

Abstract
The bioengineering of a replacement kidney has been proposed as an approach to address the growing shortage of donor kidneys for the treatment of chronic kidney disease. One approach being investigated is the recellularization of kidney scaffolds. In this study, we present several key advances toward successful re‐endothelialization of whole kidney matrix scaffolds from both rodents and humans. Based on the presence of preserved glycosoaminoglycans within the decelullarized kidney scaffold, we show improved localization of delivered endothelial cells after preloading of the vascular matrix with vascular endothelial growth factor and angiopoietin 1. Using a novel simultaneous arteriovenous delivery system, we report the complete re‐endothelialization of the kidney vasculature, including the glomerular and peritubular capillaries, using human inducible pluripotent stem cell –derived endothelial cells. Using this source of endothelial cells, it was possible to generate sufficient endothelial cells to recellularize an entire human kidney scaffold, achieving efficient cell delivery, adherence, and endothelial cell proliferation and survival. Moreover, human re‐endothelialized scaffold could, in contrast to the non‐re‐endothelialized human scaffold, be fully perfused with whole blood. These major advances move the field closer to a human bioengineered kidney.
Keywords: basic (laboratory) research/science, kidney transplantation/nephrology, regenerative medicine, stem cells, tissue/organ engineering, translational research/science, vascular biology
Short abstract
Human decellularized kidney scaffolds can be functionally re‐endothelialized using expanded human pluripotent stem cell–derived endothelial cells.
Abbreviations
- EC
endothelial cell
- ESRD
end stage renal disease
- FA
formic acid
- GAG
glycosaminoglycans
- hgMVEC
human glomerular microvascular endothelial cells
- hiPSC‐ECs
human iPSC‐derived endothelial cells
- hiPSC
human inducible pluripotent stem cell
- SEM
scanning electron microscopy
- UW
University of Wisconsin
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Kidney transplantation is at present the best therapy for patients with end‐stage renal disease (ESRD). However, there is a shortage of donor organs, while the incidence of ESRD is increasing.1 Moreover, long‐term outcomes after kidney transplantation are compromised by the effects of rejection and nephrotoxicity of immunosuppressive therapies. An autologous bioengineered kidney with patient‐derived cells would therefore constitute an attractive alternative. One proposed, but as yet unachieved, approach to generating such a replacement tissue is via the decellularization and subsequent recellularization of kidney matrix scaffolds.
Several considerations have to be taken into account when developing a clinically applicable bioengineered kidney using such an approach. Recellularization should be performed with human cells, preferably patient‐derived or HLA‐matched. In addition, large numbers of cells are required for whole organ recellularization, and hence scalability is a major challenge. Human inducible pluripotent stem cell (hiPSC)–derived kidney and endothelial cells (ECs) are an interesting cell source for bioengineering, given their potential for expansion and possibility for autologous transplantation.2, 3, 4 However, the ultimate success of the approach will also rest with the capacity for a decellularized scaffold to support and instruct the delivered cells.
The first studies investigating the use of decellularized kidney scaffolds showed that the vascular compartment of rat kidney scaffolds can be recellularized with either primary ECs or mouse embryonic stem cells, some of which showed differentiation into endothelial phenotypes.5, 6, 7, 8, 9 Such studies were performed with mouse and rat scaffolds and cells6; however, to date no bioengineering attempts have been performed using human kidney scaffolds recellularized with human cells.
This reflects the need for novel strategies to enhance endothelial coverage and the translation toward a human bio‐engineered kidney. For the lung, Ren et al described an extensive re‐endothelialization study of scaffolds using human iPSC‐derived endothelial cells (hiPSC‐ECs) where combining arterial and venous infusion of both ECs and pericytes resulted in 75% re‐endothelialization.10 However, as the microvascular diameter in the glomeruli is low, combined infusion of ECs and relatively large pericytes in the kidney would most likely lead to obstructed glomerular arterioles and is therefore probably less feasible for the kidney.
The major function of pericytes is the support of ECs via the local production of growth factors such as vascular endothelial growth factor (VEGF) and angiopoietin 1 (Ang‐1).10, 11, 12 These factors are well known for their instructiveness for endothelial cell adherence, survival, and differentiation10, 11, 12 and often share the requirement of glycosaminoglycans (GAG) as extracellular matrix (ECM) components to exert their effects.13, 14 Based on this knowledge, we hypothesized that, were GAGs maintained within the decellularized kidney matrix, preloading the kidney scaffolds with growth factors would provide a novel strategy to enhance endothelial localization without the need for co‐infusion of pericytes.
Here, we show an extensive characterization of the GAG landscape of the decellularized kidney, including the functional importance of these GAGs in growth factor binding. Human kidney scaffolds loaded with the growth factors VEGF and Ang‐1 did result in improved EC adherence and survival. Moreover, we describe several new optimized, scalable strategies to bioengineer the kidney vasculature from human iPSC‐ECs in a custom‐made perfusion‐culture system and provide a proof of concept for scaleup via the re‐endothelialization of a complete human decellularized kidney scaffold. Finally, we also show some functionality of the re‐endothelialized scaffold with whole blood perfusion.
2. METHODS
2.1. Whole organ decellularization of rat and human kidneys
All animal experiments were approved by the animal care and use committee of the Leiden University Medical Center. Research consent was given for all human kidneys. Rat and human kidneys were decellularized adapted from a protocol described previously.15
In short, 12‐week‐old Lewis rats (Charles River) were systemically heparinized with 5000 U heparin (Leo Pharma, Balledrup, Denmark) and the kidneys were flushed with University of Wisconsin (UW) cold storage solution (Bridge to Life, Elkhorn, WI) containing heparin (Leo Pharma). Kidneys were explanted and cannulated via the renal artery and renal vein with a 24G Neoflon (Becton Dickinson Medical, Erembodegem, Belgium), and ureter with a 26G Neoflon (Becton Dickinson Medical) and perfused with 1% sodium dodecylsulfate (SDS) (w/v) (Sigma Aldrich, Zwijndrecht, the Netherlands) in PBS containing 2% penicillin/streptomycin (Life Technologies, Europe Bleiswijk, the Netherlands), 6 μg/mL ciprofloxacin (Fresenius Kabi, Huis ter Heide, the Netherlands), 0.1% fungizone (w/v) (Bristol‐Myers Squibb, Utrecht, the Netherlands), and 2500E DNAse (pulmozyme, Roche, Woerden, the Netherlands) for 12 hours under a constant pressure of max. 50 mm Hg. Afterwards the kidneys were washed for 15 minutes with dH2O and perfused with 1% Triton‐X 100 (v/v) (Sigma Aldrich) for 30 minutes. Then the kidneys were perfused with phosphate‐buffered saline (PBS) containing antibiotic/antimycotic for at least 48 hours and kept in this solution until use. Patency of both arterial and venous vascular beds was confirmed by methylene blue perfusion (Figure S1A).
Human transplant grade kidneys discarded mainly for surgical reasons were flushed with UW cold storage solution containing heparin directly after procurement and were stored on ice. Within 30 hours, kidneys were again flushed with PBS with heparin (500 IE/L) and the perirenal fat and kidney capsule were removed. The renal artery was cannulated with a Luer‐lock connector (Cole‐Parmer, Barendrecht, the Netherlands) and the kidney was perfused with 1% SDS in PBS containing antibiotics, antimycotics, and DNAse as used with the rat kidneys, under a constant pressure of maximal 75 mm Hg for 5 days. Afterwards the kidneys were perfused overnight with dH2O with antibiotics, antimycotics, and DNAse. Then the kidneys were flushed with 1% (v/v) Triton‐X 100 (Sigma) for 1 day and afterwards flushed with PBS with antibiotics and antimycotics for at least 5 days, all under a constant pressure of max. 75 mm Hg. Kidneys were stored at 4°C in PBS until further use. Samples of ≈1 cm3 were taken and either snap‐frozen in liquid nitrogen, fixed for paraffin embedding, or for electron microscopy.
2.2. Proteomic analysis
Decellularized ECM samples were resolved by SDS–polyacrylamide gel electrophoresis (4% stacking and 10% running gel) and visualized by Coomassie staining. Protein bands were manually excised from the gel and subjected to in‐gel digestion by using a MassPREP digestion robot (Waters, Manchester, UK). Briefly, a solution of 50 mM ammonium bicarbonate in 50% v/v acetonitrile (ACN) was used for destaining. Cysteines were reduced with 10 mM dithiothreitol in 100 mM ammonium bicarbonate for 30 minutes followed by an alkylation reaction using 55 mM iodoacetamide in 100 mM ammonium bicarbonate for 20 minutes. Samples were washed with 100 mM ammonium bicarbonate and subsequently dehydrated with 100% ACN. Trypsin (6 ng/mL, Promega, Benelux Leiden, the Netherlands) in 50 mM ammonium bicarbonate was added and incubated at 37°C for 5 hours. Peptides were extracted using 1% v/v formic acid (FA) supplemented with 2% v/v ACN. A second extraction step was performed using 1% v/v FA supplemented with 50% v/v ACN.
Peptides were separated on a Thermo Scientific (Waltham, MA; Dionex) Ultimate 3000 Rapid Separation UHPLC system equipped with an Acclaim PepMap C18 analytical column (2 μm, 100Å, 75 μm × 150 mm). Peptide samples were first desalted on an online installed C18 trapping column. After desalting, peptides were separated on the analytical column with a 90‐minute linear gradient from 5% to 35% ACN with 0.1% FA at a 300 nL/min flow rate. The UHPLC system was coupled to a Q Exactive HF mass spectrometer (Thermo Scientific) using full MS scan between 350 and 1650 m/z at a resolution of 120 000. Subsequent MS/MS scans of the top 15 most intense ions were done at a resolution of 15 000. The obtained spectra were analyzed with the Proteome Discoverer (PD) software (version 2.2, Thermo Scientific) using the Sequest search engine and the SwissProt Human database (TaxID = 9606, v2017‐10‐25). The database search was performed with the following settings: trypsin digestion with a maximum of 2 missed cleavages, minimum peptide length of 6, precursor mass tolerance of 10 ppm, fragment mass tolerance of 0.02 Da, dynamic modifications of methionine oxidation and protein N‐terminus acetylation, and static modification of cysteine carbamidomethylation.
2.3. Immunofluorescence stainings
Biopsies of de‐ and recellularized kidneys were fixed overnight in 4% formaldehyde (Klinipath, Duiven, the Netherlands), stored in 70% ethanol and embedded in paraffin. Four‐micrometer‐thick sections were cut and following rehydration of the tissue samples, antigen retrieval was performed by heating the slides in citrate buffer (pH 6). Primary antibodies against laminin (Sigma‐Aldrich, Zwijndrecht, the Netherlands), collagenIV (NovusBio, Abingdon, UK), fibronectin (Abcam, Cambridge, UK), CD31 (Pharmingen), VEGF (Biovision, Zutphen, the Netherlands), and angiopoietin‐1 (Santa‐Cruz, Huissen, The Netherlands), were used. GAGs were shown with the lectins WGA (Invitrogen, Carlsbad, CA), LEA (Sigma‐Aldrich), heparan sulfate specific antibodies (clone 10E4 [Amsbio]), and JM403 (Dr. J. van der Vlag, RadboudUMC, Nijmegen), and a fluorescent‐labeled hyaluronan‐specific binding protein neurocan‐GFP (custom made, adapted from previous published protocol16). As secondary antibodies, anti‐mouse, anti‐rat, and anti‐rabbit Alexa Fluor 488, 568, and 647 (Molecular Probes) were used. Nuclei were counterstained using Hoechst 33258 (ThermoFisher). Sections were imaged using SP8‐WLL confocal microscopy and displayed using LAS‐X software (Leica).
2.4. Cell culture human glomerular microvascular endothelial cells
Human glomerular microvascular endothelial cells (hgMVECs) (Bio‐connect, Huissen, the Netherlands) were cultured in EGM2 (Lonza, Breda, the Netherlands) in gelatin‐coated (Sigma‐Aldrich) T175 culture flasks in a density of 2 million cells per flask.
2.5. EC differentiation from hiPSCs and expansion method
Human pluripotent stem‐cell‐derived ECs were generated and expanded as described previously2 with minor adaptation for large‐scale culture. In brief, hiPSCs (NCRM‐1) were cultured in TeSR‐E8 medium (StemCell Technologies, Cologne, Germany) and passaged at day 4. For large‐scale culture the cells were passaged to T175 cell culture flasks. Medium was refreshed at day 3 and at day 0 the differentiation started by Mesoderm induction medium consisting of B(P)EL medium containing 30 ng/mL BMP4 (Miltenyi Biotec, Leiden, the Netherlands), 25 ng/mL Activin A (Miltenyi Biotec), 1.5 μM CHIR99021 (Tocris Bioscience, Abingdon, UK), and 50 ng/mL VEGF165 (R&D Systems, Abingdon, UK). At day 3, medium was replaced by vascular induction medium consisting of B(P)EL containing 50 ng/mL VEGF and 10 μM SB431542 (Tocris Bioscience) and was refreshed again at days 7 and 9. At day 10, ECs were isolated with CD31‐dynabeads (Invitrogen) and the CD31‐positive fraction was cultured in EC‐SFM medium (GIBCO, Carlsbad, CA) on gelatin‐coated (Sigma‐Aldrich) T175 cell culture flasks. Cells were either replated (1:3) or cryopreserved when the flask is fully confluent after isolation. CD31, VE‐CAD, Tie2, and VEGFR2 (R&D Systems) immunofluorescence was stained as described above on hiPSC‐ECs cultured on coverslips and fixed with 4% paraformaldehyde (PFA). Before recellularization experiments, cells were thawed using standard protocols and reseeded in gelatin‐coated T175 cell culture flasks with EC‐SFM. Recellularization was performed with hiPS‐EC at passage 2‐3.
2.6. Acetylated low‐density lipoprotein assay
HiPSC‐ECs and hgMVECs were cultured in gelatin coated 6‐well plates for 12 hours until they reached 30%‐40% confluency. HiPSC‐ECs and hgMVECs were serum starved for 48 hours in SFM‐EC (without supplemental growth factors but with 0.3% bovine serum albumin [BSA]) and EGM (without serum but with 0.3% BSA), respectively. Afterwards, the labeled low‐density lipoprotein (LDL) was diluted in wash buffer (150 mg BSA in HBSS) to a final working concentration of 6.7 μg/mL and added to the medium used for cell incubation. After 4 hours, cells were washed with wash buffer and fixed with 4% PFA and nuclei were counterstained using Hoechst 33258 (ThermoFisher). Sections were imaged using SP8‐WLL confocal microscopy and displayed using LAS‐X software (Leica). Quantification was performed using ImageJ software by calculating the average number of positive particles per cell.
2.7. Growth factor loading scaffolds
Slices (20 μm) of cryopreserved tissue of 3 different human decellularized kidneys were made with a microtome and placed on a 18‐mm coverslip in 24‐well plates (Corning, Amsterdam, the Netherlands). All experiments were performed with 3 slices per kidney of 3 different donor kidneys. When indicated, scaffold slices were incubated with either 100 UI/mL hyaluronidase (Sigma‐Aldrich) or 1/0.6 UI/mL Heparinase I/HeparinaseIII (Sigma‐Aldrich) and protease inhibitor (ThermoFisher) for 4 hours at 37°C. Samples were washed 3 times with PBS and coated with, respectively 0, 10, 100, and 1000 ng/mL VEGFA 165 (PreproTech, London, UK) or Ang1 (R&D systems) in PBS overnight at 4°C. Samples were fixed with 4% paraformaldehyde (Klinipath, Duiven, the Netherlands) in PBS before staining. Scaffolds were stained for, respectively VEGF, heparan sulfates (10E4), Ang1, and laminin as described above and images of 2 areas per slide of 3 different kidney scaffolds were taken with confocal microscopy (Leica TSC SP8 WLL). Maximum intensity was analyzed in LAS‐X software (Leica).
2.8. EC adherence to human kidney scaffold
hgMVECs and iPS‐ECs were incubated on human kidney slices placed on coverslips in a concentration of 50 000 cells per well in a 24‐well plate. hgMVECs were cultured in EBM (Lonza) and iPS‐ECs were cultured in EC‐SFM without VEGF. Cells were incubated on scaffold slices for 24 hours and afterwards fixed with 4% paraformaldehyde (Klinipath) in PBS for 10 minutes and washed 3 times with PBS. Slices were stained for CD31 and laminin, analyzed with fluorescence microscopy (Zeiss LSM500) (3 areas per slide), and the CD31/laminin ratio was determined in triplicates (Image J).
2.9. Whole organ recellularization set‐up
The recellularization set‐up contained a sterilized custom‐made air‐tight organ chamber, sterilized perfusion tubes (LS13 for rat kidneys and LS25 for human kidneys) (Masterflex [Metrohm], Barendrecht, the Netherlands), a pressure probe (Edwards Lifesciences, Breda, the Netherlands), and a custom‐made pressure controller. Pressure‐driven perfusion was achieved with a Masterflex pump. For gas exchange, 3.35/4.65‐mm silicon tubes (Helixmark, Freudenberg Medical, Würselen, Germany) were used. At the end of the silicon tubing a filter was attached as a bubble trapper (Millex‐FG, 0.20 μm, Merck, Amsterdam, the Netherlands). The cannulated kidney artery, vein, and ureter were connected to 3‐way taps (Discofix, Braun, Oss, the Netherlands). A 3‐way tap with an inlet tube (LS‐13) for the cells was placed. A negative pressure of −30 mm Hg was achieved by attaching a filter (Millex‐FG, 0.20 μm, Merck) and LS‐25 (Masterflex) to the airtight chamber with a pressure probe (Edwards Lifesciences) and a custom‐made pressure controller.
2.10. Re‐endothelialization of rat kidney scaffolds with hgMVEC
Rat decellularized kidneys were placed in custom‐made organ chambers and perfused overnight with 1000 ng/mL VEGF in EBM2 basal medium (Lonza) in a recellularization loop. Afterwards kidneys were flushed with fresh EGM2. During recellularization, the organ chamber pressure was set to 30 mm Hg via a pressure‐controlled Masterflex pump. For arterial infusion (n = 1), 30 million cells were resuspended in 15 mL medium and placed in an infusion bag (Transfer pack, 600 mL, Fenwall Inc. [Baxter] Utrecht, the Netherlands) on a shaker plate and infused into the renal artery at an average flow of 90 μL/min into the recirculation loop under a maximum pressure of 175 mm Hg. The bag was flushed with 5 mL medium and 30 mL of medium was added to the organ chamber. Medium was recirculated. For venous infusion (n = 1), 30 million cells were infused via the renal vein at a flow of 1.2 mL/min at a maximum pressure of 20 mm Hg. For arterial and venous infusion (n = 4), 20 million cells were resuspended in 7.5 mL EGM2 and infused into the renal vein at an average flow of 600 μL/min under a maximum pressure of 20 mm Hg. The bag was flushed with 5 mL medium. Cells were recirculated through the renal vein for 3 hours and then 10 million cells were infused in 5 mL EGM2 over the renal artery with a maximum pressure of 75 mm Hg and recirculated followed by 5 mL of EGM2. Medium was refreshed daily and the maximum pressure was kept at 100 mm Hg. Recellularized kidneys were perfused with live cell Hoechst (ThermoFisher) and harvested after 48 hours. Half of the kidney was fixed in PFA and placed in paraffin. The other half was snap frozen in liquid nitrogen. When indicated, hgMVECS were first labeled with Qtracker625 and Qtracker705 according to manufacturer's protocol (ThermoFisher) and imaged using second harmonics generation (SHG) on a multiphoton microscope (Zeiss, Breda, the Netherlands).
2.11. Re‐endothelialization of rat kidney scaffolds with hiPSC‐derived ECs
The protocol used for whole organ recellularization with hiPSC‐derived EC is comparable to the combined arterial and venous recellularization with hgMVECs with the exception that instead of EGM2, the kidney is perfused with EC‐SFM. Kidneys were perfused either under a constant pressure of maximal 75 mm Hg or when the flow was >1.2 mL/min a constant flow of 1.2 mL/min.
Medium was refreshed daily and recellularized kidneys were harvested either 48 hours (n = 3), 5 days (n = 2), or 13 days (n = 2) after recellularization as indicated.
2.12. Re‐endothelialization of human kidney scaffold with hiPSC‐derived ECs
A decellularized human kidney was perfused overnight with 1 μg/mL VEGF and 1 μg/mL Ang1 in EC‐SFM at a pressure of 50 mm Hg, resulting in a flow of 27 mL/min. Afterwards the kidney was perfused with fresh EGM2. Next the chamber pressure was set to −30 mm Hg and 60 million cells were infused via the renal vein at 20 mL/min flow and kept in a recirculation loop for 3 hours. Afterwards 70 million cells were infused via the renal artery at a 20 mL/min flow. The chamber pressure was changed to 0 mm Hg and the kidney was perfused via the renal artery at a flow of 20 mL/min for 24 hours, resulting in a pressure of 35‐50 mm Hg. Biopsies of 1 cm3 were taken out of 10 different areas of the kidney cortex, medulla, and papilla and were either snap frozen or fixed in 4% paraformaldehyde and paraffinized.
2.13. Quantification of endothelial coverage and proliferation
All recellularized kidneys were deparaffinized and immunohistochemically stained with CD31 and Ki‐67 (BD Pharmingen) followed by rabbit anti‐mouse‐HRP (DAKO, Heverlee, Belgium) and NovaRed (Vector Labs, Burlingame, CA) and imaged in a slide scanner (3D Histech). From the recellularized rat kidneys, 1 complete slide per kidney was analyzed. From the human recellularized kidneys, slides of 4 different areas of the kidney cortex were analyzed. From the normal human kidney, control slides of 3 different areas were analyzed. For Ki‐67 quantification, all positive Ki‐67 nuclei were annotated and counted manually. Percentages are given as % of all Hoechst + cells.
For CD31 quantification, all glomeruli within the cortex slides were annotated manually and the amount of filled glomeruli per kidney is depicted in the graph column as % of all glomeruli present. The % of CD31+ area within these filled glomeruli was then calculated using HistoQuant software.
For cortex, medulla, and papilla CD31+ area analysis, the entire subsequent area was annotated and % CD31+ was calculated as percentage of total annotation area using HistoQuant software.
2.14. Perfusion of human re‐endothelialized scaffolds with whole blood
Human whole blood (150 mL) was collected from a venous line and anticoagulated with natrium citrate (0.11 M). The tubing was coated with heparin (1000 U/mL) and human serum albumin (10%) (2 hours), fixated with glutaraldehyde 10% (20 minutes), and washed with PBS (2 hours) before use. Calcium (10 mM CaCl2) was added to the blood right before perfusion. Blood was perfused through both kidneys with a constant flow of 15 mL/min, while the pressure (mean arterial pressure in mm Hg) was constantly measured. Afterwards, 1‐cm3 samples were taken out of different areas of the kidney cortex, medulla, and papilla and were either snap frozen or fixated in 4% paraformaldehyde. Paraffinized samples were deparaffinized and immunohistochemically stained with Movat‐Pentachrome staining.
2.15. Transmission electron microscopy
Small pieces (≈1 mm2) of recellularized kidney scaffold were fixed overnight at 4°C in 1.5% glutaraldehyde (Electron Microscopy Sciences) in 0.1 M sodium‐cacodylate buffered solution, with a pH of 7.4. Afterwards, the tissue blocks were washed twice with sodium cacodylate buffer, then fixed in 1% osmium tetroxide (Electron Microscopy Sciences) in 0.1 M sodium cacodylate buffer for 1 hour. Samples were further rinsed twice with 0.1 M sodium‐cacodylate buffer, followed by dehydration steps of 70% ethanol (overnight), 80% ethanol (10 minutes), 90% ethanol (10 minutes), and 100% ethanol absolute (2 × 15 minutes; 1 × 30 minutes).
The probes were subsequently infiltrated with a mixture of 1:1 Epon LX‐112 (Ladd Research, Williston, VT) and propylene oxide (Electron Microscopy Sciences) for 1 hour, then with pure Epon for 2 hours, embedded in pure Epon, mounted in BEEM plastic capsules (Agar Scientific, Essex, UK), and polymerized for 48 hours at 60°C. Ultrathin sections of 100 nm were collected on copper slot grids (Storck Veco B.V., Eerbeek, The Netherlands), covered with formvar film and a 7‐nm carbon layer. The sections were stained afterwards with an aqueous solution of 7% uranyl acetate (20 minutes), followed by Reynold's lead citrate (10 minutes). Samples were analyzed at an acceleration voltage of 120 kV with an FEI Tecnai 12 transmission electron microscope (FEI, Eindhoven, The Netherlands), equipped with a FEI 4k Eagle CCD camera.
2.16. Scanning electron microscopy
Tissue samples were fixed and dehydrated using the same protocol as for transmission electron microscopy. The 100% ethanol absolute dehydration step was followed by critical point drying. Afterwards, the probes were further mounted on 0.5” scanning electron microscopy (SEM) pin stubs (Agar Scientific, Essex, UK) covered with conductive carbon discs (Agar Scientific), and coated with gold‐palladium. Images were acquired with a JEOL JSM‐6700F Field Emission Scanning Electron Microscope (JEOL Europe BV, Nieuw‐Vennep, the Netherlands).
2.17. Statistical analysis
Statistical analysis was performed using ANOVA analysis with Bonferroni's post hoc comparison. Differences were considered statistically significant when P < .05. Values in graphs are presented as means with standard deviation. Data analysis was performed using GraphPad Prism, version 7.02 (Graphpad Prism San Diego, CA).
3. RESULTS
3.1. Perfusion‐decellularized human kidneys show preservation of microarchitecture and major ECM components including glycosaminoglycans
Rat and human transplant grade kidneys were decellularized via the renal artery with 1% SDS followed by 1% Triton‐X100 as described previously15 (Figure 1A). Tissue micro‐architecture was preserved as shown with SEM (Figure 1B,C). The ECM proteome was preserved as indicated by the mass spectrometry analysis performed on human decellularized transplant grade kidneys. Ninety‐two proteins comprising 49 core matrisome proteins (21 ECM glycoproteins, 23 collagens, and 5 proteoglycans) and 43 matrisome‐associated proteins (17 ECM‐regulators, 24 extracellular space, and 2 secreted factors) were identified (Table 1). In addition, histology of the decellularized human kidneys showed preservation of ECM components collagen IV, fibronectin, and laminin with complete removal of nuclei (Figure 1D). Moreover, GAGs including heparan sulfates and hyaluronan were preserved (Figure 1E). GAGs are expressed on several different compartments in the kidney including the extracellular membranes but also within the tubular brush borders. Because of the latter, the total amount of GAG expression seen after decellularization diminishes due to the removal of the tubular brush borders. Glycosaminoglycans contain different epitopes, which can be demonstrated by staining with different antibodies (such as 10E4 and JM403). JM403 specifically recognize the nonsulfated domain, whereas 10E4 is specific for epitopes of the core protein.17, 18, 19 Similar histology results were obtained with rat decellularized kidneys (Figure S1).
Figure 1.
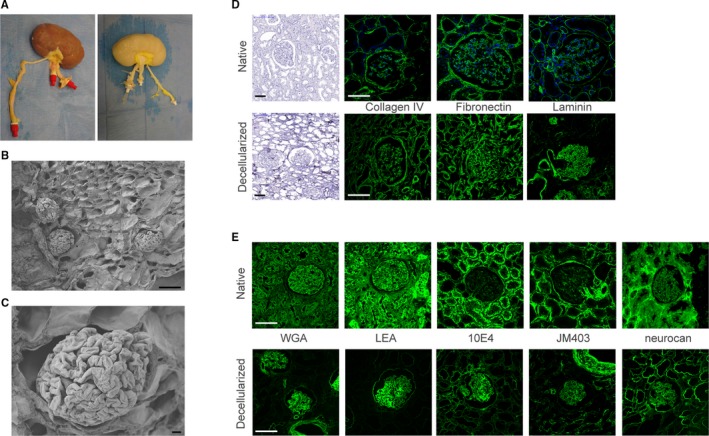
Decellularized human kidneys show intact integrity and glycosaminoglycan (GAG) landscape. A, Macroscopic pictures of human kidney before (left) and after (right) decellularization. B, Scanning electron microscopy (SEM) images of decellularized human kidney show preservation of tissue architecture after decellularization. C, Detailed SEM image of a decellularized glomerulus. D, Extracellular matrix components are preserved as shown by hematoxylin, collagen IV, fibronectin, and laminin staining of human decellularized kidneys (lower panels) compared to native kidney (upper panels). E, The GAG landscape of the human kidney is preserved after decellularization as depicted by WGA, LEA, 10E4 (heparan sulfates), JM403 (heparan sulfates), and neurocan‐eGFP (hyaluronan) stainings. eGFP, enhanced green fluorescent protein; GAG, glycosaminoglycan; LEA, lectin from Lycopersicon esculentum; WGA, wheat germ agglutinin. Scale bar B, C 10 μm; D, E 100 μm [Color figure can be viewed at wileyonlinelibrary.com]
Table 1.
Matrisome analysis of a decellularized human kidney scaffold shows preservation of both major ECM compounds as well as matrisome‐associated proteins and proteoglycans
| Core matrisome | Matrisome‐associated | ||
|---|---|---|---|
| Glycoproteins | Collagens | ECM‐regulator | Extracellular space |
| AZGP1 | COL1A1 | AMBP | AGRN |
| DPT | COL1A2 | APOA4 | ANXA1 |
| EFEMP1 | COL21A1 | CPB2 | ANXA2 |
| ELN | COL3A1 | F9 | APCS |
| EMILIN1 | COL4A1 | ITIH3 | C4BPA |
| FBLN2 | COL4A2 | LTF | CLEC18B |
| FBN1 | COL4A3 | LYZ | CLU |
| FGA | COL4A4 | MGP | DCD |
| FN1 | COL4A5 | PRSS1 | DEFA1/B |
| HRG | COL4A6 | PRSS23 | IGHG4 |
| LAMAS | COL5A1 | SERPINA3 | IGKC |
| LAMB1 | COL5A2 | SERPINE2 | INHBC |
| LAMB2 | COL5A3 | SERPINB12 | INHBE |
| LAMC1 | COL6A1 | SLPI | LTBP1 |
| MFAP4 | COL6A2 | SPP2 | MFAP2 |
| NIDI | COL6A3 | THSD4 | MFAP5 |
| NID2 | COL7A1 | TIMP3 | MFGE8 |
| POSTN | COL8A1 | NPNT | |
| TGFBI | COL10A1 | PDGFB | |
| TNC | COL11A1 | PPIA | |
| VTN | COL12A1 | PROM1 | |
| COL14A1 | PSAP | ||
| Proteoglycans | COL18A1 | SEMA3G | |
| BGN | TNFSF12/3 | ||
| FLNC | |||
| LUM | Secreted factors | ||
| PRELP | S100A9 | ||
| HSPG2 | S100A8 | ||
ECM, extracellular matrix.
3.2. Decellularized human kidney matrix can be loaded with vascular growth factors through binding to GAGs
As GAGs are still preserved after the decellularization method used, and GAGs are important for growth factor binding, we analyzed whether vascular growth factors can still bind to these GAGs. Indeed, we were able to load both the glomerular and tubular compartment of human kidney scaffold slices from 3 different donors with recombinant VEGFA 165, which localized to the heparan sulfates in the ECM (Figure 2A,B). Moreover, removal of heparan sulfates by heparinase treatment led to lower VEGF binding to the scaffold, while hyaluronidase treatment did not influence VEGF binding (Figure 2B and Figure S2). This indicates that the binding of VEGF to the scaffold is dependent on heparan sulfates and the binding domain of VEGF.14, 20 Kidney scaffolds from 3 different donors could also be loaded with Ang‐1 (Figure 2C,D). In this case, Ang‐1 binding did not appear to be dependent upon the presence of the GAGs hyaluronan and heparan sulfate because enzymatic removal of these GAGs did not decrease Ang‐1 binding (Figure 2D).
Figure 2.
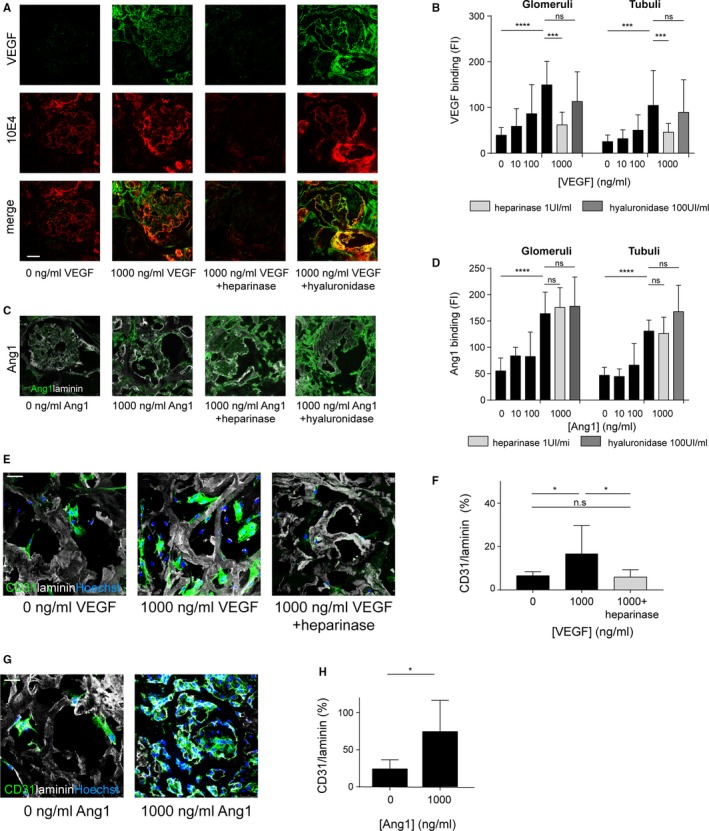
Growth factor loading of the kidney scaffolds results in increased endothelial cell adherence and survival. A, Slices of human kidney scaffold can be loaded with VEGFA 165, which is heparan sulfate dependent. B, Quantification of VEGF loading capacity shows enhanced VEGF binding after VEGF loading. C, Scaffolds can also be loaded with Ang1, independent of heparan sulfates and hyaluronan. D, Quantification of Ang1 binding. E, Representative images of CD31 staining after 24‐h culture of hgMVEC on VEGF loaded scaffolds showing enhanced endothelial cell adherence and survival after VEGF loading, which was not observed after removal of heparan sulfates. F, Quantification of re‐endothelialization after VEGF loading. G, Representative images of hgMVEC culture on Ang1‐loaded scaffolds show enhanced endothelial cell adherence and survival after Ang1 loading. H, Quantification of hgMVEC adherence and survival after Ang1 loading. Ang1, angiopoietin 1; hgMVEC, human glomerular microvascular endothelial cells; ns, nonsignificant; 10E4, heparan sulfates; VEGF, vascular endothelial growth factor A 165. *P < .05, *** P < .001, **** P < .0001. Scale bars 100 μm [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Growth factor loading is critical for EC adherence and survival on the human kidney matrix
The functional effects of growth factor loading on EC adherence and survival to the kidney matrix were analyzed. For this purpose, hgMVEC were cultured on slices of human kidney scaffolds in triplicate with or without VEGF loading (Figure 2E). An increase in EC adherence and survival was observed on VEGF‐loaded scaffolds compared to nonloaded scaffolds, as quantified in Figure 2F. Removal of heparan sulfates led to a decreased endothelial cell adherence in line with the decreased VEGF binding (Figure 2F). Ang‐1 loading also increased hgMVEC cell adherence and survival (Figure 2G,H). These data illustrate the importance of the growth factor binding capacity of the scaffold for EC adherence and survival, and are a novel strategy to enhance revascularization.
3.4. Combined arterial and venous recellularization in whole kidney scaffold is the best strategy to achieve an in situ bioengineered kidney vasculature
For whole kidney scaffold recellularization, a custom‐made, air tight, sterilizable organ culture chamber was developed. This chamber was designed in such a way that optimal recirculation of medium without air infusion could be achieved (Figure 3A,B). This organ culture chamber was connected to a pressure‐controlled perfusion system, allowing separate recellularization of the arterial, venous, and ureteric compartments (Figure 3C,D). Different infusion protocols, via the artery, the vein, or first the vein followed by the artery after 3 hours were compared. Using this device, hgMVECs were infused into either the artery or vein, or both the arterial and venous arm of the vasculature of growth factor loaded rat kidney scaffolds and scaffolds were then analyzed after 48 hours of culture. As shown in Figure 3E, arterial perfusion of the cells led to large obstructions in the afferent arterioles in the cortex, while hardly any cells were observed in the medulla and papilla (Figure 3E). In contrast, venous infusion of cells led to re‐endothelialization of the peritubular capillaries in both the cortex and medulla with some glomerular re‐endothelialization (Figure 3E). The combination of arterial and venous perfusion showed both glomerular and peritubular re‐endothelialization. No cells were observed in the tubular compartment, as shown with laminin‐CD31 double staining, confirming that the basement membrane remains intact after cell infusion. While arterial and venous infusion separately resulted in low glomerular re‐endothelialization, combined infusion increased the glomerular coverage. Compared to arterial infusion, combined infusion also resulted in an increase of medullary re‐endothelialization; however, there were still some obstructed afferent arterioles observed (Figure 3E). In order to trace hgMVECs administered into either the arterial or venous arm of the vasculature, hgMVECs were labeled with 2 different Qdots. Live‐cell imaging with second harmonics generation (SHG) showed that peritubular capillaries and part of the glomeruli were re‐endothelialized via the renal vein while the afferent arteriole and the glomeruli were re‐endothelialized via the renal artery (Figure 3F).
Figure 3.
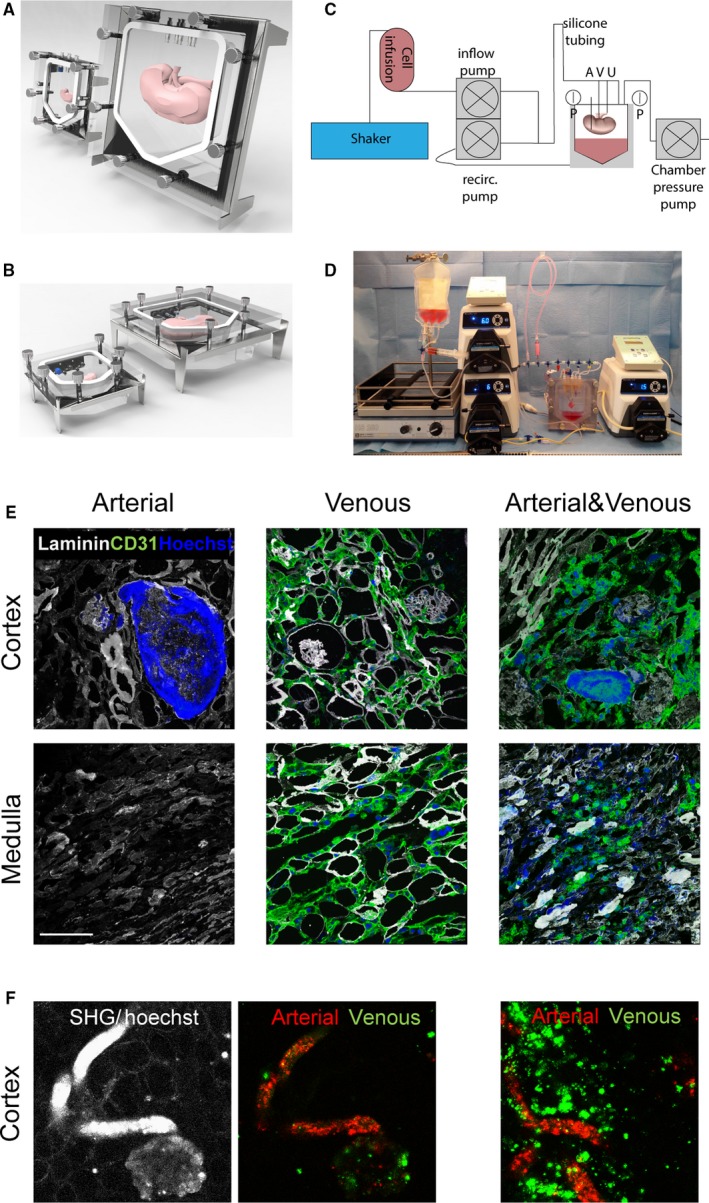
A novel arteriovenous delivery system for whole organ tissue engineering. A, Kidney matrices were cultured in custom‐made organ chamber as shown in front view and (B) side view. C, Schematic representation of recellularization set‐up. D, Image of recellularization set‐up. E, Representative images after whole organ recellularization of hgMVECs. Infusion via only the artery shows obstruction of the afferent arteriole and glomeruli, via only the vein shows re‐endothelialization of the peritubular capillaries in both the cortex and medulla and some glomeruli while combined infusion show re‐endothelialization of both peritubular capillaries and glomeruli. F, Representative live‐cell images with differential Qdot labeling and SHG show re‐endothelialization of peritubular capillaries and part of the glomeruli via the renal vein (Qdot 625, green) while the afferent arteriole and the glomeruli were re‐endothelialized via the renal artery (Qdot 705, red). SHG, second harmonics generation; recirc, recirculation; A, arterial inlet; V, venous inlet; U, ureteric inlet; p, pressure sensor. Scale bars 100 μm [Color figure can be viewed at wileyonlinelibrary.com]
3.5. Induced pluripotent derived ECs are able to adhere to and survive on growth factor loaded human kidney scaffolds
Optimal bioengineering of an autologous replacement human kidney would require large numbers of human or patient‐derived cells. In order to achieve this, we examined whether human iPSC‐ECs (hiPSC‐ECs) are a suitable candidate for re‐endothelialization of the kidney scaffold. In order to obtain large quantities of hiPSC‐ECs, a previously described hiPSC‐EC differentiation protocol2 was adjusted for large‐scale production by increasing the culture surface during differentiation from a 6‐well plate format toward the use of multiple T175 cell culture flasks (Figure 4A). The resulting cells expressed the endothelial differentiation markers CD31 and VE‐cadherin, and were positive for both VEGF receptor 2 and Tie2 (Figure 4B), suggesting that they will be responsive to VEGF and Ang‐1. Ninety‐seven percent of the cells were CD31 positive after dissociation as shown by FACS analysis in Figure S3A. Moreover, iPS‐ECs showed similar acetylated LDL uptake compared to hgMVECs as is shown in Figure S3B. In agreement, VEGF loading of the scaffold resulted in increased adherence and survival of hiPSC‐ECs on human kidney scaffold slices (Figure 4C). A similar trend was observed with Ang‐1‐loading and combined loading (Figure 4C) as quantified in Figure 4D.
Figure 4.
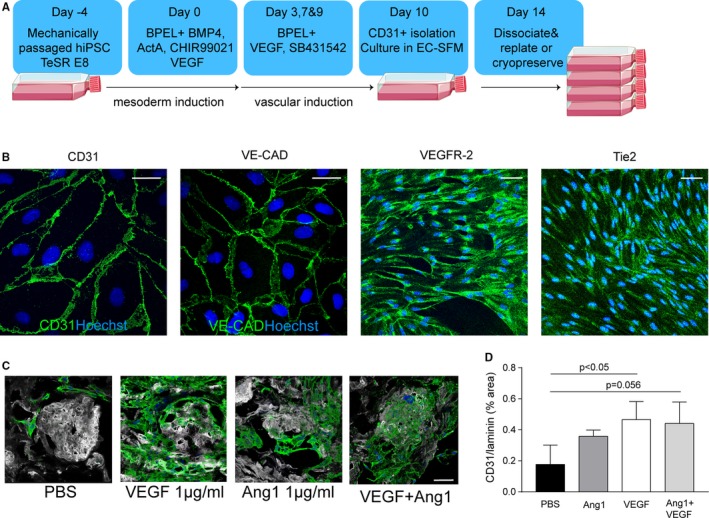
Scalability of hiPS‐EC culture and ability to adhere to growth‐factor‐loaded scaffolds. A, Adjusted hIPS‐EC differentiation protocol where large quantities of hIPS‐ECs can be obtained. B, Representative images of hiPS‐ECs show positivity for vascular endothelial markers CD31, VE‐cadherin, VEGF‐receptor 2, and Tie2. C, Representative images of hiPSC‐EC adherence and survival after VEGF, Ang‐1, or combined loading of the scaffold. D, Quantification of hiPS‐EC adherence after VEGF, Ang‐1, and combined loading. Ang‐1, angiopoietin 1; PBS, phosphate‐buffered saline; hiPS‐ECs, human inducible pluripotent stem cell–derived endothelial cells; VEGF, vascular endothelial growth factor. Scale bar 50 μm [Color figure can be viewed at wileyonlinelibrary.com]
3.6. Rat whole kidney scaffold re‐endothelialization with hiPS‐ECs
Growth factor–loaded decellularized rat kidneys were recellularized with hiPSC‐ECs via both the renal artery and vein as described above. Indeed, after 48‐hour re‐endothelialization of the glomeruli, the larger vessels and peritubular capillaries in both the cortex and medulla were observed (Figure 5A,B).
Figure 5.
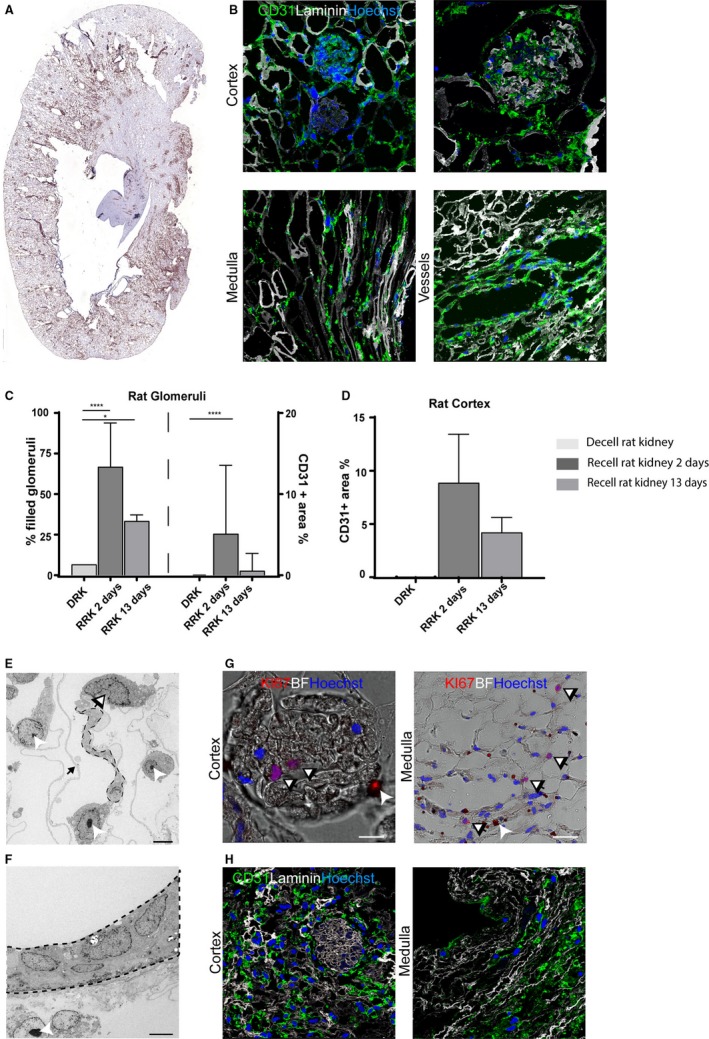
Re‐endothelialization of rat decellularized kidney matrices with hiPS‐ECs. A, Whole organ image of CD31+ in the whole re‐endothelialized rat kidney scaffold. B, Representative images after hiPS‐EC recellularization showing re‐endothelialization in the cortex, including the glomeruli, the medulla/papilla, and in the larger vessels. C, Quantification of CD31 positivity. For the recellularized rat kidneys (N = 2‐3) 1 complete slide per kidney was analyzed. Per slide all glomeruli were annotated manually (about 100 glomeruli per slide). The graph displays the amount of filled glomeruli as a % of all glomeruli per kidney (left) and the % of CD31+ area within all filled glomeruli (right) for 2 and 13 days after recellularization. ****P < .0001 *P < .05. D, For cortex analysis %CD31+ area was calculated within whole cortex area per kidney. E, TEM image of a glomerulus shows alignment of endothelial cells on the endothelial side of the GBM. F, TEM image showing alignment in a larger vessel. G, Representative image of Ki67 staining showing proliferating hiPS‐ECs in both the cortex and medulla. H, Representative image of CD31 positivity after 13 days of culture in both the cortex and medullary/papillary region. BF, bright field. Scale bar B, E, F 50 μm. Scale bar C, D 5 μm. Dashed line: aligning endothelial cell, black/white arrow: CD31 dynabeads, which are used for hiPS‐ECs isolation, black arrow: remaining extracellular matrix between foot processes on podocyte side of the GBM. GBM, glomerular basement membrane; hiPS‐ECs, human inducible pluripotent stem cell–derived endothelial cells; TEM, transmission electron microscopy
Analysis of CD31+ was performed on the re‐endothelialized rat kidneys, which showed that 70% of all the glomeruli at 2 days and 33% after 13 days contained ECs. Within these filled glomeruli, about 5% of CD31+ area was observed at 2 days (Figure 5C). The total cortex contained ≈8.9% CD31+ area after 2 days (Figure 5D). After 13 days, the amount of CD31+ area is 4.2%, which is, although not 1:1 comparable, similar in normal human kidney (3.3%) (Figure 5C,H).
Moreover, 7.8% of proliferating hiPS‐ECs could be observed 48 hours after recellularization, as illustrated by the Ki67 staining in Figure 5G. Transmission electron microscopy showed alignment of the hiPSC‐ECs on the endothelial side of the glomerular basement membrane (Figure 5E). Moreover, alignment within larger vessels was also observed (Figure 5F). Cells remained viable for the maximal duration of the current experiments (up to 13 days) where 36% of cells were still proliferating (Figure 5H).
3.7. Human whole kidney scaffold re‐endothelialization using hiPSC‐ECs
In order to evaluate whether this recellularization method would also be feasible for the much larger human kidneys, we performed 2 independent experiments where we re‐vascularized a growth factor loaded human decellularized kidney with 6 × 107 hiPSC‐ECs via the renal vein followed by 7 × 107 hiPSC‐ECs via the renal artery and cultured the kidney for 24 hours (Figure 6A). As shown in Figure 6B and C, we were able to re‐endothelialize the cortex, medulla, and papilla. Representative immunofluorescent images of EC alignment respectively in the cortex (Figure 6D), larger arteries (Figure 6E), medulla (Figure 6F), and papilla (Figure 6G) are shown. hiPSC‐ECs were able to proliferate in the human kidney scaffold (9.6% of the hiPSC‐ECs), as shown by Ki‐67 staining Figure 6H. Analysis of CD31+ in 4 biopsies taken from different locations through the kidney cortex showed that more than 80% of the glomeruli contained ECs. Within these glomeruli, 3.8% of the area was positive for CD31, which is close to the area observed in normal human kidney (6.7%; Figure 6I). The observed CD31+ area within the cortex was 3.0%, which is also similar to the normal human kidney (3.3%; Figure 6J). Obstructed blood vessels within the recellularized scaffold were minimal (area < 0.03%). This indicates that our re‐endothelialization method is scalable to the level of an entire human kidney scaffold.
Figure 6.
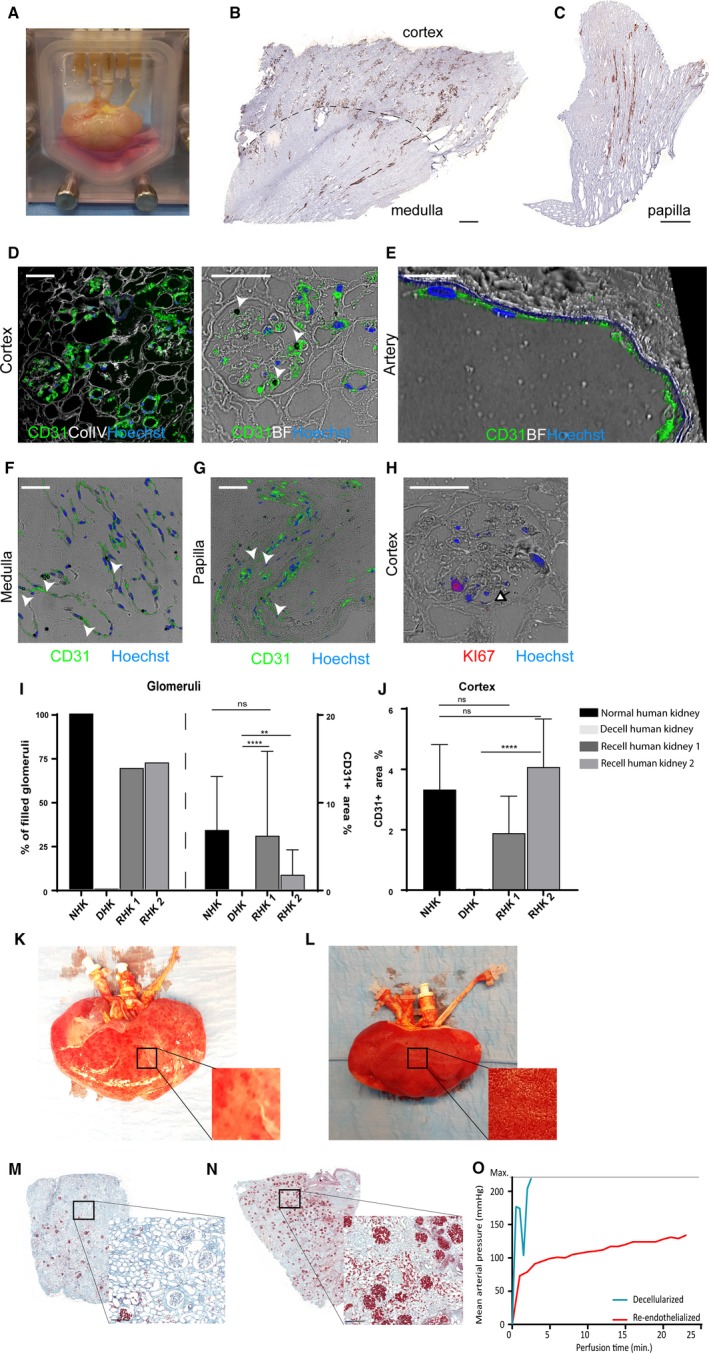
Human kidney re‐endothelialization with hiPS‐ECs. A, Image of the human re‐endothelialized kidney in the custom‐made organ chamber during culture. B, Representative immunohistochemistry images of CD31+ showing re‐endothelialization in the cortex, medulla, and (C) papilla. D, Representative CD31 immunofluorescent images show re‐endothelialization of the kidney cortex, (E) artery, (F) medulla, and (G) papilla. CD31 dynabeads used in the iPS‐EC differentiation protocol (white arrowheads) can still be observed. H, Representative image of proliferating cells in the human re‐endothelialized kidney. I, Quantification of CD31. For the recellularized human kidneys (N = 2) 4 areas per kidney were sampled and analyzed. Per slide all glomeruli were annotated manually (about 100 glomeruli per slide). The graphs display the amount of filled glomeruli as a % of all analyzed glomeruli per kidney (left) and the % of CD31+ area within all filled glomeruli (right) at 24 h after infusion of cells. ****P < .0001 **P < .01. J For cortex analysis %CD31+ area was calculated within whole cortex area per kidney. The graphs display the average of 4 different sample areas per kidney. ****P < .0001. K, Macroscopic image of a decellularized human kidney perfused with whole blood showing only some “patchy”perfused areas. L, Macroscopic image of a re‐endothelialized human kidney scaffold perfused with whole blood showing diffuse blood coverage over the whole kidney. M, Microscopic picture of a Movat's‐Pentachrome staining of a decellularized human kidney cortex scaffold perfused with whole blood shows only a few perfused areas. Because of low pressure and massive leakage (due to lack of endothelial cells), only a few red blood cells reached the glomeruli. N, When the human re‐endothelialized kidney scaffold was perfused with whole blood, high coverage of blood in the cortex was observed (shown in red), showing that perfusion of blood through the entire kidney was possible. O, Whereas the whole blood perfusion of the decellularized human kidney scaffold was stopped within 5 min because of extremely high perfusion pressures, the re‐endothelialized kidney could be perfused for more than 20 min. Scale bar B, C 500 μm; scale bar D, F, H 50 μm; scale bar E 25 μm. White arrowheads: CD31 dynabead, black/white arrowhead: proliferating cell. Scale bar M, N 200 μm. hiPS‐ECs, human inducible pluripotent stem cell–derived endothelial cells; ns, nonsignificant
3.8. Human re‐endothelialized kidney scaffold can be perfused with human whole blood
In order to show some functionality of the re‐endothelialized human kidney scaffold, both re‐endothelialized and decellularized kidney scaffolds were perfused with human recalcified whole blood. Using the decellularized kidney, the maximum mean arterial pressure measurable (>200 mm Hg) was reached within 5 minutes after whole blood perfusion and no more perfusion was possible due to clothing and massive leakage along the cannula. In contrast, the re‐endothelialized scaffold could be perfused for more than 20 minutes with only a moderate increase in mean arterial pressure (Figure 6O). In order to preserve the ECs in the scaffold for immunohistochemical analysis, the perfusion was stopped after 25 minutes when pressure stabilized. After perfusion, major differences in both macroscopic (Figure 6K and L) and microscopic (Figure 6M,N) appearance of the whole blood perfused kidneys were observed. Whereas the perfused decellularized kidney only showed some “patchy” blood‐perfused areas, the perfused re‐endothelialized kidney showed a more diffuse and complete coverage with whole blood. These results suggest that re‐endothelialization of the kidney scaffolds leads to less clotting, resulting in lower inflow pressures and a better distribution of human whole blood when perfused.
4. DISCUSSION
The current study shows that, by using hiPSC‐ECs combined with growth factor loading and a differential arteriovenous delivery system, it is possible to reconstitute both the macro‐ and microvascular compartments of a human kidney scaffold. The improved delivery via simultaneous arterial and venous infusion, coupled with enhanced cellular localization and adherence due to growth factor preloading, resulted in a significant improvement in endothelial coverage of the delivered cells within the scaffold. In combination, these represent significant advances in the use of decellularized kidney scaffolds as a platform for kidney bioengineering.
The decellularization protocol used preserved both core matrix proteins as well as matrisome proteins (Table 1). Previously Peloso et al showed preservation of some growth factors in human kidney scaffolds postdecellularization, including low levels of VEGF.21 Using our protocol, however, presence of both VEGF and Ang‐1 were low after decellularization while at the same time the GAG landscape appeared to be preserved. The latter offered the possibility to reconstitute the growth factors on the kidney ECM as a novel strategy to increase EC localization. Our data show that such a step is critical for cell adherence and survival, and it appeared to be a key step in facilitating maximal coverage which in turn improved the feasibility of scaleup for re‐endothelialization of a human‐size kidney scaffold. As both Ang‐1 and VEGF exert different effects on EC adherence, proliferation, and survival, we chose to load the kidney scaffolds with both factors in order to achieve optimal conditions for recellularization. We showed that this method resulted in sufficient endothelial coverage to accomplish whole blood perfusion of a human re‐endothelialized kidney scaffold.
Alternative strategies to enhance re‐endothelialization have also been described, including combined infusion of ECs and pericytes in the lung10 and CD31‐conjugation in the kidney.22 In the latter, CD31 antibody conjugation resulted in enhanced re‐endothelialization in porcine kidney scaffolds to similar levels as reported here. However, revascularization was performed using a murine EC line.
Our study, therefore, also underscores the potential of human pluripotent stem cells as a source of human endothelium. hIPS‐ECs could be produced from patient‐derived cells, which will most likely reduce the need for immunosuppressant therapy. In addition, their proliferative potential, which was supported by the scaffold, was a key factor in achieving the scalability for re‐endothelialization of a human kidney scaffold. Obviously, maintenance of genomic integrity during reprogramming and subsequent expansion together with improvements in the subsequent maturation of these cells into a safe transferable cellular phenotype will be critical aspects that need further development. This is the case for all proposed approaches for tissue bio‐engineering using pluripotent stem cells and is therefore at the center of attention in the stem cell field.
Next to the vascular compartment, the epithelial compartment of the kidney will also require successful recellularization in order to obtain a full functional bio‐engineered kidney. Song et al showed that, when a rat kidney scaffold was recellularized with neonatal rat kidney cells, site‐specific epithelial recellularization was observed.9 Whether hiPSC‐derived kidney progenitor cells3 show the same potential or whether recellularization with distinct subtypes of hiPSC‐derived kidney epithelial cells will be required still needs further investigation.
5. CONCLUSION
Here, we show that preservation of the GAG landscape and the possibility to load the scaffold with growth factors such as VEGF and angiopoietin‐1 are critical to EC adherence and survival on a kidney scaffold. Using a new controlled arteriovenous delivery and culture method of hiPSC‐derived ECs, we report the re‐endothelialization of the kidney vasculature, including the glomerular and peritubular capillaries. We show for the first time that this method is scalable toward the human kidney scaffold where perfusion with human whole blood could be achieved. These findings provide unique insight into a critical step to develop a human bioengineered kidney.
DISCLOSURE
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting information
ACKNOWLEDGMENTS
The research leading to these results has received funding from the European Community's Seventh Framework Program (FP7/2007‐2013) under grant agreement number 305436 (STELLAR) and from the Dutch Kidney Foundation (RECORD KID, 15RN02). We would like to thank Valeria Orlova (department of anatomy and embryology, Leiden University Medical Center) for her help with the iPS‐EC differentiation technique. We also thank the Maastricht MultiModal Molecular Imaging Institute–Division of Imaging Mass Spectrometry for performing the mass spectrometry runs.
Leuning DG, Witjas FMR, Maanaoui M, et al. Vascular bioengineering of scaffolds derived from human discarded transplant kidneys using human pluripotent stem cell–derived endothelium. Am J Transplant. 2019;19:1328‐1343. 10.1111/ajt.15200
Daniëlle G. Leuning and Franca M.R. Witjas contributed equally to this article.
REFERENCES
- 1. Levey AS, Coresh J. Chronic kidney disease. Lancet. 2012;379:165‐180. [DOI] [PubMed] [Google Scholar]
- 2. Orlova VV, Van Den Hil FE, Petrus‐Reurer S, Drabsch Y, Ten Dijke P, Mummery CL. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat Protoc. 2014;9:1514‐1531. 10.1038/nprot.2014.102. [DOI] [PubMed] [Google Scholar]
- 3. Takasato M, Pei XE, Chiu HS, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015;526:564‐568. 10.1038/nature15695. [DOI] [PubMed] [Google Scholar]
- 4. Halaidych V, Freund C, van den Hil F, et al. Inflammatory responses and barrier function of endothelial cells derived from human induced pluripotent stem cells. Stem Cell Reports. 2018;10:1642‐1656. 10.1016/j.stemcr.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bonandrini B, Figliuzzi M, Papadimou E, et al. Recellularization of well‐preserved acellular kidney scaffold using embryonic stem cells. Tissue Eng Part A. 2014;20:1486‐1498. 10.1089/ten.tea.2013.0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Remuzzi A, Figliuzzi M, Bonandrini B, et al. Experimental evaluation of kidney regeneration by organ scaffold recellularization. Sci Rep. 2017;7:43502 10.1038/srep43502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ross EA, Abrahamson DR, St. John P, et al. Mouse stem cells seeded into decellularized rat kidney scaffolds endothelialize and remodel basement membranes. Organogenesis. 2012;8:49‐55. 10.4161/org.20209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ross EA, Williams MJ, Hamazaki T, et al. Embryonic stem cells proliferate and differentiate when seeded into kidney scaffolds. J Am Soc Nephrol. 2009;20:2338‐2347. 10.1681/asn.2008111196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song JJ, Guyette JP, Gilpin SE, Gonzalez G, Vacanti JP, Ott HC. Regeneration and experimental orthotopic transplantation of a bioengineered kidney. Nat Med. 2013;19:646‐651. 10.1038/nm.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ren X, Moser PT, Gilpin SE, et al. Engineering pulmonary vasculature in decellularized rat and human lungs. Nat Biotechnol. 2015;33:1097‐1102. 10.1038/nbt.3354. [DOI] [PubMed] [Google Scholar]
- 11. Ribatti D, Nico B, Crivellato E. The role of pericytes in angiogenesis. Int J Dev Biol. 2011;55:261‐268. 10.1387/ijdb.103167dr. [DOI] [PubMed] [Google Scholar]
- 12. Caporali A, Martello A, Miscianinov V, Maselli D, Vono R, Spinetti G. Contribution of pericyte paracrine regulation of the endothelium to angiogenesis. Pharmacol Ther. 2017;171:56‐64. 10.1016/j.pharmthera.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 13. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298‐307. 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rabelink TJ, van den Berg BM, Garsen M, Wang G, Elkin M, van der Vlag J. Heparanase: roles in cell survival, extracellular matrix remodelling and the development of kidney disease. Nat Rev Nephrol. 2017;13:201‐212. 10.1038/nrneph.2017.6. [DOI] [PubMed] [Google Scholar]
- 15. Guyette JP, Gilpin SE, Charest JM, Tapias LF, Ren X, Ott HC. Perfusion decellularization of whole organs. Nat Protoc. 2014;9:1451‐1468. 10.1038/nprot.2014.097. [DOI] [PubMed] [Google Scholar]
- 16. Zhang H, Baader SL, Sixt M, Kappler J, Rauch U. Neurocan‐GFP fusion protein: a new approach to detect hyaluronan on tissue sections and living cells. J Histochem Cytochem. 2004;52:915‐922. 10.1369/jhc.3a6221.2004. [DOI] [PubMed] [Google Scholar]
- 17. Mani K, Cheng F, Sandgren S, et al. The heparan sulfate‐specific epitope 10E4 is NO‐sensitive and partly inaccessible in glypican‐1. Glycobiology. 2004;14:599‐607. 10.1093/glycob/cwh067. [DOI] [PubMed] [Google Scholar]
- 18. Raats CI, Luca ME, Bakker MA, et al. Reduction in glomerular heparan sulfate correlates with complement deposition and albuminuria in active Heymann nephritis. J Am Soc Nephrol. 1999;10:1689‐1699. [DOI] [PubMed] [Google Scholar]
- 19. Van Den Born J, van den Heuvel LP, Bakker MA, et al. Distribution of GBM heparan sulfate proteoglycan core protein and side chains in human glomerular diseases. Kidney Int. 1993;43:454‐463. [DOI] [PubMed] [Google Scholar]
- 20. Fairbrother WJ, Champe MA, Christinger HW, Keyt BA, Starovasnik MA. Solution structure of the heparin‐binding domain of vascular endothelial growth factor. Structure. 1998;6:637‐648. [DOI] [PubMed] [Google Scholar]
- 21. Peloso A, Petrosyan A, Da Sacco S, et al. Renal extracellular matrix scaffolds from discarded kidneys maintain glomerular morphometry and vascular resilience and retains critical growth factors. Transplantation. 2015;99:1807‐1816. 10.1097/tp.0000000000000811. [DOI] [PubMed] [Google Scholar]
- 22. Ko IK, Abolbashari M, Huling J, et al. Enhanced re‐endothelialization of acellular kidney scaffolds for whole organ engineering via antibody conjugation of vasculatures. Technology. 2015;2:243‐253.doi:http://www.worldscientific.com/doi/abs/10.1142/S2339547814500228 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials