

Abstract
Background
Levomilnacipran extended release (ER) is a serotonin and norepinephrine reuptake inhibitor approved for major depressive disorder (MDD) in adults. This study was designed to evaluate relapse prevention with levomilnacipran ER in patients with MDD.
Methods
Patients (≥18 years) with MDD (N = 644) received 20 weeks of open‐label treatment with levomilnacipran ER 40, 80, or 120 mg/d (8 weeks flexible dosing; 12 weeks fixed dosing). Patients with a Montgomery–Åsberg Depression Rating Scale (MADRS) total score ≤12 from the end of week 8 to week 20 were randomized to 26 weeks of double‐blind treatment with levomilnacipran ER (same dosage; n = 165) or placebo (n = 159). The primary efficacy endpoint was time to relapse, defined as insufficient therapeutic response (≥2‐point increase from randomization in Clinical Global Impression of Severity score, risk of suicide, need for hospitalization due to worsening of depression, or need for alternative antidepressant treatment as determined by the investigator) or an MADRS total score ≥18 at 2 consecutive visits.
Results
In the double‐blind intent‐to‐treat population, levomilnacipran ER‐treated patients had a significantly longer time to relapse compared with placebo (hazard ratio = 0.56; 95% CI, 0.33–0.92; P = 0.0212). Crude relapse rates were 14.5% (levomilnacipran ER) and 24.5% (placebo). Double‐blind treatment‐emergent adverse events (AEs) were reported for 58.8% and 56.0% of levomilnacipran ER and placebo patients, respectively; 3.0% and 1.3% discontinued due to AEs, and 1.2% and 0.6% had serious AEs, respectively.
Conclusion
Levomilnacipran ER (40–120 mg/d) was effective in preventing relapse in patients with MDD. Safety and tolerability results were consistent with levomilnacipran ER acute studies.
Keywords: antidepressant, depression, levomilnacipran extended release, long‐term treatment, maintenance treatment, randomized withdrawal
1. INTRODUCTION
The majority of patients with major depressive disorder (MDD) have more than one major depressive episode, and at least 50% of patients who experience one episode are likely to experience another (APA, 2010; Kessler & Walters, 1998; Kessler, Zhao, Blazer, & Swartz, 1997). A higher number of previous episodes (Berwian, Walter, Seifritz, & Huys, 2017; Kendler, Thornton, & Gardner, 2000), more residual symptoms (Berwian et al., 2017; Nierenberg et al., 2010), and failure to reach remission after acute antidepressant treatment (APA, 2010; Rush et al., 2006) are associated with a greater risk of relapse, defined as the return of depression symptoms during an index episode. In addition, the STAR*D trial showed that the more acute treatment regimens a patient requires, the higher the risk of relapse (Rush et al., 2006).
Antidepressants are effective in decreasing depressive symptoms and reducing the risk of relapse in patients with MDD (Geddes et al., 2003; Glue, Donovan, Kolluri, & Emir, 2010; Sim, Lau, Sim, Sum, & Baldessarini, 2015). Clinical guidelines recommend an additional 4 to 9 months of antidepressant treatment after initial response is achieved and at least 3 years of continual treatment for maintenance therapy, especially in patients who experience recurrent MDD (APA, 2010; Bauer et al., 2015). Meta‐analyses of relapse‐prevention studies demonstrated that longer initial and continuation treatment resulted in lower rates of relapse for antidepressants compared with placebo (Geddes et al., 2003; Glue et al., 2010; Sim et al., 2015). Continual antidepressant treatment has also been shown to reduce the risk of relapse by 70% relative to placebo (Geddes et al., 2003; Kaymaz, van Os, Loonen, & Nolen, 2008). However, no medication is effective in all patients, and relapse prevention is an area of unmet need (Stahl, Grady, Moret, & Briley, 2005).
Levomilnacipran extended release (ER) is a serotonin and norepinephrine reuptake inhibitor (SNRI) approved in the United States for the treatment of MDD in adults. Levomilnacipran ER efficacy has been evaluated in five short‐term, randomized, placebo‐controlled trials (Asnis, Bose, Gommoll, Chen, & Greenberg, 2013; Bakish et al., 2014; Gommoll, Greenberg, & Chen, 2014; Montgomery et al., 2013; Sambunaris et al., 2014). Four of these trials demonstrated statistically significant improvements versus placebo in the primary efficacy endpoint, change from baseline in Montgomery–Åsberg Depression Rating Scale (MADRS; Montgomery & Asberg, 1979) total score (Asnis et al., 2013; Bakish et al., 2014; Montgomery et al., 2013; Sambunaris et al., 2014), while one study had numerically greater improvement with levomilnacipran ER versus placebo (Gommoll et al., 2014). Long‐term safety of levomilnacipran ER (40–120 mg/d) was demonstrated in a 48‐week, open‐label extension study and a previous 24‐week levomilnacipran ER (40–120 mg/d) relapse‐prevention study (Mago, Forero, Greenberg, Gommoll, & Chen, 2013; Shiovitz, Greenberg, Chen, Forero, & Gommoll, 2014). The current study was designed to evaluate the efficacy, safety, and tolerability of levomilnacipran ER in preventing relapse in adult patients with MDD.
2. MATERIALS AND METHODS
2.1. Study design
This was a multicenter, randomized, double‐blind, placebo‐controlled, relapse‐prevention study with levomilnacipran ER in patients with MDD (NCT02288325). The study was conducted from 2014 to 2016 at 44 study centers in the United States in full compliance with the International Council on Harmonisation guideline for Good Clinical Practice and the principles of the Declaration of Helsinki. The protocol and amendments were approved by the institutional review board at each study center, and all patients provided written informed consent. This study was up to 53 weeks in duration and consisted of washout/screening; an open‐label, flexible‐dose, run‐in phase (RIP); an open‐label, fixed‐dose, stabilization phase (SP); a double‐blind treatment phase; and down‐taper (Figure 1). A 12‐week, open‐label SP is required by the US Food and Drug Administration (FDA) to consolidate treatment response after the initial run‐in open‐label treatment period.
Figure 1.
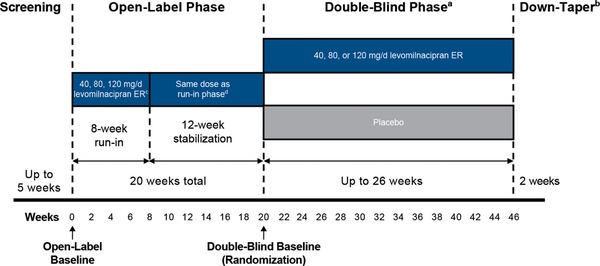
Study design.
aPatients remained on their final effective and tolerated open‐label levomilnacipran ER dose; patients randomized to placebo were down‐tapered from the levomilnacipran ER dose they received in open‐label treatment.
bIncludes patients completing double‐blind treatment or prematurely discontinuing from the study.
cDuring the run‐in phase, dose adjustments were allowed up to week 6.
dDuring the stabilization phase, no dose adjustments were allowed
2.2. Participants
The study included male and female outpatients (≥18 to ≤70 years) who met the following eligibility criteria: Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM‐5) criteria for MDD (APA, 2013), confirmed by a Mini International Neuropsychiatric Interview; ongoing major depressive episode (duration ≥8 weeks to ≤18 months); ≥3 lifetime depressive episodes (including current episode), with 2 episodes (including current) occurring within the past 5 years; MADRS total score ≥26; and body mass index between ≥18 and ≤40 kg/m2.
Patients who met any of the following criteria were excluded from study participation: DSM‐5 Axis I diagnosis of a disorder other than MDD within 6 months of screening (secondary diagnoses of comorbid generalized anxiety disorder, social anxiety disorder, and/or specific phobias were allowed); history of mania, psychotic disorder, panic disorder, obsessive–compulsive disorder, bulimia or anorexia nervosa (past 5 years), borderline or antisocial personality disorder, or neurocognitive disorder; alcohol or other substance abuse disorder (past 6 months); nonresponse to adequate treatment with ≥2 antidepressants (i.e., 8‐week duration at recommended dose) during the current episode; and suicide risk defined as suicide attempt (past 12 months), MADRS Item 10 score ≥5, or determined by the investigator based on the psychiatric interview or information collected in the Columbia‐Suicide Severity Rating Scale (C‐SSRS; Posner et al., 2011). Concomitant treatment with psychoactive medications was prohibited, except for medications for insomnia.
2.3. Treatment, randomization, and blinding
During the 8‐week, open‐label RIP, levomilnacipran ER was titrated to a dose of 40, 80, or 120 mg/d based on tolerability and response to treatment. Patients started at 20 mg/d, and the dosage was increased to 40 mg/d after 2 days. The dosage could be increased to 120 mg/d or decreased in 40‐mg increments up to the end of week 6 based on investigator judgment and dose‐limiting adverse events (AEs). Patients who completed the RIP with an MADRS total score ≤12, a threshold frequently used to define remission, and no tolerability issues entered the 12‐week, open‐label SP taking the same levomilnacipran ER dose; no dose adjustments were allowed. Patients who completed the SP and met the following randomization criteria were allowed to enter double‐blind treatment: stable clinical response (MADRS total score ≤12 at weeks 8, 10, 12, 14, 16, 18, and 20; 1 or 2 modest excursions [i.e., MADRS total score > 12 but ≤16] at weeks 10, 12, or 14 were permitted); no significant tolerability issues as determined by the investigator; and no MADRS total score ≥17 at any time during the SP. During the 26‐week double‐blind treatment, eligible patients were randomized (1:1) to placebo or levomilnacipran ER at the dose received during the SP. Randomization codes were generated electronically and implemented using an interactive web response system; identical packaging was used for all study medications. All patients, investigators, and the study sponsor were blinded to treatment assignment. Breaking the blind at the study center level disqualified the patient from further study participation. During the study, however, it was not necessary to unblind any treatment code.
2.4. Efficacy assessments
The primary efficacy parameter was time to first relapse during the double‐blind treatment phase. Relapse was defined as meeting one or more of the following criteria: insufficient therapeutic response (e.g., ≥2‐point increase from randomization in Clinical Global Impression of Severity [CGI‐S] (Guy, 1976) score, investigator‐determined risk of suicide, need for hospitalization due to worsening of depression, investigator‐determined need for alternative antidepressant treatment) or an MADRS total score ≥18 at two consecutive visits occurring within 7 to 14 days.
Additional efficacy parameters included CGI‐Improvement (CGI‐I; Guy, 1976) score relative to open‐label baseline and changes from baseline or randomization in MADRS total score, CGI‐S score, and Sheehan Disability Scale (SDS; Sheehan, Harnett‐Sheehan, & Raj, 1996) total and subscale (work/school, social, and family) scores.
2.5. Safety assessments
AEs, serious AEs (SAEs), and vital signs were recorded at all study visits. Clinical laboratory tests and electrocardiograms were administered during screening, at randomization, and at the end of double‐blind treatment. The C‐SSRS was administered at all study visits to monitor suicidal ideation or behavior.
2.6. Statistical analyses
Sample size and power calculations were based on historical relapse data, with assumed relapse rates at the end of double‐blind treatment of 30% in the placebo group and 15% in the levomilnacipran ER group, and a common discontinuation rate of 20% for both groups. It was estimated that 308 randomized patients (154 per group) would be needed for 85% power to detect a difference in the time to relapse between levomilnacipran ER and placebo using the log‐rank test at the 0.05 significance level. To attain 308 randomized patients, it was estimated that approximately 640 patients would be needed for enrollment, based on an assumption that response rates during the RIP and the SP would be 65% and 75%, respectively.
For each treatment phase, the safety population included all eligible patients who received ≥1 dose of open‐label levomilnacipran ER (open‐label phase) or double‐blind medication. Efficacy analyses used the intent‐to‐treat (ITT) population, defined for each treatment phase as patients in safety population who had ≥1 available postbaseline (open‐label) or postrandomization (double‐blind) MADRS assessment.
The primary efficacy parameter, time to relapse, was analyzed using the log‐rank test with estimates of the hazard ratio and 95% confidence interval (CI) based on the Cox proportional hazards model with treatment group as an explanatory variable. Patients who did not meet any relapse criteria were censored at study completion or upon early discontinuation for any reason other than relapse. The cumulative distribution function of time to relapse was characterized using Kaplan–Meier curves.
To additionally evaluate factors that may have affected time to relapse, post hoc analyses were conducted. To assess the possibility that the presence of a secondary anxiety disorder (agoraphobia without history of panic disorder, generalized anxiety disorder, or social anxiety disorder) may have confounded time to relapse, Kaplan–Meier estimates were generated excluding patients with these secondary diagnoses. Additionally, the potential for withdrawal symptoms to affect results during the first 28 days after randomization was evaluated by a Kaplan–Meier analysis, in which all relapses during the first 28 days after randomization were considered censored. P values were determined by the log‐rank test.
For all additional efficacy parameters, between‐group differences were analyzed using an analysis of covariance model with treatment group and pooled study center as factors and the baseline value as the covariate; missing values were imputed using a last observation carried forward (LOCF) approach. All analyses were two‐sided hypothesis tests performed at the 5% level of significance.
Safety parameters were summarized descriptively using the safety populations. AEs were classified as treatment‐emergent AEs (TEAEs) if they were not present before the first dose of open‐label treatment or if they increased in severity during open‐label treatment or thereafter. AEs were classified as newly emergent AEs (NEAEs) if they were not present before the first dose of double‐blind treatment or if they increased in severity during double‐blind treatment.
3. RESULTS
3.1. Patient disposition
Patient disposition and reasons for study withdrawal are presented in Figure 2. A total of 644 patients entered the RIP, and 499 patients completed (77.5%). A total of 429 patients entered the SP, and 331 (77.2%) patients completed. The most common reasons for open‐label discontinuation were AE (RIP), and withdrawal of consent and lack of efficacy (SP). A total of 324 patients entered double‐blind treatment (159 placebo, 165 levomilnacipran ER); 274 (84.6%) completed the study. There was no significant between‐group difference for any reason for premature discontinuation. Patient characteristics in the double‐blind safety population were generally similar between treatment groups and consistent with characteristics in the open‐label safety population (Table 1).
Figure 2.
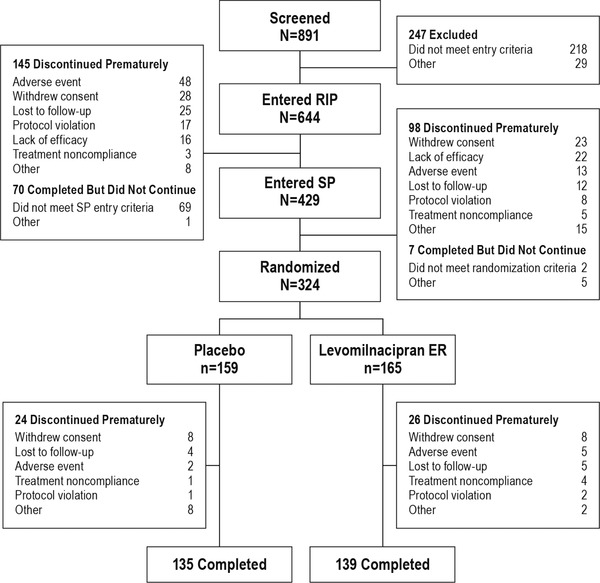
Patient disposition. All 644 patients who entered the RIP received ≥1 dose of open‐label treatment and were included in the open‐label safety population. All 324 randomized patients received ≥1 dose of double‐blind treatment and were included in the double‐blind safety population. Patients meeting one or more of the relapse criteria were considered to have completed double‐blind treatment. ER, extended release; RIP, run‐in phase; SP, stabilization phase
Table 1.
Patient characteristics at open‐label (RIP) and double‐blind baseline (safety populations)
| Open‐label | Double‐blind | ||
|---|---|---|---|
| Levomilnacipran ER | Placebo | Levomilnacipran ER | |
| n = 644 | n = 159 | n = 165 | |
| Demographics | |||
| Age, years, mean (SD) | 43.1 (13.9) | 46.2 (13.3) | 44.6 (13.6) |
| Female, n (%) | 404 (62.7) | 104 (65.4) | 114 (69.1) |
| White, n (%) | 459 (71.3) | 123 (77.4) | 114 (69.1) |
| Black/African‐American, n (%) | 144 (22.4) | 25 (15.7) | 40 (24.2) |
| BMI, kg/m2, mean (SD) | 28.8 (5.9) | 29.7 (5.9) | 28.8 (5.6) |
| Psychiatric history | |||
| Age at onset, years, mean (SD) | 26.6 (13.3) | 28.1 (12.9) | 27.0 (13.7) |
| Number of lifetime episodes, mean (SD) | 5.1 (3.8) | 5.3 (4.6) | 5.1 (3.8) |
| Duration of current episode, months, mean (SD) | 7.1 (4.5) | 6.7 (4.2) | 7.0 (4.5) |
| Prior suicide attempt, n (%) | 102 (15.8) | 18 (11.3) | 30 (18.2) |
| Antidepressant history, n (%) | |||
| Prior antidepressant treatment | 466 (72.4) | 126 (79.2) | 125 (75.8) |
| Nonresponse to treatment | 300 (46.6) | 87 (54.7) | 77 (46.7) |
| Intolerant to treatment | 83 (12.9) | 21 (13.2) | 21 (12.7) |
BMI, body mass index; ER, extended release; RIP, run‐in phase; SD, standard deviation.
3.2. Primary efficacy
Time to relapse was significantly longer in patients continuing on levomilnacipran ER compared with patients on placebo (P = 0.0212; Figure 3). Overall, relapse occurred in 24.5% (39/159) of patients in the placebo group and 14.5% (24/165) of patients in the levomilnacipran ER group. The hazard ratio (HR) of relapse in the levomilnacipran ER group was approximately half that of the placebo group (HR [95% CI] = 0.56 [0.33–0.92]). The most common reason for relapse was insufficient therapeutic response defined as a ≥2‐point increase from randomization in CGI‐S score (placebo: 79.5% [31/39]; levomilnacipran ER: 70.8% [17/24]). The need for alternative treatment or MADRS total score ≥18 at two consecutive visits was the reason for relapse of eight patients in the placebo group and seven patients in the levomilnacipran ER group. Among patients who terminated early, two placebo patients (noncompliance = 1, withdrawal of consent = 1) had an MADRS total score ≥18 as their last recorded score (last visit) but discontinued before their confirmation visit. No patients in either treatment group relapsed due to suicide risk or hospitalization for worsening of depression. Relapse rates began to diverge at approximately week 4.
Figure 3.
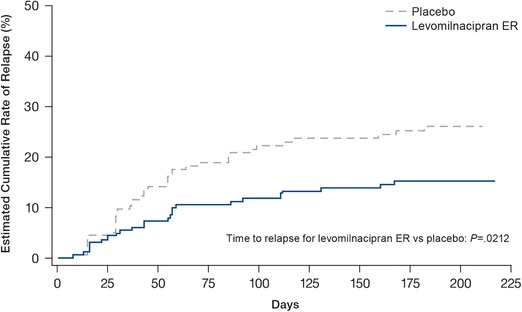
Cumulative rate of relapse (double‐blind ITT population). The 25th percentile for time to relapse was 168 days in the placebo group and was not able to be estimated for the levomilnacipran ER group using the Kaplan–Meier method. ER, extended release; ITT, intent‐to‐treat
Post hoc analyses found that the time to relapse remained statistically significant in favor of levomilnacipran ER versus placebo when patients with secondary anxiety disorders (levomilnacipran ER = 18 patients; placebo = 14 patients) were excluded from analysis (P = 0.0171). When evaluating whether early relapse events were related to withdrawal symptoms, it was noted that rates of relapse were low and similar (∼5%) for both levomilnacipran ER‐ and placebo‐treatment arms in the first few weeks following randomization (Figure 3). Post hoc Kaplan–Meier analysis found that when all relapses during the first 28 days after randomization (i.e., when withdrawal events were likely to occur) were considered censored, the significant benefit of levomilnacipran ER versus placebo in prolonging time to relapse during double‐blind treatment was maintained (P = 0.0140).
3.3. Additional efficacy
At the end of open‐label treatment with levomilnacipran ER, decreases (improvement) were observed in MADRS total score, CGI‐S score, SDS total score, and SDS subscale scores (Table 2). At the end of the 26‐week double‐blind treatment phase, greater worsening of rating scale scores was noted in the placebo group relative to the levomilnacipran ER group on all additional efficacy measures. The treatment group differences were statistically significant using LOCF analyses (P < 0.05, all outcomes).
Table 2.
Additional efficacy parameters (LOCF; ITT populations)
| Open‐label | Double‐blind | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Levomilnacipran ER | Placebo | Levomilnacipran ER | ||||||||
| n | Baseline (SD) | Mean changea (SD) | n | Baseline (SD) | LS mean changeb (SE) | n | Baseline (SD) | LS mean changeb (SE) | LSMD vs. placebo (95% CI) | |
| MADRS total score | 641 | 32.2 (4.1) | −20.5 (10.5) | 159 | 4.7 (3.6) | +5.3 (0.8) | 165 | 4.9 (3.5) | +3.0 (0.8) | −2.3 (−4.3, −0.4)* |
| CGI‐S score | 641 | 4.6 (0.6) | −2.2 (1.4) | 159 | 1.5 (0.7) | +0.6 (0.1) | 165 | 1.5 (0.7) | +0.3 (0.1) | −0.3 (−0.5, −0.1)* |
| SDS total score | 482 | 19.0 (5.2) | −12.0 (8.3) | 142 | 2.9 (3.8) | +2.1 (0.5) | 145 | 2.9 (4.4) | +0.3 (0.5) | −1.8 (−3.1, −0.4)** |
| SDS work/school subscale score | 482 | 5.8 (2.4) | −3.6 (3.2) | 142 | 0.9 (1.4) | +0.8 (0.2) | 145 | 0.9 (1.6) | +0.1 (0.2) | −0.7 (−1.1, −0.2)** |
| SDS social life subscale score | 625 | 7.0 (2.0) | −4.4 (3.2) | 159 | 1.1 (1.6) | +0.8 (0.2) | 164 | 1.1 (1.6) | +0.3 (0.2) | −0.5 (−1.0, −0.0)* |
| SDS family life subscale score | 625 | 6.4 (2.0) | −4.0 (3.2) | 159 | 0.9 (1.4) | +1.0 (0.2) | 164 | 0.8 (1.4) | +0.4 (0.2) | −0.6 (−1.1, −0.1)** |
| n | Mean score (SE) at end of treatment | n | Mean score (SE) at end of treatment | n | Mean score (SE) at end of treatment | LSMD vs. placebo (95% CI) | |
|---|---|---|---|---|---|---|---|
| CGI‐I score at end of treatment, mean (SE) | 641 | 1.9 (1.1) | 159 | 1.8 (0.1) | 165 | 1.5 (0.1) | −0.3 (−0.5, 0.1)* |
* P < 0.05; ** P < 0.01, levomilnacipran ER versus placebo during double‐blind treatment.
At the end of the open‐label phase.
At the end of the double‐blind phase.
CGI‐I, Clinical Global Impressions‐Improvement; CGI‐S, CGI‐Severity; CI, confidence interval; ER, extended release; ITT, intent‐to‐treat; LOCF, last observation carried forward; LS, least squares; LSMD, LS mean difference; MADRS, Montgomery–Åsberg Depression Rating Scale; SD, standard deviation; SDS, Sheehan Disability Scale; SE, standard error.
3.4. Safety
3.4.1. Extent of exposure
Mean treatment duration during the open‐label phase (RIP + SP) was 100.1 days; median duration was 138 days (range, 1–148 days). Patients who continued receiving levomilnacipran ER during the double‐blind phase had a mean treatment duration of 150.4 days during double‐blind treatment; median duration was 182 days (range, 4–193 days). Of note, due to various factors during the study (e.g., study visit dates that differed from targeted dates), the actual duration of open‐label treatment differed slightly from the duration of treatment that was designed for the trial (140 days [20 weeks]). However, given the low proportion of relapses during double‐blind treatment, the actual median duration of double‐blind treatment and the designed double‐blind duration of treatment were the same.
3.4.2. Adverse events
No deaths occurred during the study. During the open‐label treatment period, TEAEs were reported in 85.9% (553/644) of patients (Table 3). TEAEs reported in ≥10% of patients were nausea, headache, heart rate increased, and constipation. Most events (> 95%) were mild or moderate in severity. SAEs were reported in 9 patients; no preferred term for an SAE was reported in > 1 patient. Only one SAE (severe mania during the RIP) was considered related to levomilnacipran ER. AEs that led to premature discontinuation in > 2 patients were nausea (n = 11), headache (n = 6), anxiety and tachycardia (n = 4 each), erectile dysfunction and testicular pain (n = 3 each [male patients only]), and hyperhidrosis (n = 3).
Table 3.
Adverse events (safety populations)
| Open‐label | Double‐blind | ||
|---|---|---|---|
| Levomilnacipran ER | Placebo | Levomilnacipran ER | |
| n = 644 | n = 159 | n = 165 | |
| n (%) | n (%) | n (%) | |
| Any serious AE | 9 (1.4) | 1 (0.6) | 2 (1.2) |
| Any AE leading to discontinuation | 61 (9.5) | 2 (1.3) | 5 (3.0) |
| Any TEAE | 553 (85.9) | 89 (56.0) | 97 (58.8) |
| Newly emergent AEsa | NA | 82 (51.6) | 84 (50.9) |
| Common TEAEsb | |||
| Nausea | 174 (27.0) | 5 (3.1) | 13 (7.9) |
| Headache | 103 (16.0) | 12 (7.5) | 17 (10.3) |
| Heart rate increased | 71 (11.0) | 4 (2.5) | 4 (2.4) |
| Constipation | 69 (10.7) | 3 (1.9) | 3 (1.8) |
| Hyperhidrosis | 57 (8.9) | 2 (1.3) | 1 (0.6) |
| Erectile dysfunctionc | 19 (7.9) | 0 (0.0) | 0 (0.0) |
| Dizziness | 50 (7.8) | 4 (2.5) | 1 (0.6) |
| Tachycardia | 50 (7.8) | 3 (1.9) | 6 (3.6) |
| Upper respiratory tract infection | 46 (7.1) | 10 (6.3) | 16 (9.7) |
| Dry mouth | 37 (5.7) | 2 (1.3) | 0 (0.0) |
| Blood pressure increased | 36 (5.6) | 3 (1.9) | 5 (3.0) |
| Insomnia | 34 (5.3) | 2 (1.3) | 5 (3.0) |
| Nasopharyngitis | 25 (3.9) | 7 (4.4) | 11 (6.7) |
A newly emergent AE was an AE that was not present before the first dose of double‐blind treatment or that increased in severity during the double‐blind phase.
Reported in ≥5% of patients in any treatment group in either the open‐label or double‐blind treatment period.
Reported in male patients (n = 240).
AE, adverse event; ER, extended release; NA, not applicable; TEAE, treatment‐emergent AE.
The incidence of TEAEs in the double‐blind safety population was 56.0% and 58.8% with placebo and levomilnacipran ER, respectively. The only TEAE reported in ≥5% of levomilnacipran ER‐treated patients and at twice the rate of placebo was nausea (7.9% vs. 3.1%, respectively). The incidence of nausea reported as a NEAE was 1.9% and 4.2% with placebo and levomilnacipran ER, respectively. SAEs were reported in one patient in the placebo group (suspected overdose Tylenol PM) and two patients in the levomilnacipran ER group (transient ischemic attack [related] and Escherichia pyelonephritis [not related]); all SAEs resolved. Nausea was the only AE that led to premature discontinuation in > 1 patient (two patients) in the levomilnacipran ER group. During the double‐blind down‐taper phase, NEAEs were reported in 7 placebo‐ and 10 levomilnacipran ER‐treated patients; NEAEs were considered to be treatment‐related in one placebo patient and three levomilnacipran ER patients (diarrhea, headache, initial insomnia, cold sweat, and hot flush).
3.4.3. C‐SSRS assessments
During the open‐label treatment phase, C‐SSRS‐assessed suicidal ideation and behavior were reported in 180 (28.1%) and 12 (1.9%) patients, respectively. Suicidal ideation was reported as a TEAE in two patients. During the double‐blind treatment phase, the C‐SSRS‐assessed incidence of suicidal ideation was 17.6% and 12.1% in the placebo and levomilnacipran ER groups, respectively. One event of suicidal ideation was reported as a TEAE during double‐blind treatment period in the placebo group.
3.4.4. Other safety parameters
The mean changes in liver enzyme, metabolic, or hematologic parameters during the open‐label or double‐blind treatment phases were not clinically meaningful relative to baseline or placebo, respectively. At the end of the double‐blind treatment phase, patients in the levomilnacipran ER group had greater mean increases relative to placebo in systolic blood pressure, diastolic blood pressure, and pulse rate (Table 4). No potentially clinically significant change in blood pressure or pulse rate occurred in ≥2% of patients in either treatment group. Weight gain ≥7% from baseline was reported in 3.3% of patients during open‐label treatment and in 10.1% and 13.9% of placebo‐ and levomilnacipran ER‐treated patients, respectively, during double‐blind treatment. Mean change in heart rate was higher at the end of double‐blind treatment in the levomilnacipran ER group compared with placebo (Table 4). No patient had >60 msec change in QTcF, a clinically significant shift from a normal to abnormal ECG reading, or QTc values >500 msec during double‐blind treatment. Changes in additional ECG parameters over time were otherwise generally similar.
Table 4.
Changes in vital signs and electrocardiographic parameters during double‐blind treatment (safety population)
| Placebo | Levomilnacipran ER | |
|---|---|---|
| Mean (SD) | Mean (SD) | |
| Vital sign parameters | n = 159 | n = 165 |
| Supine systolic blood pressure, mm Hg | ||
| Baseline | 119.6 (12.0) | 118.5 (11.4) |
| Change at EOT | 2.0 (11.0) | 5.0 (12.5) |
| Supine diastolic blood pressure, mm Hg | ||
| Baseline | 75.6 (8.6) | 74.3 (8.0) |
| Change at EOT | 0.8 (8.3) | 3.9 (8.1) |
| Supine pulse rate, bpm | ||
| Baseline | 70.1 (10.9) | 72.2 (10.6) |
| Change at EOT | 1.8 (9.6) | 7.1 (10.8) |
| Electrocardiogram parameters | n = 157 | n = 163 |
|---|---|---|
| Heart rate, bpm | ||
| Baseline | 66.4 (10.7) | 65.9 (10.1) |
| Change at EOT | 3.9 (9.9) | 11.7 (10.8) |
| PR interval, msec | ||
| Baseline | 159.1 (21.8) | 157.7 (23.1) |
| Change at EOT | −2.7 (13.5) | −6.2 (15.1) |
| QRS interval, msec | ||
| Baseline | 89.2 (9.9) | 90.4 (12.5) |
| Change at EOT | −0.4 (8.3) | −1.3 (7.4) |
| QT interval, msec | ||
| Baseline | 394.1 (29.3) | 393.0 (30.7) |
| Change at EOT | −9.1 (22.7) | −21.7 (23.6) |
| QTcB, msec | ||
| Baseline | 412.3 (21.4) | 410.0 (24.7) |
| Change at EOT | 2.1 (16.0) | 9.3 (19.2) |
| QTcF, msec | ||
| Baseline | 405.7 (18.9) | 403.8 (22.4) |
| Change at EOT | −1.8 (13.1) | −1.6 (16.3) |
| RR interval, msec | ||
| Baseline | 920.8 (152.2) | 925.3 (145.7) |
| Change at EOT | −51.5 (124.9) | −133.5 (120.9) |
n = number of patients with available analysis value at both baseline and a specific time point during double‐blind treatment or double‐blind down‐taper in the double‐blind safety population.
EOT, end of double‐blind treatment; QTcB, QT interval corrected for heart rate using the Bazett formula; QTcF, QT interval corrected for heart rate using the Fridericia formula.
4. DISCUSSION
Levomilnacipran ER, an SNRI, preferentially inhibits norepinephrine reuptake at a rate two‐fold more than serotonin reuptake, whereas other SNRIs (i.e., venlafaxine, duloxetine, and desvenlafaxine) preferentially inhibit serotonin over norepinephrine reuptake (Bruno, Morabito, Spina, & Muscatello, 2016). In patients who respond to antidepressant treatment, subsequent depletion of norepinephrine in the brain is correlated with the return of depressive symptoms and increases the risk of relapse (Moret & Briley, 2011). Thus, levomilnacipran ER has the potential to be an effective antidepressant for maintenance treatment and prevention of relapse in patients who have responded to acute treatment.
In this relapse‐prevention study in adult patients with MDD, patients who responded to 20 weeks of open‐label treatment with levomilnacipran ER were randomized to 26 weeks of double‐blind treatment with levomilnacipran ER or placebo. The primary endpoint, time to relapse, was significantly longer for levomilnacipran ER‐treated patients versus placebo‐treated patients. The risk of relapse in patients in the placebo group was approximately twice that in the levomilnacipran group. The relapse rate was 14.5% in the levomilnacipran ER group compared with 24.5% in the placebo group, yielding an absolute difference of 10 points. At the end of treatment, increases in MADRS total score, CGI‐S score, SDS total, and subscale scores were significantly greater in the placebo group than in the levomilnacipran ER group; mean CGI‐I scores decreased significantly less for placebo‐ versus levomilnacipran ER‐treated patients. Collectively, these changes suggest that patients who switched to placebo at randomization had greater worsening of depressive symptoms and functional impairment compared with those who continued treatment with levomilnacipran ER.
Because a randomized withdrawal study design was used in this relapse‐prevention trial, there is a potential that the higher rate of relapse in the placebo arm may have been due to withdrawal symptoms as opposed to true relapse events. If this were so, an excess of relapse events would be expected in the placebo arm during the first few weeks of treatment when withdrawal symptoms are likely to occur. On the contrary, our study found low and similar rates of relapse in levomilnacipran ER‐ and placebo‐treated patients in the first few weeks of treatment, which suggests that withdrawal events were not an issue for placebo‐treated patients. Further, a post hoc Kaplan–Meier analysis of time to relapse in which relapse events were considered censored during the first 28 days found that the significant advantage for levomilnacipran ER over placebo in improving time to relapse was maintained.
Relapse prevention with antidepressant treatment was evaluated in meta‐analyses of 31 trials (Geddes et al., 2003) and 15 clinical trials using data submitted to the FDA between 1987 and 2012 (Borges et al., 2014). The relapse rates for levomilnacipran ER and placebo in the current study were lower than the average rates reported in the meta‐analyses. These meta‐analyses yielded relapse rates of 18% for antidepressant treatment and relapse rates of 37% to 41% for placebo, with absolute drug–placebo differences in rates ranging from 10 to 31 points. Many of the trials included in the analyses by Geddes et al. (2003) were conducted in secondary care settings in patients at a high risk of relapse. These analyses found no notable impact of total duration of treatment prior to randomization, which may be related to a wide variance in prerandomization treatment duration in the included studies (i.e., as short as 6 weeks to greater than 1 year). Additionally, the studies included in the Geddes et al. (2003) meta‐analysis did not routinely include a fixed‐dose stabilization period following response to flexible‐dose acute treatment, which is a recent requirement by FDA's Division of Psychiatry Products (Borges et al., 2014).
A prior relapse‐prevention study in levomilnacipran ER 40–120 mg was conducted before this positive study; it included a 12‐week flexible‐dose open‐label phase followed by a 24‐week double‐blind phase in which patients who responded to open‐label treatment were randomized to continued levomilnacipran ER or placebo (Shiovitz et al., 2014). Although analysis of relapse rates showed that the time to relapse was slower for levomilnacipran ER than for placebo, the treatment effect was not statistically significant for the primary parameter, time to relapse (HR = 0.68; relapse rates: levomilnacipran ER = 13.9%, placebo = 20.5%). Of note, actual relapse rates were lower than the anticipated rates in the statistical analysis plan (levomilnacipran ER = 20%; placebo = 38%), compromising the projected power to demonstrate a difference between groups. Given the reduced power to detect a significant treatment effect, as well as the low placebo relapse rate, this may more likely be characterized as a failed study than as a negative one. Additionally, the results of the first levomilnacipran ER relapse‐prevention study are inconsistent with relapse‐prevention outcomes reported in the literature for trials of other SNRIs versus placebo (e.g., venlafaxine, 28% vs. 52%; desvenlafaxine, 24% vs. 42%; duloxetine, 21.9% vs. 43.1%; Perahia et al., 2006; Rickels et al., 2010; Simon, Aguiar, Kunz, & Lei, 2004).
Unlike the prior levomilnacipran ER relapse study that included only 12 weeks of open‐label treatment, the present study included an 8‐week run‐in period followed by a 12‐week SP for patients who had achieved clinical stability, for a total of 20 weeks of open‐label treatment. Additional differences in study design included increased minimum baseline depression severity criteria (MADRS total score ≥22 in the first study to ≥26 in this study), less stringent response and relapse criteria that more closely conform to clinical practice, and a 1:1 ratio of patients randomized to placebo and levomilnacipran ER. Results showed that the relapse rates for levomilnacipran ER were similar in the current (14.5%) and previous (13.9%) studies (Shiovitz et al., 2014), while the rates for placebo were higher in the current study (24.5%) than in the previous one (20.5%). Of additional note, the relapse rates observed in the current study were similar to those reported in recently completed studies with desvenlafaxine versus placebo (13.6% vs. 28.3%; Rosenthal, Boyer, Vialet, Hwang, & Tourian, 2013) and vortioxetine versus placebo (13% vs. 26%; Boulenger, Loft, & Florea, 2012).
Long‐term treatment with levomilnacipran ER was generally well tolerated; no new or unexpected TEAEs were reported. The most common TEAEs in the open‐label phase appear to have been transient with lower incidences in the double‐blind phase. The overall safety profile of levomilnacipran ER was consistent with the completed acute (Asnis et al., 2013; Bakish et al., 2014; Gommoll et al., 2014; Montgomery et al., 2013; Sambunaris et al., 2014) and long‐term (Mago et al., 2013) levomilnacipran ER studies; nausea is one of the most commonly reported TEAEs in both short‐term and long‐term trials. The percentage of patients reporting increased heart rate as a TEAE declined between randomization and end of double‐blind treatment.
Given the long open‐label treatment period, including the 12‐week SP, a study of longer duration may have enabled us to observe more relapse events; however, because this patient population had characteristics that made relapse likely (e.g., high baseline MADRS and SDS scores, average of 5 prior depressive episodes, suicide history), enough events were observed to determine that levomilnacipran ER is effective in preventing relapse. Additionally, although the initial phases of the study were open‐label, which could be considered a limitation, they may have provided some descriptive measures of drug effectiveness; the double‐blind phase did not include an active comparator. Further, because the dose of levomilnacipran ER was optimized for each patient, no conclusions can be drawn about specific doses. Other limitations of this study included the lack of ability to generalize to a broader patient population because of the strict eligibility criteria, including the exclusion of patients with comorbid psychiatric disorders or a history of a nonresponse to antidepressant treatment. Also, while an MADRS score ≥18 is a common and appropriate criterion for determining relapse, this high cutoff score may have lacked the sensitivity to fully detect relapse in this study given the low (∼5) mean MADRS baseline score at randomization.
5. CONCLUSIONS
Levomilnacipran ER (40–120 mg/d) was effective in preventing relapse in patients with MDD who responded to acute treatment. Long‐term treatment with levomilnacipran ER was generally safe and well tolerated, and side effects were consistent with those found in shorter studies.
ACKNOWLEDGMENTS
The study was sponsored by the Forest Research Institute, an affiliate of Allergan (Madison, NJ). The sponsor was involved in the study design, collection, analysis and interpretation of data, and the decision to present these results. The authors would like to thank the investigators and subjects who participated in the study. Writing and editorial support was provided by Jill Shults, PhD, of Prescott Medical Communications Group (Chicago, IL), funding for which was provided by Allergan.
FUNDING AND FINANCIAL DISCLOSURES
M.E. Thase has served as a scientific consultant to Alkermes, Astra‐Zeneca, Bristol‐Myers Squibb Company, Eli Lilly & Company, Forest Pharmaceuticals, Inc., Gerson Lehman Group, GlaxoSmithKline, Guidepoint Global, H. Lundbeck A/S, MedAvante, Inc., Merck and Co. Inc., Neronetics, Inc., Novartis, Otsuka, Ortho‐McNeil Pharmaceuticals, Pfizer (formerly Wyeth‐Ayerst Laboratories), Shire US Inc., Sunovion Pharmaceuticals, Inc., and Takeda. Dr. Thase receives grant funding from the Agency for Healthcare Research and Quality, Eli Lilly & Company, National Institute of Mental Health, Otsuka Pharmaceuticals, PharmaNeuroboost (ended March 2013), and Roche (ended June 2013). He has equity holdings in MedAvante, Inc., and receives royalty income from American Psychiatric Foundation, Inc., Guilford Publications, Herald House, Oxford University Press, and W.W. Norton & Company. S. Durgam, C. Chen, R. Migliore, and C. Prakash are full‐time employees of Allergan.
Durgam S, Chen C, Migliore R, Prakash C, Thase ME. Relapse prevention with levomilnacipran ER in adults with major depressive disorder: A multicenter, randomized, double‐blind, placebo‐controlled study. Depress Anxiety. 2019;36:225–234. 10.1002/da.22872
REFERENCES
- APA . (2010). Practice guidelines for the treatment of patients with major depressive disorder (3rd ed.). Arlington, VA: American Psychiatric Association. [Google Scholar]
- APA . (2013). Diagnostic and statistical manual of mental disorders (5th ed.). Washington, DC: American Psychiatric Association. [Google Scholar]
- Asnis, G. M. , Bose, A. , Gommoll, C. P. , Chen, C. , & Greenberg, W. M. (2013). Efficacy and safety of levomilnacipran sustained release 40 mg, 80 mg, or 120 mg in major depressive disorder: A phase 3, randomized, double‐blind, placebo‐controlled study. Journal of Clinical Psychiatry, 74, 242–248. [DOI] [PubMed] [Google Scholar]
- Bakish, D. , Bose, A. , Gommoll, C. , Chen, C. , Nunez, R. , Greenberg, W. M. , … Khan, A. (2014). Levomilnacipran ER 40 mg and 80 mg in patients with major depressive disorder: A phase III, randomized, double‐blind, fixed‐dose, placebo‐controlled study. Journal of Psychiatry & Neuroscience, 39, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, M. , Severus, E. , Kohler, S. , Whybrow, P. C. , Angst, J. , & Moller, H. J. WFSBP Task Force on Treatment Guidelines for Unipolar Depressive Disorders. (2015). World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders. Part 2: Maintenance treatment of major depressive disorder—Update 2015. The World Journal of Biological Psychiatry, 16, 76–95. [DOI] [PubMed] [Google Scholar]
- Berwian, I. M. , Walter, H. , Seifritz, E. , & Huys, Q. J. (2017). Predicting relapse after antidepressant withdrawal—A systematic review. Psychological Medicine, 47, 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges, S. , Chen, Y. F. , Laughren, T. P. , Temple, R. , Patel, H. D. , David, P. A. , … Khin, N. A. (2014). Review of maintenance trials for major depressive disorder: A 25‐year perspective from the US Food and Drug Administration. Journal of Clinical Psychiatry, 75, 205–214. [DOI] [PubMed] [Google Scholar]
- Boulenger, J. P. , Loft, H. , & Florea, I. (2012). A randomized clinical study of Lu AA21004 in the prevention of relapse in patients with major depressive disorder. Journal of Psychopharmacology, 26, 1408–1416. [DOI] [PubMed] [Google Scholar]
- Bruno, A. , Morabito, P. , Spina, E. , & Muscatello, M. R. (2016). The role of levomilnacipran in the management of major depressive disorder: A comprehensive review. Current Neuropharmacology, 14, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes, J. R. , Carney, S. M. , Davies, C. , Furukawa, T. A. , Kupfer, D. J. , Frank, E. , & Goodwin, G. M. (2003). Relapse prevention with antidepressant drug treatment in depressive disorders: A systematic review. Lancet, 361, 653–661. [DOI] [PubMed] [Google Scholar]
- Glue, P. , Donovan, M. R. , Kolluri, S. , & Emir, B. (2010). Meta‐analysis of relapse prevention antidepressant trials in depressive disorders. Australian and New Zealand Journal of Psychiatry, 44, 697–705. [DOI] [PubMed] [Google Scholar]
- Gommoll, C. P. , Greenberg, W. M. , & Chen, C. (2014). A randomized, double‐blind, placebo‐controlled study of flexible doses of levomilnacipran ER (40‐120 mg/day) in patients with major depressive disorder. Journal of Drug Assessment, 3, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy, W. (1976). The clinician global severity and impression scales ECDEU assessment manual for psychopharmacology (pp. 218–222). Rockville, MD: National Institute of Mental Health. DHEW Publication No. 76‐338. [Google Scholar]
- Kaymaz, N. , van Os, J. , Loonen, A. J. , & Nolen, W. A. (2008). Evidence that patients with single versus recurrent depressive episodes are differentially sensitive to treatment discontinuation: A meta‐analysis of placebo‐controlled randomized trials. Journal of Clinical Psychiatry, 69, 1423–1436. [DOI] [PubMed] [Google Scholar]
- Kendler, K. S. , Thornton, L. M. , & Gardner, C. O. (2000). Stressful life events and previous episodes in the etiology of major depression in women: An evaluation of the “kindling” hypothesis. American Journal of Psychiatry, 157, 1243–1251. [DOI] [PubMed] [Google Scholar]
- Kessler, R. C. , & Walters, E. E. (1998). Epidemiology of DSM‐III‐R major depression and minor depression among adolescents and young adults in the National Comorbidity Survey. Depression and Anxiety, 7, 3–14. [DOI] [PubMed] [Google Scholar]
- Kessler, R. C. , Zhao, S. , Blazer, D. G. , & Swartz, M. (1997). Prevalence, correlates, and course of minor depression and major depression in the National Comorbidity Survey. Journal of Affective Disorders, 45, 19–30. [DOI] [PubMed] [Google Scholar]
- Mago, R. , Forero, G. , Greenberg, W. M. , Gommoll, C. , & Chen, C. (2013). Safety and tolerability of levomilnacipran ER in major depressive disorder: Results from an open‐label, 48‐week extension study. Clinical Drug Investigation, 33, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery, S. A. , & Asberg, M. (1979). A new depression scale designed to be sensitive to change. British Journal of Psychiatry, 134, 382–389. [DOI] [PubMed] [Google Scholar]
- Montgomery, S. A. , Mansuy, L. , Ruth, A. , Bose, A. , Li, H. , & Li, D. (2013). Efficacy and safety of levomilnacipran sustained release in moderate to severe major depressive disorder: A randomized, double‐blind, placebo‐controlled, proof‐of‐concept study. Journal of Clinical Psychiatry, 74, 363–369. [DOI] [PubMed] [Google Scholar]
- Moret, C. , & Briley, M. (2011). The importance of norepinephrine in depression. Neuropsychiatric Disease and Treatment, 7, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierenberg, A. A. , Husain, M. M. , Trivedi, M. H. , Fava, M. , Warden, D. , Wisniewski, S. R. , … Rush, A. J. (2010). Residual symptoms after remission of major depressive disorder with citalopram and risk of relapse: A STAR*D report. Psychological Medicine, 40, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perahia, D. G. , Gilaberte, I. , Wang, F. , Wiltse, C. G. , Huckins, S. A. , Clemens, J. W. , … Detke, M. J. (2006). Duloxetine in the prevention of relapse of major depressive disorder: Double‐blind placebo‐controlled study. British Journal of Psychiatry, 188, 346–353. [DOI] [PubMed] [Google Scholar]
- Posner, K. , Brown, G. K. , Stanley, B. , Brent, D. A. , Yershova, K. V. , Oquendo, M. A. , … Mann, J. J. (2011). The Columbia‐Suicide Severity Rating Scale: Initial validity and internal consistency findings from three multisite studies with adolescents and adults. American Journal of Psychiatry, 168, 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickels, K. , Montgomery, S. A. , Tourian, K. A. , Guelfi, J. D. , Pitrosky, B. , Padmanabhan, S. K. , … Brisard, C. (2010). Desvenlafaxine for the prevention of relapse in major depressive disorder: Results of a randomized trial. Journal of Clinical Psychopharmacology, 30, 18–24. [DOI] [PubMed] [Google Scholar]
- Rosenthal, J. Z. , Boyer, P. , Vialet, C. , Hwang, E. , & Tourian, K. A. (2013). Efficacy and safety of desvenlafaxine 50 mg/d for prevention of relapse in major depressive disorder: A randomized controlled trial. Journal of Clinical Psychiatry, 74, 158–166. [DOI] [PubMed] [Google Scholar]
- Rush, A. J. , Trivedi, M. H. , Wisniewski, S. R. , Nierenberg, A. A. , Stewart, J. W. , Warden, D. , … Fava, M. (2006). Acute and longer‐term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. American Journal of Psychiatry, 163, 1905–1917. [DOI] [PubMed] [Google Scholar]
- Sambunaris, A. , Bose, A. , Gommoll, C. P. , Chen, C. , Greenberg, W. M. , & Sheehan, D. V. (2014). A phase III, double‐blind, placebo‐controlled, flexible‐dose study of levomilnacipran extended‐release in patients with major depressive disorder. Journal of Clinical Psychopharmacology, 34, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan, D. V. , Harnett‐Sheehan, K. , & Raj, B. A. (1996). The measurement of disability. International Clinical Psychopharmacology, 11(Suppl 3), 89–95. [DOI] [PubMed] [Google Scholar]
- Shiovitz, T. , Greenberg, W. M. , Chen, C. , Forero, G. , & Gommoll, C. P. (2014). A randomized, double‐blind, placebo‐controlled trial of the efficacy and safety of levomilnacipran ER 40–120 mg/day for prevention of relapse in patients with major depressive disorder. Innovations in Clinical Neuroscience, 11, 10–22. [PMC free article] [PubMed] [Google Scholar]
- Sim, K. , Lau, W. K. , Sim, J. , Sum, M. Y. , & Baldessarini, R. J. (2015). Prevention of relapse and recurrence in adults with major depressive disorder: Systematic review and meta‐analyses of controlled trials. The International Journal of Neuropsychopharmacology, 19, pyv076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, J. S. , Aguiar, L. M. , Kunz, N. R. , & Lei, D. (2004). Extended‐release venlafaxine in relapse prevention for patients with major depressive disorder. Journal of Psychiatric Research, 38, 249–257. [DOI] [PubMed] [Google Scholar]
- Stahl, S. M. , Grady, M. M. , Moret, C. , & Briley, M. (2005). SNRIs: Their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectrums, 10, 732–747. [DOI] [PubMed] [Google Scholar]