

Abstract
Cardiomyocytes are highly coordinated cells with multiple proteins organized in micro domains. Minor changes or interference in subcellular proteins can cause major disturbances in physiology. The cardiac sodium channel (NaV1.5) is an important determinant of correct electrical activity in cardiomyocytes which are localized at intercalated discs, T‐tubules and lateral membranes in the form of a macromolecular complex with multiple interacting protein partners. The channel is tightly regulated by post‐translational modifications for smooth conduction and propagation of action potentials. Among regulatory mechanisms, phosphorylation is an enzymatic and reversible process which modulates NaV1.5 channel function by attaching phosphate groups to serine, threonine or tyrosine residues. Phosphorylation of NaV1.5 is implicated in both normal physiological and pathological processes and is carried out by multiple kinases. In this review, we discuss and summarize recent literature about the (a) structure of NaV1.5 channel, (b) formation and subcellular localization of NaV1.5 channel macromolecular complex, (c) post‐translational phosphorylation and regulation of NaV1.5 channel, and (d) how these phosphorylation events of NaV1.5 channel alter the biophysical properties and affect the channel during disease status. We expect, by reviewing these aspects will greatly improve our understanding of NaV1.5 channel biology, physiology and pathology, which will also provide an insight into the mechanism of arrythmogenesis at molecular level.
Keywords: CaMKII, Fyn kinase, macromolecular complex, NaV1.5, phosphorylation, PKA, PKC
1. INTRODUCTION
The NaV1.5 channel is the major isoform of the population of sodium channels in human heart responsible for the depolarizing phase of the action potential and conduction of the cardiac impulse. NaV1.5 is encoded by the SCN5A gene, located on the shorter arm of chromosome 3p21.1 The reported half‐life of NaV1.5 is within the range of 17‐35 hours,2, 3 and during its life cycle NaV1.5 interacts with multiple protein partners forming a macromolecular complex. These interacting partners regulate gene transcription, protein synthesis, trafficking, membrane incorporation, channel function and finally degradation. Post‐translational modifications, especially phosphorylation, play a crucial role throughout the lifecycle of NaV1.5 channels. Multiple kinases phosphorylate and regulate NaV1.5 channel physiology and pathology. Cyclic AMP‐dependent protein kinase (PKA), protein kinase C (PKC) and calcium/calmodulin‐dependent kinase II (CaMKII) are among the most abundant kinases expressed in the left ventricle of the heart, according to proteomic studies.4 NaV1.5 channel function and its regulation are in themselves complex processes, becoming ever more complex as new interacting protein partners are identified. In this review, we summarize structure and function of the NaV1.5 channel, formation of the macromolecular complex, its subcellular distribution and modulation by phosphorylation.
2. STRUCTURE AND FUNCTION
The cardiac sodium channel consists of one α‐ (NaV1.5) and one or more auxiliary β‐subunits in a 1:1 ratio. The NaV1.5 adult or canonical isoform is composed of 2016 amino acid residues with a molecular mass of about 260 kDa.5, 6, 7 Five different β‐subunits (β1‐β4 and β1B) are expressed in cardiac tissue. The β‐subunits share a common membrane topology including an extracellular N‐terminal that adopts an immunoglobulin fold, a transmembrane domain and an intracellular C‐terminal domain. The subunit β1B is an exception that is a splice variant of β1 which lacks a transmembrane domain. The β1 and β3‐subunits associate with the NaV1.5 channel α‐subunit non‐covalently, while β2 and β4‐subunits are linked covalently by disulfide bonds.5, 8 These non‐pore forming β‐subunits are implicated in the physiology and pathology of the α‐subunit and play an important role in regulating the kinetics, gating, surface expression and voltage dependence of the NaV1.5 channel.5, 9
NaV1.5 α‐subunit RNA is a product of 28 different exons. Exon 1 and part of exon 2 encode the 5′‐untranslated region; the protein‐coding region spans exons 2‐28, while the 3′‐untranslated region is encoded by exon 28.1 Alternative splicing results in the production of several NaV1.5 RNA transcripts which can be categorized into functional (NaV1.5a, NaV1.5c, NaV1.5d, NaV1.5e and hH1c) and non‐functional (NaV1.5b, NaV1.5f and C‐terminal splice variant) splice variants.7, 10 NaV1.5 channel protein has a modular structure consisting of four domains (DI‐DIV), which are connected by intracellular connecting loops (ICLI‐II, ICLII‐III, and ICLIII‐IV). In addition to intracellular connecting loops, both carboxyl terminus (C‐terminus) and amino terminus (N‐terminus) are also located intracellularly. Each domain is further comprised of six transmembrane segments (S1‐S6), which are connected by short, alternating, intra‐ and extracellular loops.11 The transmembrane subunit S4 of each domain contains positively charged amino acids at every third or fourth position and serves as a voltage sensor.12 The S5 and S6 subunits of each domain constitute the pore lining, and are connected by loops called P‐loops which curve back into the pore and form the selectivity filter (a group of four amino acid residues: aspartic acid, glutamic acid, lysine and alanine; DEKA arrangement). Of these four amino acids, lysine in DIII is vital for differentiation between monovalent Na+ and divalent Ca++ ions (Figure 1).13, 14, 15
Figure 1.
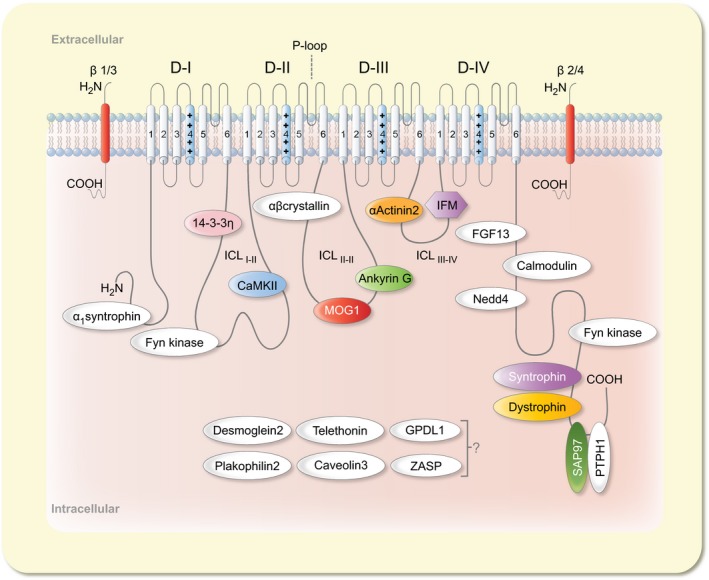
Schematic representation of cardiac sodium channel. The NaV1.5 α‐subunit consists of four domains (DI‐DIV), connected by intracellular loops (ICLI ‐ II‐ICLIII ‐ IV). Each domain is further comprised of six transmembrane subunits (S1‐S6). The S4 subunit of each domain constitutes the voltage sensor while IFM motif in ICLIII ‐ IV plays a critical role in channel inactivation. The extracellular loop between S5 and S6 of each domain form P‐loop which acts as a selectivity filter. Various NaV1.5 protein partners are shown in different colours which correspond to the formation of macromolecular complex as shown in Figure 2. The position of these protein partners in IC‐loops, N‐ and C‐terminal is according to their approximate interaction site with the NaV1.5 α‐subunit
Voltage‐dependent gating is a process by which alteration in membrane potential, results in structural conformations causing the ion channels to open (conductive) or close (nonconductive). NaV1.5 channels are activated (opened) by the outward movement of voltage sensor S4 and allow Na+ influx. Initially, Na+ permeability increases rapidly during phase 0 of the action potential and then decreases due to NaV1.5 channel inactivation (closed), which renders NaV1.5 refractory until repolarization is completed.6 The kinetics of inactivation can be subdivided into slow inactivation which develops over several seconds and regulates excitability, while fast inactivation which occurs within milliseconds is important in action potential repolarization.16, 17 The process of slow inactivation is not well understood; involvement of P‐loops and various conformational states are assumed to lead the channel into slow inactivation.16, 18 The mechanism of fast inactivation on the other hand, is well established and an inactivation gate comprising the amino acid residues isoleucine, phenylalanine and methionine (IFM), has been identified in ICLIII‐IV. Scanning mutation analysis has identified several amino acids in the short intracellular connecting loops of transmembrane segments S4 and S6 in DIII and DIV, which serve as docking sites for inactivation and closing of the channel pore.13, 15 The C‐terminus is also known to modulate NaV1.5 channel inactivation by stabilizing and minimizing channel reopening.19 NaV1.5 channel activation derives its voltage dependence from outward movement of the voltage sensor, S4, in response to alteration in membrane potential. This outward movement of the S4 subunit also initiates inactivation, thus deriving its voltage dependence by coupling with the process of activation.6, 20 NaV1.5 channels can be activated again during phase 4 of the action potential after recovering from inactivation, although, some channels (< 1%) may reactivate during phase 2 and 3 of the action potential and generate small late sodium current (late INa), also called persistent non‐inactivating current. This small inward current is usually less than 0.5% of the peak INa but it flows approximately 300‐400 milliseconds longer thereby maintaining action potential plateau and playing an important role in Na+ loading. Increased intracellular Na+ levels also increase Ca++ levels via the Na+/Ca++‐exchanger hence also affecting contraction and relaxation.21 Late INa has minimal contribution to the action potential under physiological conditions, but plays an important role in the pathological context. Late INa is increased in acquired disease conditions like heart failure (HF), hypertrophy and diabetes mellitus (DM) or under congenital cardiac disorders like long‐QT syndromes (LQTS).21, 22 This increased Late INa can trigger early afterdepolarizations by prolonging action potential duration or delayed afterdepolarizations by increasing intracellular Ca++ levels thus contributing to arrythmogenesis.21, 23, 24
Alterations in the SCN5A gene can lead to NaV1.5 channel dysfunction resulting in either gain‐of‐function or loss‐of‐function effects. These mutations in the NaV1.5 channel affect structure, function, trafficking, interaction with other protein partners and formation of the macromolecular complex. NaV1.5 channel variants have been associated with several congenital cardiac disorders such as atrial standstill, atrial fibrillation (AF), Burgada syndrome (BrS), cardiac conduction disease (CCD), dilated cardiomyopathy (DCM), LQTS and sudden infant death syndrome (SIDS).25, 26 Gain‐of‐function mutations in NaV1.5 result in increased persistent current which may lead to long‐QT syndrome type 3 (LQTS‐3), while loss‐of‐function mutations result in decreased peak INa and are implicated in BrS and sick sinus syndrome (SSD). The phenotype of these NaV1.5 channel mutations depends on several factors which may include genetic, transcriptional, translational and post‐translational modifiers. Discussion of these modifiers affecting the phenotype of disease‐causing mutant channels is beyond the scope of this review and we refer interested readers to some excellent review articles for further reading.9, 26, 27
3. MACROMOLECULAR COMPLEX AND SUBCELLULAR DISTRIBUTION
Cardiac myocytes are rod‐shaped cells, approximately 100 μm in length and 20 μm in width, expressing myriad of proteins in different membrane compartments. These proteins are precisely localized indicating their distinct functional roles. Depending on subcellular localization, NaV1.5 channels are known to be arranged in three different compartments on plasma membrane, namely intercalated discs, lateral membranes and T‐tubules.28 Intercalated discs (IDs) are highly coordinated structures located between the ends of myocytes along their 20 μm breadth, and comprising both communication (gap junction) and anchoring complexes (desmosomes and adherens junctions).29 Mutations or acquired diseases that disrupt ID components are known to contribute in arrythmogenesis. The second and third populations of NaV1.5 channels reside at lateral membranes which also comprise invaginations called T‐tubules. This population of sodium channels ensures propagation of electrical impulse both in longitudinal and transverse directions (Figure 2).28
Figure 2.
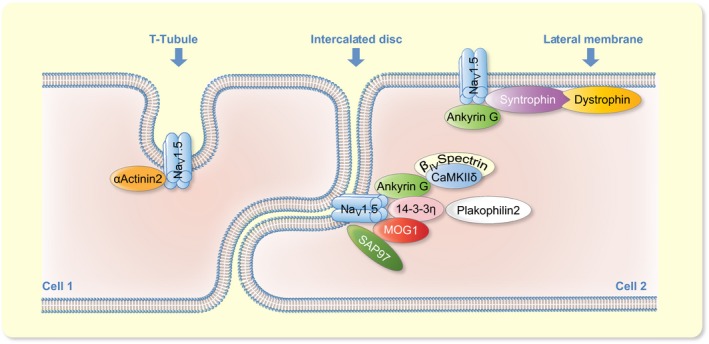
NaV1.5 channel macromolecular complex. Representative figure showing formation of cardiac sodium channel macromolecular complex at T‐tubules, intercalated discs and lateral membranes. The clustering of interacting protein partners at respective sites is known from the literature
The NaV1.5 channel contains several binding motifs for auxiliary proteins which interact to regulate its intracellular transport, cellular localization, gating and degradation (Figure 1). These auxiliary proteins can be broadly categorized as adaptor proteins (anchor NaV1.5 channel to cytoskeleton for trafficking and targeting to specific compartments of plasma membrane), enzymes (modify NaV1.5 by post‐translational modifications) and regulatory proteins (modulate gating).9 NaV1.5 ICLI‐II contains several sites for phosphorylation and an interaction motif for a dimeric cytosolic adaptor protein 14‐3‐3η, a 30 kDa protein widely distributed in tissues of different organisms. Seven isoforms of the 14‐3‐3 protein are expressed in mammalian cells that interact with other protein partners by two consensus sequences known as “mode‐1” (RXXpS/pTXP) and “mode‐2” (RXXXpS/pTXP) binding motifs.30 In cardiomyocytes 14‐3‐3η is co‐localized with NaV1.5 at the ID and its interaction with NaV1.5 shifts steady‐state inactivation towards hyperpolarization, with slowed recovery from inactivation.31 Recently it has been shown that 14‐3‐3η facilitates NaV1.5 channel α‐α dimerization and mediates coupled gating.32 These findings are quite intriguing given the widely held hypothesis that sodium channels exist as a single α‐subunit in macromolecular complexes.
Ankyrin‐G and a cofactor of NaV1.5, multi‐copy suppressor of GSP1 (MOG1), interact with ICLII‐III. Both ankyrin‐G and MOG1 are co‐localized with NaV1.5 at the ID. MOG1 is considered a cofactor for optimal expression of NaV1.5,33 while ankyrin‐G organizes trafficking of NaV1.5 at IDs and T‐tubules.34 Ankyrin‐G is known to interact via the residue sequence 1047‐VPIAVAESD‐1055 in ICLII‐III,34 and coordinates a functional macromolecular complex at IDs with NaV1.5, βIV‐spectrin and CaMKII.35 βIV‐spectrin interacts with the C‐terminal domain of CaMKIIδ and transgenic mice lacking this interaction show reduced levels of CaMKIIδ at intercalated discs, while levels in T‐tubules remain unaffected, indicating distinct trafficking mechanisms at each subcellular compartment. Recruitment of CaMKII by βIV‐spectrin at IDs in the vicinity of NaV1.5 is followed by sodium channel phosphorylation, while disruption of this interaction, as observed in qv 3J transgenic mice, results in an increased INa, rightward shift in fast inactivation and decreased late INa.36 Cardio‐specific ankyrin‐G knockout mice exhibit reduced INa, decreased expression and localization of NaV1.5 specifically at IDs of cardiomyocytes. At the same time, expression and localization of CaMKIIδ and βIV spectrin to IDs was decreased resulting in disruption of late INa regulation by ankyrin‐G, βIV spectrin, CaMKIIδ and the NaV1.5 macromolecular complex.28, 35 Ankyrin‐G also interacts with plakophilin‐2 and connexin43 (desmosomal and gap junction proteins),37 and is thus apparently an important player in coordinating mechanical and electrical signalling at IDs in cardiomyocytes (Figure 2).38 ICLIII‐IV is known to interact with α‐actinin2 which is an F‐actin cross‐linking protein. Both proteins co‐localize at T‐tubules and the interaction increases surface expression of NaV1.5 without any changes in channel gating.39, 40
The last three amino acids (SIV) of the NaV1.5 C‐terminus constitute a PDZ domain binding site which interacts with syntrophins, synapse associated protein 97 (SAP97) and protein tyrosine phosphatase 1 (PTPH1).41 Syntrophins form a macromolecular complex with dystrophins and coordinate localization of Nav1.5 at lateral membranes (Figure 2).42 Dystrophin knockout mice exhibit conduction defects and reduced NaV1.5 expression,42 while SAP97 together with ankyrin‐G, ensures correct surface expression of NaV1.5 at IDs.28, 39 Genetically modified NaV1.5 (ΔSIV) mice, in which binding of syntrophin and SAP97 has been disrupted, demonstrate downregulation of NaV1.5 channels at lateral membranes, with slowed cardiac impulse conduction.43 PTPH1 interaction affects NaV1.5 gating by shifting the availability curve towards hyperpolarized potentials.44 A calmodulin (CaM) binding IQ‐motif is also present at the C‐terminus of the NaV1.5 channel.45 CaM is a small ubiquitous 17 kDa protein which binds/senses calcium ions. Its interaction with NaV1.5 by binding through the IQ‐motif results in enhancing the slow inactivation process and a hyperpolarizing shift in the I‐V curve.45, 46 Recently, two additional interaction sites in ICLIII‐IV have been reported for CaM which modulates the NaV1.5 channel by destabilizing the inactivated state and promoting faster recovery from inactivation.47 The size of the NaV1.5 channel population is determined by a balance between synthesis and degradation. An ubiquitin protein ligase (NEDD4‐2) binding pY‐motif has been identified at the C‐terminus of the NaV1.5 channel.48 E3 ubiquitin‐protein ligase NEDD4‐2 ubiquitinates the NaV1.5 channel, thus giving the signal for internalization and finally degradation.48 Co‐expression of NEDD4‐2 with NaV1.5 in Xenopus oocytes, decreased ionic currents by up to 40‐65% with significant reduction in membrane expression. Conversely, an inactive NEDD4‐2 analogue increased both INa and NaV1.5 membrane expression.48, 49 Several modulators such as 14‐3‐3 protein, MAPKs, PKA and serine/threonine kinase SGK regulate NEDD4.27, 50 Recently, αB‐crystallin was reported to interact with NaV1.5 via ICLII‐III and C‐terminus. αB‐crystallin co‐localizes with NaV1.5 and increases INa by decreasing the ubiquitination.51 Additionally, fibroblast growth factor homologous factors like FGF12, FGF13 and FGF14 also interact with the C‐terminus of NaV1.5 affecting the expression, trafficking and gating of the channel.52, 53
Lastly, the role of the N‐terminus of NaV1.5 is not fully understood yet; however, certain missense mutations in the N‐terminus lead to degradation of the channel and exert a negative effect on wild‐type (WT) channels. Also co‐expression of the N‐terminus peptide fragment with WT channels resulted in a twofold increase in surface expression and INa compared to WT channel alone. This indicates that α‐subunits of Nav1.5 channel interact via the N‐terminus by unknown mechanisms.54 Recently, it has been shown that an internal PDZ‐like domain, present in the N‐terminus of NaV1.5 interacts with α1‐syntrophin, exerting a chaperone‐like effect to positively modulate NaV1.5, Kir2.1 and Kir2.2 channels in cardiomyocytes.55 Moreover, several phosphorylation sites for Fyn kinase have also been identified in the N‐terminus which may contribute to a depolarizing shift of fast inactivation, produced by Fyn kinase interaction with NaV1.5 channel.56 Caveolin‐3, desmoglein‐2, glycerophosphoryl diester phosphodiesterase‐like protein 1 (GPDL1), plakophilin‐2, telethonin and Z‐band alternatively spliced PDZ‐motif protein (ZASP), are also reported to interact with NaV1.5 channel through unidentified sites (Figure 1).39 The specific purpose of these distinct pools is not well established, but based on recent studies the proportions of current generated by these pools can be speculated. Sodium channels present on T‐tubules generate around 20%, while NaV1.5 channels residing on lateral membranes about 30% of INa. Thus, the remaining 50% of INa may be attributed to NaV1.5 channels localized on IDs.28
4. MODULATION OF KINETICS AND TRAFFICKING OF NAV1.5 CHANNEL BY PHOSPHORYLATION
4.1. PKA
PKA, first described in 1968,57 is a well‐studied protein kinase. It is a holoenzyme existing as a heterotetramer with two catalytic and two regulatory subunits. When a second messenger cAMP binds to these regulatory subunits, a conformational change takes place which releases and activates the catalytic subunits.58 PKA is targeted to different sub‐cellular locations by a scaffolding protein called A‐kinase anchoring protein (AKAP), where upon activation, it phosphorylates the target proteins by transferring γ‐phosphate of ATP.59 PKA phosphorylates myriad of proteins including the NaV1.5 channel.60 The generalized consensus motifs in substrate proteins for phosphorylation by PKA include R/KXS/T, RRXS/T and R/KXXS/T.61 Initial evidence for PKA mediated modulation of the NaV1.5 channel was observed by stimulation of the β‐adrenergic (β‐AR) system with isoproterenol which decreased INa and produced a hyperpolarizing shift in steady state inactivation by increasing levels of cAMP.62, 63 Both decreased upstroke velocity of action potential and decreased INa has been reported in neonatal rat and adult guinea pig ventricular myocytes through PKA and G‐protein‐regulated pathways.62, 63, 64, 65 Contrarily, increased INa has also been reported in guinea pig myocytes,66 rabbit myocytes67 and NaV1.5 expressed in Xenopus oocytes.68 This increase in INa was attributed either to phosphorylation of NaV1.5 by PKA or activation of G‐protein (Gsα), which was observed at hyperpolarized potentials (negative to ‐75 mV) without any effect on NaV1.5 channel gating kinetics.67 Some possible explanations for these initial conflicting reports include different voltage protocols used (depolarized vs hyperpolarized holding potentials),62 or different concentrations of isoproterenol used in different experimental settings.69, 70 This discrepancy in INa by cAMP or β‐AR stimulation was further elaborated in experiments showing a test pulse of ‐50 mV at a holding potential of ‐150 mV increased INa, while a test pulse of +30 mV at a holding potential of ‐90 mV decreased INa. However, no effect was observed when sodium currents were elicited by a test pulse of +30 mV at a holding potential of ‐150 mV.71 Later, with the help of biochemical studies, it was demonstrated that PKA phosphorylates serine residues at positions 526 and 529 in ICLI‐II of the rat NaV1.5 channel. However, no functional data for the biophysical characteristics of NaV1.5 channel for these phosphorylated sites were provided by the authors.72 The involvement of ICLI‐II in modulation by PKA, was also confirmed in human NaV1.5 channel (hH1 variant), where injection of cAMP in Xenopus oocytes expressing NaV1.5 channel increased the conductance of channel without affecting half maximal activation or inactivation.73 The increase in INa by PKA stimulation develops slowly without reaching saturation over a time period of one hour, suggesting the involvement of an additional mechanism other than direct modulation of the channel by phosphorylation.74 Whole cell patch clamp experiments in rat myocytes demonstrated that the increased INa observed upon β‐AR stimulation was due to the increased number of functional sodium channels, rather than activation by PKA and Gsα, which does not affect gating or open probability of the channel.75 Further evidence for the involvement of PKA in NaV1.5 channel trafficking was obtained by incubating NaV1.5 channel expressing cells with chloroquine and monensin. Both the drugs interfere in recycling of membrane proteins and pre‐incubation with these two drugs does not increase INa on PKA stimulation.74 These reports clearly indicate that PKA mediated phosphorylation increases INa by promoting forward trafficking and increasing total number of functional NaV1.5 channels.76, 77 In the rat NaV1.5 channel, the involvement of two serine residues at positions 526 and 529 was reported earlier to increase INa upon PKA activation.72 Mutation of these conserved serine residues at positions 525 and 528 to alanine (Figure 3; Table 1), in human NaV1.5 channel (hH1 variant), abolished the PKA‐mediated increase in INa suggesting the same mechanism may also play a role in modulation of other NaV1.5 channel isoforms.78 In ICLI‐II there are three endoplasmic reticulum (ER) retention signals (RXR), R479KR481, R533RR535 and R659QR661 which are either up‐ or downstream of the PKA‐phosphorylated serine residues. Addition of a phosphate group on serine 525 or 528 imparts large negative charge on these residues which may mask the ER retention signals, thus promoting forward trafficking of the NaV1.5 channel. ER retention signals play an important role in trafficking of membrane proteins, since newly formed proteins are retained in the ER and released when these ER signals are masked by binding of another protein.78 Among the three ER‐retention sites, the R533RR535 site plays a major role in PKA‐mediated increased INa; it could be argued, that phosphorylation of serine 525 and 528 could mask this site and promote forward trafficking of the NaV1.5 channel.78 This forward trafficking redistributes NaV1.5 channels from intracellular reservoirs like the ER and caveolae in the plasma membrane, thus increasing NaV1.5 channel recruitment up to 45%.77
Figure 3.
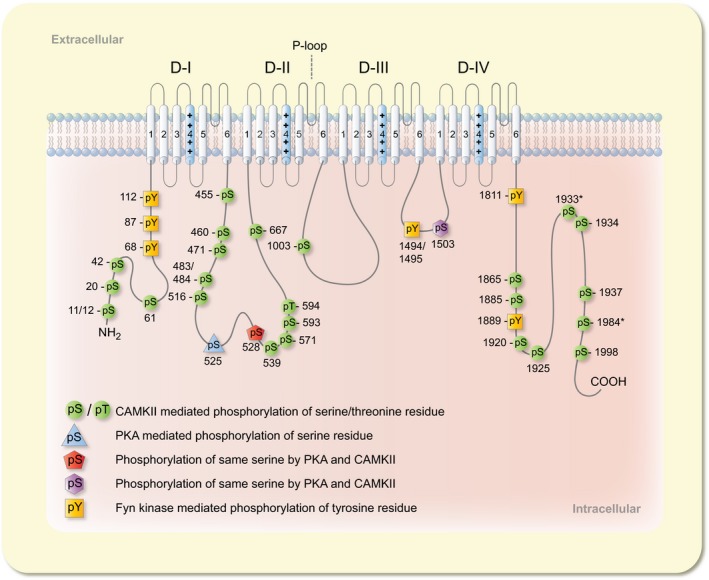
Phosphorylation of NaV1.5 channel. Diagrammatic presentation of phosphorylation of the NaV1.5 channel α‐subunit at serine, threonine and tyrosine residues by PKA, PKC, CaMKII and Fyn kinase with respective colours. The amino acids numbering is according to NaV1.5c isoform (Uniprot identifier # Q14524‐1), while the amino acids with asterisk signs113 correspond to NaV1.5 hH1c isoform numbering
Table 1.
Modulation and phosphorylation of NaV1.5 channel. Phosphorylation of NaV1.5 channel serine, threonine and tyrosine residues by PKA, PKC, CaMKII, Fyn and SGK with the method of identification and the most commonly reported effects on NaV1.5 channel by the respective kinases
| Kinase | Phosphorylated residue | Effect on NaV1.5 channel | Method of indentification | Reference |
|---|---|---|---|---|
| PKA |
|
|
|
62, 63, 66, 67, 68, 72, 77, 78 |
| PKC |
|
|
|
87, 88, 89, 91, 92, 93, 94, 95, 96 |
| CaMKII |
|
|
|
36, 103, 106, 112, 113, 114, 115 |
| Fyn |
|
|
|
56, 130, 131 |
| PI3Kα and PKB/Act | N/A |
|
|
133, 134 |
| PDK1 | N/A |
|
|
136 |
| SGK |
|
|
|
138, 139, 140 |
| β1‐AR stimulation |
|
N/A |
|
141 |
| Unknown kinase |
|
N/A |
|
115 |
| Native phosphorylated residues |
|
N/A |
|
113, 114, 115 |
PKA is a downstream effector of the β‐AR signalling pathway and plays an important role in cardiac excitation‐contraction coupling.79 In cardiomyocytes, β‐AR couples with stimulatory Gs protein and stimulation of the adrenergic system produces cAMP which activates PKA. β‐AR stimulation modulates NaV1.5 channel directly by PKA‐mediated phosphorylation or indirectly by Gsα signalling pathway.75 PKA activation can fractionally increase INa in epicardial border zone cardiomyocytes of infarcted canine heart.80 Moreover, a missense mutation (R526H) in ICLI‐II of NaV1.5 channel resulted in a BrS phenotype with markedly reduced INa. This mutation resides in the PKA recognition site which inhibits PKA‐mediated phosphorylation of the NaV1.5 channel, resulting in reduced incorporation of functional channel into the plasma membrane. Activation of PKA does not increase the INa by rescuing R526H mutant channels, which also underpins the importance of PKA‐mediated phosphorylation of the channel.81 Apart from the NaV1.5 channel, PKA also phosphorylates CaV1.2, phospholamban, troponin I and C, ryanodine receptors and several other proteins, and so plays important role in cardiac physiology.82 However the direct involvement of PKA in cardiac pathophysiology or the use of PKA to improve cardiac pathology is not well described; further studies are required to characterize the PKA‐NaV1.5 interaction as a potential target for drug discovery and to reveal its potential role in cardiac pathophysiology.
4.2. PKC
PKC consists of a single polypeptide with a regulatory N‐terminus and catalytic C‐terminus. This family of kinases transduces a diverse range of signals and since the identification of the first member in the 1980s, 11 different isozymes have been identified and categorized into three classes: calcium‐dependent conventional PKCs (α, βI, βII and γ), calcium‐independent novel PKCs (δ, ε, η, θ and μ) and atypical PKCs (ζ and λ).83 PKC is a downstream effector of several circulating hormones such as angiotensin II (Ang‐II), endothelin and norepinephrine, which upon stimulation cause phosphorylation of several cardiac proteins, activate other kinases and alter gene expression. These processes influence impulse conduction and EC‐coupling implicated both in normal physiology and pathological conditions.84 Like PKA, PKC phosphorylates serine and threonine residues in substrate proteins, but compared to PKA it displays less specificity.85 Initially it was reported that PKC activators like TPA (phorbol ester) and 1,2‐dioctanoglycerol (diacylglycerol analogue) increased single channel sodium currents and rate of current decay in neonatal rat ventricular myocytes.86 Later it was shown that activation of PKC by OAG (1‐pleoyl‐2‐acetyl‐sn‐glycerol) decreases INa and creates a hyperpolarizing shift in steady‐state inactivation both in neonatal rat ventricular myocytes and rat cardiac sodium channel (rNaV1.5), stably expressed in the Chinese hamster lung 1610 cell line. This decrease in INa was attributed to a decreased open probability in single‐channel studies.87 Subsequently, the same authors reported that serine 1505 in ICLIII‐IV was involved in modulation of rNaV1.5 by PKC; upon replacement of this serine by alanine, PKC activation did not decrease INa nor did it mediate a hyperpolarizing shift in fast inactivation.88 This serine residue is conserved in several sodium channel isoforms and plays a role in modulation by PKC; however, the authors did not provide any direct evidence for phosphorylation of serine 1505. Similarly, in Xenopus oocytes transiently expressing human NaV1.5 (hH1 variant), activation of PKC by PMA (phorbol 12‐myristate 13‐actate) or OAG decreased INa without any hyperpolarizing shift in steady‐state inactivation. The decrease in INa was in part attributed to phosphorylation of serine 1503 (homologous to serine 1505 in rNaV1.5) in ICLIII‐IV because mutation of this serine did not completely abolish the effect of PKC activation (Figure 3; Table 1).89 PKC activation by phorbol esters or diacylglycerol derivatives exhibit diverse effects on cardiomyocytes. These agonists are nonspecific and have multiple targets besides PKC which may also exert nonspecific effects on NaV1.5 channel. To investigate these differential effects a peptide PKCP was used to specifically activate endogenous PKC and observe effects on the NaV1.5 channel in rat ventricular myocytes. This peptide blocks the auto‐regulatory region of native PKC and exposes the catalytic site to bring about phosphorylation of substrate proteins. Activation of PKC by PKCP peptide caused a dose dependent depolarizing shift in half maximal inactivation of the NaV1.5 channel, while no effect was observed on half maximal activation or peak INa. This depolarizing shift in inactivation was reversed by PKC inhibitors such as chelerythrine chloride or saturosporine. Besides the depolarizing shift in the inactivation curve, PKC activation also slows INa decay and channel inactivation, while recovery from inactivation is enhanced.90
Human cardiomyocytes express nine different isoforms of PKCs irrespective of normal or diseased states; however, levels of these isoforms may vary in different conditions.84 PKC activators such as phorbol ester or diacylglycerol analogues are nonspecific in nature and it is highly likely that different PKC isoforms act differently providing a reason for differential effects reported in various studies of activation of PKC.86, 87, 90 By using peptide activators or inhibitors of PKC isoforms it was described that specific activation of εPKC decreases INa in rat ventricular myocytes and the human NaV1.5 channel (hH1 isoform) heterologously expressed in Xenopus oocytes, without affecting voltage dependence of activation or inactivation. However the blockade of εPKC did not completely abolish the effect of PMA which suggests the involvement of other PKC isoforms as evidenced in another study where activation of PKCα decreased the INa.91, 92 With the development of specific PKC activators it was described that activation of PKCα decreased INa without any effect on gating. This decrease in INa was slow in development and non‐saturable which was supposedly because of internalization or re‐distribution of functional hNaV1.5 channels away from the plasma membrane.92, 93 This modified intracellular trafficking of hNaV1.5 channels was attributed to PKC‐mediated phosphorylation of the channel and ROS.93 Moreover, the authors also described that in pre‐blocked cPKCs, low concentrations of PKC activators such as PMA (1 nM) and thymeleatoxin (50 nM) cross activate PKA which slightly increases INa and explains initial conflicting results on PKC activation.92 Renin‐angiotensin signalling is known to activate several PKC isozymes and in several cardiovascular disorders this pathway is chronically activated. In transgenic mice overexpressing cardio‐specific angiotensin‐II type 1 receptors (AT1R), QRS complex widening and slower action potential was observed because of decreased INa. This AngII‐AT1R pathway stimulation activates PKCα and co‐localizes it with NaV1.5 at the plasma membrane where their interaction results in decreased INa.94
Recently a metabolic pathway for PKC activation has been described where the elevated levels of NADH activate PKCδ, resulting in a decrease in INa.95 Interestingly, this decrease in INa was not because of any change in surface expression but rather channel conductance was decreased directly by phosphorylation of serine 1503 in NaV1.5 channels and indirectly by increasing ROS production in mitochondria.95, 96 The PKCδ antagonism completely reversed both decrease in INa and ROS production from the mitochondria while specific inhibition of PKCα could partially recover INa without any effect on ROS production, thus indicating the involvement of more than one PKC isozyme. By mutational analysis it was described that both phosphorylation of serine 1503 in NaV1.5 channels and ROS production in mitochondria are required for PKCδ‐mediated modulation of the NaV1.5 channel.95 Alteration in sodium current is detrimental in underlying disease conditions and may trigger arrhythmias leading to sudden cardiac death.94 As discussed above, activation of several PKC isozymes are reported to influence the INa and the reported effects on NaV1.5 are contradictory, which might be because of involvement of different PKC isozymes. Activation of PKC results in phosphorylation of a conserved serine residue in ICLIII‐IV which alters NaV1.5 channel trafficking, but besides this serine residue phosphorylation of other serine residues is also likely. Additional detailed studies are warranted which might unravel further aspects of PKC signalling and may explain the differential effects reported on NaV1.5 channel by PKC activation.
4.3. CaMKII
CaMKII is a multifunctional enzyme class consisting of four isoforms. The α‐ and β‐ isoforms are mainly expressed in neuronal tissues while γ‐ and δ‐ are expressed ubiquitously.97 Alternative splicing of primary transcripts results in generation of multiple variants. Six splice variants of the CaMKIIδ isoform have been identified in the heart.98 As the name indicates, CaMKII activity is dependent on Ca2+ and/or CaM, which after binding induces conformational changes in CaMKII to autophosphorylate at threonine 287. Autophosphorylation keeps CaMKII active after dissociation of Ca2+/CaM.99 CaMKII is also activated in a Ca2+ independent manner by ROS‐mediated oxidation of methionine residues at positions 281/282. This mode of activation requires initial binding of Ca2+/CaM to expose the potential methionine residues followed by oxidation. Oxidative stress is observed in HF, after myocardial infarction (MI) and with increased levels of Ang‐II.100 Moreover levels of CaMKIIδC are also increased during pressure overload, HF and sustained β1‐AR stimulation thus both factors contributing in outcomes of adverse cardiac events.101, 102, 103 Hence, CaMKII Inhibition appears to be cardio‐protective after MI by reducing apoptosis and remodelling associated with excessive stimulation of β‐AR signalling and Ang‐II.100, 104
Initial evidence for CaMK‐mediated modulation of NaV1.5 channel was obtained indirectly using CaMK inhibitors such as KN‐93 and autocamtide‐2 related inhibitory peptide (AIP). KN‐93 slowed NaV1.5 channel current decay, produced a depolarizing shift in fast inactivation and slowed entry into inactivated states. Interestingly, AIP the specific inhibitor of CaMKII did not exhibit any effects on NaV1.5 channels and the authors suggested the involvement of CaMKIV in modulation of NaV1.5 channels.105 Direct evidence for CAMKII interaction with NaV1.5 channel was obtained from transgenic mice expressing CaMKIIδC and in rabbit ventricular myocytes with acute overexpression of CaMKIIδC. Overexpression of CaMKIIδC increased the phosphorylation of NaV1.5 channels and in both experimental models CaMKIIδC interaction slowed NaV1.5 channel fast inactivation, created a hyperpolarizing shift in steady‐state inactivation, increased late INa and number of NaV1.5 channels undergoing intermediate inactivation along with slowed recovery from inactivation. Together these effects could be arrhythmogenic (prolonged QT and QRS intervals) as has been observed in transgenic mice overexpressing CaMKIIδC.106 CaMKII inhibition by KN‐93 reduced late INa in ventricular myocytes from normal and experimental canine model of chronic HF.107 CaMKII inhibition by KN‐93 under basal physiological conditions in rat ventricular myocytes reduced peak INa and late INa, produced a hyperpolarizing shift in fast inactivation, and augmented intermediate inactivation with slowed recovery from both fast and slow inactivation. It also reduced membrane excitability by decreasing upstroke velocity during action potential.108 These results are not consistent with overexpression of CaMKII,106 which caused a hyperpolarizing shift in inactivation and slowed recovery from inactivation. In guinea pig ventricular myocytes CaMKIIα increased peak INa, produced a depolarizing shift in fast inactivation, accelerated recovery from inactivation, increased late INa and decreased the fraction of channels undergoing intermediate inactivation. CaMKIIα also increased action potential duration while blockade by KN‐93 shortened it. Moreover, CaMKIIα‐mediated phosphorylation was observed in the ICLI‐II and C‐terminal domains of the NaV1.5 channel.46 These results differ in certain aspects from rabbit ventricular myocytes overexpressing CaMKIIδC, which demonstrated hyperpolarizing shift in fast inactivation, slowed recovery from inactivation and increased intermediate inactivation.106 These differences can be accounted for by different methodological approaches, different CaMKII (α vs δ) isoforms, altered phosphorylation of NaV1.5 channel due to overexpression of CaMKIIδC and association of the chronic overexpression of CaMKIIδC with cellular and structural remodelling. The effects of CaMKII on NaV1.5 remained disputed; however, there is a general consensus on increased late INa, but sodium channel gating and kinetics are debatable. Late INa is increased in certain pathological conditions like LQTS‐3, cardiac ischemia or HF. Increased late INa overloads cellular calcium and sodium levels, which may play a key role in diastolic dysfunction and arrythmogenesis. Late INa prolongs action potential duration by reducing repolarization reserves and may trigger early after depolarizations, while sodium and calcium overload may lead to delayed after depolarizations both in atrial and ventricular myocytes.109 Pathological or drug‐induced, increased late INa elevates Na+ levels in cardiomyocytes, which in turn elevates Ca+2 levels by sodium calcium exchanger thus paradoxically activating CaMKII.21, 23, 24, 110 Inhibition of late INa by ranolazine prevents CaMKII activation and acts as a cardio‐protective agent.111
CaMKII interaction with NaV1.5 in myocytes has been established by co‐immunoprecipitation experiments and immunostaining which indicates co‐localization of CaMKII and NaV1.5 channels at IDs and T‐tubules.36, 106, 108 These co‐immunoprecipitation experiments and co‐localization of CaMKII with NaV1.5 channels strongly suggest direct interaction of CaMKII with the NaV1.5 channel. There are several CaMKII‐mediated phosphorylation motifs (RXXS/T) in NaV1.5 channel and alanine‐scanning suggested phosphorylation of serine residue at position 571 which was also validated by site‐specific antibodies. In transgenic qv 3J mice, decreased phosphorylation of serine 571 was observed following disruption of interaction of CaMKII with the NaV1.5 channel.36 Activated CaMKIIδC interacted stably with ICLI‐II of NaV1.5, the loop which has been shown to contain more than one phospho‐acceptor sites for CaMKII. In vitro phosphorylation of NaV1.5 by CaMKIIδC showed phosphorylation of serine 483/484, 516 and serine 593/threonine 594, but the authors112 detected no phosphorylation on serine 57135, 36 as previously reported. Further biochemical tests established that only phosphorylation on serine 516, 593 and threonine 594 are involved in CaMKIIδC‐mediated modulation of NaV1.5 with preference order of 516 > 594 > 593. Alanine mutagenesis of serine 516, 571 and threonine 594, but not serine 593, prevented a CaMKIIδC mediated hyperpolarizing shift in sodium channel availability and intermediate inactivation, indicating their phosphorylation‐dependent role in regulation of NaV1.5 by CaMKIIδC.112 Mass‐spectrometry based analysis of in situ immunopurified NaV1.5 channel revealed eleven phosphorylated residues, of which 10 reside in ICLI‐II (serine 457, 460, 483, 484, 497, 510, 524/525, 571, 664, 667), and one resides in the N‐terminus (serine 36/39/42/threonine38). Several interacting protein partners, including CaMKII ‐β, ‐δ, ‐γ subunits, CAM, and FGF‐13 were also identified by mass‐spectrometry based analysis from immunoprecipitated NaV1.5 of mouse ventricular myocytes by the same authors.113 Recently, nine more NaV1.5 phosphorylated serine and threonine residues (486, 499, 516, 539, 1012, 1888, 1937, 1938 and 1989) have been reported by the same research group investigating WT and transgenic mice overexpressing CaMKIIδC. Among these, phosphorylation of serine residues at position 1938 and 1989 was increased by CaMKIIδC overexpression. Both serine residues (1938 and 1989) are conserved in the human NaV1.5 channel and when the orthologous serine residues at position 1933 and 1984 in human NaV1.5 (hH1c) are mutated, interaction of FGF‐13 with NaV1.5 channels is disrupted; this alters fast inactivation, increases late INa and decreases channel availability.114 In another recent study, mass‐spectrometry based analysis of immunopurified and CaMKIIδC‐mediated in vitro phosphorylation of human NaV1.5 expressed in HEK‐293 cells revealed 31 serine and three threonine phosphorylated residues.115 Fifty percent of these phosphorylation sites were located in ICLI‐II, underpinning the importance of the first IC‐loop in modulation of NaV1.5 by post‐translational modifications. Of these phosphorylated residues 17 were present at baseline while 23 residues were phosphorylated by CaMKIIδC. Phosphorylated amino acids were more scattered in the ICLI‐II, while they were clustered together in the N‐ and C‐termini.115 At the N‐terminus, serine 11, 12, 20, 42 and 61 were phosphorylated by CaMKIIδC, and of these, serine 42 was also phosphorylated at baseline while threonine 17 was phosphorylated by an unknown kinase. Eighteen phosphorylated residues were present in ICLI‐II and seven of them (serine 460, 471, 484, 516, 528, 539, 571, 667 and threonine 455) were phosphorylated by CaMKIIδC. Serine 460, 483, 484, 497, 510, 516, 577 and threonine 570 were also phosphorylated at baseline, while serine 457, 464, 499 and 664 were phosphorylated by an unknown kinase. ICLII‐III and ICLIII‐IV contained one phosphorylated residue each, with serine 1003 and 1503, respectively. Eight phosphorylated residues were identified in the C‐terminus and seven of them (serine 1865, 1885, 1920, 1925, 1934, 1937 and 1998) were phosphorylated by CaMKIIδC. Two serine residues 1937 and 2007 were also phosphorylated at baseline (Figure 3; Table 1).115
The majority of these identified phosphorylation sites are not characterized functionally; however, several are well described in pathological processes. Secondly, it may be emphasized that the phosphorylation status can differ on the same residue under normal physiological or pathological conditions and the same residue can be phosphorylated by multiple kinases.113 Phosphorylation at S516 is decreased in HF patients and it is also known that methylation at arginine 513 decreases phosphorylation of serine at position 516 indicating intriguing crosstalk between methylation and phosphorylation.115, 116, 117 CaMKII‐mediated phosphorylation of serine 571 specifically regulates the late INa component of NaV1.5 without affecting channel availability and recovery from inactivation. This serine 571 mediated increased late INa prolongs APD and QT interval with increased susceptibility to arrhythmias. Ventricular myocytes from transgenic mice with hyperphosphorylation at position 571 show increased late INa and β‐AR stress induced afterdepolarizations with repolarization abnormalities. Moreover, phosphorylation at S571 contributes in HF, but not in pressure overload hypertrophy.118 Two proarrhythmic NaV1.5 variants (A572D and Q573E) were initially identified in extensive genetic analysis of LQTS probands.119, 120, 121 Both of these variants reside near the CaMKII phosphorylation site serine 571. Although these variants do not have any effect on serine 571, this substitution introduces negative charge which simulates a phosphorylated residue. Both these variants exhibited increased late INa which was reversed by ranolazine and produced hyperpolarizing shift in fast inactivation with slowed recovery from inactivation. Lengthening of action potential duration was also observed with afterdepolarizations, which were eliminated by ranolazine.122 Effects produced by these variants recapitulate CaMKII mediated effects, and computational models indicate that both variants show structural and electrostatic features similar to the serine 571 phosphorylated channel. Phosphorylated CaMKII and CaMKII mediated increased phosphorylation of serine 571 was observed in ventricular myocytes of non‐ischaemic HF patients and in canine post infarct border zone, but not in transgenic mice AC3‐I expressing CaMKII inhibitor.122 CaMKII‐mediated NaV1.5 channel phosphorylation and regulation exhibits both gain‐of‐function (increased late INa) and loss‐of‐function (decreased availability) effects, indicating complex regulation by phosphorylation. Moreover, further detailed studies are required to link CaMKII‐mediated phosphorylated residues with pathological process of cardiac disorders and their role in disease progression.
4.4. Fyn kinase
Besides phosphorylation of serine and threonine, the NaV1.5 channel is also modulated by phosphorylation of tyrosine residues. Initial evidence for tyrosine phosphorylation of the NaV1.5 channel was reported indirectly by the use of protein tyrosine kinase inhibitors (genistein, AG957, PP2 and ST638), which all decreased INa while genistein and AG657 also produced a hyperpolarizing shift in steady‐state fast inactivation and prolonged recovery from inactivation.123 Similarly, co‐expression of PTPH1 with NaV1.5 also shifts steady‐state inactivation towards hyperpolarized potentials.44
Fyn tyrosine kinase is a member of the non‐receptor Src family of tyrosine kinases which is expressed ubiquitously.124 Fyn kinase consists of four domains (SH1 ‐ SH4). The SH1 domain possesses tyrosine kinase activity and is located in the C‐terminal domain. The SH2 domain recognizes phosphorylated tyrosine while the SH3 domain is non‐catalytic and binds target proteins through proline‐rich regions.124 The generalized proline‐rich recognition motifs of the SH3 domain are categorized as class I or class II and have the sequence R/KXXPXXPX and PXXPXR/K, respectively.125, 126 The SH4 domain constitutes a palmitoylation and myristoylation sequence for membrane anchoring and is located at the N‐terminus.124 In cardiomyocytes Fyn kinase regulates stability of adherens junctions and is also localized at caveolae along with NaV1.5 channels.127, 128, 129 Interaction of Fyn kinase with NaV1.5 has been established where it co‐immunoprecipitates and phosphorylates the NaV1.5 channel.130 This phosphorylation creates a depolarizing shift in steady‐state fast inactivation, increases recovery from inactivation and decreases rate of entry into slow inactivation.131 Interaction of Fyn kinase with NaV1.5 is complex and involves multiple steps. Binding of Fyn kinase to proline‐rich regions in the ICLI‐II domain and C‐terminus is followed by phosphorylation of nearby tyrosine residues in the N‐terminus (Y68, Y87, and Y112), ICLIII‐IV (Y1494, Y1495)130, 131 domain and C‐terminus (Y1811, Y1889), indicating a complex and multistep modulation.56
4.5. Phosphoinositide 3‐kinase signalling
Phosphoinositide 3‐kinases (PI3K) are a group of kinases that phosphorylate the 3‐hydroxyl group of inositol in phosphoinositides. They are categorized into three classes (class‐1, class‐2 and class‐3) depending upon structure, subunits and substrate specificity. The mammalian heart expresses different isoforms from the three classes of PI3Ks but most of the studies have focused on describing the role of class‐1 PI3Ks in cardiac electrophysiology. Class‐1 PI3Ks are heterodimers consisting of a catalytic subunit (PI3Kα, PI3Kβ, PI3Kγ or PI3Kδ) bound to a regulatory subunit.132 Specific inhibition of PI3Kα in mouse ventricular myocytes increased action potential duration by prolonging the QT interval, decreasing peak INa and increasing the late INa, while no effect was observed on the blockade of other three catalytic subunits.133, 134 The downstream effectors of PI3K pathway are protein kinase Akt/PKB and 3‐phosphoinositide dependent protein kinase 1 (PDK1). Downregulation of Akt is observed in diabetes as PI3K signalling is reduced due to decreased insulin levels which increase QT interval and late INa.135 Activation of PI3K translocates PDK1 to the plasma membrane which then activates atypical PKC isoforms and serum and glucocorticoid inducible kinase (SGK).132 PDK1 is an important member of the AGC family of protein kinases and it acts an upstream protein kinase to several members of AGC family, including SGK. Mice with conditional knockout of PDK1 exhibit lower heart rate with QRS and QTc interval prolongation, due to reduction of peak INa and reduced surface expression of sodium channels. NaV1.5 channel gating was also changed with a moderate hyperpolarizing shift both in activation and inactivation curves. This decreased surface expression of NaV1.5 channel was attributed to PDK1 mediated activation of Foxo1 pathway.136
SGK is a serine and threonine kinase, which is transcriptionally regulated by gluco‐ and mineralocorticoids. It is a downstream target of PI3K pathway and phosphorylated by PDK1 which activates SGK.137 Three isoforms of this kinase have been identified and named as SGK1, SGK2 and SGK3. Among these, SGK1 and SGK3 are expressed in cardiac tissues and interact with the NaV1.5 channel. The co‐expression of SGK1 or SGK3 with NaV1.5 channel (hH1 variant) in Xenopus oocytes increases INa and additionally SGK3 also creates a depolarizing shift in the inactivation curve and a hyperpolarizing shift in the activation curve. These effects have been attributed to the phosphorylation of two serine residues at positions 483 and 664 in the NaV1.5 channel, identified by mutational analysis (Table 1).138, 139 SGK1 also controls sodium transport in the kidneys, thus acting as an important contributor in HF and arrhythmia. In pressure overload after transverse aortic constriction (TAC), its acute activation proves to be cardio‐protective however its chronic activation in TAC induced HF acts conversely. Both systolic and diastolic dysfunction has been observed in transgenic mice expressing cardio‐specific, catalytically active SGK1, while transgenic mice expressing the catalytically inactive variant of SGK1 showed normal cardiac structure and function. When additional stress was introduced by TAC, the baseline cardiac dysfunction in catalytically active SGK1 mice was markedly exacerbated, while WT and transgenic mice expressing catalytically inactive SGK1 better tolerated TAC, suggesting that SGK1 inhibition prevents fibrosis, cardiac hypertrophy and development of HF after pressure overload. Moreover, transgenic mice expressing the catalytically active variant of SGK1 exhibit ECG abnormalities, action potential prolongation along with spontaneous ventricular tachycardia. These effects were attributed to increased peak INa, late INa and changes in NaV1.5 channel gating including a hyperpolarizing shift both in the activation and inactivation curves, thus increasing the window current. Surface expression of NaV1.5 channels was also increased by SGK1 activation since SGK1 inhibits Nedd4‐2, thus preventing NaV1.5 channel internalization.139 Recently, a threonine residue at position 1590 in the C‐terminal domain of the NaV1.5 channel has been described as a candidate site for phosphorylation by SGK1; replacing this threonine with alanine almost completely abolished SGK1‐mediated increase in INa.140
Interestingly, PI3K‐mediated effects on INa appear to be opposite to that of its downstream effector SGK. Moreover Akt and SGK also behave oppositely, suggesting that phosphorylation of the NaV1.5 channel is the underlying cause of this differential modulation. PI3K pathway inhibition prolongs QT interval as observed with nilotinib which increases QT interval through inhibition of PI3K pathway.133 Besides the NaV1.5 channel, PI3K signalling affects several cardiac ion channels,132 rendering description of its effect on cardiac electrophysiology complicated and warranting further studies to describe its specific role.
5. CONCLUDING REMARKS
Cardiac disorders such as structural heart diseases and arrhythmogenic conduction defects are major public health problems both in developed and developing countries. Use of ion channel blockers can treat arrhythmias but still cannot reduce mortality rates, thus stressing the need for new suitable treatment options. An appealing approach which has emerged recently is antagonizing the protein kinases implicated in pathology of the cardiac disease. One such example is CaMKII which is upregulated in HF and hypertrophy and pharmacological inhibition of which in experimental animal models has proven to be cardio‐protective in a setting of arrhythmias and HF. Kinases and their mediated pathways are also gaining attention as biomarkers for early detection of cardiac disorders. Phosphorylation and dephosphorylation reactions are carried out by more than 500 kinases and 100 phosphatases. Mass‐spectrometry based proteomic studies are unveiling information regarding phosphorylated residues but still a lot of information is missing regarding the sites, sequences, order of phosphorylation, cross talk between the modified residues and how these complicated events unfold to modulate physiological or pathological processes. This missing information strongly requires a concerted effort in the form of future studies to address these questions in order to understand these pathways for the discovery of new drug targets.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Dr. Susan Tzotzos for her helpful comments.
Iqbal SM, Lemmens‐Gruber R. Phosphorylation of cardiac voltage‐gated sodium channel: Potential players with multiple dimensions. Acta Physiol. 2019;225:e13210 10.1111/apha.13210
REFERENCES
- 1. Wang Q, Li Z, Shen J, Keating MT. Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics. 1996;34:9‐16. [DOI] [PubMed] [Google Scholar]
- 2. Sherman SJ, Chrivia J, Catterall WA. Cyclic adenosine 3’:5’‐monophosphate and cytosolic calcium exert opposing effects on biosynthesis of tetrodotoxin‐sensitive sodium channels in rat muscle cells. J Neurosci. 1985;5:1570‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maltsev VA, Kyle JW, Mishra S, Undrovinas A. Molecular identity of the late sodium current in adult dog cardiomyocytes identified by Nav1.5 antisense inhibition. Am J Physiol Heart Circ Physiol. 2008;295:H667‐H676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aye TT, Scholten A, Taouatas N, et al. Proteome‐wide protein concentrations in the human heart. Mol BioSyst. 2010;6:1917‐1927. [DOI] [PubMed] [Google Scholar]
- 5. Brackenbury WJ, Isom LL. Na channel beta subunits: overachievers of the ion channel family. Front Pharmacol. 2011;2:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeMarco KR, Clancy CE. Cardiac Na channels: structure to function. Curr Top Membr. 2016;78:287‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schroeter A, Walzik S, Blechschmidt S, Haufe V, Benndorf K, Zimmer T. Structure and function of splice variants of the cardiac voltage‐gated sodium channel Nav1.5. J Mol Cell Cardiol. 2010;49:16‐24. [DOI] [PubMed] [Google Scholar]
- 8. Rook MB, Evers MM, Vos MA, Bierhuizen MF. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc Res. 2012;93:12‐23. [DOI] [PubMed] [Google Scholar]
- 9. Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011;108:884‐897. [DOI] [PubMed] [Google Scholar]
- 10. Makielski JC, Ye B, Valdivia CR, et al. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93:821‐828. [DOI] [PubMed] [Google Scholar]
- 11. Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage‐gated sodium channels. J Physiol. 1998;508:647‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stuhmer W, Conti F, Suzuki H, et al. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597‐603. [DOI] [PubMed] [Google Scholar]
- 13. Denac H, Mevissen M, Scholtysik G. Structure, function and pharmacology of voltage‐gated sodium channels. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:453‐479. [DOI] [PubMed] [Google Scholar]
- 14. Heinemann SH, Terlau H, Stuhmer W, Imoto K, Numa S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 1992;356:441‐443. [DOI] [PubMed] [Google Scholar]
- 15. Catterall WA. Structure and function of voltage‐gated sodium channels at atomic resolution. Exp Physiol. 2014;99:35‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ulbricht W. Sodium channel inactivation: molecular determinants and modulation. Physiol Rev. 2005;85:1271‐1301. [DOI] [PubMed] [Google Scholar]
- 17. Mangold KE, Brumback BD, Angsutararux P, et al. Mechanisms and models of cardiac sodium channel inactivation. Channels (Austin, Tex). 2017;11:517‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kass RS. Sodium channel inactivation goes with the flow. J Gen Physiol. 2004;124:7‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goldin AL. Mechanisms of sodium channel inactivation. Curr Opin Neurobiol. 2003;13:284‐290. [DOI] [PubMed] [Google Scholar]
- 20. Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage‐gated sodium channels. Neuron. 2000;26:13‐25. [DOI] [PubMed] [Google Scholar]
- 21. Makielski JC, Kyle JW. Late I(Na) in the heart: physiology, pathology, and pathways. Circulation. 2015;132:553‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Makielski JC. Late sodium current: a mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc Med. 2016;26:115‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Antzelevitch C, Nesterenko V, Shryock JC, Rajamani S, Song Y, Belardinelli L. The role of late I Na in development of cardiac arrhythmias. Handb Exp Pharmacol. 2014;221:137‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grandi E, Herren AW. CaMKII‐dependent regulation of cardiac Na(+) homeostasis. Front Pharmacol. 2014;5:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Remme CA. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol. 2013;591:4099‐4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zaklyazminskaya E, Dzemeshkevich S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim Biophys Acta. 1863;1799–1805:2016. [DOI] [PubMed] [Google Scholar]
- 27. Liu M, Yang KC, Dudley SC Jr. Cardiac sodium channel mutations: why so many phenotypes? Nat Rev Cardiol. 2014;11:607‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shy D, Gillet L, Abriel H. Targeting the sodium channel NaV1.5 to specific membrane compartments of cardiac cells: not a simple task! Circ Res. 2014;115:901‐903. [DOI] [PubMed] [Google Scholar]
- 29. Stroemlund LW, Jensen CF, Qvortrup K, Delmar M, Nielsen MS. Gap junctions‐guards of excitability. Biochem Soc Trans. 2015;43:508‐512. [DOI] [PubMed] [Google Scholar]
- 30. Park DJ, Freitas TA, Wallick CJ, Guyette CV, Warn‐Cramer BJ. Molecular dynamics and in vitro analysis of Connexin43: a new 14‐3‐3 mode‐1 interacting protein. Protein Sci. 2006;15:2344‐2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Allouis M, Le Bouffant F, Wilders R, et al. 14‐3‐3 is a regulator of the cardiac voltage‐gated sodium channel Nav1.5. Circ Res. 2006;98:1538‐1546. [DOI] [PubMed] [Google Scholar]
- 32. Clatot J, Hoshi M, Wan X, et al. Voltage‐gated sodium channels assemble and gate as dimers. Nat Commun. 2017;8:2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu L, Yong SL, Fan C, et al. Identification of a new co‐factor, MOG1, required for the full function of cardiac sodium channel Nav 1.5. J Biol Chem. 2008;283:6968‐6978. [DOI] [PubMed] [Google Scholar]
- 34. Mohler PJ, Rivolta I, Napolitano C, et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin‐G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci USA. 2004;101:17533‐17538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Makara MA, Curran J, Little SC, et al. Ankyrin‐G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ Res. 2014;115:929‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hund TJ, Koval OM, Li J, et al. A β(IV)‐spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508‐3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sato PY, Coombs W, Lin X, et al. Interactions between ankyrin‐G, Plakophilin‐2, and Connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mohler PJ, Hund TJ. Membrane‐select regulation of cardiac Na(v) channel isoforms. Heart Rhythm. 2011;8:1931‐1932. [DOI] [PubMed] [Google Scholar]
- 39. Shy D, Gillet L, Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta. 2013;1833:886‐894. [DOI] [PubMed] [Google Scholar]
- 40. Ziane R, Huang H, Moghadaszadeh B, Beggs AH, Levesque G, Chahine M. Cell membrane expression of cardiac sodium channel Na(v)1.5 is modulated by alpha‐actinin‐2 interaction. Biochemistry. 2010;49:166‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Albesa M, Ogrodnik J, Rougier JS, Abriel H. Regulation of the cardiac sodium channel Nav1.5 by utrophin in dystrophin‐deficient mice. Cardiovasc Res. 2011;89:320‐328. [DOI] [PubMed] [Google Scholar]
- 42. Gavillet B, Rougier JS, Domenighetti AA, et al. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99:407‐414. [DOI] [PubMed] [Google Scholar]
- 43. Chen‐Izu Y, Shaw RM, Pitt GS, et al. Na+ channel function, regulation, structure, trafficking and sequestration. J Physiol. 2015;593:1347‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jespersen T, Gavillet B, van Bemmelen MX, et al. Cardiac sodium channel Na(v)1.5 interacts with and is regulated by the protein tyrosine phosphatase PTPH1. Biochem Biophys Res Commun. 2006;348:1455‐1462. [DOI] [PubMed] [Google Scholar]
- 45. Tan HL, Kupershmidt S, Zhang R, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442‐447. [DOI] [PubMed] [Google Scholar]
- 46. Aiba T, Hesketh GG, Liu T, et al. Na+ channel regulation by Ca2 + /calmodulin and Ca2 + /calmodulin‐dependent protein kinase II in guinea‐pig ventricular myocytes. Cardiovasc Res. 2010;85:454‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johnson CN, Potet F, Thompson MK, et al. A Mechanism of Calmodulin Modulation of the Human Cardiac Sodium Channel. Structure (London, England). 2018;26:683‐694 e683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Bemmelen MX, Rougier JS, Gavillet B, et al. Cardiac voltage‐gated sodium channel Nav1.5 is regulated by Nedd4‐2 mediated ubiquitination. Circ Res. 2004;95:284‐291. [DOI] [PubMed] [Google Scholar]
- 49. Abriel H, Kamynina E, Horisberger JD, Staub O. Regulation of the cardiac voltage‐gated Na+ channel (H1) by the ubiquitin‐protein ligase Nedd4. FEBS Lett. 2000;466:377‐380. [DOI] [PubMed] [Google Scholar]
- 50. Snyder PM. Down‐regulating destruction: phosphorylation regulates the E3 ubiquitin ligase Nedd4‐2. Sci Signal. 2009;2:pe41. [DOI] [PubMed] [Google Scholar]
- 51. Huang Y, Wang Z, Liu Y, et al. alphaB‐Crystallin Interacts with Nav1.5 and Regulates Ubiquitination and Internalization of Cell Surface Nav1.5. J Biol Chem. 2016;291:11030‐11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu CJ, Dib‐Hajj SD, Renganathan M, Cummins TR, Waxman SG. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J Biol Chem. 2003;278:1029‐1036. [DOI] [PubMed] [Google Scholar]
- 53. Wang C, Hennessey JA, Kirkton RD, et al. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ Res. 2011;109:775‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Clatot J, Ziyadeh‐Isleem A, Maugenre S, et al. Dominant‐negative effect of SCN5A N‐terminal mutations through the interaction of Na(v)1.5 alpha‐subunits. Cardiovasc Res. 2012;96:53‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matamoros M, Perez‐Hernandez M, Guerrero‐Serna G, et al. Nav1.5 N‐terminal domain binding to alpha1‐syntrophin increases membrane density of human Kir2.1, Kir2.2 and Nav1.5 channels. Cardiovasc Res. 2016;110:279‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iqbal SM, Aufy M, Shabbir W, Lemmens‐Gruber R. Identification of phosphorylation sites and binding pockets for modulation of NaV 1.5 channel by Fyn tyrosine kinase. FEBS J. 2018;285:2520‐2530. [DOI] [PubMed] [Google Scholar]
- 57. Walsh DA, Perkins JP, Krebs EG. An adenosine 3’,5’‐monophosphate‐dependant protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243:3763‐3765. [PubMed] [Google Scholar]
- 58. Turnham RE, Scott JD. Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Gene. 2016;577:101‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Taylor SS, Yang J, Wu J, Haste NM, Radzio‐Andzelm E, Anand G. PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259‐269. [DOI] [PubMed] [Google Scholar]
- 60. Gordon D, Merrick D, Wollner DA, Catterall WA. Biochemical properties of sodium channels in a wide range of excitable tissues studied with site‐directed antibodies. Biochemistry. 1988;27:7032‐7038. [DOI] [PubMed] [Google Scholar]
- 61. Shabb JB. Physiological substrates of cAMP‐dependent protein kinase. Chem Rev. 2001;101:2381‐2411. [DOI] [PubMed] [Google Scholar]
- 62. Schubert B, Vandongen AM, Kirsch GE, Brown AM. Inhibition of cardiac Na+ currents by isoproterenol. Am J Physiol. 1990;258:H977‐H982. [DOI] [PubMed] [Google Scholar]
- 63. Ono K, Kiyosue T, Arita M. Isoproterenol, DBcAMP, and forskolin inhibit cardiac sodium current. Am J Physiol. 1989;256:C1131‐C1137. [DOI] [PubMed] [Google Scholar]
- 64. Schubert B, VanDongen A, Kirsch G, Brown A. Beta‐adrenergic inhibition of cardiac sodium channels by dual G‐protein pathways. Science. 1989;245:516‐519. [DOI] [PubMed] [Google Scholar]
- 65. Hisatome I, Kiyosue T, Imanishi S, Arita M. Isoproterenol inhibits residual fast channel via stimulation of beta‐adrenoceptors in guinea‐pig ventricular muscle. J Mol Cell Cardiol. 1985;17:657‐665. [DOI] [PubMed] [Google Scholar]
- 66. Tytgat J, Vereecke J, Carmeliet E. A combined study of sodium current and T‐type calcium current in isolated cardiac cells. Pflugers Arch ‐ Eur J Physiol. 1990;417:142‐148. [DOI] [PubMed] [Google Scholar]
- 67. Matsuda JJ, Lee H, Shibata EF. Enhancement of rabbit cardiac sodium channels by beta‐adrenergic stimulation. Circ Res. 1992;70:199‐207. [DOI] [PubMed] [Google Scholar]
- 68. Frohnwieser B, Weigl L, Schreibmayer W. Modulation of cardiac sodium channel isoform by cyclic AMP dependent protein kinase does not depend on phosphorylation of serine 1504 in the cytosolic loop interconnecting transmembrane domains III and IV. Pflugers Arch ‐ Eur J Physiol. 1995;430:751‐753. [DOI] [PubMed] [Google Scholar]
- 69. Herzig JW, Kohlhardt M. Na+ channel blockade by cyclic AMP and other 6‐aminopurines in neonatal rat heart. J Membr Biol. 1991;119:163‐170. [DOI] [PubMed] [Google Scholar]
- 70. Kirstein M, Eickhorn R, Kochsiek K, Langenfeld H. Dose‐dependent alteration of rat cardiac sodium current by isoproterenol: results from direct measurements on multicellular preparations. Pflugers Arch. 1996;431:395‐401. [DOI] [PubMed] [Google Scholar]
- 71. Ono K, Fozzard HA, Hanck DA. Mechanism of cAMP‐dependent modulation of cardiac sodium channel current kinetics. Circ Res. 1993;72:807‐815. [DOI] [PubMed] [Google Scholar]
- 72. Murphy BJ, Rogers J, Perdichizzi AP, Colvin AA, Catterall WA. cAMP‐dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J Biol Chem. 1996;271:28837‐28843. [DOI] [PubMed] [Google Scholar]
- 73. Frohnwieser B, Chen LQ, Schreibmayer W, Kallen RG. Modulation of the human cardiac sodium channel alpha‐subunit by cAMP‐dependent protein kinase and the responsible sequence domain. J Physiol. 1997;498:309‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou J, Yi J, Hu N, George AL Jr, Murray KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res. 2000;87:33‐38. [DOI] [PubMed] [Google Scholar]
- 75. Lu T, Lee H‐C, Kabat JA, Shibata EF. Modulation of rat cardiac sodium channel by the stimulatory G protein α subunit. J Physiol. 1999;518:371‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wagner S, Maier LS. Modulation of cardiac Na(+) and Ca(2 + ) currents by CaM and CaMKII. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S26‐S33. [DOI] [PubMed] [Google Scholar]
- 77. Hallaq H, Yang Z, Viswanathan PC, et al. Quantitation of protein kinase A‐mediated trafficking of cardiac sodium channels in living cells. Cardiovasc Res. 2006;72:250‐261. [DOI] [PubMed] [Google Scholar]
- 78. Zhou J, Shin HG, Yi J, Shen W, Williams CP, Murray KT. Phosphorylation and putative ER retention signals are required for protein kinase A‐mediated potentiation of cardiac sodium current. Circ Res. 2002;91:540‐546. [DOI] [PubMed] [Google Scholar]
- 79. Aromolaran AS, Chahine M, Boutjdir M. Regulation of cardiac voltage‐gated sodium channel by kinases: roles of protein kinases A and C. Handb Exp Pharmacol. 2018;246:161‐184. [DOI] [PubMed] [Google Scholar]
- 80. Baba S, Dun W, Boyden PA. Can PKA activators rescue Na+ channel function in epicardial border zone cells that survive in the infarcted canine heart? Cardiovasc Res. 2004;64:260‐267. [DOI] [PubMed] [Google Scholar]
- 81. Aiba T, Farinelli F, Kostecki G, et al. A mutation causing Brugada syndrome identifies a mechanism for altered autonomic and oxidant regulation of cardiac sodium currents. Circ Cardiovasc Genet. 2014;7:249‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xiang Y, Kobilka BK. Myocyte adrenoceptor signaling pathways. Science. 2003;300:1530‐1532. [DOI] [PubMed] [Google Scholar]
- 83. Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298:E395‐E402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shin HG, Barnett JV, Chang P, et al. Molecular heterogeneity of protein kinase C expression in human ventricle. Cardiovasc Res. 2000;48:285‐299. [DOI] [PubMed] [Google Scholar]
- 85. Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495‐28498. [DOI] [PubMed] [Google Scholar]
- 86. Moorman JR, Kirsch GE, Lacerda AE, Brown AM. Angiotensin II modulates cardiac Na+ channels in neonatal rat. Circ Res. 1989;65:1804. [DOI] [PubMed] [Google Scholar]
- 87. Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA. Modulation of cardiac Na+ channels expressed in a mammalian cell line and in ventricular myocytes by protein kinase C. Proc Natl Acad Sci USA. 1994;91:3289‐3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Qu Y, Rogers JC, Tanada TN, Catterall WA, Scheuer T. Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. J Gen Physiol. 1996;108:375‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Murray KT, Hu N, Daw JR, et al. Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circ Res. 1997;80:370‐376. [DOI] [PubMed] [Google Scholar]
- 90. Watson CL, Gold MR. Modulation of Na+ current inactivation by stimulation of protein kinase C in cardiac cells. Circ Res. 1997;81:380‐386. [DOI] [PubMed] [Google Scholar]
- 91. Xiao G‐Q, Qu Y, Sun Z‐Q, Mochly‐Rosen D, Boutjdir M. Evidence for functional role of εPKC isozyme in the regulation of cardiac Na+ channels. Am J Physiol Cell Physiol. 2001;281:C1477‐C1486. [DOI] [PubMed] [Google Scholar]
- 92. Shin HG, Murray KT. Conventional protein kinase C isoforms and cross‐activation of protein kinase A regulate cardiac Na+ current. FEBS Lett. 2001;495:154‐158. [DOI] [PubMed] [Google Scholar]
- 93. Hallaq H, Wang DW, Kunic JD, George AL Jr, Wells KS, Murray KT. Activation of protein kinase C alters the intracellular distribution and mobility of cardiac Na+ channels. Am J Physiol Heart Circ Physiol. 2012;302:H782‐H789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mathieu S, El Khoury N, Rivard K, et al. Reduction in Na(+) current by angiotensin II is mediated by PKCα in mouse and human‐induced pluripotent stem cell‐derived cardiomyocytes. Heart Rhythm. 2016;13:1346‐1354. [DOI] [PubMed] [Google Scholar]
- 95. Liu M, Shi G, Yang KC, et al. Role of protein kinase C in metabolic regulation of the cardiac Na+ channel. Heart Rhythm. 2017;14:440‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liu M, Sanyal S, Gao G, et al. Cardiac Na(+) Current Regulation by Pyridine Nucleotides. Circ Res. 2009;105:737‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tobimatsu T, Fujisawa H. Tissue‐specific expression of four types of rat calmodulin‐dependent protein kinase II mRNAs. J Biol Chem. 1989;264:17907‐17912. [PubMed] [Google Scholar]
- 98. Mayer P, Mohlig M, Idlibe D, Pfeiffer A. Novel and uncommon isoforms of the calcium sensing enzyme calcium/calmodulin dependent protein kinase II in heart tissue. Basic Res Cardiol. 1995;90:372‐379. [DOI] [PubMed] [Google Scholar]
- 99. Hudmon A, Schulman H. Structure‐function of the multifunctional Ca2 + /calmodulin‐dependent protein kinase II. Biochem J. 2002;364:593‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Erickson JR, Joiner ML, Guan X, et al. A dynamic pathway for calcium‐independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kirchhefer U, Schmitz W, Scholz H, Neumann J. Activity of cAMP‐dependent protein kinase and Ca2 + /calmodulin‐dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999;42:254‐261. [DOI] [PubMed] [Google Scholar]
- 102. Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta‐isoforms of the multifunctional Ca2 + /calmodulin‐dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713‐721. [DOI] [PubMed] [Google Scholar]
- 103. Zhu WZ, Wang SQ, Chakir K, et al. Linkage of beta1‐adrenergic stimulation to apoptotic heart cell death through protein kinase A‐independent activation of Ca2 + /calmodulin kinase II. J Clin Invest. 2003;111:617‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zhang R, Khoo MS, Wu Y, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409‐417. [DOI] [PubMed] [Google Scholar]
- 105. Deschenes I, Neyroud N, DiSilvestre D, Marban E, Yue DT, Tomaselli GF. Isoform‐specific modulation of voltage‐gated Na(+) channels by calmodulin. Circ Res. 2002;90:E49‐E57. [DOI] [PubMed] [Google Scholar]
- 106. Wagner S, Dybkova N, Rasenack EC, et al. Ca2 + /calmodulin‐dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127‐3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2 + , calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597‐H1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yoon JY, Ho WK, Kim ST, Cho H. Constitutive CaMKII activity regulates Na+ channel in rat ventricular myocytes. J Mol Cell Cardiol. 2009;47:475‐484. [DOI] [PubMed] [Google Scholar]
- 109. Couchonnal LF, Anderson ME. The role of calmodulin kinase II in myocardial physiology and disease. Physiology (Bethesda). 2008;23:151‐159. [DOI] [PubMed] [Google Scholar]
- 110. Herren AW, Bers DM, Grandi E. Post‐translational modifications of the cardiac Na channel: contribution of CaMKII‐dependent phosphorylation to acquired arrhythmias. Am J Physiol Heart Circ Physiol. 2013;305:H431‐H445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yao L, Fan P, Jiang Z, et al. Nav1.5‐dependent persistent Na+ influx activates CaMKII in rat ventricular myocytes and N1325S mice. Am J Physiol Cell Physiol. 2011;301:C577‐C586. [DOI] [PubMed] [Google Scholar]
- 112. Ashpole NM, Herren AW, Ginsburg KS, et al. Ca2 + /calmodulin‐dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem. 2012;287:19856‐19869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Marionneau C, Lichti CF, Lindenbaum P, et al. Mass spectrometry‐based identification of native cardiac Nav1.5 channel alpha subunit phosphorylation sites. J Proteome Res. 2012;11:5994‐6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Burel S, Coyan FC, Lorenzini M, et al. C‐terminal phosphorylation of NaV1.5 impairs FGF13‐dependent regulation of channel inactivation. J Biol Chem. 2017;292:17431‐17448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Herren AW, Weber DM, Rigor RR, Margulies KB, Phinney BS, Bers DM. CaMKII phosphorylation of Na(V)1.5: novel in vitro sites identified by mass spectrometry and reduced S516 phosphorylation in human heart failure. J Proteome Res. 2015;14:2298‐2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Beltran‐Alvarez P, Feixas F, Osuna S, Diaz‐Hernandez R, Brugada R, Pagans S. Interplay between R513 methylation and S516 phosphorylation of the cardiac voltage‐gated sodium channel. Amino Acids. 2015;47:429‐434. [DOI] [PubMed] [Google Scholar]
- 117. Beltran‐Alvarez P, Pagans S, Brugada R. The cardiac sodium channel is post‐translationally modified by arginine methylation. J Proteome Res. 2011;10:3712‐3719. [DOI] [PubMed] [Google Scholar]
- 118. Glynn P, Musa H, Wu X, et al. Voltage‐gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation. 2015;132:567‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Paulussen A, Matthijs G, Gewillig M, Verhasselt P, Cohen N, Aerssens J. Mutation analysis in congenital Long QT Syndrome–a case with missense mutations in KCNQ1 and SCN5A. Genet Test. 2003;7:57‐61. [DOI] [PubMed] [Google Scholar]
- 120. Mank‐Seymour AR, Richmond JL, Wood LS, et al. Association of torsades de pointes with novel and known single nucleotide polymorphisms in long QT syndrome genes. Am Heart J. 2006;152:1116‐1122. [DOI] [PubMed] [Google Scholar]
- 121. Napolitano C, Priori SG, Schwartz PJ, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294:2975‐2980. [DOI] [PubMed] [Google Scholar]
- 122. Koval OM, Snyder JS, Wolf RM, et al. Ca2 + /calmodulin‐dependent protein kinase II‐based regulation of voltage‐gated Na+ channel in cardiac disease. Circulation. 2012;126:2084‐2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wang Y, Wagner M, Kumar R, Cheng J, Joyner R. Inhibition of fast sodium current in rabbit ventricular myocytes by protein tyrosine kinase inhibitors. Pflugers Arch ‐ Eur J Physiol. 2003;446:485‐491. [DOI] [PubMed] [Google Scholar]
- 124. Resh MD. Fyn, a Src family tyrosine kinase. Int J Biochem Cell Biol. 1998;30:1159‐1162. [DOI] [PubMed] [Google Scholar]
- 125. Saksela K, Permi P. SH3 domain ligand binding: what's the consensus and where's the specificity? FEBS Lett. 2012;586:2609‐2614. [DOI] [PubMed] [Google Scholar]
- 126. Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. Structural basis for the binding of proline‐rich peptides to SH3 domains. Cell. 1994;76:933‐945. [DOI] [PubMed] [Google Scholar]
- 127. Hsu KL, Fan HJ, Chen YC, et al. Protein kinase C‐Fyn kinase cascade mediates the oleic acid‐induced disassembly of neonatal rat cardiomyocyte adherens junctions. Int J Biochem Cell Biol. 2009;41:1536‐1546. [DOI] [PubMed] [Google Scholar]
- 128. Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431‐467. [DOI] [PubMed] [Google Scholar]
- 129. Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin‐rich membrane domains: regulation of sodium current amplitude. Circ Res. 2002;90:443‐449. [DOI] [PubMed] [Google Scholar]
- 130. Iqbal SM, Andavan GS, Lemmens‐Gruber R. Differential modulation of fast inactivation in cardiac sodium channel splice variants by Fyn tyrosine kinase. Cell Physiol Biochem. 2015;37:825‐837. [DOI] [PubMed] [Google Scholar]
- 131. Ahern CA, Zhang JF, Wookalis MJ, Horn R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ Res. 2005;96:991‐998. [DOI] [PubMed] [Google Scholar]
- 132. Ballou LM, Lin RZ, Cohen IS. Control of cardiac repolarization by phosphoinositide 3‐kinase signaling to ion channels. Circ Res. 2015;116:127‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lu Z, Wu CY, Jiang YP, et al. Suppression of phosphoinositide 3‐kinase signaling and alteration of multiple ion currents in drug‐induced long QT syndrome. Sci Transl Med. 2012;4:131ra150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Yang T, Meoli DF, Moslehi J, Roden DM. Inhibition of the alpha‐subunit of phosphoinositide 3‐kinase in heart increases late sodium current and is arrhythmogenic. J Pharmacol Exp Ther. 2018;365:460‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lu Z, Jiang YP, Wu CY, et al. Increased persistent sodium current due to decreased PI3K signaling contributes to QT prolongation in the diabetic heart. Diabetes. 2013;62:4257‐4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Han Z, Jiang Y, Yang Y, et al. Deletion of PDK1 causes cardiac sodium current reduction in mice. PLoS ONE. 2015;10:e0122436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Park J, Leong MLL, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid‐inducible kinase (SGK) is a target of the PI 3‐kinase‐stimulated signaling pathway. EMBO J. 1999;18:3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Boehmer C, Wilhelm V, Palmada M, et al. Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc Res. 2003;57:1079‐1084. [DOI] [PubMed] [Google Scholar]
- 139. Das S, Aiba T, Rosenberg M, et al. Pathological role of serum‐ and glucocorticoid‐regulated kinase 1 in adverse ventricular remodeling. Circulation. 2012;126:2208‐2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Bezzerides VJ, Zhang A, Xiao L, et al. Inhibition of serum and glucocorticoid regulated kinase‐1 as novel therapy for cardiac arrhythmia disorders. Sci Rep. 2017;7:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lundby A, Andersen MN, Steffensen AB, et al. In vivo phosphoproteomics analysis reveals the cardiac targets of beta‐adrenergic receptor signaling. Sci Signal. 2013;6:rs11. [DOI] [PubMed] [Google Scholar]