of MSC for lung cancer detection showed 87% sensitivity and 81% specificity with a five‐fold reduction of LDCT false‐positive rate.9 A serum miRNA signature, called miR‐Test, based on 13 circulating miRNAs with high sensitivity and specificity (77.8% and 74.8%, respectively) for early detection of lung cancer was also reported.10
Nowadays, the two tests, miR‐test and MSC, are being independently validated in prospective screening trials enrolling high‐risk subjects.
Interestingly, the classifier was able to identify a risk profile up to two years before a significant finding by LDCT. This observation leads us to hypothesize that circulating miRNAs may not therefore primarily originate from tumor tissue but rather from the lung microenvironment reflecting a shift toward a tumor‐supporting milieu as a result of continuous exposure to inflammatory and damaging stimuli due to cigarette smoking.11
Hematopoietic and stromal cells are among the major contributors of miRNAs in the circulatory system12 and changes in their amount or phenotype in relation to specific stress conditions might alter their miRNA profile and consequently circulating miRNA levels.
Cell‐specific miRNA expression and the role of miRNAs in the lung microenvironment have not been extensively analyzed so far. The intrinsic cellular heterogeneity of tissues makes it difficult to unequivocally identify cellular origins for different miRNAs species.13 Nonetheless, the knowledge of cell type‐restricted miRNA expression has extreme biological relevance for the planning of appropriate functional studies and for the development of new miRNA‐based therapeutic strategies.
In the present study we aimed to identify the cells of origin and their contribution to the circulating miRNAs composing our lung cancer risk classifier. To this purpose: (i) we assessed modulation of circulating miRNAs in plasma samples of lung cancer patients before and after surgery, (ii) evaluated miRNAs expression levels and their secretion in conditioned medium of different cell lineages and (iii) analyzed cell‐specific localization of miRNAs with in situ hybridization (ISH) in lung tissues. Finally, the potential role of specific circulating miRNAs to induce a pro‐tumorigenic lung microenvironment in heavy‐smokers by the modulation of the immune contexture was assessed.
Materials and Methods
Clinical specimens
Tissue and blood were collected from high‐risk heavy smoker volunteers (age‐range: 50 to 75 years) including current or former smokers with a minimum pack/year index of 20, enrolled in two independent LDCT screening trials performed at our Institution.14 For miRNA analysis, lung tissue samples from twenty lung cancer patients from the Multicentric Italian Lung Detection (MILD) trial were selected;15 in addition, whole blood samples from subjects from the BioMILD trial (a biomarker‐directed follow‐up trial of the MILD trial) were selected for blood cells isolation and polarization. Tissue and blood specimens were obtained according to the Internal Review and the Ethics Boards of the Fondazione IRCCS Istituto Nazionale Tumori of Milan. All patients provided informed consent.
Cell lines and primary cell cultures
Immortalized bronchial‐epithelial cells and their genetically modified variants (HBEC3KT: hTERT+Cdk4; HBEC3KT‐p53: hTERT+Cdk4 + shp53; HBEC3KT‐p53/KRAS: hTERT+Cdk4 + sh‐p53 + KRASV12; HBEC3KT‐p53/KRAShigh: hTERT+Cdk4 + sh‐p53 + KRASV12high) were obtained from Prof J. Minna (UT Southwestern, TX) and were described previously.16 Human lung cancer cell lines (A549, Calu‐1, H1299 and H460) were obtained from the American Type Culture Collection (ATCC, LGC Standards), primary lung cancer cell line LT73 was established in our laboratory, whereas the human primary cells HUVEC (Human Umbelical Vascular Endothelial Cells), NHBE (Normal Human Bronchial Epithelial Cells), SAEC (Small Airway Epithelial Cells), SkMC (Skeletal Muscle Cells) and BSMC (Bronchial Smooth Muscle Cells) were obtained from Lonza (Lonza, Basel, Switzerland). All immortalized cell lines were authenticated by DNA short tandem repeat (STR) profiling and confirmed to be mycoplasma negative. Cultures of primary fibroblasts from different areas of surgical specimens, including lung cancer (CAF, cancer associated fibroblasts) and normal tissue (NF, normal fibroblasts) were isolated in our laboratory. These cultures have been characterized immunophenotypically for expression of mesenchymal markers (CD90, CD105, CD73, CD166) to confirm their stromal origin and for activation markers (alpha‐SMA, FAP). For the in vitro modulation of miRNA endothelial cells were incubated in hypoxia (5% oxygen incubator) for 48 h.
MiRNA transfection
Macrophages were transfected with mirVana mimics (50 nM) using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions.
miRCURY LNA inhibitor (anti‐miR‐320, Exiqon Denmark), or scrambled LNA were transfected into macrophages 24 h before adding PMNs CM to assess the transfer of mir‐320 into these cells.
Flow Citometry
Macrophages were stained with CD163‐PE or CD206‐FITC (Biolegend, San Diego) for 15 min at room temperature in the dark; then cells were washed, resuspended in PBS and analyzed. Total events were analyzed using BD FACSCalibur with FlowJo software.
Cell proliferation
Five thousand A549‐GFP cells were spotted on a 96‐wells plate and CM from different treated macrophages was added to cells. GFP signal was measured every 24 h until 72 h with the Infinite M1000 (Tecan Trading AG, Switzerland).
miRNAs and gene expression analysis
RNA from cells and relative CM (600ul) was extracted using mirVana Paris kit (Thermo Fisher Scientific, Massachusetts, USA). Retro‐transcription reaction and pre‐amplification step were performed accordingly with the “Protocol for Creating Custom RT and Pre‐amplification Pools with TaqMan MicroRNA Assays” (Thermo Fisher Scientific) as previously described.17 Cellular miRNA expression was analyzed with custom microfluidic cards using U6 as normalizer (Thermo Fisher Scientific). Digital PCR for CM analysis was performed as described by Conte et al.18 For gene expression analysis, cDNA synthesis was performed using 250 ng of total RNA. The relative quantification of the analyzed genes was performed using Taqman assay (Thermo Fisher Scientific) and GAPDH was used as endogenous control.
Hierarchical clustering of genes and samples was performed using BRB‐ArrayTools. Genes were centered and scaled, using one minus correlation as metric and average linkage. Centered correlation was adopted for cluster analysis of samples.
ISH analysis of miRNAs
miRNA‐in situ hybridization was performed on FFPE tissue sections and PBMCs cell‐blocks as previously described by Gualeni et al.19 The protocol is based on combination of double DIG‐conjugated mirCURY locked nucleic acid (LNA) probes (Exiqon,Vedbæk, Denmark) and an automatic DAB‐chromogenic detection system, that together enable specific and sensitive detection of miRNAs.
Probes selected for ISH analysis are listed in Supporting Information Table 1. A scramble probe (sequence with no homology to any known miRNA or mRNA sequences) was used as negative control.
Prior to use, each probe was pre‐denatured at 90 °C for 4 min and diluted with ISH buffer (Exiqon,Vedbæk, Denmark) to final concentrations of 100 nM for miRNA specific probe and 40 nM for scrambled‐negative control probe.
In brief, FFPE sections (5 μm thick) were first deparaffinized in xylene, rehydrated on alcohol descending scale and treated with Proteinase‐K (Sigma‐Aldrich, Saint Louis, Missouri, USA) diluted 1:200, for 30 or 15 min for tissue or cell block sections respectively, at 37 °C on a Dako Hybridizer (Dako, Glostrup, Denmark). Slides were washed in PBS 1X, dehydrated in alcohol increasing scale and air‐dried.
Samples were hybridized with probe mixture for 2 h, in the Dako Hybridizer, at specific probe hybridization temperature (RNA Tm − 30 °C) (Supporting Information Table 1). Slides were then stringently washed in 5X SSC, 1X SSC, 0.2X SSC at the hybridization temperature for 5 min each and in 0.2X SSC at room temperature for 5 min. after a wash in distilled water and one in Reaction Buffer 1X (Ventana Medical Systems, Arizona, USA), the miRNA expression was automated detected with the Ventana BenchMark ULTRA instrument using the OptiviewDAB Detection Kit (Ventana Medical Systems Arizona, USA).
For image analysis, stained sections were examined by optical microscope and scanned with Aperio Scanscope XT (Leica Biosystems, Nussloch, Germany), that enables to view, manage and analyze the slides with high image quality. For the analysis of positivity, we excluded tissue macrophages form these analyses due to their nonspecific binding. miRNAs signals were quantified in terms of positive cells on total cells by counting three random field for each slides. An arbitrary cut‐off (above 20% of positive cells) was set to define positivity.
In silico analysis
Targetscan predictions are ranked based on the predicted efficacy of targeting as calculated using cumulative weighted context++ scores of the sites20 and also ranked by their probability of conserved targeting (PCT).21 STAT4 was identified with an exact match to positions 2–8 of the mature miRNA for a conserved binding site with cumulative weighted context++ of −0.24 and PCT of −0.24. Mirwalk performed comparative platform of miRNA binding site predictions within 3′‐UTR region identifying STAT4 binding sites for mir‐320 in three different algorithms.
Luciferase assays
To investigate whether STAT4 is a direct target of mir‐320, the 3′ untranslated region (UTR) of STAT4 was purchased from Switchgear Genomics. Conserved binding sites in STAT4 3′UTR at position 87–93 was identified using TargetScan (http://www.targetscan.org). An empty vector was used as control. Furthermore, the predicted target site for miR‐320 was mutated by direct mutagenesis of the pLightSwitch_STAT4 3′UTR vector, using the PCR‐based QuikChange II XL site‐directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions and the after primers:
5′‐CCACATTTTATTTCTTCCAACCTGTAAATACCAGGTTC‐3′;
Rev 5′‐ GAACCTGGTATTTACAGGTTGGAAGAAATAAAATGTGG‐3′.
The presence of the mutations was confirmed by sequencing (Eurofins Genomics). The different luciferase constructs were transfected into HEK293 cells together with miR‐320 or a scrambled oligonucleotide sequence (mim‐ctr). Cells were cultured for 48 h and assayed with the Luciferase Reporter Assay System (Switchgear Genomics).
ELISA
VEGF secretion in CM of macrophages were analyzed using Human Angiogenesis ELISA Strip I kit according to manufacturer's instructions (Signosis, Inc. Santa Clara, CA).
Western blot analysis
Western blot analysis was performed as described in Fortunato et al.22 using the after antibodies: rabbit anti‐STAT4 (1:1000, Cell Signaling), mouse anti‐actin (1:2000, Sigma Aldrich) and goat anti‐rabbit or goat anti‐mouse secondary antibodies conjugated to horseradish peroxidase (1:5000, GE Healthcare). Signal detection was performed via chemiluminescence reaction (ECL, GE Healthcare) using MINI HD9 Western Blot Imaging System (Cleaver Scientific Ltd, United Kingdom). WB quantification was performed using ImageJ software analysis.
Migration and invasion assay
Migratory and invasive ability of A549 treated cells with CM of macrophages were assessed as previously described.22
Immunohistochemistry and immunofluorescence staining
Cell expression markers were investigated by immunohistochemistry methods. Briefly sections 2.5/3 μm‐thick were cut from paraffin blocks, dried, de‐waxed, rehydrated, and unmasked with Dako PT‐link (EnVision FLEX Target Retrieval Solution, High pH‐ 15 min). Anti‐α‐SMA monoclonal antibody (1:800), CD31 (1:200), CD3 (1:400), TTF1 (1:2000) and pan‐cytokeratin (1:100) (all Dako, Denmark), were incubated with a commercially available detection kit (EnVision FLEX+, Dako, Denmark) in an automated immunostainer (Dako Autostainer System). Immunofluorescence staining was performed by macrophages with appropriately dilution of anti‐CD206 (1:100, Biolegend San Diego CA, USA) for 1 h at RT, followed by secondary fluorescence‐conjugated antibodies for 1 h at room temperature. Image acquisition was carried out with Olympus BX51 microscopy (Tokyo, Japan).
In vivo tumor growth
Animal studies were performed according to the Ethics Committee for Animal Experimentation of the Fondazione IRCCS Istituto Nazionale dei Tumori, according to institutional guidelines.23 All experiments were carried out with female SCID mice, 7–10 weeks old (Charles River Laboratories). Twenty thousand A549 cells and macrophages (1:3), were subcutaneously injected with Matrigel/RPMI (1:1) and tumor growth was measured weekly using caliper.
Conditioned medium
Cells (tumor, normal bronchial and small airway epithelial, fibroblasts, blood cells, smooth and skeletal muscle and HUVEC) were seeded at 1 × 106 cells/mL and serum free‐conditioned medium (CM) was obtained after 48 h.
Statistical analysis
The Sign Test was applied considering positive or negative the miRNA ratios exceeding or not the respective cut‐offs previously reported by Sozzi et al.9 Only miRNAs composing at least 3 ratios of the signatures of “presence of disease” (PD) and “presence of aggressive disease” (PAD) were included in the analysis. The two‐tailed p‐value for the Sign test was considered significant if p < 0.01 and was calculated using the GraphPad QuickCalcs online tool (www.graphpad.com/quickcalcs) with a probability of 0.5: number of experiments = Calls at surgery; number of “successes” = Calls at surgery – Calls post‐surgery.
The differences between two groups for each experiments were assessed using the two‐tailed Student's T test. Differences were regarded as significant at p < 0.05.
Results
Analysis of miRNA expression and release by different cellular components of the lung microenvironment
To investigate at the lung tissue level the potential origin of plasma circulating miRNAs, we analyzed miRNAs expression using real‐time PCR in tumor and normal lung tissues from lung cancer patients (n = 20). A significantly decreased expression of all 24‐miRNAs in tumors compared to normal tissues was observed (p < 0.05) except for mir‐21 that was up‐regulated in tumor samples (Supporting Information Fig. 1A). These changes do not recapitulate miRNAs modulations observed in plasma indicating that interactions between different cellular components cooperate in producing risk‐associated plasma signatures.
To identify the cellular origin of the 24‐miRNAs composing the classifier, we therefore analyzed by Real‐time PCR their expression levels in various cell types that recapitulate the lung microenvironment. In particular, we selected normal alveolar and bronchial epithelial cells, immortalized bronchial epithelial cells16 and lung cancer cell lines from different histological subtypes which could be related to different cells of origin.24 As representative of the stromal microenvironment we analyzed miRNA expression in lung fibroblasts primary cultures, endothelial cells, skeletal muscle cells and bronchial smooth‐muscle cells.
Our results identified a set of miRNAs quite abundant in all epithelial cell types (mir‐16‐17‐19b‐30b‐106) whereas other miRNAs (mir‐101‐148‐140‐3p‐28‐3p‐660‐15b) were expressed at lower levels (Supporting Information Tables 2–3). Lung tumor cells generally showed lower expression of the 24 miRNAs compared to the normal or immortalized epithelial cells (miRNAs reduction average: 70% and 50% compared to normal and immortalized epithelial cells respectively). Interestingly, mir‐142‐3p, mir‐133 and mir‐451 expression was absent in all epithelial cells (Fig. 1 a and Supporting Information Fig. 1B).
Figure 1.
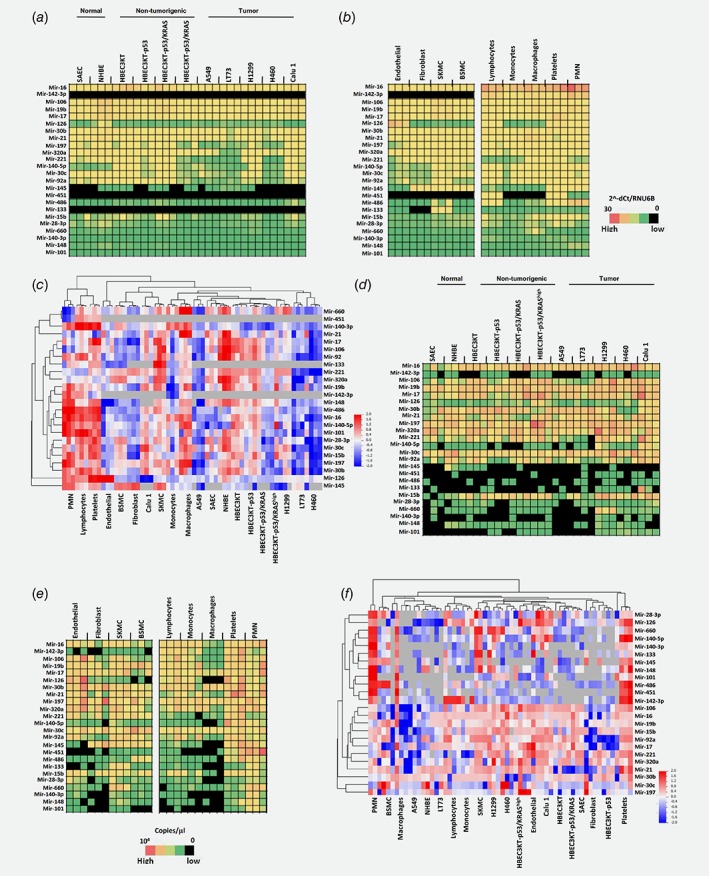
miRNA expression and release profiles across different cell types of lung microenvironment. Heatmaps showing levels of miRNA expression in lung epithelial (a) or stromal (b) cells. (c) Bi‐cluster of intracellular microRNA expression. Heatmaps describing miRNAs secretion in conditioned medium of lung epithelial (d) or stromal (e) cells. (f) Hierarchical clustering of microRNAs secreted by different cell types. Black boxes indicate no miRNA detection. Cellular expression profiles were performed using qPCR whereas miRNA secretion using digital PCR (n = 3 replicates for each cell types). [Color figure can be viewed at wileyonlinelibrary.com]
In the stromal compartment we observed high levels of mir‐126 exclusively in endothelial cells, mir‐145 showed increased expression in fibroblasts and muscle cells whereas mir‐133 and mir‐486 were present in skeletal muscle cells only, as already described in literature.25 Two miRNAs (mir‐142‐3p and mir‐451) were not expressed by stromal cells (Fig. 1 b ; Supporting Information Table 4; Supporting Information Fig. 1C). The analysis of miRNA expression in different subsets of hematopoietic cells isolated from heavy‐smoker volunteers identified a group of miRNAs highly expressed in all blood cell types (mir‐16‐17‐19b‐30b‐106 and mir‐142‐3p) whereas mir‐451 expression was restricted to lymphocytes, platelets and granulocytes. Overall, granulocytes and platelets were the blood components with higher content of miRNAs (Fig. 1 b and Supporting Information Table 5).
Bi‐clustering analysis on the cellular miRNAs data revealed that hematopoietic cells have higher amounts of miRNAs whereas epithelial cells compartment expressed low levels of all the 24 miRNAs (Fig. 1 c).
Furthermore, we analyzed by digital PCR the release of the 24 miRNAs in conditioned medium (CM) from each cell type and correlated miRNAs secretion with intracellular expression (Figs. 1 d and 1 e and Supporting Information Fig. 1D‐E). We confirmed that mir‐126 and mir‐133 were exclusively released by endothelial and skeletal muscle cells, respectively (Supporting Information Tables 6–9 and Supporting Information Fig. 1E). In the hematopoietic lineage, we found that platelets and granulocytes were the components that contributed mostly to miRNAs release. A good degree of correlation between cellular and secreted miRNAs levels for each cell types was observed (Pearson correlation range: 0.59–0.81) (Supporting Information Table 10).
Bi‐clustering approach allowed us to confirm that granulocytes and platelets released higher amount of miRNAs (Fig. 1 f).
Some discrepancies observed between miRNA secretion and their cellular levels could be likely due to the different sensitivity of the detection methods (digital versus real time PCR), however we cannot exclude that some miRNAs are enriched in extracellular vesicles as already described.26
ISH analysis of miRNAs origin
To confirm at the cell/tissue level the results obtained by Real‐Time PCR we performed ISH on FFPE specimens of tumor and normal lung tissues and cell‐blocks containing blood cells obtained from heavy‐smokers individuals (n = 3 for each miRNA).
To establish the miRNAs expression threshold detectable by ISH, we first generated by real‐time PCR a standard curve using serial dilutions of mir‐660 mimic transfected into H460 cells and compared expression values (2−dCt) with miRNA ISH detection and its quantification (Supporting Information Fig. 2). We estimated that miRNA expression levels with 2−dCt values lower than 0.2 when normalized to RNU6B (e.g. approximately five fold less than RNU6B expression) could not be detected by ISH. Therefore, based on PCR results in tissues we excluded the evaluation of some miRNAs (mir‐660, mir‐28‐3p, mir‐197, mir‐101, mir‐133, mir‐140‐3p, mir‐148).
As shown in Figure 2 a ISH staining for mir‐451 was restricted to lung interstitial alveolar wall circumscribed by normal pneumocytes. Mir‐451 expression was observed in erythrocytes and lymphocytes confirming real‐time PCR data obtained in primary cells. Similarly, mir‐126 expression, which was found in HUVEC cells, was restricted to CD31 positive endothelial cells. Mir‐145 was largely expressed by stromal fibroblasts in all tissues analyzed and it was positive in the lung muscularis fibers of the bronchial interstitial space as confirmed by alfa‐smooth muscle actin (α‐SMA) staining. The expression of mir‐142‐3p in lung tissues mirrored that of mir‐145 being present in CD3 lymphocytes, in α‐SMA positive fibroblasts and muscularis fibers of the bronchial interstitial space. Mir‐320a was expressed by different cell types of the microenvironment but its expression was absent in epithelial cells both from normal and tumor tissues (Fig. 2 a and Supporting Information Table 11).
Figure 2.
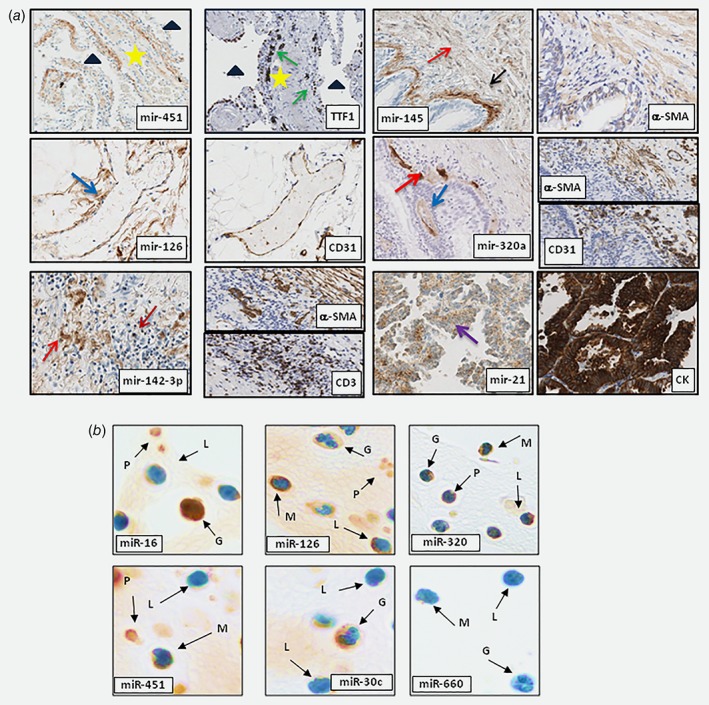
In situ miRNAs localization in human lungs and hematopoietic cells. (a) Localization of selected miRNAs in human lungs . mir‐451 expression is localized in lung interstitial alveolar cells (star) (triangle for air space original magnification: ×20). TTF1 IHC identified properly interstitial space circumscribed by normal pneumocytes (green arrows); Tissue vessels defined as CD31+ cells were positive for mir‐126 staining (blue arrow; original magnification: ×10). mir‐142‐3p were expressed by α‐SMA fibroblast (red arrow), CD3+ lymphocytes (dark red arrow) (Original magnification: ×20). mir‐145 was expressed by fibroblast (red arrow) and smooth muscles cells in lungs as confirmed by α‐SMA staining (black arrow; original magnification: ×10). Mir‐320a staining is localized in lung stromal components. CD31 and α‐SMA staining confirmed specific cellular expression (Original magnification: ×10). mir‐21 is over‐expressed in lung tumors compared to normal epithelial cells (purple arrow; original magnification: ×20). Tumoral cells were defined as pan‐cytokeratin+ cells. IHC on consecutive slides identified cell‐specific microRNAs expression (b) Representative images of miRNAs detection in cell‐blocks of peripheral blood cells. mir‐16, mir‐126 and mir‐320a were positive in all blood cells. mir‐451 was expressed in monocytes, platelets and granulocytes. mir‐30c expression is localized only in granulocytes whereas mir‐660 was positive in lymphocytes. Letters and arrows indicate G = granulocyte; L = lymphocyte; M = monocyte; P = platelet; Original magnification: ×100. n = 3 tissues or PBMC samples for each miRNAs. [Color figure can be viewed at wileyonlinelibrary.com]
ISH analysis confirmed that mir‐21, mir‐221 and mir‐16 were expressed by tumor cells and in particular mir‐21 was up‐regulated in tumor compared to normal epithelial cells (Fig. 2 a and Supporting Information Table 11). The percentage of miRNAs positive cells for each tissues compartment was determined and shown in Supporting Information Figure 3 a.
From miRNA ISH on circulating cells we observed that platelets expressed the majority of the miRNAs, with the exception of mir‐197, mir‐30c and mir‐660 that were not detected (Supporting Information Table 12).
Granulocytes displayed a consistent expression of miRNAs, with the exception of mir‐145 and mir‐660, which proved to be negative and of mir‐221 that stained faintly rare mature granulocyte forms. Notably, miRNA expression in the granulocytes was mainly cytoplasmatic though mir‐16, mir‐142‐3p and mir‐21 resulted enriched in the nuclear compartment. Monocytes were generally characterized by a low expression of miRNAs, being consistently positive only for mir‐21, mir‐126, mir‐145 and mir‐451, while lymphocytes had a rather heterogeneous expression (Fig. 2 b and Supporting Information Table 12).
Overall, mir‐16, mir‐126 and miR‐320a were expressed in most peripheral blood circulating cells, mir‐451 was homogeneously expressed in circulating myeloid cells, while mir‐30c and mir‐660 were detected at low levels in granulocytes and monocytes, respectively (Fig. 2 b and Supporting Information Table 12).
ISH data matched the expression profiles of the same cell types analyzed by real time PCR as described above. The few discrepancies observed of mir‐451, mir‐140‐5p, mir‐30c expression in circulating and tissue‐based lymphocytes could be at least partially explained by a modulation of miRNAs levels during the activation of extravasated lymphocytes. For each blood subpopulations we also quantified the number of miRNAs positive cells (Supporting Information Fig. 3B).
Modulation of circulating miRNAs in post‐surgery plasma samples of lung cancer patients
The analysis of plasma samples during patients’ follow‐up was then used to further distinguish “tumor‐related miRNAs” (linked to interactions between the cancer lesion and the microenvironment), which are expected to return to normal levels after cancer removal, from “host‐related” miRNAs that could remain deregulated during follow‐up reflecting a risk profile dictated from the host and the lung microenvironment.
To this purpose we used plasma samples from 26 lung cancer patients of the MILD trial27 longitudinally collected before and after curative surgery for lung cancer with a median time elapsed from tumor resection of 1.3 years (IQR = 1.4). We analyzed which miRNAs composing the classifier exceeded the established risk cut‐offs9 at the time of diagnosis and were afterward modulated during follow‐up. These patients did not receive any treatment post‐surgery. The results showed that among the miRNAs with greater relevance in the classifier, miR‐142‐3p, miR‐197, miR‐106a, miR‐17, miR‐660, miR‐140‐5p and miR‐19b could be classified as tumor‐associated and significantly declined to basal levels after tumor resection (p < 0.01). On the other hand, miR‐126, miR‐92a, miR‐320a, miR‐28‐3p, miR‐486‐5p, miR‐451 and miR‐16 remained deregulated in plasma after tumor resection suggesting a “host‐related” origin and likely indicating the persistence of a risk profile (Supporting Information Tables 13–14).
Circulating miRNA de‐regulation reflects an immunosuppressive and pro‐tumorigenic microenvironment
We then reasoned that dynamic miRNA alterations observed in plasma and related to disease risk could therefore reflect the formation of systemic/local microenvironments conductive to cancer progression. To investigate if some of the observed alterations could be related to specific changes in hematopoietic and stromal cells phenotypes, we profiled cellular miRNA after in vitro treatments known to induce changes associated with cancer progression and correlated them to changes observed in plasmatic miRNAs of 69 lung cancer patients and 870 disease free individuals previously analyzed9 (Fig. 3 a). Among cellular contributors to pro‐tumorigenic microenvironments,28 we investigated in detail polymorphonuclear leukocytes (PMNs), macrophages, T‐cells, platelets, endothelial cells and fibroblasts. (Fig. 3 and Supporting Information Fig. 4).
Figure 3.
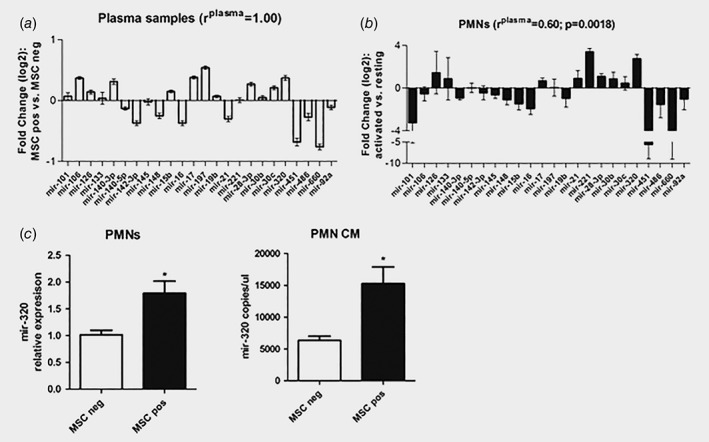
‘Risk’ miRNAs modulation in plasma of CT‐screening subjects is recapitulated by changes observed during immunosuppressive conversion of polymorphonuclear cells (PMNs). ( a ) Graph shows 24 miRNAs modulation in plasma of MSC positive individuals (high risk) vs. MSC negative individuals (low risk) (n = 216). (b ) Scatter plots represent miRNAs changes during PMNs activation (n = 3). ( c ) Mir‐320a levels in granulocytes isolated from heavy smokers with different MSC profile (n = 5 for each group). Levels were measured in the cellular compartment (left panel) and in conditioned medium (CM, right panel). Data are expressed as mean + SEM.
Comparing activated vs. resting PMNs (defined as low density granulocytes),29 17 out of the 24 (71%) miRNAs were concordantly up‐ (n = 9) or down‐ (n = 8) regulated compared to lung cancer patients’ plasma samples. Moreover, all these modulations were consistent with the observed de‐regulation of miRNAs in plasma samples of patients (Pearson r plasma = 0.60, p = 0.0018), indicating a potential role of activated PMNs in determining plasma miRNA profile associated with lung cancer risk (Fig. 3 b and Supporting Information Fig. 4A).
When macrophages were polarized in vitro toward a pro‐tumorigenic M2 phenotype,30 we found an increased expression of miR‐15b, miR‐197 and miR‐320a and a down modulation of miR‐142‐3p and miR‐451, resulting in a Pearson r plasma = 0.28 (Supporting Information Fig. 4B). On the contrary, an anti‐tumorigenic M1 polarization mediated a general down‐regulation of the 24 miRNAs and an inverse nonsignificant correlation (Pearson r plasma = −0.27) (Supporting Information Fig. 4C).
The majority of the 24 miRNAs were up‐regulated in Foxp3+ Tregs obtained from naïve CD4+CD25−Foxp3− T (Tconv) of healthy donors,31 showing a Pearson r plasma = 0.01 (Supporting Information Fig. 4D). Conversely, activated T cells stimulated with anti‐CD3 mAb showed a down‐regulation of all the 24‐miRNAs (Pearson r plasma = 0.30) (Supporting Information Fig. 4E).
A higher amount of the 24 miRNAs was observed in the CM of thrombin activated platelets, known to mediate several inflammatory and immunomodulatory activities32 compared to untreated platelets (Pearson r plasma = 0.25) (Supporting Information Fig. 4F).
Recent studies have established the hypoxia/HIF signaling pathway as a master regulator of the vascular system.33 Interestingly, by culturing HUVEC in hypoxic conditions we found that mir‐126 was significantly upregulated and secreted, in agreement with the increased miRNA levels observed in plasma of patients (Supporting Information Fig. 4G).
Finally, since fibroblasts have been shown to participate in lung tumorigenesis by promoting tumor growth and invasiveness34 and regulating metastatic potential of lung cancer35 we evaluated miRNA expression in cancer‐associated (CAF) compared to normal (NF) lung fibroblasts primary cultures established from lung cancer patients. A significantly higher mir‐145 expression (n = 5 pairs) and secretion (n = 10 pairs) was detected in CAFs compared to NF (Supporting Information Fig. 4H).
In conclusion we show that miRNA changes associated with modulation of lung cancer risk correlate to ‘pro‐tumorigenic’ conversions of different cell‐types indicating they may be involved in the generation of a tumor promoting microenvironment.
Mir‐320a‐induced M2‐polarization of macrophages
To clarify the potential role of circulating miRNAs in molding a pro‐tumorigenic microenvironment we next focused our attention on miRNAs whose levels remained deregulated after tumor removal in cancer patients and potentially reflected immunosuppressive changes.
As a result of miRNAs screening of different cell types, we observed that higher amount of the 24 miRNAs were secreted by PMNs compared to other cells. More importantly, we focused on this immune cell population and its activation as published literature had described them as having a critical role in lung cancer development.36, 37 During PMNs activation, our observation indicated mir‐320a as being the most over‐expressed and in agreement with its level in plasma of MSC positive subjects, therefore, we hypothesized that the increased circulating miR‐320a levels in lung cancer patients could be mainly contributed by PMNs. A statistical significant correlation between mir‐320a levels and neutrophils count was observed in 18 lung cancer patients (Supporting Information Tables 15 and 16) sustaining our hypothesis. Furthermore, an increased expression and secretion of mir‐320a expression in CM from PMNs isolated from high risk (MSC positive) compared to low risk (MSC negative) subjects (n = 5 for each MSC groups; fold increase vs. MSCneg: 1.7 in cells and 2.4 in CM) was observed, supporting the contribution of PMNs to increased circulating levels of mir‐320a in plasma of high‐risk subjects (Fig. 3 h).
Since it has been reported that PMNs can promote the activation of other immune cells such as T cells, macrophages and platelets38 and in particular, macrophages were described as fundamental in lung cancer progression,39 we transfected macrophages from heavy‐smokers subjects, with some of the miRNAs found up‐regulated during M2 polarization such as miR‐17, miR‐197 and mir‐320a (Supporting Information Fig. 5A) and analyzed IL‐10, IL‐6, TNF‐a and IL‐12 cytokine expression to evaluate skewing toward Th1 or Th2 responses.40 A miRNA negative sequence (M SCR) was used as control. In mir‐320a over‐expressing macrophages (M mim‐320) intracellular IL‐10 up‐regulation and IL‐6, TNF‐a and IL‐12 down‐modulation were observed (similar to IL‐4‐treated M2‐polarized macrophages, Fig. 4 a). Furthermore, M mim‐320 have similar expression of CD163, CD206 and CSF1R mRNA to M2 macrophages confirming the immunosuppressive phenotype of these cells (Fig. 4 b).
Figure 4.
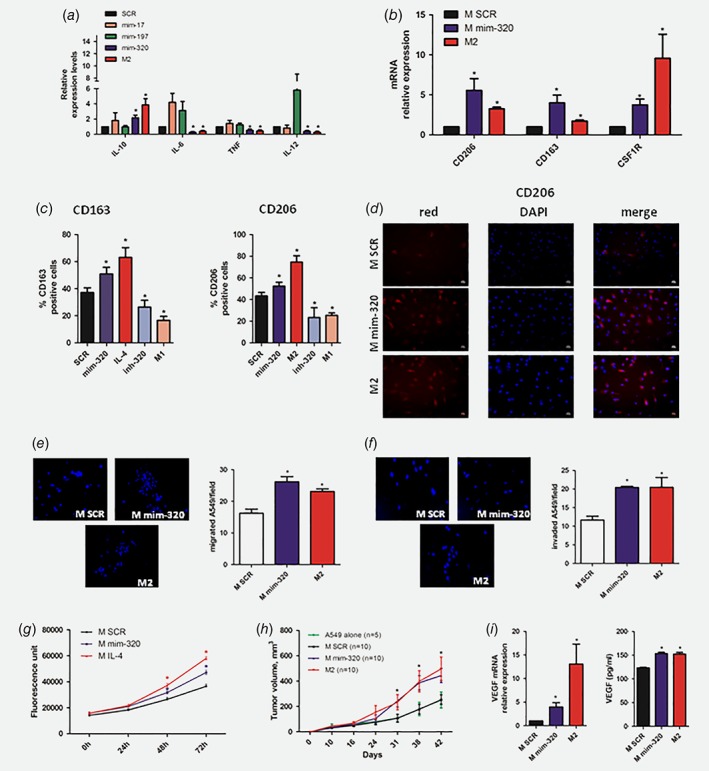
mir‐320a induces M2 polarization of macrophages. ( a ) Intracellular expression of M2 phenotype marker by qPCR (n = 5 independent experiments) after miRNA modulation ( b ) Additional M2 markers expression on mir‐320a or IL‐4‐treated macrophages (n = 5) ( c ) CD163 and CD206 expression on the surface of macrophages over‐expressing mir‐320a or IL‐4 treated compared to control (n = 5 independent experiments). (d ) Representative images of CD206 positive macrophages after miRNA modulation. Representative images of A549 migrated ( e ) or invaded ( f ) after macrophages CM treatment. Bar graphs show A549 migrated or invaded after CM treatment (n = 3 independent experiments). ( g ) Graphs show A549 proliferation after CM treatment (n = 5 independent experiments). (h ) In vivo tumor growth of A549 co‐injected with mir‐320a over‐expressing macrophages (n = 10) or controls (n = 10 for MSCR and n = 5 for A549 alone). ( i ) VEGF expression and secretion in macrophages after miRNA over‐expression (n = 5 for mRNA and n = 3 for ELISA). Data are expressed as mean + SEM. The differences between two groups were assessed using Student's t test (* = p < 0.05 vs. M SCR). [Color figure can be viewed at wileyonlinelibrary.com]
Flow cytometry and immunofluorescence staining of M mim‐320 also showed a CD163+ and CD206+ expression similar to M2 macrophages (Figs. 4 c and 4 d). Interestingly, inhibition of mir‐320a reverted the effects on macrophages resulting in M1 phenotype polarization (Fig. 4 c and Supporting Information Fig. 5B). Based on these findings and on published literature describing the role of mir‐320a in the activation of macrophages,41 we decided to further focus on the functional role of this miRNA in macrophages.
CM of mir‐320a over‐expressing macrophages increased migratory (Fig. 4 e) and invasive (Fig. 4 f) capacity of A549 lung cancer cells similarly to M2 controls. To assess the effects of M2‐like polarization induced by mir‐320a expression on tumor growth, A549 lung cancer cells were treated with CM of M mim‐320. A significant increase in A549 cell proliferation in vitro similar to that observed in IL‐4‐induced M2 macrophages was observed (Fig. 4 g). Furthermore, subcutaneous co‐injection of M mim‐320 with A549 cells in immunodeficient mice showed that M mim‐320 cells significantly boosted in vivo tumor growth compared to controls exerting the same effect of IL4‐ induced M2 macrophages (Fig. 4 h).
To explain the tumor promoting activity of mim‐320 macrophages, we analyzed the expression of VEGF, a factor known to increase A549 proliferation42 and observed increased VEGF levels both at mRNA and protein levels similar to M2 macrophages (fold increase for mRNA: 13.1 and 3.95 for M2 and M mim‐320, respectively; fold increase for VEGF protein in CM: 1,24 for both compared to M SCR) (Fig. 4 i).
STAT4 is a direct target of mir‐320a and is responsible for M2 profile
In silico analysis using two bio‐informatics programs (Targetscan and mirWalk), identified putative binding sites for mir‐320a in the 3′UTR of the STAT4 gene (Fig. 5 a), a transcription factor down‐modulated after IL‐4 stimulus in macrophages.43 To prove that STAT4 is a direct target of mir‐320a, we performed a luciferase reporter assay and observed a down‐modulation (40% of reduction) of the luciferase activity when HEK‐293 cells were co‐transfected with mir‐320a (Fig. 5 a). Target specificity was validated either using a 3′UTR EMPTY vector or by site‐directed mutagenesis in the putative mir‐binding sites where we did not detected any change in luciferase activity (Fig. 5 a).
Figure 5.
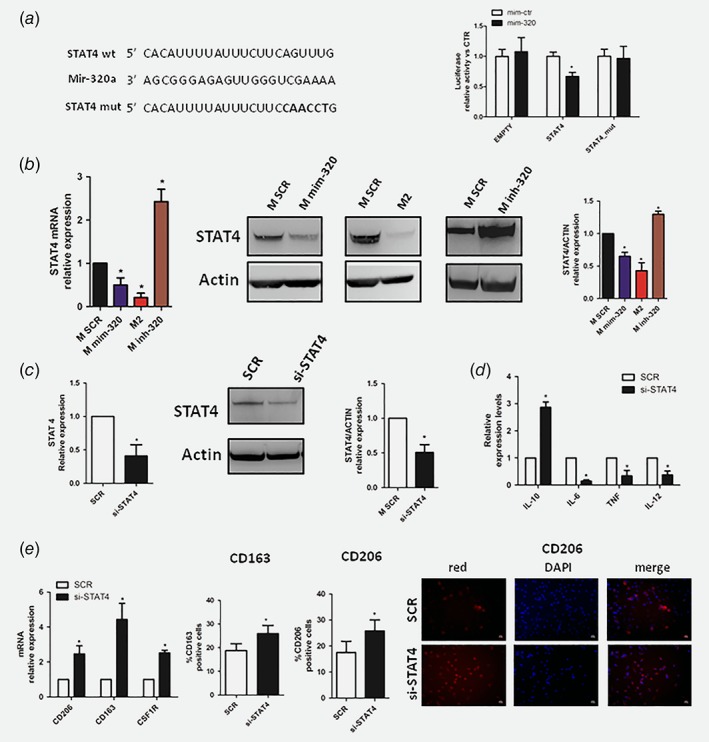
STAT4 is a direct target of mir‐320a in macrophages. (a) Predicted STAT4 3′UTR‐binding site for mir‐320a. The figures show alignment of the mir‐320a seed sequence with STAT4 3′UTR (left). Bar graphs showing average luciferase activity compared to control. Reporter systems were transfected in HEK293 with STAT4 wild type or mutated and EMPTY 3′UTR in combination with mir‐320a mimics (mim‐320) or control (mim‐ctr) (n = 5). (b) Real time PCR (left panel), western blot bands (center panel) and quantification (right panel) showing STAT4 down‐modulation after mir‐320a over‐expression compared to M2‐like macrophages. Inhibition of mir‐320a (inh‐320) resulted in STAT4 over‐expression (n = 5 for each assay). (c) The efficiency of si‐STAT4 silencing was confirmed both by mRNA (left) and western blot bands and quantification (center and right) (n = 5 for mRNA and n = 3 for WB independent experiments) (d) M2 markers characterization of human macrophages silencing with siRNA STAT4 by qPCR (n = 5). (e) Additional M2 markers in si‐STAT4 macrophages (left). CD163 and CD206 flow citometry analysis (center) and CD206 immunofluorescence in STAT4‐silenced macrophages (right) (n = 3 for each assay). Data are expressed as mean + SEM. The differences between si‐STAT4 and SCR were assessed using Student's t test (* = p < 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
We analyzed STAT4 expression in macrophages after mir‐320a over‐expression and we observed a down‐modulation of this transcription factor in mir‐320a and M2 macrophages compared to controls both at mRNA and protein levels (Fig. 5 b) (% of reduction vs. SCR: 35% and 57% for M mim‐320 and M2 respectively). Interestingly, inhibition of mir‐320a resulted in increased STAT4 levels both as mRNA and protein (Fig. 5 b).
In order to demonstrate that mir‐320a is able to induce M2 phenotype through STAT4 modulation we silenced STAT4 inside macrophages and analyzed their phenotype. As shown in Figure 5 c STAT4 inhibition increased intracellular IL‐10 expression and down‐modulated IL‐6, TNF and IL‐12 (Fig. 5 d). Furthermore, to better elucidate the importance of STAT4 in M2 polarization, we analyzed additional M2 markers such as CD206, CD163 and CSF1R mRNA, and CD163, CD206 protein by flow cytometry and immunofluorescence staining, in STAT4‐silenced macrophages and we observed upregulation of these markers (Fig. 5 e).
PMNs from high‐risk individuals induce macrophage M2 polarization through mir‐320a
Finally, to model the potential mechanism that could occur in high risk heavy‐smokers individuals (MSCpos), we decided to treat cultured macrophages with CM from PMNs isolated from individuals with different risk to develop lung cancer.
CM from PMNs increased the amount of CD163+/CD206+ cells and more importantly the number of CD163+/CD206+ macrophages was higher using CM isolated from MSC pos individuals (Fig. 6 a). Up‐regulation of CD206 expression assessed by immunofluorescence after treatment with CM from PMNs further confirmed the induction of M2‐like phenotype (Fig. 6 b). Furthermore, an increase of mature mir‐320a inside macrophages after 24 h without upregulation of its pri‐miRNA was observed, supporting the transfer of exogenous mature mir‐320a without endogenous mir‐320a biogenesis (Fig. 6 c). Of note, the transfer of miRNA was higher after treatment with CM derived from PMNs of high‐risk compared to low risk individuals. Interestingly, exogenous mir‐320a transferred from PMNs induced M2 polarization, evaluated as an increase of M2 markers (IL‐10, CD206,CD163, CSF1R) expression and down‐modulation of IL‐6, TNF and IL‐12, with concomitant down‐modulation of the target STAT4 mRNA (Fig. 6 c). Overall, these data demonstrate that PMNs isolated from MSC‐pos subjects are more efficient in inducing M2 polarization compared to low risk individuals.
Figure 6.
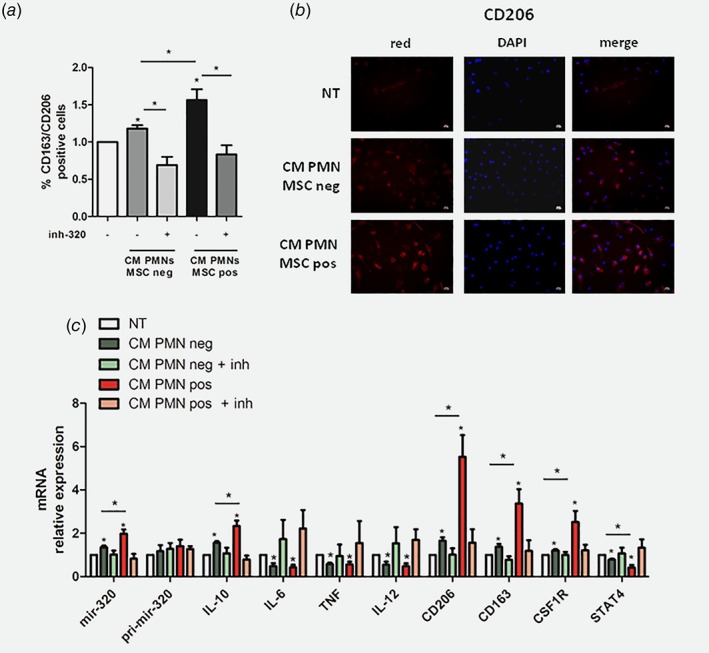
PMNs‐derived mir‐320a is transferred into macrophages and mediates M2 polarization. (a) CD163 and CD206 expression in PMN CM‐treated macrophages (n = 4 independent experiments). (b) Representative images of CD206 expression on CM treated macrophages. (c ) Relative expression of miR‐320a levels and M2 markers (mRNA) in macrophages after PMN's CM treatment (n = 5 independent experiments). Data are expressed as mean + SEM. The differences between two groups were assessed using Student's t test (* = p < 0.05 vs. NT or MSC neg vs. MSC pos). [Color figure can be viewed at wileyonlinelibrary.com]
Furthermore, all the immunosuppressive effects observed with CM PMNs were completely abrogated using an anti‐mir‐320a Locked Nucleic Acid transfected in macrophages before adding CM confirming the fundamental role of mir‐320a transferred from PMNs to macrophages (Figs. 6 a–6 c).
Discussion
The rationale behind the investigation of miRNAs as potential tools in anti‐cancer therapy is based on the observation that miRNA expression is often reduced in tumor compared to normal tissues.44 However in carcinomas some of the reported changes such as downregulation of mir‐451 or mir‐126 are difficult to explain as cancer‐cell specific alterations given that these miRNAs are specifically expressed by hematopoietic and endothelial cells respectively.13 These data suggest therefore that the apparently frequent reduction of miRNAs expression in epithelial tumors might reflect their cellular composition (expansion of the epithelial compartment) with a concomitant reduction in the stromal compartment (well represented in the ‘control’ normal tissue). Therefore, studies based on the replacement of these miRNAs in tumor cells have doubtful significance. Knowledge of cell‐type restricted miRNA expression is thus crucial to perform miRNA functional studies in appropriate cellular types and context.
We previously showed that a combination of 24 circulating miRNAs predicts lung cancer development and is able to identify tumor aggressiveness.7 Due to the complex nature of the observed signatures, composed of both upregulated and downregulated miRNAs and their potential to anticipate disease occurrence up to two years before CT‐detected nodules, we hypothesized that the lung microenvironment could be involved in the generation of diagnostic miRNA signatures possibly reflected in the bloodstream and representing powerful risk biomarkers of lung cancer development.
In the present study, we focused on miRNAs cellular localization to possibly understand the source of circulating miRNAs found de‐regulated in high‐risk individuals. Our initial findings identified a specific topography of these miRNAs in normal and pathological lung tissues exemplified by mir‐126 expression (restricted to endothelial vessels) and mir‐451 (detectable in the fibro‐monocytic infiltrate of the alveolar lung interstitial wall). Among miRNAs composing the risk signatures only mir‐21 appeared over‐expressed in tumor cells compared to normal epithelial cells, as already described.45
Furthermore, ISH and PCR analysis of blood cells from heavy‐smokers indicated that the majority of circulating miRNAs are highly expressed and release by blood cells. Even if miRNA profile in plasma represents the collective miRNA contribution of various cell types, our data suggest that granulocytes and platelets could be the major contributors to miRNA release since they are the more represented cell populations in blood.
We focused therefore on the potential origin of ‘risk‐miRNAs’ from cells of the immune system either reflecting or inducing their immunosuppressive and pro‐tumorigenic conversion.
Evidences of complex interactions between immune cells and (pre)neoplastic cells have been accumulating in the past decades and in particular it is now well recognized that the immune system could have both positive or negative effects on tumor biology, from development to metastasis through mechanisms that facilitate or control tumor growth and progression.46 Among the major players it is now clear that immune cells from the myeloid lineage, defined as myeloid derived suppressor cells (MDSCs) are engaged in tumor development by regulating different aspects of immune response.47
It is well known that PMNs promote the activation of innate immune cells like such as T cells, macrophages and platelets.38 An interesting study showed that PMNs were the most abundant infiltrating immune cell type identified in NSCLC tissues,36 a finding consistent with previous clinical observations pointing out that an increased number of neutrophils and macrophages in the lungs of smokers correlated with the severity of airway inflammation.48
Our data suggest a potential modulation of the 24 miRNAs composing the MSC risk classifier during the activation process of PMNs: this process closely mirrors changes observed in circulating miRNAs in high‐risk individuals. Furthermore, we describe higher expression and secretion of PNMs‐derived mir‐320a in MSC positive individuals supporting the contribution of PMNs as a risk factor for lung cancer development. Moreover, in MSC positive individuals, mir‐320a levels were 1.3 fold higher compared to MSC negative individuals indicating that miRNAs modulation observed in patients could mainly reflect neutrophils activation, being neutrophils the predominant immune cells population present in NSCLC.36
Macrophages are known to infiltrate the tumor microenvironment and can be polarized from an anti‐tumorigenic M1 to a pro‐tumorigenic M2 phenotype.30 Our results show that the modulation of our 24‐miRNAs was different in these lineages and are in keeping with previous studies highlighting the importance of these miRNAs in the polarization of macrophages with a fundamental role in tumor immune escape.49
Mir‐320a is de‐regulated in several cancer types such as gastric,50 breast,51 colon and lung cancer.52 Recent works demonstrated the role of mir‐320a as tumor suppressor in NSCLC by directly regulating IGF‐1R53 or VDAC152 expression suggesting this miRNA as a potential therapeutic target. Furthermore, mir‐320a was also involved in lung metastasis formation through down‐regulation of STAT354 and NRP‐1.55 Here describe a novel role of mir‐320a in the cross‐talk among immune cells in lung microenvironment to promote tumor growth.
Recently, an interesting work demonstrated that exosomal mir‐221/320 transfer from lung epithelial cells to macrophages induced a general activation of macrophages in terms of migration and cytokine secretion.41
Consistent with a central role of mir‐320a in regulating polarization, its over‐expression in heavy‐smoker macrophages induced an M2 phenotype confirmed by up‐regulation of macrophage scavenger receptor CD163 and CD206 and indicating an alternative and pro‐tumorigenic activation of these cells.56
STAT‐proteins appear to be crucial factors involved in macrophages polarization, in particular it was already described an important role of STAT1 and STAT6 in M1 and M2 phenotype respectively.57 Our data revealed that another component of this family, STAT4 is involved in M2 macrophages polarization. This transcription factor was already described in the monocyte lineage and shown to be negatively regulated by IL‐4;58 based on the consideration that macrophages phenotypes depend on the balance between Th1 and Th2 cytokines we suggest that mir‐320a is a new modulator of M2 switch through down‐regulation of STAT4. Finally, we demonstrated that mir‐320a can be transferred from PMNs, isolated from subjects at high‐risk to develop lung cancer, to macrophages inducing an M2 polarization and may be involved in lung cancer development.
Therapies using immunocheckpoint inhibitors are emerging as new tools for the management of lung cancer59 but a rapid disease progression has been reported in a small group of patients, suggesting the need to find circulating biomarkers for the selection of patients who could benefit of immunotherapy.60 Our study describes miRNA modulation in immune cells such as T regulatory cells, M2 macrophages and activated PMNs that could potentially promote an immunosuppressive lung microenvironment. These changes are in agreement with modulation of these miRNAs in plasma of high risk individuals suggesting their potential use as biomarkers for the selection of patients that could benefit from immunotherapy. In the same setting our observations also potentially pave the way for clinical applications. On the basis of our findings a potential reprogramming of macrophage phenotype toward an anti‐tumor polarization through mir‐320a inhibition either to block tumor growth or to induce immune response against malignant cells could be envisaged.
Supporting information
Appendix S1: Supplementary material.
Suppl. Figure 1 24‐miRNAs expression in lung tissues. A)Bar graphs show miRNA expression levels in lung cancer tissues compared to normal lung (n = 20). The differences between Normal and Tumor tissues were assessed using Student's T test (* = p < 0.05). Data are expressed as mean ± SEM. Heatmaps showing levels of miRNA expression and secretion normalized by Z‐scores in the epithelial (B‐D), stromal and hematopoietic compartment (C‐E).
Suppl. Figure 2 miRNA ISH sensitivity compared to qPCR. A) Representative images of miRNAs ISH serial dilution using mimic mir‐660 oligonucleotide transfected into H460 cells B) ISH quantification as number of mir‐660 positive cells/total cells. C) mir‐660 relative expression (2^‐dCt) analyzed by qPCR.
Suppl. Figure 3 miRNAs quantification in lung tissues and hematopoietic cells. A) Histograms show the number of miRNA positive cells/total cells for the specific lung tissues compartment. B) Hematopoietic miRNAs positive cells on the total of blood cell subpopulation. For each miRNAs five random fields were counted. Data are expressed as mean ± SEM.
Suppl. Figure 4 Risk’ miRNAs modulation in plasma of CT‐screening subjects is recapitulated by changes observed during immunosuppressive conversion. A) Representative gating strategy for PMNs purity. Scatter plots represent miRNAs changes during PMNs activation M2 (n = 3) (B) or M1 (n = 3) (C) macrophages polarization D) After 5 days of in vitro culture in the presence of anti‐CD3/CD28 microbeads and IL2 ± TGF‐β, CD4+CD25− Tconv cells were stained with a probe to distinguish live cells and with anti‐ CD3, ‐CD4, ‐CD25 and ‐Foxp3 mAbs. Treg frequency, evaluated as the percentage of Foxp3+CD25high cells inside CD3+CD4+ live lymphocytes, was compared between untreated (No TGF‐β) and TGF‐β stimulated T cells (left). The percentage of CD25hiFoxp3+ cells is reported in the dot plots (one representative of 5). Histograms show the fold increase expression of Treg markers between untreated (No TGF‐β) and TGF‐β stimulated T cells (n = 5 donors)(center). Scatter plot for miRNA modulation during Treg conversion (n = 3) (right) E) miRNA modulaton in activated T cell (n = 3). F) Activation of platelets was confirmed by CD62P positive cells increase (n = 4) and miRNAs secretion was represented (n = 3). G) Hypoxia treatment up‐regulated mir‐126 expression (left) and secretion (middle) in endothelial cells. Induction of hypoxia was demonstrated by increase of HIF‐1a expression (right). H) mir‐145 expression analysis in CAF and their normal counterpart using qPCR (left) (n = 5 pairs) and mir‐145 secretion in CAF and NF analyzed using dPCR (n = 10)(right). Data are expressed as mean ± SEM.
Suppl. Figure 5 Phenotipic characterization of miRNAs over‐expressing macrophages. A) Bar graphs show expression levels after miRNA mimics compared to control and M2 macrophages. (n = 3 for each miRNA). B) Histrograms revealed that inhibition of mir‐320a induced M1 polarization (n = 5). Data are expressed as mean ± SEM. The differences between controls and treated macrophages were assessed using Student's T test.
Acknowledgements
The authors thank Dr. Gloghini for the support for miRNA in situ hybridization. The work was supported by the Italian Association for Cancer Research [Investigator Grants No. 15928 to UP, 14318 and 18812 to GS, 16847 to LR and 12162 to UP, GS and LR (Special Program “Innovative Tools for Cancer Risk Assessment and early Diagnosis”, 5x1000)]; the Italian Ministry of Health [RF‐2010] and NIH/U01CA166905. O.F. was supported by Cariplo Foundation Young Investigator Grant 2015 n.2015‐0901. MB was supported by Fondazione Umberto Veronesi Fellowship.
O.F designed the study, acquired, analyzed and drafted the article; C.B.; G.C.; D.C.; F.F.; M.B.; C.V.; M.Mo.; L.C.;F.A.; V.H.; A.C.; C.Cam.; V.C. acquired and analyzed data; revised critically the article; M.Mi.; L.R.; C.T.; C.Cas.; U.P.; G.S. designed the study, interpreted data, revised critically the article. All the authors approved the final version of the study.
References
- 1. Malvezzi M, Carioli G, Bertuccio P, et al. European cancer mortality predictions for the year 2017, with focus on lung cancer. Ann Oncol 2017;28:1117–23. [DOI] [PubMed] [Google Scholar]
- 2. Fumagalli C, Bianchi F, Raviele PR, et al. Circulating and tissue biomarkers in early‐stage non‐small cell lung cancer. Ecancermedicalscience 2017;11:717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Massion PP. Biomarker‐driven programs for lung cancer screening (abstract). J Thorac Oncol 2013;8:S6. [Google Scholar]
- 4. Barger JF, Nana‐Sinkam SP. MicroRNA as tools and therapeutics in lung cancer. Respir Med 2015;109:803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schwarzenbach H, Nishida N, Calin GA, et al. Clinical relevance of circulating cell‐free microRNAs in cancer. Nat Rev Clin Oncol 2014;11:145–56. [DOI] [PubMed] [Google Scholar]
- 6. Halvorsen AR, Sandhu V, Sprauten M, et al. Circulating microRNAs associated with prolonged overall survival in lung cancer patients treated with nivolumab. Acta Oncol 2018;57:1225–31. [DOI] [PubMed] [Google Scholar]
- 7. Boeri M, Verri C, Conte D, et al. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc Natl Acad Sci USA 2011;108:3713–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bianchi F, Nicassio F, Marzi M, et al. A serum circulating miRNA diagnostic test to identify asymptomatic high‐risk individuals with early stage lung cancer. EMBO Mol Med 2011;3:495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sozzi G, Boeri M, Rossi M, et al. Clinical utility of a plasma‐based miRNA signature classifier within computed tomography lung cancer screening: a correlative MILD trial study. J Clin Oncol 2014;32:768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Montani F, Marzi MJ, Dezi F, et al. miR‐test: a blood test for lung cancer early detection. J Natl Cancer Inst 2015;107:djv063. [DOI] [PubMed] [Google Scholar]
- 11. Walser T, Cui X, Yanagawa J, et al. Smoking and lung cancer: the role of inflammation. Proc Am Thorac Soc 2008;5:811–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pritchard CC, Kroh E, Wood B, et al. Blood cell origin of circulating microRNAs: a cautionary note for cancer biomarker studies. Cancer Prev Res (Phila) 2012;5:492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kent OA, McCall MN, Cornish TC, et al. Lessons from miR‐143/145: the importance of cell‐type localization of miRNAs. Nucleic Acids Res 2014;42:7528–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pastorino U, Bellomi M, Landoni C, et al. Early lung‐cancer detection with spiral CT and positron emission tomography in heavy smokers: 2‐year results. Lancet 2003;362:593–7. [DOI] [PubMed] [Google Scholar]
- 15. Pastorino U, Rossi M, Rosato V, et al. Annual or biennial CT screening versus observation in heavy smokers: 5‐year results of the MILD trial. Eur J Cancer Prev 2012;21:308–15. [DOI] [PubMed] [Google Scholar]
- 16. Sato M, Larsen JE, Lee W, et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol Cancer Res 2013;11:638–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fortunato O, Boeri M, Verri C, et al. Assessment of circulating microRNAs in plasma of lung cancer patients. Molecules 2014;19:3038–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Conte D, Verri C, Borzi C, et al. Novel method to detect microRNAs using chip‐based QuantStudio 3D digital PCR. BMC Genomics 2015;16:849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gualeni AV, Volpi CC, Carbone A, et al. A novel semi‐automated in situ hybridisation protocol for microRNA detection in paraffin embedded tissue sections. J Clin Pathol 2015;68:661–4. [DOI] [PubMed] [Google Scholar]
- 20. Agarwal V, Bell GW, Nam JW, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015;4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friedman RC, Farh KK, Burge CB, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009;19:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fortunato O, Boeri M, Moro M, et al. Mir‐660 is downregulated in lung cancer patients and its replacement inhibits lung tumorigenesis by targeting MDM2‐p53 interaction. Cell Death Dis 2014;5:e1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Workman P, Aboagye EO, Balkwill F, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer 2010;102:1555–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Giangreco A, Groot KR, Janes SM. Lung cancer and lung stem cells: strange bedfellows? Am J Respir Crit Care Med 2007;175:547–53. [DOI] [PubMed] [Google Scholar]
- 25. Ludwig N, Leidinger P, Becker K, et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res 2016;44:3865–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lawson J, Dickman C, MacLellan S, et al. Selective secretion of microRNAs from lung cancer cells via extracellular vesicles promotes CAMK1D‐mediated tube formation in endothelial cells. Oncotarget 2017;8:83913–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sestini S, Boeri M, Marchiano A, et al. Circulating microRNA signature as liquid‐biopsy to monitor lung cancer in low‐dose computed tomography screening. Oncotarget 2015;20:32868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schmielau J, Finn OJ. Activated granulocytes and granulocyte‐derived hydrogen peroxide are the underlying mechanism of suppression of t‐cell function in advanced cancer patients. Cancer Res 2001;61:4756–60. [PubMed] [Google Scholar]
- 30. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012;122:787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Camisaschi C, Casati C, Rini F, et al. LAG‐3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol 2010;184:6545–51. [DOI] [PubMed] [Google Scholar]
- 32. Herter JM, Rossaint J, Zarbock A. Platelets in inflammation and immunity. J Thromb Haemost 2014;12:1764–75. [DOI] [PubMed] [Google Scholar]
- 33. Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev 2007;26:281–90. [DOI] [PubMed] [Google Scholar]
- 34. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- 35. Bertolini G, D'Amico L, Moro M, et al. Microenvironment‐modulated metastatic CD133+/CXCR4+/EpCAM‐ lung cancer‐initiating cells sustain tumor dissemination and correlate with poor prognosis. Cancer Res 2015;75:3636–49. [DOI] [PubMed] [Google Scholar]
- 36. Kargl J, Busch SE, Yang GH, et al. Neutrophils dominate the immune cell composition in non‐small cell lung cancer. Nat Commun 2017;8:14381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Masubuchi T, Koyama S, Sato E, et al. Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Pathol 1998;153:1903–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kumar V, Sharma A. Neutrophils: Cinderella of innate immune system. Int Immunopharmacol 2010;10:1325–34. [DOI] [PubMed] [Google Scholar]
- 39. Gong L, Cumpian AM, Caetano MS, et al. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol Cancer 2013;12:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8:958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee H, Zhang D, Zhu Z, et al. Epithelial cell‐derived microvesicles activate macrophages and promote inflammation via microvesicle‐containing microRNAs. Sci Rep 2016;6:35250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koyama S, Sato E, Tsukadaira A, et al. Vascular endothelial growth factor mRNA and protein expression in airway epithelial cell lines in vitro. Eur Respir J 2002;20:1449–56. [DOI] [PubMed] [Google Scholar]
- 43. Fukao T, Frucht DM, Yap G, et al. Inducible expression of Stat4 in dendritic cells and macrophages and its critical role in innate and adaptive immune responses. J Immunol 2001;166:4446–55. [DOI] [PubMed] [Google Scholar]
- 44. Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med 2012;4:143–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu XG, Zhu WY, Huang YY, et al. High expression of serum miR‐21 and tumor miR‐200c associated with poor prognosis in patients with lung cancer. Med Oncol 2012;29:618–26. [DOI] [PubMed] [Google Scholar]
- 46. Pollard JW. Tumour‐educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 2004;4:71–8. [DOI] [PubMed] [Google Scholar]
- 47. Kumar V, Patel S, Tcyganov E, et al. The nature of myeloid‐derived suppressor cells in the tumor microenvironment. Trends Immunol 2016;37:208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pesci A, Balbi B, Majori M, et al. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur Respir J 1998;12:380–6. [DOI] [PubMed] [Google Scholar]
- 49. Quatromoni JG, Eruslanov E. Tumor‐associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res 2012;4:376–89. [PMC free article] [PubMed] [Google Scholar]
- 50. Wang Y, Zeng J, Pan J, et al. MiR‐320a inhibits gastric carcinoma by targeting activity in the FoxM1‐P27KIP1 axis. Oncotarget 2016;7:29275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bronisz A, Godlewski J, Wallace JA, et al. Reprogramming of the tumour microenvironment by stromal PTEN‐regulated miR‐320. Nat Cell Biol 2012;14:159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang G, Jiang G, Wang C, et al. Decreased expression of microRNA‐320a promotes proliferation and invasion of non‐small cell lung cancer cells by increasing VDAC1 expression. Oncotarget 2016;7:49470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang J, Shi C, Wang J, et al. MicroRNA‐320a is downregulated in non‐small cell lung cancer and suppresses tumor cell growth and invasion by directly targeting insulin‐like growth factor 1 receptor. Oncol Lett 2017;13:3247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lv Q, Hu JX, Li YJ, et al. MiR‐320a effectively suppresses lung adenocarcinoma cell proliferation and metastasis by regulating STAT3 signals. Cancer Biol Ther 2017;18:142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu H, Jiang X, Zhou X, et al. Neuropilin‐1 regulated by miR‐320 contributes to the growth and metastasis of cholangiocarcinoma cells. Liver Int 2018;38:125–35. [DOI] [PubMed] [Google Scholar]
- 56. Gordon S. Alternative activation of macrophages. Nat Rev Immunol 2003;3:23–35. [DOI] [PubMed] [Google Scholar]
- 57. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 2011;11:750–61. [DOI] [PubMed] [Google Scholar]
- 58. Frucht DM, Aringer M, Galon J, et al. Stat4 is expressed in activated peripheral blood monocytes, dendritic cells, and macrophages at sites of Th1‐mediated inflammation. J Immunol 2000;164:4659–64. [DOI] [PubMed] [Google Scholar]
- 59. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus Docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med 2015;373:123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Champiat S, Ferrara R, Massard C, et al. Hyperprogressive disease: recognizing a novel pattern to improve patient management. Nat Rev Clin Oncol 2018;15:748–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary material.
Suppl. Figure 1 24‐miRNAs expression in lung tissues. A)Bar graphs show miRNA expression levels in lung cancer tissues compared to normal lung (n = 20). The differences between Normal and Tumor tissues were assessed using Student's T test (* = p < 0.05). Data are expressed as mean ± SEM. Heatmaps showing levels of miRNA expression and secretion normalized by Z‐scores in the epithelial (B‐D), stromal and hematopoietic compartment (C‐E).
Suppl. Figure 2 miRNA ISH sensitivity compared to qPCR. A) Representative images of miRNAs ISH serial dilution using mimic mir‐660 oligonucleotide transfected into H460 cells B) ISH quantification as number of mir‐660 positive cells/total cells. C) mir‐660 relative expression (2^‐dCt) analyzed by qPCR.
Suppl. Figure 3 miRNAs quantification in lung tissues and hematopoietic cells. A) Histograms show the number of miRNA positive cells/total cells for the specific lung tissues compartment. B) Hematopoietic miRNAs positive cells on the total of blood cell subpopulation. For each miRNAs five random fields were counted. Data are expressed as mean ± SEM.
Suppl. Figure 4 Risk’ miRNAs modulation in plasma of CT‐screening subjects is recapitulated by changes observed during immunosuppressive conversion. A) Representative gating strategy for PMNs purity. Scatter plots represent miRNAs changes during PMNs activation M2 (n = 3) (B) or M1 (n = 3) (C) macrophages polarization D) After 5 days of in vitro culture in the presence of anti‐CD3/CD28 microbeads and IL2 ± TGF‐β, CD4+CD25− Tconv cells were stained with a probe to distinguish live cells and with anti‐ CD3, ‐CD4, ‐CD25 and ‐Foxp3 mAbs. Treg frequency, evaluated as the percentage of Foxp3+CD25high cells inside CD3+CD4+ live lymphocytes, was compared between untreated (No TGF‐β) and TGF‐β stimulated T cells (left). The percentage of CD25hiFoxp3+ cells is reported in the dot plots (one representative of 5). Histograms show the fold increase expression of Treg markers between untreated (No TGF‐β) and TGF‐β stimulated T cells (n = 5 donors)(center). Scatter plot for miRNA modulation during Treg conversion (n = 3) (right) E) miRNA modulaton in activated T cell (n = 3). F) Activation of platelets was confirmed by CD62P positive cells increase (n = 4) and miRNAs secretion was represented (n = 3). G) Hypoxia treatment up‐regulated mir‐126 expression (left) and secretion (middle) in endothelial cells. Induction of hypoxia was demonstrated by increase of HIF‐1a expression (right). H) mir‐145 expression analysis in CAF and their normal counterpart using qPCR (left) (n = 5 pairs) and mir‐145 secretion in CAF and NF analyzed using dPCR (n = 10)(right). Data are expressed as mean ± SEM.
Suppl. Figure 5 Phenotipic characterization of miRNAs over‐expressing macrophages. A) Bar graphs show expression levels after miRNA mimics compared to control and M2 macrophages. (n = 3 for each miRNA). B) Histrograms revealed that inhibition of mir‐320a induced M1 polarization (n = 5). Data are expressed as mean ± SEM. The differences between controls and treated macrophages were assessed using Student's T test.