

Abstract
Receptor occupancy (RO) is a translational biomarker for assessing drug efficacy and safety. We aimed to apply a physiologically based pharmacokinetic (PBPK) modeling approach to predict the brain dopamine D2 RO time profiles of antipsychotics. Clozapine and risperidone were modeled together with their active metabolites, norclozapine and paliperidone, First, in PK‐Sim a rat PBPK model was developed and optimized using literature plasma PK data. Then, blood‐brain barrier parameters including the expression and efflux transport kinetics of P‐glycoprotein were optimized using literature microdialysis data on brain extracellular fluid (brainECF), which were further adapted when translating the rat PBPK model into the human PBPK model. Based on the simulated drug and metabolite concentrations in brainECF, drug‐D2 receptor binding kinetics (association and dissociation rates) were incorporated in MoBi to predict RO. From an extensive literature search, 32 plasma PK data sets (16 from rat and 16 from human studies) and 23 striatum RO data sets (13 from rat and 10 from human studies) were prepared and compared with the model predictions. The rat PBPK‐RO model adequately predicted the plasma concentrations of the parent drugs and metabolites and the RO levels. The human PBPK‐RO model also captured the plasma PK and RO levels despite the large interindividual and interstudy variability, although it tended to underestimate the plasma concentrations and RO measured at late time points after risperidone dosing. The developed human PBPK‐RO model was successfully applied to predict the plasma PK and RO changes observed after risperidone dose reduction in a clinical trial in schizophrenic patients.
Keywords: antipsychotics, central nervous system (CNS), dopamine receptors, modeling and simulation, PBPK, receptor occupancy
From lead generation to clinical trials, the assessment of in vivo target engagement plays an important role in drug discovery and development.1, 2 It serves as a biomarker that reflects the portion of an endogenous target that is bound to the drug at a specific drug dose, which is determined together by the pharmacokinetics (PK) at the target site and drug‐target binding kinetics.3 More importantly, it allows the translation of preclinical dose‐efficacy findings to clinical settings. Its translational value has been demonstrated for different drug targets, ranging from G protein–coupled receptors to ligand‐gated ion channels and transporters.2 A well‐known example is the dopamine D2 receptor occupancy (RO) of antipsychotics. A therapeutic window of 60% to 80% D2 RO in striatum is preserved across species, from rodents4, 5 to human schizophrenic patients, in which the desired antipsychotic effects can be achieved without excessive extrapyramidal side effects.6, 7 Therefore, knowledge on RO can provide guidance on achieving optimal outcomes in the pharmacotherapy of antipsychotics.
Direct assessment of RO for receptors in the human central nervous system (CNS) is challenging. Despite the advancement of positron emission tomography and single photon emission computed tomography, measuring RO with imaging techniques remain resource and logistically demanding. Another approach is to indirectly estimate RO based on the drug concentration at brain extracellular fluid (brainECF), which is the driving force for RO for membrane receptors such as D2 receptors, along with the drug's affinity to the receptor. Microdialysis is currently the only method that is able to measure PK in brainECF,8 but for obvious ethical reasons it is seldom used in clinical trials or in the routine clinical setting. Drug concentration in the cerebrospinal fluid (CSF) can provide an indication of brainECF concentrations, but it is not always reliable due to the differences between blood‐brain barrier (BBB) and blood‐CSF barrier, as well as intrabrain distribution kinetics.9 Many drugs, including some antipsychotics, are substrates of active transporters such as P‐glycoprotein (P‐gp) that modulate drug transport across the BBB and blood‐CSF barrier.8, 10 Consequently, the PK profile in brainECF can be very different from that in plasma or CSF, and prediction of CNS RO based on plasma PK might not be accurate.
Human CNS PK data are not available in most cases, which makes a direct assessment of the relationship between plasma PK and RO time profiles challenging. Often, rats and mice are used for measuring in vivo CNS RO during preclinical drug development. Since both CNS PK and RO data can be collected from rats, mathematical PK‐RO models can first be constructed and validated in rats, and subsequently be extrapolated from rats to humans, particularly with the physiologically based PK (PBPK) approach. PBPK analysis uses models that combine physiology, population and drug characteristics to mechanistically describe the PK and/or pharmacodynamic behaviors of a drug. PBPK models are different from the empirical PK models, primarily because they use more accurate representations of the various body compartments. By adapting the species‐specific physiological parameters, the animal PBPK model can be translated to a human PBPK model. The aim of the present study is to establish a PBPK‐RO model that can predict D2 RO levels of antipsychotics. Recently, an example of applying PBPK‐RO modeling to predict human CNS D2 RO of antipsychotics is reported.11 Using the commercial PBPK software GastroPlus, the predicted RO level of quetiapine was comparable with data observed in a clinical trial on healthy subjects.12 However, there is much room for improvement. First, active drug transport across the BBB was not incorporated, although in vivo evidence from rat studies did indicate the involvement of active transports for quetiapine.13, 14 In fact, the histamine H1 RO values in the cortex did not correlate with the plasma quetiapine concentrations in humans,15 indicating the limitation of predicting CNS RO based solely on plasma PK. Second, the contribution of active metabolites to the RO level was not considered. Third, translation from the animal PBPK model to the human PBPK model was not reported. Fourth, the maximal response and half maximal effective concentration values for RO prediction had to be estimated by the PBPK model using prior RO data from another clinical trial. Fifth, for validation of the predictive power of this model only a single data set was used.
In the present study, to establish a PBPK‐RO model that could predict D2 RO of antipsychotics, 2 antipsychotics, clozapine and risperidone, were selected because (1) they are efficacious16 and are commonly used in patients with schizophrenic disorders,17 (2) they have different binding kinetics toward the D2 receptor,18 and (3) literature data on the plasma PK and D2 RO are available in both rats and humans. Furthermore, the parent drugs and their active metabolites are all P‐gp substrates.19 Our translational approach was to first develop a rat PBPK‐RO model using multiple rat data sets and to subsequently develop the human PBPK‐RO model based on the rat model with the use of multiple clinical trial data sets.
Methods
Software
Models and simulations were created with the aid of the Computational Systems Biology Software Suite that comprises the software tools PK‐Sim (Version 6.2.2, Bayer Technology Services, Leverskusen, Germany) and MoBi (Version 6.2.2, Bayer Technology Services, Leverskusen, Germany). Moreover, the PK‐Sim Express Gene Expression Database (Version 5.6.3, Bayer Technology Services, Leverskusen, Germany) was used for the quantification of cytochrome P450 (CYP) expression in the human PBPK model. Data extraction from graphic information was performed with WebPlotDigitizer (Version 3.8, http://www.arohatgi.info/WebPlotDigitizer/app/). Data analysis and visualization were performed with R Studio (Integrated Development for R. RStudio, Inc., Boston, Massachusetts).
PK‐Sim is a PBPK software tool that allows whole‐body PBPK modeling for humans and the most common laboratory animals. MoBi is a systems biology software tool that enables the user to explore and modify the model structure, mathematical equations, and parameters underlying the PBPK models in PK‐Sim. Unless otherwise described, the default anatomic and physiological parameters for rats and humans defined in the software were used.
Workflow for Developing the PBPK Models and Model Structure
The workflow for development of the rat and human PBPK and PBPK‐RO models is shown in Figure 1, which will be elaborated in the following section. In brief, PBPK models for the drugs and their metabolites were first developed for rats and then translated to humans. Subsequently, in these models the D2 receptors in the brain were incorporated. These PBPK‐RO models were used to simulate the D2 RO levels after clozapine or risperidone dosing. The physicochemical, absorption, distribution, metabolism, elimination, and D2 receptor–binding parameters used for the PBPK‐RO models are summarized in Table 1.
Figure 1.
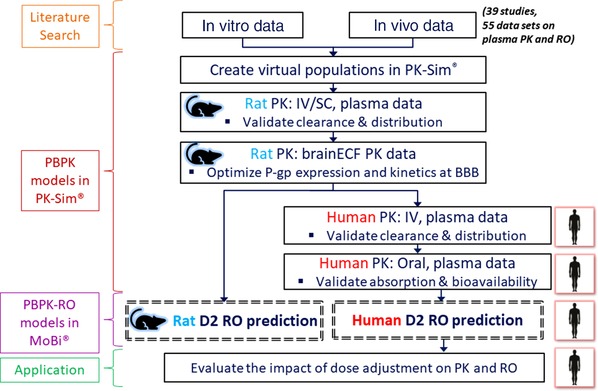
Workflow for developing the PBPK‐RO models. IV, intravenous; RO, receptor occupancy; SC, subcutaneous.
Table 1.
Parameters Used for the Rat and Human PBPK Models
| Parameters | Units | Clozapine | Norclozapine | Risperidone | Paliperidone |
|---|---|---|---|---|---|
| Parameter shared by both the rat and human modelsa | |||||
| Molecular weight | g/mol | 326.8 | 312.8 | 410.5 | 426.5 |
| Log P | 3.44100 | 3.07100 | 2.5101 | 2.4102 | |
| pKa1 | 3.69103 | NA | 3.11104 | 8.2102 | |
| pKa2 | 7.57103 | NA | 8.24104 | NA | |
| Vmax,pgp | nmol/min/nmol | 2.1119 | 27b | 2.8619 | 15b (10c) |
| Km,pgp | μM | 5819 | 0.5b | 12.419 | 12.4b |
| Kd,D2 | nM | 82105 (218.7818)d | 165.9618 | 7.2418 | 6.1718 |
| koff,D2 | 1/s | 0.023105 (0.04218)d | 0.04818 | 0.00518 | 0.00518 |
| Rat model‐specific parameters | |||||
| fu,plasma | 0.06126 | 0.1126 | 0.118102 | 0.285102 | |
| t1/2,absorption | min | 40 (87e)b | 160b | 15 (23e)b | 15b |
| CLHepatic | ml/min/kg | 35 (100%f)b | 30b | 25 (4%f)b | 10b |
| CLRenal | ml/min/kg | 10b | 10b | 15b | 10b |
| Human model‐specific parameters | |||||
| fu,plasma | 0.0540 | 0.09740 | 0.1106 | 0.226106 | |
| Dissolution time/shape | Min | 100/0.92b , g | NA | 10/0.92104 , g | NA |
| CLHepatic | mL/min/kg | NA | 4.6b | NA | 1.04b |
| CLRenal | mL/min/kg | 0 | 0 | 0 | 1.04b |
| CYP1A2,Vmax | pmol/min/pmol | 4.4107 | NA | NA | NA |
| CYP1A2,Km | μM | 18107 | NA | NA | NA |
| CYP2D6,Vmax | pmol/min/pmol | NA | NA | 2.3108 | NA |
| CYP2D6,Km | μM | NA | NA | 1.1108 | NA |
| CYP3A4,Vmax | pmol/min/pmol | 5.4107 | NA | 15108 | NA |
| CYP3A4,Km | μM | 304107 | NA | 61108 | NA |
| CYP3A5,Vmax | pmol/min/pmol | NA | NA | 15108 | NA |
| CYP3A5,Km | μM | NA | NA | 200108 | NA |
CLHepatic, total plasma clearance in liver; CLRenal, total plasma clearance in kidney; CYP, cytochrome P450; fu,plasma, fraction unbound in plasma; Kd,D2, affinity (dissociation constant) at D2 receptor; koff,D2, dissociation rate at D2 receptor; Km,pgp, substrate concentration at which the transport rate is half of Vmax,pgp; NA, not applicable; t1/2,absorption: absorption half‐life after subcutaneous injection; Vmax,pgp: maximum rate of the P‐gp mediated efflux transport at blood‐brain barrier administration.
The rat model and the human model used the same parameter values unless specified.
Values were estimated in PK‐Sim.
Vmax,pgp in the human model (10 nmol/min/nmol) was reduced to two‐third of those in the rat model (15 nmol/min/nmol). Further explanations on the Vmax,pgp and Km,pgp values are provided in Table S8.
The K d,D2R and k off,D2R values, used in both the rat and human models, were provided by Sahlholm et al.18 The only exception was that for clozapine in the rat model the values were provided by Kapur and Seeman105 instead, which had improved the model prediction.
The estimated t1/2,absorption values could adequately capture the subcutaneous PK profiles in all rat studies except for those in the Cremers et al26 and Culot et al.27 For these two studies the absorption rate from the subcutaneous injection site was slower than the others and a higher t1/2,absorption value was needed to adequately capture the observed plasma and brainECF PK profiles. Possibly, a distinct formulation and/or injection site was applied in these studies. Specifically, 20% cyclodextrin was used for the clozapine injection in Culot's study, and the strong complexation between the drug and cyclodextrin could have slowed down the absorption rate.109
The percentage indicates the portion of total plasma clearance in liver (CLHepatic) of the parent drug that generates the metabolite norclozapine and paliperidone.
Weibull function, with dissolution time (50% dissolved) and a shape parameter as input, was used to simulate the dissolution of the oral tablet. The in vitro dissolution profile from Saibi et al104 was used as the input for risperidone tablet. For clozapine, in vitro tablet dissolution rate was not reported and thus it was estimated in PK‐Sim.
Schematic diagrams showing the model structures in relation to the input parameters and the data sets used to estimate these parameters for the parent drugs and the metabolites are provided in Figures S1 (for clozapine) and S2 (for risperidone).
Collection of Plasma PK and RO Datasets From Literature
A digital database search was conducted in PubMed and Google Scholar on literature and patents from 1950 to February 2017. Forward and backward citation searching were performed to identify additional literature. Abstracts were primarily scanned for relevance and, if appropriate, the full content was evaluated further. Rat and human data regarding the 2 antipsychotics risperidone and clozapine and the 2 active metabolites paliperidone (also known as 9‐hydroxyrisperidone) and norclozapine (also known as N‐desmethylclozapine) were summarized, including (1) in vivo plasma PK data, (2) in vivo D2 RO data in striatum (caudate/putamen); (3) in vitro drug dissolution profiles; and (4) in vitro drug‐protein interaction parameters such as plasma protein binding, CYP metabolism kinetics, P‐gp transport kinetics, and drug‐D2 receptor–binding kinetics. PK and RO data from subjects administered with the metabolite (paliperidone or norclozapine) alone were also collected. The main exclusion criteria were (1) formulations other than the conventional intravenous (IV) and subcutaneous dosage forms for rats and conventional IV and oral dosage forms for humans; (2) data from anesthetized rats, since anesthesia can cause dramatic changes in the PK20, 21 and RO time profile22; (3) data from special patient populations (eg, pediatrics and elderly); (4) publications that were not fully accessible; (5) studies that did not report drug formulation, administration route, or dose; and (6) studies providing RO data without specifying the time at which the measurements were performed with respect to the drug administration. The details of each included study are summarized in Tables S1 to S6.
Development of the PBPK Model for Rats
The rat PBPK models were developed using the Rat Population module in PK‐Sim. Experimentally determined physicochemical properties (such as log P and pKa) of the drugs and the metabolites were used as input. If such information was not available, then values estimated from software programs were used.
Regarding the absorption, since subcutaneous administration was not available in PK‐Sim, together with the fact that in most literature the exact site of subcutaneous injection was not reported, drug administration into the muscle plasma compartment with a first‐order absorption rate into plasma was used as a surrogate, and the corresponding absorption half‐life was estimated based on the in vivo data. All IV and subcutaneous formulations were assumed to be fully dissolved before administration.
Regarding the tissue distribution, the 5 methods available in PK‐Sim for the calculation of tissue‐to‐plasma partition coefficient were compared in terms of their accuracy in describing the observed plasma PK after an IV bolus dose. The most accurate methods were utilized for further simulations in both the rat and human PBPK models, which were the Berezhkovskiy method (a modification of the method proposed by Poulin and Theil) for risperidone, paliperidone, and norclozapine, and the Schmitt method (proposed by Schmitt) for clozapine.23, 24
Drug distribution from plasma to the brainECF is determined by passive diffusion in conjunction with active efflux transport from brain to plasma by P‐gp at the BBB. Passive diffusion parameters were calculated by PK‐Sim based on the compound's physicochemical properties, which were adopted without manual modification. There was one identified citation on the P‐gp transport kinetics for clozapine, risperidone, and verapamil (a well‐characterized substrate for P‐gp), which were calculated from an in vitro adenosine triphosphatase assay using Michaelis‐Menten kinetics (substrate concentration at which the transport rate is half of maximum velocity [Km] and maximum velocity [Vmax]).19 The concentration of P‐gp at BBB was estimated to be 4 μM in the rat PBPK model using verapamil brainECF data from the rat microdialysis study of Nagaya et al25 in combination with verapamil's P‐gp transport kinetics. The certainty of this P‐gp concentration value was further confirmed by validation with additional rat brainECF data from Nagaya et al25 (risperidone and paliperidone), Cremers et al26 (clozapine, norclozpaine, risperidone), and Culot et al27 (clozpaine and risperidone). For the human PBPK model, this value was reduced from 4 μM to 1 μM, based on the approximately 4‐fold difference in P‐gp expression at isolated brain microvessels between rat (19.1 fmol/μg protein) and human (3.98‐6.06 fmol/μg protein).28, 29, 30 This value was comparable to the value calculated based on the P‐gp concentration measured in isolated human brain microvessels29, 30 (which is 0.94‐1.43 μM, and the calculation is explained in Table S7). Such approach of translating lower P‐gp level to lower BBB efflux is further supported by the observation that while the P‐gp level at the BBB of cynomolgus monkeys is similar to that in humans,31 the in vivo brainECF‐to‐unbound plasma ratios of risperidone and paliperidone in cynomolgus monkeys were 3.9‐ and 4.8‐fold higher than those in rats.32 For the 2 metabolites norclozapine and paliperidone, since information regarding P‐gp transport kinetics was not available, these parameters were optimized based on the values of the parent compounds19 and rat brainECF data.25, 26 Details on the optimization of the Km and Vmax values of the metabolites are provided in Table S8.
The eliminations the parent drugs and metabolites were modeled by linear hepatic and renal clearance, which were estimated by fitting the plasma PK data observed after a single IV dose to the PBPK model. The hepatic conversion of the parent drugs clozapine and risperidone into the specific metabolites, norclozapine and paliperidone respectively, was also estimated using the observed IV data. Simulations of plasma PK time profile by the rat PBPK model were performed individually for each study (Tables S1, S3, and S4), and the hepatic and renal clearance values were automatically adjusted within PK‐Sim according to rat body weight reported in that study.
Development of the PBPK‐RO Model for Rats
After developing a PBPK model in PK‐Sim that could adequately predict the plasma PK profiles observed in multiple data sets, the model was then extended by adding D2 receptors to the brain and incorporating drug‐receptor binding in MoBi. The RO‐time profile was simulated using the following equation:
in which dN/dt represents the change in the amount of drug‐receptor complex over time, koff is the first‐order dissociation rate constant of the drug‐receptor complex, Kd is the affinity of the drug to the receptor, koff/Kd gives the second‐order association rate constant (kon) of the drug to the receptor (koff and Kd were fixed at values obtained from in vitro binding kinetics studies, as listed in Table 1), AD2R‐unbound is remaining amount of unbound D2 receptor (not bound to the parent drug or the metabolite) in the brainECF that is still available for drug binding, AD2R‐drug is the amount of drug‐receptor complex in the brainECF, Cdrug is the brainECF drug concentration, and Kwater/brain‐ECF is the partition coefficient that corrects for the partition of the drug between water and protein within the brainECF (Kwater/brainECF was calculated by PK‐Sim based on the physicochemical properties of the drug). The density of D2 receptors in both rat and human models was fixed to 25 nM based on the receptor density measured in striatum.33 RO was calculated as the percentage of the receptor that was binding to the drug at a particular time point. After the dosing of the parent drug, RO of the parent drug and the active metabolite were simulated in the same manner simultaneously, and the sum of the drug RO and metabolite RO gave the total RO.
In PK‐Sim, the rat model represents an average adult animal of the species and is not strain specific. Wistar and Sprague Dawley were the only strains that had been used in the rat PK (Table S1) and RO (Tables S3 and S4) studies involved in the present PBPK‐RO analysis. A recent quantitative proteomic analysis found no difference between these 2 strains in the protein levels of P‐gp and also claudin‐5 (a major component of tight junction proteins at the rodent BBB) at the BBB.28 Moreover, in vitro radioligand binding studies identified similar D2 receptor expression in dopaminergic regions including striatum and also similar binding affinity toward radiolabeled D2 antagonists for these 2 strains.34, 35 It thus could be reasonably assumed that the BBB transport and subsequent drug‐receptor binding processes are comparable between these 2 strains.
Development of the PBPK and PBPK‐RO Models for Humans
After the development of the rat PBPK and PBPK‐RO models, the human PBPK and PBPK‐RO models were developed in a similar fashion. A major characteristic of the human model was the application of an in vitro to in vivo extrapolation approach for the biotransformation of the parent drugs risperidone and clozapine to their metabolites paliperidone and norclozapine, respectively. In the rat model, the generation of these metabolites was parameterized by an empirical nonspecific hepatic clearance process, while in the human model the Michaelis‐Menten kinetics based on the in vitro kinetics data (Km and Vmax) of specific CYP isoenzymes was implemented. The PK‐Sim library includes 3 human CYP gene‐expression databases that describe the expression of CYP enzymes in different body tissues: reverse transcription polymerase chain reaction–derived gene expression estimates from literature, whole genome expression arrays from ArrayExpress (European Informatics Institute), and expressed sequence tags from UniGene (National Center for Biotechnology Information).36 Preliminary simulations suggested that the reverse transcription polymerase chain reaction database gave the most accurate human plasma PK profiles of the parent drug and the metabolite. Therefore, this database was used in all the subsequent simulations. Information is limited regarding the elimination pathways for the metabolites. Following administration of a single 1‐mg dose of oral solution 14C‐paliperidone (a dosage form that had approximately 100% oral bioavailability) to healthy volunteers, renal excretion of the unchanged paliperidone accounted for around 50% of the total clearance of paliperidone. Other than renal excretion, 4 metabolic pathways were identified as being involved in the elimination of paliperidone, each of which accounted for up to a maximum of 6.5% of the administered dose.37 In vitro studies suggested a role for CYP2D6 and CYP3A4 in the metabolism of paliperidone.38 As enzyme kinetics data are not available, in the human PBPK model the total plasma clearance of paliperidone was modeled by linear liver clearance and kidney clearance, each accounting for 50% of the total clearance. In contrast, renal elimination of risperidone and clozapine accounts for only <4% of the total clearance of the corresponding drugs,39, 40, 41 and therefore in the human PBPK model we assumed zero renal clearance. For norclozapine, the relative contribution of renal elimination to total clearance has not been reported, and we assumed that norclozapine was eliminated by linear hepatic clearance.
Simulations with the human PBPK model were performed for each individual study (Tables S2, S5, and S6). Human populations were recreated with the aid of the ethnicity‐specific models in PK‐Sim. If the exact population option was not available, the topographically closest ethnic group was chosen instead. Demographic characteristics (age, weight, height, and body mass index) were defined in the population toolbox. For subjects receiving chronic dosing, the simulated RO time profile was obtained on the 15th day of dosing, based on the fact that steady‐state plasma concentrations would be achieved within 6 days for risperidone and 10 days for clozapine after repeated administrations.42 In the simulations, the time of dosing and RO time profile predictions were set according to the chronological scheme stated in the corresponding publications (Tables S5 and S6), and the total daily dose was administered as a once‐daily dosing unless otherwise specified.
Evaluation of Model Performance
The model‐predicted drug concentration in plasma and RO levels were compared with those observed in the literature, and the bias was evaluated by calculating the median absolute percentage error.43 The absolute percentage error (APE) was calculated for every time point as follows:
where ValueIPRED,t and ValueOBS,t represent the individual predicted and observed value for drug concentration or RO at time t, respectively. Goodness‐of‐fit plots were also used for the graphic analysis of model results.
Application of the Human PBPK‐RO Model and Extension to Non‐D2 Receptors
To demonstrate the predictive value of the developed human PBPK‐RO model, the model was used to simulate the human plasma PK and RO for D2 and 5‐hydroxytryptamine 2A (5‐HT2A) according to the settings of the clinical trial by Nyberg et al44 in 1999, which investigated a reduction of the risperidone dose from 6 mg/day to 3 mg/day in schizophrenic patients. The simulations were then compared with the observed data. In addition, the RO time profiles of other non‐D2 receptors including alpha‐1A, alpha‐2 and histamine H1 receptors were also simulated to explore the receptor selectivity toward D2 receptor versus non‐D2 receptors during chronic risperidone treatment.
Results
Characteristics of the PK and RO Data Collected From Literature
Altogether, 55 data sets (29 rat data sets, 26 human data sets) were collected from 39 studies that met the inclusion criteria. Thirty‐one data sets were collected for plasma PK and 23 data sets were collected for striatum D2 RO. The details of each study are summarized in Tables S1 to S6. The settings of these studies varied considerably in terms of, for example, the rat species/human ethnic populations studied, healthy versus diseased subjects, dose amount and single dose vs chronic doses, which contributed to the variability in the observed plasma PK and D2 RO data. While all the rat data were collected from nondiseased male rats receiving a single dose, human data were collected from a more heterogeneous group of subjects (involving healthy volunteers and schizophrenic patients from both sexes) receiving either a single dose or repeated doses. Nevertheless, even among rat studies variations in the experimental design were considerable. For instance, difference in the recovery period from anesthesia and surgery for cannulation (or vs rats that did not receive any surgery) could have led to deviation in plasma PK of the parent drugs and the metabolites.21
Variations in assay sensitivity is a particular issue of concern. As shown in Table S2, in human PK studies the lower limit of quantification (LLOQ) for clozapine and norclozapine in plasma (or serum) ranged from 0.1 ng/mL45 to 15 ng/mL, 46 which represented a 150‐fold difference in assay sensitivity. For risperidone and paliperidone, the LLOQ was in the range of 0.1 to 1 ng/mL (which is around 0.24‐2.4 nM). If a high proportion of samples had concentrations below LLOQ and if samples below LLOQ were simply discarded from the study report, the overall observed concentrations and terminal half‐life could be artificially inflated. This could be particularly relevant for samples collected at the later time points during which the drug concentrations became very low, and might indeed be a possible cause of the deviation between model prediction and observation for the plasma concentrations of risperidone and paliperidone (refer to the subsection Prediction Accuracy of Plasma PK in the Human PBPK Model).
Variations in the conditions of the subjects were also found to be prominent. While all the collected risperidone PK studies were performed on healthy volunteers (Table S2), most of the risperidone RO studies were performed on patients with schizophrenic disorders (Table S5). Both CNS and non‐CNS conditions could alter the drug PK. For instance, an increase in CNS dopaminergic activity and stress are suggested to modify multiple hormone systems that regulate the expression of CYPs in liver.47, 48 Systemic inflammation and infection have been shown to increase the plasma concentrations of risperidone and the metabolite paliperidone by >20% and the plasma concentration of clozapine by up to 300%, probably via modification of plasma binding to alpha‐1 acid glycoprotein (an acute‐phase protein elevated by several‐fold during inflammation) and suppression of CYP activity.49, 50 Therefore, higher variability was expected in the observed RO data than the PK data.
Prediction Accuracy of Plasma PK in the Rat PBPK Model
A total of 16 data sets (9 for clozapine/norclozapine and 7 for risperidone/paliperidone) from 9 studies were processed and compared to the simulated rat plasma PK. The model adequately predicted the plasma PK of clozapine and norclozapine (following norclozapine administration as well as metabolite formed following clozapine administration) (Figure 2A) and risperidone and its metabolite paliperidone (following paliperidone administration as well as metabolite formed following risperidone administration) (Figure 2B). Nevertheless, the model underestimated the relatively high paliperidone plasma concentrations observed in the study of Sun et al51 in which a high IV bolus dose of paliperidone was administered. Noteworthy is that in the rat PBPK model linear, nonsaturable clearance in liver and kidney was applied to all compounds due to a lack of information about the enzyme kinetics in rats. The actual in vivo clearance of paliperidone, however, can be nonlinear (eg, Michaelis‐Menten kinetics) and saturable at high concentrations.
Figure 2.
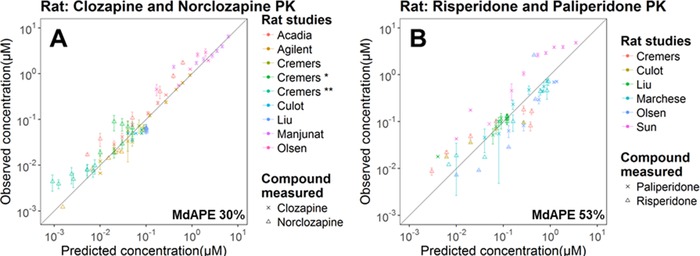
Prediction performance of the rat PBPK model on plasma concentrations of clozapine and norclozapine (A) and risperidone and paliperidone (B) in multiple studies. The gray diagonal line is the unity line. The standard deviation of the observed data, if available, is shown as the error bar. Details of each study are summarized in Table S1.
Prediction Accuracy of D2 RO in the Rat PBPK‐RO Model
A total of 13 data sets (4 for clozapine/norclozapine and 9 for risperidone/paliperidone) from 7 studies were processed and compared to the simulated rat D2 RO levels. The rat model reasonably predicted the total D2 RO (the sum of RO contributed by the parent drug and the metabolite) after administration of clozapine (Figure 3A), risperidone, and paliperidone (Figure 3B). Nevertheless, for both risperidone and clozapine, the model underestimated the relatively high RO observed in Barth.52 Barth's study was the only RO study in which IV administration rather than subcutaneous administration was applied (Tables S3 and S4), and the sudden surge in plasma concentration might have saturated the in vivo drug clearance, thus possibly inflating the observed plasma concentration and RO.
Figure 3.
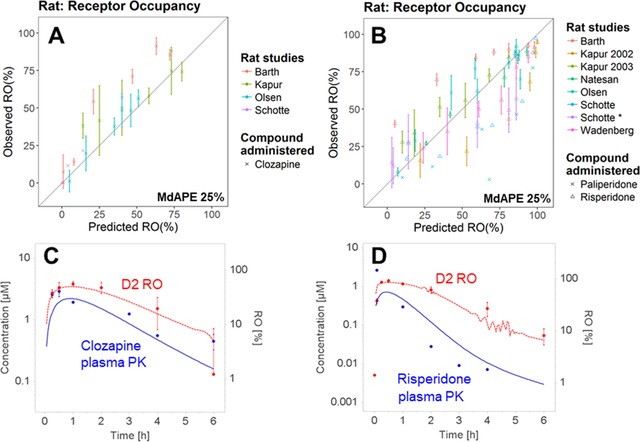
Prediction performance of the rat PBPK model on D2 receptor occupancy (RO) after administration of clozapine (A) and risperidone and paliperidone (B) in multiple studies. Two examples are shown in (C) and (D). Observed (red dots) and predicted (red broken lines) D2 RO time profiles were obtained after subcutaneous administration of clozapine (C) and risperidone (D) according to the study of Olsen et al.53 The observed (blue dots) and predicted (blue solid lines) plasma concentration time profiles are also presented. Details of each study are summarized in Table S3 (risperidone) and Table S4 (clozapine). In (D) the oscillation of the simulated RO values at around Time = 4 hours could be caused by a numerical instability, which is further explained and with suggestion provided in Figure S3.
The performance of the rat PBPK‐RO model was demonstrated with the representative examples in the study by Olsen et al,53 which reported the time profiles of both the plasma PK and D2 RO time profiles after clozapine (Figure 3C) and risperidone (Figure 3D) administration. A MoBi file that contains the PBPK‐RO model is provided in the supplemental materials.
Prediction Accuracy of Plasma PK in the Human PBPK Model
A total of 16 data sets (8 for clozapine/norclozapine and 8 for risperidone/paliperidone) from 16 studies were processed and compared to the simulated human plasma PK.
For clozapine (Figure 4A) and the metabolite norclozapine (Figure 4C), the human PBPK model adequately predicted their plasma PK. Nevertheless, the model overestimated the clozapine concentration and underestimated the norclozapine plasma PK observed in Fadiran et al,46 which contained only 1 subject. A possible explanation is that the subject had a relatively fast metabolism of clozapine to norclozapine.
Figure 4.
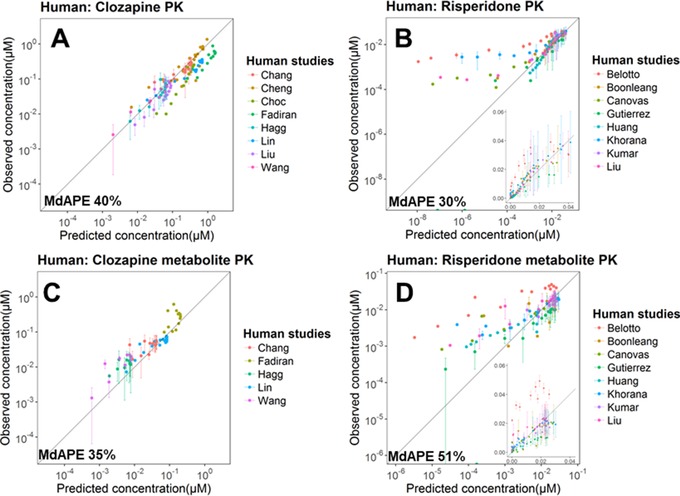
Prediction performance of the human PBPK model on plasma concentrations (in log scale) of clozapine (A), risperidone (B) and the active metabolites norclozapine (C) and paliperidone (D) in multiple studies. In (B) and (D) the data are also shown in normal scale in the inserts. Details of each study are summarized in Table S2.
For risperidone (Figure 4B) and the metabolite paliperidone (Figure 4D), the human PBPK model reasonably predicted the plasma concentration. However, at the very low concentrations (especially when <3 nM), there were underestimations for both risperidone and paliperidone. These plasma concentrations were mostly measured at a late time point (>10 hours after the single‐dose administration). In addition, the model underestimated the concentrations of both the parent risperidone and its metabolite paliperidone in Belotto's study in Brazil.54 A possible explanation is that the absorption of the orally administered risperidone and the paliperidone formed in intestine was enhanced due to a suppressed P‐gp efflux activity at the intestine. Concentration of P‐gp in intestinal epithelial cells is linked to a C3435T polymorphism of the human MDR1 gene and is substantially lower in people with the T/T genotype (higher frequency in whites) than those with the C/C genotype (higher frequency in Africans).55 The Brazil population is mainly composed of whites (52%), followed by browns (ie, mixed‐race, 36%) and blacks (11%).56 Unfortunately, the exact ethnic background of the subjects was not mentioned in Belotto's publication. In addition to P‐gp, CYP2D6 is also highly polymorphic. Poor metabolizers of CYP2D6 (frequency ranges from 5.4% in Europeans, 2.5% in Africans/blacks to 0.4% in East Asians57) were found to have increased plasma risperidone levels,58 which might also contribute to the higher‐than‐expected plasma concentrations observed in some subjects/studies. Nevertheless, the CYP2D6 genotype or phenotype was not reported in most of the clinical studies.
The performance of the model was demonstrated with the representative examples of 2 clinical trials, which reported the plasma PK profiles of both the parent drug and the metabolite after clozapine (Hägg et al41; Figure 5A) and risperidone (Cánovas et al59; Figure 5B) administration. For clozapine/norclozapine, the mean predicted time to maximum plasma concentration (Tmax) and maximum plasma concentration (Cmax) (normalized by clozapine dose) were 2.4/4.7 hours and 5.7/1.0 nM, which were in good agreement with those observed in the 8 collected PK studies: the observed Tmax and Cmax (mean, range) were (2.3, 1.0‐3.0)/(3.2, 2.0‐4.7) hours and (5.6, 2.9‐8.1)/(1.6, 1.0‐2.2) nM, respectively. For risperidone/paliperidone, the predicted PK parameters were also mostly within the range reported from the clinical trials. After oral administration of 2 mg risperidone, the mean predicted Tmax and Cmax were 1.1/1.9 hours and 32.5/17.6 nM, and the observed Tmax and Cmax (mean, range) were (1.1, 0.9‐1.5)/(4.8, 4.0‐5.9) hours and (37.3, 29.8‐40.1)/(29.7, 22.7‐49.2) nM, respectively. Although the prediction slightly underestimated the plasma concentrations of risperidone/paliperidone as discussed above, the predicted half‐life of 8.9/18.1 hours were close to the observed half‐life (mean, range) of (6.4, 4.7‐8.6)/(18.5, 11.5‐29.4) hours. The observed PK parameters are summarized in Table S2.
Figure 5.
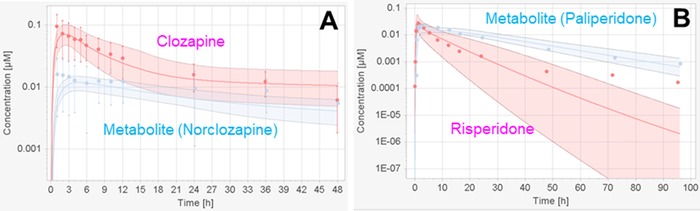
Two examples to show the prediction performance of the human PBPK model on plasma concentrations. Observed (red dots for parent drugs, blue dots for the generated metabolites) and predicted (arithmetic mean as solid lines with 95% confidence interval) plasma concentration time profiles were obtained after oral administration of clozapine (A) and risperidone (B) according to the studies of Hägg et al41 and Cánovas et al,59 respectively.
Prediction Accuracy of D2 RO in the Human PBPK‐RO Model
A total of 10 data sets (5 for clozapine/norclozapine and 5 for risperidone/paliperidone) from 7 studies were processed and compared to the simulated human D2 RO.
For clozapine (Figure 6A), the model reasonably predicted the RO observed in all studies except the one by Catafau et al60 in which the subjects were allowed to smoke. The systemic clearance of clozapine and norclozapine in smokers is around 50% higher than that in nonsmokers,61 which is at least partly due to the induction of CYP1A2 activity by smoking.62 This would have lowered the observed plasma concentration and D2 RO.
Figure 6.
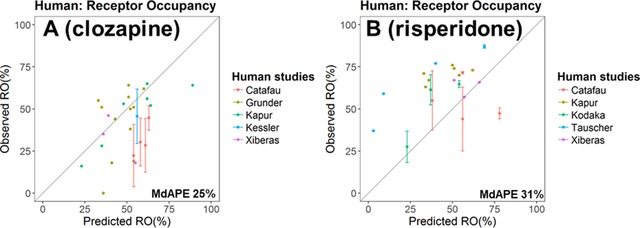
Prediction performance of the human PBPK model on D2 receptor occupancy (RO) after administration of clozapine (A) and risperidone (B) in multiple studies. Details of each study are summarized in Table S5 (risperidone) and Table S6 (clozapine).
For risperidone (Figure 6B), the model could capture the general trend of the RO levels; however, a certain degree of underestimation was apparent. Most of the RO was measured between 12 to 72 hours after the last dose (Table S5). As mentioned above, the human PBPK model might have underestimated the plasma concentrations of risperidone and paliperidone for time points later than 10 hours after the dose, which might explain the underestimation of the RO.
In the human PBPK‐RO model, we assumed that the same P‐gp efflux kinetics at BBB and drug‐receptor–binding kinetics values were shared by all the ethnic populations because we could not identify any reports providing data on the quantitative differences in these 2 kinetic processes among different populations. Certain genetic variations (such as polymorphism in ANKK1) are associated with the D2 receptor availability in brain, and the frequency of these variants is population dependent.63, 64 However, there is no report on how such genetic variations affect the D2 RO level of an antipsychotic drug. Moreover, in most of the PK and RO clinical studies involved in this PBPK work, the genotypes of the subjects were not reported. Because the clinical trial settings differed markedly in terms of, for example, the duration of the chronic drug dosing and disease status at the time of RO evaluation, any interstudy difference in the observed RO values could be attributed not only to the population but also to genetic variations, disease, and RO assay methods.
Application of the Human PBPK‐RO Model and Extension to Non‐D2 Receptors
The clinical trial by Nyberg et al44 reported the changes in human plasma PK and RO time profile after reducing the daily risperidone dose by half from 6 mg/day to 3 mg/day in schizophrenic patients. The simulations generated by the human PBPK‐RO model adequately predicted the plasma PK (Figure 7A) and D2 RO levels (Figure 7B), but again with a slight underestimation in D2 RO, as mentioned in the previous section. Our simulations are in good agreement with the observations from another clinical trial on schizophrenic patients dosed with risperidone also at 6 mg/day, of which the trough plasma concentration and D2 RO at steady state were 26 ng/mL (risperidone+paliperidone) and 64%, respectively.65 The simulation was extended to the serotonin 5‐HT2A receptor by replacing the D2‐kon and D2‐koff values with those measured with 5‐HT2A receptors (Table S9). The simulation captured the high 5‐HT2A RO, which persisted even after the risperidone dose was halved to 3 mg/day (Figure 7C), consistent with the observations from other clinical trials showing that 5‐HT2A RO is >90% when the risperidone dose is ≥3 mg/day.66 The RO of other non‐D2 receptors including alpha‐1A, alpha‐2, and histamine H1 receptors were also simulated (Figure 7C). Such simulation provided insights on the in vivo receptor selectivity of the drug after chronic dosing. For instance, it can be observed that the histamine H1 RO (gray line) is comparable to D2 RO (black line). The considerable binding to H1 receptor has been suggested to be responsible for the sedative effects of antipsychotics.15
Figure 7.
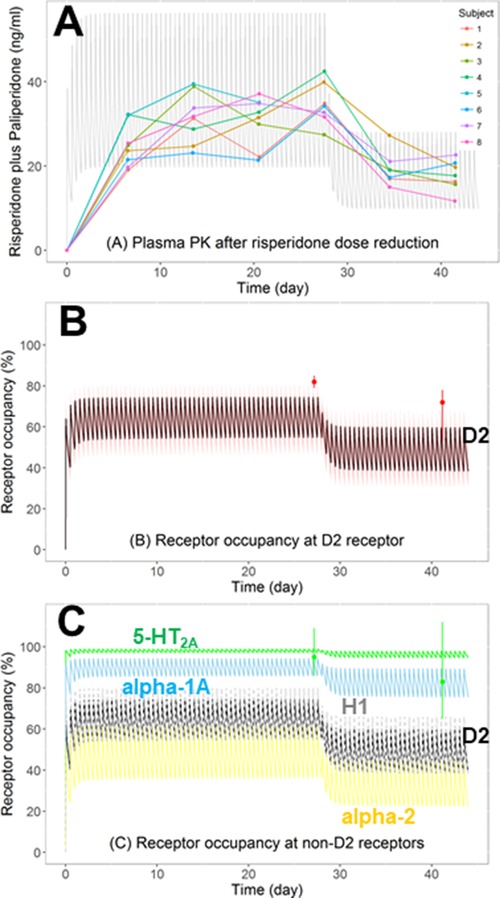
Predicting the impact of dose adjustment on plasma PK and receptor occupancy (RO) according to the clinical trial of Nyberg et al. Eight schizophrenic patients first received risperidone oral dose at 6 mg/day (2 times daily) for 28 days, which was then reduced to 3 mg/day (2 times daily) for 14 days. (A) Observed (dots) and predicted (arithmetic mean as gray lines) plasma concentration‐time profiles of the total active moiety (risperidone plus paliperidone). (B) Observed (dots, mean ± range) and predicted (arithmetic mean as black line with 95% confidence interval) D2 RO. (C) Predicted RO‐time profiles on D2 (black line) and non‐D2 receptors (serotonin 5‐HT2A, green; adrenergic alpha‐1A, blue; alpha‐2, yellow; histamine H1, gray) in the brain. The observed RO of 5‐HT2A (green dots, mean ± range) was also shown.
Discussion
In this work, we developed predictive whole‐body PBPK‐RO models for the 2 commonly used antipsychotics risperidone and clozapine for both rats and humans, which represented an important step toward model‐based prediction of RO of antipsychotics in the management of CNS disorders. Knowledge from rats on the systemic and CNS disposition of these drugs allowed the construction of a rat PBPK‐RO model. By adopting a cross‐species translational approach, with species differences in physiological and pharmacological parameters taken into account, the rat model was then successfully scaled to a human model, which enabled the prediction of the in vivo D2 RO level during antipsychotic treatment.
The PBPK approach allows the incorporation of the active metabolites, which is crucial in the assessment of total RO and pharmacodynamic outcomes of risperidone and clozapine. In our PBPK‐RO models, we explicitly incorporated the active metabolites paliperidone and norclozapine and their characteristic pharmacological properties (such as the systemic elimination rate, P‐gp transport kinetics at BBB, and the binding kinetics to D2). Using the developed PBPK models, we simulated the plasma unbound and brainECF drug concentrations (Figure S4). In rats, after risperidone and clozapine administration, the simulated brainECF concentrations of the parent drugs were at least several‐fold higher than that of the metabolites, which is consistent with the in vivo observations in microdialysis studies.26, 67 In contrast, the simulated human brain unbound concentrations of the parent drug risperidone were several‐fold lower than that of the metabolite paliperidone. Our simulation is in line with the observations from a PK‐RO study in monkeys that after repeated dosing of risperidone only paliperidone but not risperidone was detected in the CSF, while the striatum D2 RO measured by positron emission tomography was as high as 80%.68 In fact, while the simulated total D2 RO in Nyberg's clinical trial was about 60% on average (Figure 7B), about 50% was contributed by the metabolite paliperidone and only 10% was contributed by the parent risperidone (Figure S5). This illustrates the importance of taking into account the contribution of active metabolites in in silico prediction of brain RO levels, which is particularly relevant to the marketed antipsychotics since many of them generate active metabolites whose affinity to D2 (and other neurotransmitter receptors) and antipsychotic potency are comparable to or even higher than that of the parent drugs.69, 70 In fact, paliperidone has been marketed as an antipsychotic drug in its own right, and norclozapine has demonstrated antipsychotic effects in animal studies.71
The PBPK approach facilitates the translation of animal findings to humans. The clear separation of drug‐specific and system‐specific (species‐specific) parameters allows cross‐species extrapolation of the PK model according to species difference in, for example, physiological parameters and transporter expression, which is mechanistically more realistic than allometric scaling. Model validation first by (the more abundant) rat data can enhance confidence in the later human model. Recently, Johnson et al72 reported a 5‐compartment PK‐RO model first in rats and then applied it to predict the human D2 RO of antipsychotics. The model underestimated the observed human D2 RO of haloperidol. The authors suggested that this could be due to in vivo metabolic conversion of haloperidol metabolites back to haloperidol in brain tissue and also active influx of haloperidol across the BBB, which had not been considered in their model. If sufficient in vitro or in vivo information about these pharmacological processes are available, they can be incorporated into the PBPK‐RO model relatively easily.
New antipsychotic drugs are needed to address the unmet clinical needs, especially in the areas like improvement in negative symptoms, cognition, and safety.73 While there are more than 20 US Food and Drug Administration–approved first‐ and second‐generation antipsychotics, the management of schizophrenic disorders with antipsychotics is far from satisfactory. A recent meta‐analysis on 167 double‐blind randomized controlled trials conducted in the past 6 decades in patients with acute exacerbations of schizophrenia indicated that while approximately twice as many patients improved with antipsychotics as with placebo, only a minority experienced a good response.74 The value of the model‐informed approach at different stages of drug discovery and development for improving efficiency and decision making has been demonstrated in recent years.75 When implementing model‐informed drug development for CNS disorders76 such as schizophrenia,77 RO is an indispensable component of the quantitative pharmacology models (population PK‐pharmacodynamic models, PBPK models, and systems pharmacology models). Our PBPK‐RO model can be further extended to incorporate clinical outcomes such as the responses in Positive and Negative Syndrome Scale or Brief Psychiatric Rating Scale7, 78 (therapeutic outcome), Simpson‐Angus Scale or Extrapyramidal Symptom Rating Scale7, 78 and hyperprolactinemia79 (side effect outcomes), in which quantitative relationships between RO level and the pharmacodynamic outcome have been established. A PBPK‐RO‐pharmacodynamic model that connects antipsychotic dose, PK, RO, and clinical outcomes can serve as a dose selection tool to aid the development of new antipsychotics.
The application of the PBPK‐RO model can be extended from D2 to other CNS receptors. All of the marketed antipsychotics have appreciable affinity to non‐D2 CNS receptors,80 and drug candidates that interact with different subtypes of serotonin receptors and acetylcholine receptors in addition to dopamine receptors are now in phase 2 and phase 3 trials.81 It is clinically relevant to assess the receptor selectivity. For instance, clozapine is one of the antipsychotics that is most prone to induce metabolic disturbance such as weight gain and diabetes,16 which could be attributed to its high affinity to both serotonin 5‐HT2C and hypothalamic histamine H1 receptors.82, 83 Also, RO at cortical H1 receptors contributes to the prominent sedative effect of clozapine.15, 16 Clozapine's high affinity to both muscarinic M1 and 5‐HT2A receptors could have contributed to its low propensity to induce motor side effects.84 By incorporating the drug‐receptor binding kinetics values (kon and koff), the PBPK‐RO model would allow simultaneous estimation of the RO of the drug candidate on different receptors, as demonstrated by our simulation of the RO time profiles of serotonin 5‐HT2A, alpha‐1A, alpha‐2, and histamine H1 after risperidone administration (Figure 7C). It is worth noting that our simulation captured the persistently high RO at 5‐HT2A (>90%) when the RO at D2 was decreasing after dose reduction,44, 85 which further demonstrated the utility of the model in the evaluation of receptor selectivity.
To accurately predict the drug concentration at the brain target site, which is the driving force for drug‐receptor binding, the present PBPK model for the brain needs to be further developed. In PK‐Sim, the brain is represented by the brain vessel plasma, brainECF, and brain intracellular compartments and the BBB. In the past decade, several PBPK and semi‐PBPK models that incorporate additional brain distribution compartments and physiological parameters have been reported.8 Particularly, drug in the brainECF distributes into the ventricle CSF and is carried by CSF to spinal cord and then returns to systemic circulation.9 Therefore, compartments related to the ventricular‐CSF system (lateral ventricle, third and fourth ventricles, cisterna magna, and subarachnoid space) and the blood‐CSF barrier (where both passive and active drug transports present) are necessary for accurate prediction of drug concentrations within the brain. After the development of our models, PBPK CNS drug distribution models that incorporate these compartments have been published, and they adequately predict the drug concentration in brainECF, brain tissue, and CSF in both rats and humans.86, 87 Because the focus of this work was on predicting the D2 RO level, which is driven by the drug concentration at the brainECF, creating additional brain compartments relating to CSF in PK‐Sim and validating them with CSF PK data were outside the scope of this work. Nevertheless, future PBPK models should consider incorporating these CSF compartments and the blood‐CSF barrier.
A prominent hurdle of applying a pure bottom‐up PBPK approach, that is, to rely solely on in vitro data to predict the in vivo drug transport across the BBB and blood‐CSF barrier, is the lack of reliable quantitative information on the amount of active transporters present in the CNS. P‐gp expression and localization at BBB, blood‐CSF barrier, and brain parenchymal cells (neurons and glial cells) all affect drug distribution to and within the brain, and they need to be explored with better proteomic studies, taking into account species differences and the effects of diseases.88 Currently, intra‐ and interlaboratory variations in transporter quantification by the proteomics methods remain large.89 Moreover, heterogenous P‐gp expression among different brain regions have been reported in both rats and humans,90, 91 and this should be taken into account to allow region‐specific prediction of drug concentrations in brainECF and subsequent RO levels. Furthermore, maturation of P‐gp expression with postnatal age had been reported for some regions of the rat brain and was shown to be region dependent.92 However, this was not yet considered in the present rat PBPK model because data on striatum region are not available. In spite of that, after risperidone administration, the D2 RO levels in striatum and extrastriatal regions were found to be similar in well‐controlled imaging studies in humans and monkeys.68, 93 Besides P‐gp expression, transport kinetics of P‐gp also need to be explored with better in vitro cellular transport studies and mathematical models.94 Standardization of the aforementioned expression and transport assays is needed to provide reliable input to the PBPK model.
In our model, P‐gp is expressed only at the BBB but not at other body tissues (such as kidney, liver, intestine, and bile duct), which might have impaired the accuracy in PK prediction, especially for risperidone and paliperidone, which are strong P‐gp substrates. For instance, enterohepatic circulation could prolong the drug residence time in systemic circulation, as observed in other P‐gp substrates such as digoxin.95 P‐gp at proximal renal tubules, which facilitates drug excretion into urine, are also suggested to be contributing to the renal clearance of P‐gp substrates.96 Human ABCB1 (the gene encoding P‐gp) polymorphism is associated with altered oral bioavailability and renal clearance of digoxin,97 although clinical data regarding its impact on the PK of risperidone and paliperidone remain inconclusive.98 These factors might, at least in part, explain the underestimation of plasma PK of risperidone and paliperidone of our human PBPK model. If the P‐gp expression values in these tissues become available, they could be easily incorporated into the PBPK model.99
Conclusions
The present study demonstrated that PBPK modeling is a useful approach to predict RO time profiles, particularly for CNS drugs since the PBPK‐RO model could incorporate the active transport processes at the BBB and also the multiple CNS receptors that the drugs interact with. The PBPK approach also allows the translation of knowledge from preclinical studies to the clinical setting and enhance the confidence in the final human PBPK‐RO model. The rat and human PBPK‐RO models developed could be applied to estimate the RO at D2 receptors and non‐D2 receptors of current antipsychotics at different doses. The models can also provide guidance to dose selection, efficacy, and safety assessments and decision making during the preclinical and clinical development of new antipsychotics.
Declaration of Conflicting Interests
The authors declare no conflict of interest. Y.C.W. is a former employee of Leiden University and is currently a full‐time employee of UCB Pharma Ltd. M.C. is a former master student of Leiden University. E.C.M.d.L. is currently a full‐time employee of Leiden University. Y.C.W. and E.C.M.d.L. have been part of the K4DD consortium, which is supported by the Innovative Medicines Initiative Joint Undertaking under grant agreement no 115366. The Innovative Medicines Initiative Joint Undertaking is a project supported by the EU's Seventh Framework Programme (FP7/2007‐2013) and the European Federation of Pharmaceutical Industries and Associations.
Supporting information
FigureS1
FigureS2
FigureS3
FigureS4
FigureS5
Figure S1. Schematic diagram showing the major components of the PBPK models for clozapine, input parameters, and the data sets (obtained from published rat or human data) used to estimate these parameters.
Figure S2. Schematic diagram showing the major components of the PBPK models for risperidone, input parameters, and the data sets (obtained from published rat or human data) used to estimate these parameters.
Figure S3. Improving the simulation accuracy by adapting the simulation solver setting, and the simulation in Figure 3D is used as an example.
Figure S4. Comparison between plasma unbound (Cu,plasma) and brain extracellular fluid unbound (Cu,brain) drug concentrations in rat and human models. Simulations were made based on the experimental settings in Olsen et al53 (subcutaneous administration of risperidone (A) and clozapine (B) to rats) and Cánovas et al58 and Hagg et al41 (oral administration of risperidone (C) and clozapine (D), respectively, to humans). Mean absolute percentage difference between Cu,brain versus Cu,plasma is represented in the tables.
Figure S5. Contribution of the metabolite to the total D2 RO after repeated dosing of risperidone to schizophrenic patients. The total RO and individual RO contributed by the parent drug risperidone and the metabolite paliperidone in the clinical trial by Nyberg et al are simulated.
Table S1. Data From Rat Pharmacokinetic Studies
Table S2. Data From Human Pharmacokinetic Studies
Table S3. Data From Rat D2 Receptor Occupancy Studies for Risperidone
Table S4. Data From Rat D2 Receptor Occupancy Studies for Clozapine
Table S5. Data From Human D2 Receptor Occupancy Studies for Risperidone
Table S6. Data From Human D2 Receptor Occupancy Studies for Clozapine
Table S7. Calculation of P‐Glycoprotein (P‐gp) Concentration Based on the Blood‐Brain Barrier (BBB) Physiology of Humans and Rats
Table S8. Optimization of the Efflux Transport Kinetics Values (Km, Vmax) of P‐Glycoprotein (P‐gp) at Blood‐Brain Barrier (BBB)
Tables S9. Binding Kinetics of Risperidone and Paliperidone to Non‐D2 Receptors (5‐HT2A, Alpha‐1A, Alpha‐2, and Histamine H1)
supporting information
supporting information
Acknowledgments
The authors thank Bayer Technology Services and Dr. Tobias Kanacher for providing academic licenses to use the software packages PK‐Sim and MoBi and for their technical support.
Data Accessibility Statement
The data used in this manuscript are not accessible.
This work was presented in part at the following meetings:
Abstract and poster presentation at the 2017 FIP Pharmaceutical Sciences World Congress (PSWC), Stockholm, Sweden, May 21‐24, 2017.
Abstract and oral presentation at the 2017 Kinetics for Drug Discovery (K4DD) Meeting, “Binding Kinetics: Time Is of the Essence,” Berlin, Germany, October 16‐18, 2017.
References
- 1. Durham TB, Blanco M‐J. Target engagement in lead generation. Bioorg Med Chem Lett. 2015;25(5):998–1008. [DOI] [PubMed] [Google Scholar]
- 2. Grimwood S, Hartig PR. Target site occupancy: emerging generalizations from clinical and preclinical studies. Pharmacol Ther. 2009;122(3):281–301. [DOI] [PubMed] [Google Scholar]
- 3. Danhof M, Alvan G, Dahl SG, Kuhlmann J, Paintaud G. Mechanism‐based pharmacokinetic—pharmacodynamic modeling—a new classification of biomarkers. Pharm Res. 2005;22(9):1432–1437. [DOI] [PubMed] [Google Scholar]
- 4. Wadenberg MLG, Kapur S, Soliman A, Jones C, Vaccarino F. Dopamine D2 receptor occupancy predicts catalepsy and the suppression of conditioned avoidance response behaviour in rats. Psychopharmacology (Berl). 2000;150(4):422–429. [DOI] [PubMed] [Google Scholar]
- 5. Wadenberg M, Soliman A, VanderSpek SC, Kapur S. Dopamine D2 receptor occupancy is a common mechanism underlying animal models of antipsychotics and their clinical effects. Neuropsychopharmacology. 2001;25(5):633–641. [DOI] [PubMed] [Google Scholar]
- 6. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2‐receptor occupancy. Eur Psychiatry. 2007;22(5):267–275. [DOI] [PubMed] [Google Scholar]
- 7. Uchida H, Takeuchi H, Graff‐Guerrero A, Suzuki T, Watanabe K, Mamo DC. Dopamine D2 receptor occupancy and clinical effects: a systematic review and pooled analysis. J Clin Psychopharmacol. 2011;31(4):497–502. [DOI] [PubMed] [Google Scholar]
- 8. Yamamoto Y, Danhof M, de Lange ECM. Microdialysis: the key to physiologically based model prediction of human CNS target site concentrations. AAPS J. 2017;19(4):891–909. [DOI] [PubMed] [Google Scholar]
- 9. de Lange ECM. Utility of CSF in translational neuroscience. J Pharmacokinet Pharmacodyn. 2013;40(3):315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sanchez‐Covarrubias L, Slosky LM, Thompson BJ, Davis TP, Ronaldson PT. Transporters at CNS barrier sites: obstacles or opportunities for drug delivery? Curr Pharm Des. 2014;20(10):1422–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alqahtani S, Kaddoumi A. Development of a physiologically based pharmacokinetic/pharmacodynamic model to predict the impact of genetic polymorphisms on the pharmacokinetics and pharmacodynamics represented by receptor/transporter occupancy of central nervous system drugs. Clin Pharmacokinet. 2016;55(8):957–969. [DOI] [PubMed] [Google Scholar]
- 12. Nord M, Nyberg S, Brogren J, Jucaite A, Halldin C, Farde L. Comparison of D2 dopamine receptor occupancy after oral administration of quetiapine fumarate immediate‐release and extended‐release formulations in healthy subjects. Int J Neuropsychopharmacol. 2011;14(10):1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Loryan I, Melander E, Svensson M, et al. In‐depth neuropharmacokinetic analysis of antipsychotics based on a novel approach to estimate unbound target‐site concentration in CNS regions: link to spatial receptor occupancy. Mol Psychiatry. 2016;21(11):1527–1536. [DOI] [PubMed] [Google Scholar]
- 14. Carreño F, Paese K, Silva CM, Guterres SS, Dalla Costa T. Pharmacokinetic investigation of quetiapine transport across blood–brain barrier mediated by lipid core nanocapsules using brain microdialysis in rats. Mol Pharm. 2016;13(4):1289–1297. [DOI] [PubMed] [Google Scholar]
- 15. Sato H, Ito C, Hiraoka K, et al. Histamine H1 receptor occupancy by the new‐generation antipsychotics olanzapine and quetiapine: a positron emission tomography study in healthy volunteers. Psychopharmacology (Berl). 2015;232(19):3497–3505. [DOI] [PubMed] [Google Scholar]
- 16. Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple‐treatments meta‐analysis. Lancet. 2013;382(9896):951–962. [DOI] [PubMed] [Google Scholar]
- 17. Ó Hálfdánarson, H Zoëga, Aagaard L, et al. International trends in antipsychotic use: a study in 16 countries, 2005–2014. Eur Neuropsychopharmacol. 2017;27(10):1064–1076. [DOI] [PubMed] [Google Scholar]
- 18. Sahlholm K, Zeberg H, Nilsson J, Ögren SO, Fuxe K, Århem P. The fast‐off hypothesis revisited: a functional kinetic study of antipsychotic antagonism of the dopamine D2 receptor. Eur Neuropsychopharmacol. 2016;26(3):467–476. [DOI] [PubMed] [Google Scholar]
- 19. Boulton DW, De Vane CL, Liston HL, Markowitz JS. In vitro P‐glycoprotein affinity for atypical and conventional antipsychotics. Life Sci. 2002;71(2):163–169. [DOI] [PubMed] [Google Scholar]
- 20. Wong YC, Ilkova T, van Wijk RC, Hartman R, de Lange ECM. Development of a population pharmacokinetic model to predict brain distribution and dopamine D2 receptor occupancy of raclopride in non‐anesthetized rat. Eur J Pharm Sci. 2018;111:514–525. [DOI] [PubMed] [Google Scholar]
- 21. Wong YC, Qian S, Zuo Z. Pharmacokinetic comparison between the long‐term anesthetized, short‐term anesthetized and conscious rat models in nasal drug delivery. Pharm Res. 2014;31(8):2107–2123. [DOI] [PubMed] [Google Scholar]
- 22. Müller CP, Pum ME, Amato D, Schüttler J, Huston JP, Silva MADS. The in vivo neurochemistry of the brain during general anesthesia. J Neurochem. 2011;119(3):419–446. [DOI] [PubMed] [Google Scholar]
- 23. Berezhkovskiy LM. Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J Pharm Sci. 2004;93(6):1628–1640. [DOI] [PubMed] [Google Scholar]
- 24. Schmitt W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol Vitr. 2008;22(2):457–467. [DOI] [PubMed] [Google Scholar]
- 25. Nagaya Y, Nozaki Y, Takenaka O, et al. Investigation of utility of cerebrospinal fluid drug concentration as a surrogate for interstitial fluid concentration using microdialysis coupled with cisternal cerebrospinal fluid sampling in wild‐type and Mdr1a(−/−) rats. Drug Metab Pharmacokinet. 2016;31(1):57–66. [DOI] [PubMed] [Google Scholar]
- 26. Cremers TIFH, Flik G, Hofland C, Stratford RE. Microdialysis evaluation of clozapine and N‐desmethylclozapine pharmacokinetics in rat brain. Drug Metab Dispos. 2012;40(10):1909–1916. [DOI] [PubMed] [Google Scholar]
- 27. Culot M, Fabulas‐da Costa A, Sevin E, et al. A simple method for assessing free brain/free plasma ratios using an in vitro model of the blood brain barrier. PLoS One. 2013;8(12):e80634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoshi Y, Uchida Y, Tachikawa M, Inoue T, Ohtsuki S, Terasaki T. Quantitative atlas of blood‐brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J Pharm Sci. 2013;102(9):3343–3355. [DOI] [PubMed] [Google Scholar]
- 29. Shawahna R, Uchida Y, Declèves X, et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm. 2011;8(4):1332–1341. [DOI] [PubMed] [Google Scholar]
- 30. Uchida Y, Ohtsuki S, Katsukura Y, et al. Quantitative targeted absolute proteomics of human blood‐brain barrier transporters and receptors. J Neurochem. 2011;117(2):333–345. [DOI] [PubMed] [Google Scholar]
- 31. Ito K, Uchida Y, Ohtsuki S, et al. Quantitative membrane protein expression at the blood‐brain barrier of adult and younger cynomolgus monkeys. J Pharm Sci. 2011;100(9):3939–3950. [DOI] [PubMed] [Google Scholar]
- 32. Doran AC, Osgood SM, Mancuso JY, Shaffer CL. An evaluation of using rat‐derived single‐dose neuropharmacokinetic parameters to project accurately large animal unbound brain drug concentrations. Drug Metab Dispos. 2012;40(11):2162–2173. [DOI] [PubMed] [Google Scholar]
- 33. Cumming P. Absolute abundances and affinity states of dopamine receptors in mammalian brain: a review. Synapse. 2011;65(9):892–909. [DOI] [PubMed] [Google Scholar]
- 34. Zamudio S, Fregoso T, Miranda A, De La Cruz F, Flores G. Strain differences of dopamine receptor levels and dopamine related behaviors in rats. Brain Res Bull. 2005;65(4):339–347. [DOI] [PubMed] [Google Scholar]
- 35. Luedtke RR, Artymyshyn RP, Monks BR, Molinoff PB. Comparison of the expression, transcription and genomic organization of D2 dopamine receptors in outbred and inbred strains of rat. Brain Res. 1992;584(1‐2):45–54. [DOI] [PubMed] [Google Scholar]
- 36. Meyer M, Schneckener S, Ludewig B, Kuepfer L, Lippert J. Using expression data for quantification of active processes in physiologically based pharmacokinetic modeling. Drug Metab Dispos. 2012;40(5):892–901. [DOI] [PubMed] [Google Scholar]
- 37. Vermeir M, Naessens I, Remmerie B, et al. Absorption, metabolism, and excretion of paliperidone, a new monoaminergic antagonist, in humans. Drug Metab Dispos. 2008;36(4):769–779. [DOI] [PubMed] [Google Scholar]
- 38. Janssen Inc. Invega (paliperidone extended‐release tablets) product monograph. Ontario, Canada: Janssen Inc.; 2018. [Google Scholar]
- 39. Huang ML, Van Peer A, Woestenborghs R, et al. Pharmacokinetics of the novel antipsychotic agent risperidone and the prolactin response in healthy subjects. Clin Pharmacol Ther. 1993;54(3):257–268. [DOI] [PubMed] [Google Scholar]
- 40. Schaber G, Stevens I, Gaertner HJ, Dietz K, Breyer‐Pfaff U. Pharmacokinetics of clozapine and its metabolites in psychiatric patients: plasma protein binding and renal clearance. Br J Clin Pharmacol. 1998;46(5):453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hägg S, Spigset O, Mjörndal T, Granberg K, Persbo‐Lundqvist G, Dahlqvist R. Absence of interaction between erythromycin and a single dose of clozapine. Eur J Clin Pharmacol. 1999;55(3):221–226. [DOI] [PubMed] [Google Scholar]
- 42. Grundmann M, Kacirova I, Urinovska R. Therapeutic drug monitoring of atypical antipsychotic drugs. Acta Pharm. 2014;64(4):387–401. [DOI] [PubMed] [Google Scholar]
- 43. Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm. 1981;9(4):503–512. [DOI] [PubMed] [Google Scholar]
- 44. Nyberg S, Eriksson B, Oxenstierna G, Halldin C, Farde L. Suggested minimal effective dose of risperidone based on PET‐measured D2 and 5‐HT2A receptor occupancy in schizophrenic patients. Am J Psychiatry. 1999;156(6):869–875. [DOI] [PubMed] [Google Scholar]
- 45. Wang C‐Y, Zhang Z‐J, Li W‐B, et al. The differential effects of steady‐state fluvoxamine on the pharmacokinetics of olanzapine and clozapine in healthy volunteers. J Clin Pharmacol. 2004;44(7):785–792. [DOI] [PubMed] [Google Scholar]
- 46. Fadiran EO, Leslie J, Fossler M, Young D. Determination of clozapine and its major metabolites in human serum and rat plasma by liquid chromatography using solid‐phase extraction and ultraviolet detection. J Pharm Biomed Anal. 1995;13(2):185–190. [DOI] [PubMed] [Google Scholar]
- 47. van den Brink WJ, van den Berg DJ, Bonsel FEM, et al. Fingerprints of CNS drug effects: a plasma neuroendocrine reflection of D2 receptor activation using multi‐biomarker pharmacokinetic/pharmacodynamic modelling. Br J Pharmacol. 2018;175(19):3832–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Konstandi M, Johnson EO, Lang MA. Consequences of psychophysiological stress on cytochrome P450‐catalyzed drug metabolism. Neurosci Biobehav Rev. 2014;45:149–167. [DOI] [PubMed] [Google Scholar]
- 49. Clark SR, Warren NS, Kim G, et al. Elevated clozapine levels associated with infection: a systematic review. Schizophr Res. 2018;192:50–56. [DOI] [PubMed] [Google Scholar]
- 50. Hefner G, Shams MEE, Unterecker S, Falter T, Hiemke C. Inflammation and psychotropic drugs: the relationship between C‐reactive protein and antipsychotic drug levels. Psychopharmacology (Berl). 2016;233(9):1695–1705. [DOI] [PubMed] [Google Scholar]
- 51. Sun F, Su Z, Sui C, et al. Studies on the acute toxicity, pharmacokinetics and pharmacodynamics of paliperidone derivatives: comparison to paliperidone and risperidone in mice and rats. Basic Clin Pharmacol Toxicol. 2010;107(2):656–662. [DOI] [PubMed] [Google Scholar]
- 52. Barth VN. Typical and Atypical Antipsychotics: Relationships Between Rat In Vivo Dopamine D(2) Receptor Occupancy Assessed Using LC/MS and Changes in Neurochemistry and Catalepsy [dissertation]. Bloomington: Indiana University; 2006.
- 53. Olsen CK, Brennum LT, Kreilgaard M. Using pharmacokinetic‐pharmacodynamic modelling as a tool for prediction of therapeutic effective plasma levels of antipsychotics. Eur J Pharmacol. 2008;584(2‐3):318–327. [DOI] [PubMed] [Google Scholar]
- 54. Belotto KCR, Raposo NRB, Ferreira AS, Gattaz WF. Relative bioavailability of two oral formulations of risperidone 2 mg: a single‐dose, randomized‐sequence, open‐label, two‐period crossover comparison in healthy Brazilian volunteers. Clin Ther. 2010;32(12):2106–2115. [DOI] [PubMed] [Google Scholar]
- 55. Schaeffeler E, Eichelbaum M, Brinkmann U, et al. Frequency of C3435T polymorphism of MDR1 gene in African people. Lancet. 2001;358(9279):383–384. [DOI] [PubMed] [Google Scholar]
- 56. Loveman M, Muniz JO, Bailey SR. Brazil in black and white? Race categories, the census, and the study of inequality. Ethn Racial Stud. 2012;35(8):1466–1483. [Google Scholar]
- 57. Gaedigk A, Sangkuhl K, Whirl‐Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19(1):69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Puangpetch A, Vanwong N, Nuntamool N, Hongkaew Y, Chamnanphon M, Sukasem C. CYP2D6 polymorphisms and their influence on risperidone treatment. Pharmgenomics Pers Med. 2016;9:131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cánovas M, Delgadillo J, Torres F, et al. Bioequivalence evaluation of two strengths of risperidone tablet formulations in healthy volunteers. Int J Clin Pharmacol. 2009;47(2):124–131. [DOI] [PubMed] [Google Scholar]
- 60. Catafau AM, Penengo MM, Nucci G, et al. Pharmacokinetics and time‐course of D(2) receptor occupancy induced by atypical antipsychotics in stabilized schizophrenic patients. J Psychopharmacol. 2008;22(8):882–894. [DOI] [PubMed] [Google Scholar]
- 61. Li L, Shang D, Li W, et al. Population pharmacokinetics of clozapine and its primary metabolite norclozapine in Chinese patients with schizophrenia. Acta Pharmacol Sin. 2012;33(11):1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dobrinas M, Cornuz J, Oneda B, Kohler Serra M, Puhl M, Eap CB. Impact of smoking, smoking cessation, and genetic polymorphisms on CYP1A2 activity and inducibility. Clin Pharmacol Ther. 2011;90(1):117–125. [DOI] [PubMed] [Google Scholar]
- 63. Gluskin BS, Mickey BJ. Genetic variation and dopamine D2 receptor availability: a systematic review and meta‐analysis of human in vivo molecular imaging studies. Transl Psychiatry. 2016;6(3):e747–e747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Eum S, Lee AM, Bishop JR. Pharmacogenetic tests for antipsychotic medications: clinical implications and considerations. Dialogues Clin Neurosci. 2016;18(3):323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Catafau AM, Bullich S, Nucci G, et al. Contribution of SPECT measurements of D2 and 5‐HT2A occupancy to the clinical development of the antipsychotic SB‐773812. J Nucl Med. 2011;52(4):526–534. [DOI] [PubMed] [Google Scholar]
- 66. Stone JM, Davis JM, Leucht S, Pilowsky LS. Cortical dopamine D2/D3 receptors are a common site of action for antipsychotic drugs–an original patient data meta‐analysis of the SPECT and PET in vivo receptor imaging literature. Schizophr Bull. 2009;35(4):789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shimizu S, den Hoedt SM, Mangas‐Sanjuan V, et al. Target‐site investigation for the plasma prolactin response: mechanism‐based pharmacokinetic‐pharmacodynamic analysis of risperidone and paliperidone in the rat. Drug Metab Dispos. 2017;45(2):152–159. [DOI] [PubMed] [Google Scholar]
- 68. Muly EC, Votaw JR, Ritchie J, Howell LL. Relationship between dose, drug levels, and D2 receptor occupancy for the atypical antipsychotics risperidone and paliperidone. J Pharmacol Exp Ther. 2012;341(1):81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wong YC, Qian S, Zuo Z. Regioselective biotransformation of CNS drugs and its clinical impact on adverse drug reactions. Expert Opin Drug Metab Toxicol. 2012;8(7):833–854. [DOI] [PubMed] [Google Scholar]
- 70. Scott Obach R. Pharmacologically active drug metabolites: Impact on drug discovery and pharmacotherapy. Pharmacol Rev. 2013;65(2):578–640. [DOI] [PubMed] [Google Scholar]
- 71. Lameh J, Burstein ES, Taylor E, Weiner DM, Vanover KE, Bonhaus DW. Pharmacology of N‐desmethylclozapine. Pharmacol Ther. 2007;115(2):223–231. [DOI] [PubMed] [Google Scholar]
- 72. Johnson M, Kozielska M, Pilla Reddy V, et al. Translational modeling in schizophrenia: predicting human dopamine D2 receptor occupancy. Pharm Res. 2016;33(4):1003–1017. [DOI] [PubMed] [Google Scholar]
- 73. Fellner C. New schizophrenia treatments address unmet clinical needs. P T. 2017;42(2):130–134. [PMC free article] [PubMed] [Google Scholar]
- 74. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo‐controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta‐analysis, and meta‐regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927–942. [DOI] [PubMed] [Google Scholar]
- 75. Visser SAG, Aurell M, Jones RDO, et al. Model‐based drug discovery: implementation and impact. Drug Discov Today. 2013;18(15‐16):764–775. [DOI] [PubMed] [Google Scholar]
- 76. de Lange ECM, van den Brink W, Yamamoto Y, de Witte WEA, Wong YC. Novel CNS drug discovery and development approach: model‐based integration to predict neuro‐pharmacokinetics and pharmacodynamics. Expert Opin Drug Discov. 2017;12(12):1207–1218. [DOI] [PubMed] [Google Scholar]
- 77. Nucci G, Gomeni R, Poggesi I. Model‐based approaches to increase efficiency of drug development in schizophrenia: a can't miss opportunity. Expert Opin Drug Discov. 2009;4(8):837–856. [DOI] [PubMed] [Google Scholar]
- 78. de Greef R, Maloney A, Olsson‐Gisleskog P, Schoemaker J, Panagides J. Dopamine D2 occupancy as a biomarker for antipsychotics: quantifying the relationship with efficacy and extrapyramidal symptoms. AAPS J. 2011;13(1):121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Iwata Y, Nakajima S, Caravaggio F, et al. Threshold of dopamine D2/3 receptor occupancy for hyperprolactinemia in older patients with schizophrenia. J Clin Psychiatry. 2016;77(12):e1557–e1563. [DOI] [PubMed] [Google Scholar]
- 80. Li P, r GL Snyde, Vanover KE. Dopamine targeting drugs for the treatment of schizophrenia: past, present and future. Curr Top Med Chem. 2016;16(29):3385–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Konstantinos D, Rohit S. Pharmacological treatment of schizophrenia—a review of progress. Prog Neurol Psychiatry. 2016;20(3):28–35. [Google Scholar]
- 82. Montastruc F, Palmaro A, Bagheri H, Schmitt L, Montastruc J‐L, Lapeyre‐Mestre M. Role of serotonin 5‐HT2C and histamine H1 receptors in antipsychotic‐induced diabetes: a pharmacoepidemiological‐pharmacodynamic study in VigiBase. Eur Neuropsychopharmacol. 2015;25(10):1556–1565. [DOI] [PubMed] [Google Scholar]
- 83. He M, Deng C, HuangX‐F. The role of hypothalamic H1 receptor antagonism in antipsychotic‐induced weight gain. CNS Drugs. 2013;27(6):423–434. [DOI] [PubMed] [Google Scholar]
- 84. Nguyen TTH, Pariente A, Montastruc J‐L, et al. An original pharmacoepidemiological‐pharmacodynamic method: application to antipsychotic‐induced movement disorders. Br J Clin Pharmacol. 2017;83(3):612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tauscher J, Jones C, Remington G, Zipursky RB, Kapur S. Significant dissociation of brain and plasma kinetics with antipsychotics. Mol Psychiatry. 2002;7(3):317–321. [DOI] [PubMed] [Google Scholar]
- 86. Yamamoto Y, Välitalo PA, Huntjens DR, et al. Predicting drug concentration‐time profiles in multiple CNS compartments using a comprehensive physiologically‐based pharmacokinetic model. CPT Pharmacometrics Syst Pharmacol. 2017;6(11):765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yamamoto Y, Välitalo PA, Wong YC, et al. Prediction of human CNS pharmacokinetics using a physiologically‐based pharmacokinetic modeling approach. Eur J Pharm Sci. 2018;112:168–179. [DOI] [PubMed] [Google Scholar]
- 88. Morris ME, Rodriguez‐Cruz V, Felmlee MA. SLC and ABC transporters: expression, localization, and species differences at the blood‐brain and the blood‐cerebrospinal fluid barriers. AAPS J. 2017;19(5):1317–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wegler C, Gaugaz FZ, Andersson TB, et al. Variability in mass spectrometry‐based quantification of clinically relevant drug transporters and drug metabolizing enzymes. Mol Pharm. 2017;14(9):3142–3151. [DOI] [PubMed] [Google Scholar]
- 90. Seegers U, Potschka H, Löscher W. Expression of the multidrug transporter P‐glycoprotein in brain capillary endothelial cells and brain parenchyma of amygdala‐kindled rats. Epilepsia. 2002;43(7):675–684. [DOI] [PubMed] [Google Scholar]
- 91. Cordon‐Cardo C, O'Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P‐glycoprotein) in human normal and tumor tissues. J Histochem Cytochem. 1990;38(9):1277–1287. [DOI] [PubMed] [Google Scholar]
- 92. Matsuoka Y, Okazaki M, Kitamura Y, Taniguchi T. Developmental expression of P‐glycoprotein (multidrug resistance gene product) in the rat brain. J Neurobiol. 1999;39(3):383–392. [DOI] [PubMed] [Google Scholar]
- 93. Ito H, Arakawa R, Takahashi H, et al. No regional difference in dopamine D2 receptor occupancy by the second‐generation antipsychotic drug risperidone in humans: a positron emission tomography study. Int J Neuropsychopharmacol. 2009;12(05):667. [DOI] [PubMed] [Google Scholar]
- 94. Nagar S, Tucker J, Weiskircher EA, Bhoopathy S, Hidalgo IJ, Korzekwa K. Compartmental models for apical efflux by P‐glycoprotein—part 1: evaluation of model complexity. Pharm Res. 2014;31(2):347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Malik MY, Jaiswal S, Sharma A, Shukla M, Lal J. Role of enterohepatic recirculation in drug disposition: cooperation and complications. Drug Metab Rev. 2016;48(2):281–327. [DOI] [PubMed] [Google Scholar]
- 96. Ivanyuk A, Livio F, Biollaz J, Buclin T. Renal drug transporters and drug interactions. Clin Pharmacokinet. 2017;56(8):825–892. [DOI] [PubMed] [Google Scholar]
- 97. Kurata Y, Ieiri I, Kimura M, et al. Role of human MDR1 gene polymorphism in bioavailability and interaction of digoxin, a substrate of P‐glycoprotein. Clin Pharmacol Ther. 2002;72(2):209–219. [DOI] [PubMed] [Google Scholar]
- 98. Saiz‐Rodríguez M, Belmonte C, Román M, et al. Effect of ABCB1 C3435T polymorphism on pharmacokinetics of antipsychotics and antidepressants. Basic Clin Pharmacol Toxicol. 2018;123(4):474–485. [DOI] [PubMed] [Google Scholar]
- 99. Edginton AN, Zimmerman EI, Vasilyeva A, Baker SD, Panetta JC. Sorafenib metabolism, transport, and enterohepatic recycling: physiologically based modeling and simulation in mice. Cancer Chemother Pharmacol. 2016;77(5):1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. AAEl Ela, S Härtter, Schmitt U, Hiemke C, Spahn‐Langguth H, Langguth P. Identification of P‐glycoprotein substrates and inhibitors among psychoactive compounds ‐ implications for pharmacokinetics of selected substrates. J Pharm Pharmacol. 2004;56(8):967–975. [DOI] [PubMed] [Google Scholar]
- 101. Badhan KR, Chenel M, Penny IJ. Development of a physiologically‐based pharmacokinetic model of the rat central nervous system. Pharmaceutics. 2014;6(1):97–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Loryan I, Sinha V, Mackie C, et al. Mechanistic understanding of brain drug disposition to optimize the selection of potential neurotherapeutics in drug discovery. Pharm Res. 2014;31(8):2203–2219. [DOI] [PubMed] [Google Scholar]
- 103. Novartis Pharmaceuticals Canada Inc . CLOZARIL® Product Monograph. 2015. http://www.ask.novartispharma.ca. Updated August 26, 2015. Accessed January 1, 2017.
- 104. Saibi Y, Sato H, Tachiki H. Developing in vitro–in vivo correlation of risperidone immediate release tablet. AAPS PharmSciTech. 2012;13(3):890–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kapur S, Seeman P. Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action. J Psychiatry Neurosci. 2000;25(2):161–166. [PMC free article] [PubMed] [Google Scholar]
- 106. Mannens G, Meuldermans W, Snoeck E, Heykants J. Plasma protein binding of risperidone and its distribution in blood. Psychopharmacology (Berl). 1994;114(4):566–572. [DOI] [PubMed] [Google Scholar]
- 107. Eiermann B, Engel G, Johansson I, Zanger UM, Bertilsson L. The involvement of CYP1A2 and CYP3A4 in the metabolism of clozapine. Br J Clin Pharmacol. 1997;44(5):439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Okubo M, Morita S, Murayama N, Akimoto Y, Goto A, Yamazaki H. Individual differences in in vitro and in vivo metabolic clearances of antipsychotic risperidone from Japanese subjects genotyped for cytochrome P450 2D6 and 3A5. Hum Psychopharmacol Clin Exp. 2016;31(2):93–102. [DOI] [PubMed] [Google Scholar]
- 109. Tokihiro K, Arima H, Tajiri S, Irie T, Hirayama F, Uekama K. Improvement of subcutaneous bioavailability of insulin by sulphobutyl ether beta‐cyclodextrin in rats. J Pharm Pharmacol. 2000;52(8):911–917. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FigureS1
FigureS2
FigureS3
FigureS4
FigureS5
Figure S1. Schematic diagram showing the major components of the PBPK models for clozapine, input parameters, and the data sets (obtained from published rat or human data) used to estimate these parameters.
Figure S2. Schematic diagram showing the major components of the PBPK models for risperidone, input parameters, and the data sets (obtained from published rat or human data) used to estimate these parameters.
Figure S3. Improving the simulation accuracy by adapting the simulation solver setting, and the simulation in Figure 3D is used as an example.
Figure S4. Comparison between plasma unbound (Cu,plasma) and brain extracellular fluid unbound (Cu,brain) drug concentrations in rat and human models. Simulations were made based on the experimental settings in Olsen et al53 (subcutaneous administration of risperidone (A) and clozapine (B) to rats) and Cánovas et al58 and Hagg et al41 (oral administration of risperidone (C) and clozapine (D), respectively, to humans). Mean absolute percentage difference between Cu,brain versus Cu,plasma is represented in the tables.
Figure S5. Contribution of the metabolite to the total D2 RO after repeated dosing of risperidone to schizophrenic patients. The total RO and individual RO contributed by the parent drug risperidone and the metabolite paliperidone in the clinical trial by Nyberg et al are simulated.
Table S1. Data From Rat Pharmacokinetic Studies
Table S2. Data From Human Pharmacokinetic Studies
Table S3. Data From Rat D2 Receptor Occupancy Studies for Risperidone
Table S4. Data From Rat D2 Receptor Occupancy Studies for Clozapine
Table S5. Data From Human D2 Receptor Occupancy Studies for Risperidone
Table S6. Data From Human D2 Receptor Occupancy Studies for Clozapine
Table S7. Calculation of P‐Glycoprotein (P‐gp) Concentration Based on the Blood‐Brain Barrier (BBB) Physiology of Humans and Rats
Table S8. Optimization of the Efflux Transport Kinetics Values (Km, Vmax) of P‐Glycoprotein (P‐gp) at Blood‐Brain Barrier (BBB)
Tables S9. Binding Kinetics of Risperidone and Paliperidone to Non‐D2 Receptors (5‐HT2A, Alpha‐1A, Alpha‐2, and Histamine H1)
supporting information
supporting information