

Abstract
Tolvaptan is the first approved drug treatment to slow kidney function decline in adults at risk of rapidly progressing autosomal dominant polycystic kidney disease (ADPKD). The objective is to develop (1091 subjects, 7335 observations) and validate (678 subjects, 3012 observations) a population pharmacokinetic model to describe tolvaptan pharmacokinetics in ADPKD subjects. The final model was evaluated with a bootstrapping method. The final model was internally and externally evaluated using visual predictive checks (VPC). Pharmacokinetics was best described by a 1‐compartmental model with 0‐order absorption, nonlinear relative bioavailability (F1), and first‐order elimination. Accounting for changes in F1 significantly improved the model: as the dose increased from 15 mg to 120 mg, F1 decreased by 36%. Population estimates for clearance/F (CL/F), volume of distribution/F (Vd/F), duration of absorption (D1), the highest dose at which F1 is lowest, and the amount of dose at which F1 is 50% were 12.6 L·h‐1, 110 L, 0.58 hour, 182 mg, and 166 mg, respectively. The interindividual variability was 64% in CL/F, 70% in Vd/F, and 238% in D1. Residual variability was described by a combined‐error model. The VPC (500 data sets simulated) showed that 76% to 92% of the observed data fell within the 90% prediction intervals. The model stability assessed by a 1000‐run bootstrap analysis showed that the mean parameter estimates of data were within 10% of those obtained with the final model. The developed model is robust and stable. Internal and external validation confirmed the model ability to describe the data optimally.
Keywords: ADPKD, tolvaptan, population pharmacokinetics, model development, validation, prediction
Otsuka Pharmaceutical Company discovered tolvaptan, an orally effective, nonpeptide arginine vasopressin (AVP) V2 receptor antagonist. Tolvaptan has been approved as an oral aquaretic agent for the treatment of fluid volume‐overload conditions1, 2, 3 and hyponatremia.4, 5, 6 Tolvaptan is also approved as the first drug treatment to slow kidney function decline in adults at risk of rapidly progressing autosomal dominant polycystic kidney disease (ADPKD).7, 8 The mechanism of these effects is proposed to involve inhibition of the AVP V2 receptor and the subsequent decrease in adenosine cyclic 3’,5’‐monophosphate (cAMP) concentrations in the kidney. Elevated cAMP in the kidney is thought to promote cyst growth by secretion of fluid into the cyst lumen.9 Subjects with ADPKD have elevated plasma AVP concentrations or exaggerated response of AVP to sodium challenge, and their cyst fluid cAMP levels are elevated by AVP, suggesting that similar mechanisms may be responsible for disease progression across species and causative mutations. Several trials have shown that human ADPKD subjects respond to tolvaptan with potent vasopressin inhibition as indicated by increased urine output and decreased urine osmolality.10 In a phase 3, multicenter, double‐blind, placebo‐controlled, 3‐year trial, tolvaptan (n = 961) exhibited a significantly lower rate of growth in total kidney volume, a lower rate of worsening kidney pain, and slower rate of decline in kidney function relative to the placebo group (n = 483).11, 12 The objectives of the population (Pop) pharmacokinetic (PK) analysis of tolvaptan in subjects with ADPKD were to develop a Pop PK model to describe tolvaptan PK in ADPKD subjects following oral administration of tolvaptan and to perform external validation to determine the predictive performance of the model.
Methods
Study Design and Patient Population for Model Development
The data used for Pop PK model development are from 5 phase 2 trials10: ClinicalTrials.gov Identifier NCT00413777, NCT01210560 (data on file) and a phase 3b, multicenter, international, open‐label, extension trial, ClinicalTrials.gov Identifier NCT01214421, using oral tolvaptan tablets in subjects with ADPKD. The protocols for these trials were approved by local institutional review boards/independent ethics committees, and the studies were conducted in accordance with the International Conference of Harmonisation Good Clinical Practice Guidelines. In 5 phase 2 trials, the dose was administered either as a single dose of 15 mg, 30 mg, 60 mg, or 120 mg or daily split‐dosage regimens of 15/15 mg or 30 mg QD or 30/15 mg or 30/30 mg or 90/30 mg. In the open‐label trial, tolvaptan was administered as daily split‐dose regimens of 45/15 mg, 60/30 mg, or 90/30 mg. Tolvaptan split dosage regimens were administered as am/pm doses: the morning dose was administered at approximately 8 am and the second dose 8 to 9 hours later. Food intake was not recorded in the phase 3b trial. Data included for the model development are from 1091 subjects with 7335 observations split into dense and sparse data at a ratio of 1:5. Tolvaptan plasma concentrations without dosing information or time or below the limit of quantification (1.2%) were excluded from analysis. Missing continuous covariate values were either imputed to the closest nonmissing value within the same subject or to the median value corresponding to the same category of sex, country, and race in absence of covariate information for the given individual.
The number of patients taking 1 or more concomitant medications is not uncommon in phase 3 studies. The concomitant medications taken were grouped on the basis of CYP3A inhibitor or inducer effect. Tolvaptan is a sensitive CYP3A4 substrate.13 The usage of concomitant medication is not continuous: it is a covariate that changes with each record within an individual, so it is a time‐dependent covariate (see equation 2 below). There were 131 subjects with at least 1 instance of strong, moderate, or weak CYP3A inhibitor (Inh) coadministration and 22 subjects with at least 1 instance of strong, moderate, or weak CYP3A4 inducer coadministration.
Analytical Methods for PK Analysis
Plasma tolvaptan concentrations were assayed using high‐performance liquid chromatography with tandem mass spectrometry methods described previously.14 This method used for the determination of tolvaptan in plasma had adequate linearity, specificity, sensitivity, and accuracy. The assay analysis range was 5.00–1000 ng/mL. Samples with concentrations greater than the upper limit of quantitation were handled by dilution procedure. Plasma concentrations below the lower limit of quantitation were excluded from Pop PK analysis.
Pop PK Modeling
The Pop PK model was developed using software package NONMEM, version 7.3 (ICON, Hanover, Maryland). R version 3.4.0 (R Foundation for Statistical Computing, Vienna, Austria) was used for data preparation, graphical analysis, model diagnostics, and statistical summaries. Exploratory data analyses and data visualization techniques were used to understand the data and search for potential outliers. Stochastic approximation expectation maximization (SAEM) estimation method with important sampling was used to identify a stable base model and was used in all stages of the model development process. The SAEM setting produces first‐order approximation standard errors, that is, MATRIX = S type, but not a proper objective function for hypothesis testing. Hence, the SAEM estimation process was followed by an evaluation step using important sampling to obtain an objective function value (OFV) that can be used for model comparison.15 One‐ and 2‐compartmental models with first‐ or 0‐order absorption and first‐order elimination were evaluated to determine the base model. The interindividual variability and residual variability were included within the error model as random effects. Four different residual error models were tested, including additive, proportional, exponential, and combined additive and proportional error. The base model was first developed using dense data. Sparse data were then added to the data set.
Covariate Model Building
The covariate analysis consisted of a stepwise forward addition (α‐risk of 0.05) and a stepwise backward elimination (α‐risk of 0.01) approach. A significance level of 0.05 corresponds to a 3.84 decrease in OFV for 1 degree of freedom for forward addition. The forward addition was continued until no covariate could be declared statistically significant. The model obtained is the full model. As discussed by Joerger,16 forward addition of covariates was guided based on pathophysiological rationale rather than empirical importance. In the backward elimination step, the parameter‐covariate relationships within the full model were eliminated one by one, and the resulting OFV were compared. The parameter‐covariate relationship with the least increase in OFV was removed from the model. This procedure was performed until an increase of OFV higher than 6.635 for 1 degree of freedom (ie, chi‐squared distribution with α‐risk 0.01) was obtained for all covariates included in the reduced model issued from the full model. The model obtained at this step was considered as a final model. The model selection was based on the goodness‐of‐fit plots, precision of parameter estimation, OFV, and individual fits.
Covariates were modeled according to equations 1 to 3. For continuous covariates,
| (1) |
where MU_1 is a function of θ, and Covi represents the covariate of the i‐th individual.
Time‐dependent covariates such as estimated glomerular filtration rate (eGFR; calculated by serum creatinine concentration and the chronic kidney disease epidemiology collaboration formula)17 or covariates changing with each record within an individual such as Inh cannot be part of the MU equation, so they were included outside the MU_equation as shown below.
| (2) |
where i is the value of the PK parameter for the i‐th individual, θtypical is the typical value of the PK parameter for an individual having the covariate equal to the median covariate value, Covi is the value of the covariate for the individual, and θcov is the value of the effect of the covariate on the PK parameter.
The effect of categorical covariates (which take the value 0 or 1,) on typical values of PK parameters was introduced into the model using a linear model as follows:
| (3) |
where Icov is the categorical covariate indicator variable (taking the value 0 or 1), and θtypical is the typical value of the PK parameter for an individual with Icov = 0, and θcov is the value of the effect of the covariate on the PK parameter.
Model Diagnostics and Evaluations
The accuracy, stability, and robustness of the final model were assessed using a visual predictive check (VPC) and nonparametric bootstrapping using features available in Perl speaks NONMEM.18 In VPC, 500 data sets were simulated using the final model estimates. The 5th, 50th, and 95th percentiles of the simulated concentrations were constructed and compared against the observed concentrations. Bootstrapping was conducted with 1000 data sets generated by randomly resampling with replacement from the original data set. The bootstrap estimates with 95% confidence intervals were compared with the estimates from the original data set.
Validation Data
Plasma concentrations from a second phase 3b, multicenter, randomized‐withdrawal, placebo‐controlled, double‐blind, parallel‐group trial11 (ClinicalTrials.gov Identifier: NCT02160145) in ADPKD patients were used for external validation of the final Pop PK model. Subjects in the tolvaptan treatment arm who had at least 1 quantifiable tolvaptan concentration following tolvaptan administration were included in the analysis. The data set (with sparse PK sampling) consisted of 678 subjects and 3012 observations. Tolvaptan was administered as daily split‐dose regimens of 60/30 or 90/30 mg consisting of a 60‐ or 90‐mg dose on waking up and a 30‐mg dose approximately 8 to 9 hours later, with down‐titrations to 45/15 and 30/15 mg as needed for tolerability. Food intake was not recorded in this phase 3b trial.
Assessment of Predictive Performance
The final Pop PK model discussed was used to generate normalized prediction distribution errors (NPDE) for the validation data set.19 A cumulative distribution was assembled for each observation with 500 simulated concentrations. The null hypothesis of NPDE is that it obeys a standard normal distribution N(0,1). The NPDE results were summarized graphically and statistically using the R package. NPDE values were generated by setting MAXEVAL = 0 and specifying NPDE as an output variable for the Pop PK model and applying it to the validation data set. The Pop PK model was used to generate 500 simulated data sets for the population in the validation data. The simulated data set was used to plot VPC. The 5th, 50th, and 95th percentiles of the simulated concentrations were constructed and compared against the validation data set observed concentrations. Model performance was assessed through VPC and NPDE plots.
Results
Population Pharmacokinetic Modeling
Base Model
A 1‐compartment model with 0‐order absorption, first‐order elimination, and a combined additive and proportional error structure effectively described the dense PK data (75 subjects, 1968 observations). This model was then applied to both dense and remaining sparse data. Based on noncompartmental analysis it was observed (data on file) that an increase in dose results in disproportional increase of tolvaptan PK parameters such as clearance/F (CL/F) and volume of distribution (Vd/F). This change was considered due to differences in relative bioavailability (F1) of different doses. Hence, before full covariate analysis, the effect of dose was investigated on tolvaptan F1. Both a stepwise effect and a nonlinear effect approach were tested to investigate the effect of dose on tolvaptan F1. This investigation led to the development of a nonlinear F1 model (equation 4), where Dosemax is the dose level at which F1 is minimum, and Dose50 is the dose at which F1 is 50%. The effect of dose on tolvaptan F1 was explained with respect to a 15‐mg dose. The F1 parameter is fixed to 1 for the lowest dose, and other doses are relatively scaled. As shown in equation 4, F1 can be calculated for any dose using Dosemax and Dose50. Addition of nonlinear F1 to the model significantly improved the model with an OFV change by 132 points. The model with inclusion of F1 was considered as the base model (equation 5). The estimated Dosemax and Dose50 are 182 and 160 mg, respectively. The bounds for the parameterization of the Dosemax and Dose50 are within the dose range.
| (4) |
| (5) |
Covariate Model Building
Data used for covariate analysis are summarized in Tables 1 and 2. Body weight (WT), eGFR, and Inh were identified as statistically significant covariates on CL/F, and WT was identified as a statistically significant covariate on Vd/F (Table S1). The parameters of the final model are presented in Table 3. The impact of covariates included in the final model is presented in the PK forest plot (Figure 1). The effect of eGFR at the 5th and 95th percentiles on CL/F exceeds the 80% to 125% range. The effect of median eGFR suggests that for subjects with the extremes of eGFR (5th and 95th percentiles were 32 and 110 mL/[min·1.73 m2], respectively), CL/F would be 33% lower and 27% higher, respectively, than the median eGFR of 69 mL/(min·1.73 m2). The effect of WT at the 5th and 95th percentiles on CL/F does not exceed the 80% to 125% range. The effect of median WT suggests that for subjects with the extremes of WT (5th and 95th percentiles were 56 kg and 113 kg, respectively), CL/F would be 20% higher and 17% lower, respectively, than the median WT 79 kg. Similarly the effect of WT at the 5th and 95th percentiles on Vd/F does not exceed the 80% to 125% range. The effect of median WT suggests that for subjects with the extremes of WT, Vd/F would be 8% lower and 8% higher, respectively, than the median WT of 79 kg. Inh reduced CL/F by 23.2%.
Table 1.
Distribution of Continuous Covariates
| Descriptive Statistics | Age (y) | WT (kg) | BMI (kg·m−2) | eGFR (mL/[min·1.73 m2]) |
|---|---|---|---|---|
| Median | 42 | 79.5 | 25.9 | 69.4 |
| [min;max] | [18;62] | [42.3;164.5] | [17.31;90.8] | [13.62;137.37] |
BMI indicates body mass index; eGFR, estimated glomerular filtration rate (Serum creatinine concentration and the chronic kidney disease epidemiology collaboration [CKD‐EPI] formula were used to calculate eGFR); WT, body weight.
Table 2.
Distribution of Categorical Covariates
| Sex: Male/Female (n, Ratio) | Site: Japan/Non‐Japan (n, % Japan Subjects) | Number of Observations With CYP3A4 Inhibitors Coadministered (Yes/Total Observations, %) | Number of Observations With CYP3A4 Inducers Coadministered (Yes/No, %) |
|---|---|---|---|
| 526/565 | 18/1091a | 255/7355 | 64/7355 |
| 0.93 | 1.64% | 3.46% | 0.87% |
Among 1091 subjects, 1073 subjects enrolled outside Japan, including 1024 whites, 14 blacks, 1 Hispanic, 9 Asians, and 25 others.
Table 3.
Summary of Pop PK Model Parameter Estimates and Bootstrap Results
| Bootstrap Value (n = 1000) | |||||
|---|---|---|---|---|---|
| Parameter | Base Model Estimates Mean %RSE | Final Model Estimates Mean (%RSE) | Mean Estimate | 5th Percentile | 95th Percentile |
| CL/F (L/h) | 11.4 | 12.6 | 12.7 | 11.8 | 13.4 |
| Power coefficient eGFR on CL/F | … | 0.52 | 0.52 | 0.49 | 0.54 |
| Power coefficient WT on CL/F | … | −0.53 | −0.53 | −0.71 | −0.35 |
| Fractional change of CL/F with Inha | … | −0.23 | −0.21 | −0.27 | −0.19 |
| Vd/F (L) | 109 | 110 | 111 | 99.8 | 120 |
| Power coefficient WT on Vd/F | … | 0.23 | 0.24 | 0.012 | 0.44 |
| D1 (h) | 0.55 | 0.58 | 0.60 | 0.39 | 0.78 |
| Dose50 (mg) | 152 | 166 | 158 | 142 | 190 |
| Dosemax (mg) | 175 | 182 | 180 | 161 | 203 |
| Interindividual variability | Mean (CV) | Mean (CV) | |||
| CL/F | 0.43 (6.2) | 0.40 (6.3) | 0.41 | 0.35 | 0.46 |
| Covariance CL/F on Vd/F | −0.21 (15.1) | −0.25 (12.7) | −0.25 | −0.32 | −0.17 |
| Vd/F | 0.46 (13.6) | 0.49 (13.1) | 0.51 | 0.37 | 0.66 |
| Covariance CL/F on D1 | 1.06 (10.2) | 1.13 (9.1) | 1.1 | 0.89 | 1.33 |
| Covariance V/F on D1 | −1.42 (12.2) | −1.51 (11.2) | −1.49 | −1.86 | −1.09 |
| D1 | 5.67 (13.4) | 5.67 (12.7) | 5.62 | 4.26 | 7.03 |
| Dose50 | 0.12 (22.8) | 0.086 (33.6) | 0.082 | 0.018 | 0.152 |
| Dosemax | 0.007 (121.2) | 0.019 (77.1) | 0.019 | 0.004 | 0.042 |
| Residual variability | Mean (%CV) | Mean (%CV) | … | … | |
| Additive | 4.88 (15.5) | 5.3 (15.1) | 5.46 | 3.41 | 8.28 |
| Proportional | 0.19 (1.3) | 0.18 (1.3) | 0.18 | 0.17 | 0.20 |
CL/F indicates total body clearance of drug from plasma following extravascular administration; %CV, percentage coefficient of variation; D1, duration of absorption; Dosemax, the highest dose at which F1 is lowest; Dose50, the dose at which F1 is 50%; eGFR, estimated glomerular filtration rate; F1, relative bioavailability; %RSE, percentage relative standard error; Vd/F, apparent central volume of distribution; WT, body weight.
CYP3A inhibitors coadministered.
Figure 1.
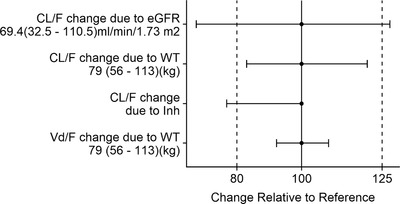
Covariate impacts on CL/F and Vd/F. The bars represent the percentage change of each pharmacokinetic parameter from the base to the 5th and 95th percentile range of the covariate. The dashed lines represent the 80th and 125th percentiles, respectively. CL/F indicates total body clearance of the drug from plasma following extravascular administration; eGFR, estimated glomerular filtration rate; Inh, coadministration of an inhibitor of CYP3A4; Vd/F, apparent volume of administration; WT, body weight.
Model Diagnostics and Evaluations
Diagnostic plots for the final model adequately described the observed data (Figure S1), and significant trends within these scatterplots were not observed. Moreover, the final model was much improved relative to that of the base model (data on file). The subjects involved in this trial were titrated to 1 of 3 split‐dose regimens (45 mg am/15 mg pm [45/15 mg], 60 mg am/30 mg pm [60/30 mg], or 90 mg am/30 mg pm [90/30 mg]), with the am dose on waking up and the second dose of the day 8–9 hours after the am dose of tolvaptan using multiples of either 15‐mg or 30‐mg tablets. The majority of observations (97%) are distributed between the split‐dose regimens of 60/30 mg and 90/30 mg. The predictive performance was assessed by VPC stratified by dose regimen for the above‐mentioned 2 dose strengths. The final model showed acceptable predictive performance. For the dose‐stratified VPCs of 60/30 mg, 90/30 mg split‐dose regimens the median and the 5th and the 95th percentiles of the observed concentration‐time profiles were well within the respective model‐based simulated 90% prediction intervals (Figure S2). The observed medians and simulated medians are close, which indicates that the model adequately described the data.
Bootstrap analysis was performed to determine the model stability. The bootstrap success rate was 99.7%. The model stability assessed by a 1000‐run bootstrap analysis showed that the mean parameter estimates of data were within 10% of those obtained with the final model and fell within 95%CI (Table 3). Hence, the model was confirmed to be stable, robust, and accurate.
Assessment of Predictive Performance
The subjects included in the external validation data set had observations (93.7%) distributed between the split‐dose regimens of 60/30 mg and 90/30 mg. As shown in the VPC plot (Figure 2), the observed and predicted medians are close, which indicates the optimal predictive performance of the final model.
Figure 2.
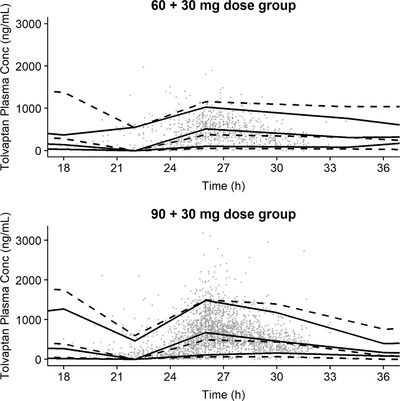
External validation. A visual predictive check of the final population pharmacokinetic model. Asterisks indicate observations; black solid lines, observed median and 5th and 95th percentiles; dashed black lines, model‐predicted median and 5th and 95th percentiles; X + Y mg, pm dose of Y mg taken 8 to 9 hours after am dose of X mg; Time represents time in hours from the first dose on the i‐th day.
Normal Q‐Q plots for NPDE and histograms of NPDE (Figure 3) show that there is minimal skewness. The 3 central moments of the distribution of the NPDE—mean, variance, and skewness—are 0.16, 1.3, and 0.15, respectively.
Figure 3.
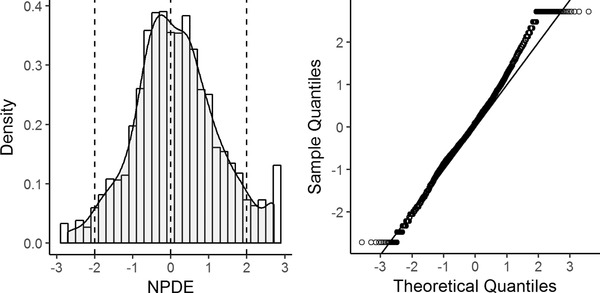
Histogram of the NPDE (left panel) and quantile‐quantile plots (right panel). Histogram of the normalized prediction distribution errors (NPDE) with the density of the standard normal distribution overlaid (left panel) and quantile‐quantile plots of the NPDE vs the expected standard normal distribution (right panel).
Discussion
This is the first published Pop PK model to evaluate tolvaptan PK in ADPKD subjects. Various PK models were explored to describe the data, but a 1‐compartment model, with 0‐order absorption and first‐order elimination described the model optimally. The effect of dose as a covariate on F1 was investigated on the complete data set instead of the dense data alone because of the availability of a wide dose range in the complete data set. Physiologically, dose does not decrease the absorption rate constant; instead, an increase in dose affects F1 due to the low solubility of tolvaptan. Tolvaptan is classified as a low‐solubility compound in the biopharmaceutical classification system and the biopharmaceutical drug disposition classification system.20, 21 Decreased F1 at high doses is not uncommon for low‐solubility compounds because the drug does not dissolve readily and residence time at the absorption site may be insufficient, and in such instances, F1 tends to be low at high dose. Accounting for changes in F1 significantly improved the model: as the dose increased from 15 mg to 120 mg, F1 decreased by 36%.
The most significant covariate after dose is eGFR. It has been determined that renal impairment can affect the PK of drugs that are predominantly eliminated by nonrenal processes such as metabolism by cytochrome P450 isozymes and/or active transport.22 Earlier studies indicated that chronic kidney disease was associated with a decrease in the expression of specific liver P450 isoforms; the main hypothesis appears to be the accumulation of uremic toxins, which can modulate CYP activity.23 CL/F decreased with decrease in eGFR, which subsequently is expected to result in an increase in tolvaptan exposure. CL/F decreased with increase in WT. The impact of WT on CL/F ranged from 10.2 L/h for a 113‐kg subject to 14.7 L/h for a 56‐kg subject as compared with the typical value of 12.3 L/h for a typical 79.5‐kg subject.
Tolvaptan is a sensitive CYP3A4 substrate,13 so it is expected that coadministration of CYP3A inhibitors and inducers would effect tolvaptan CL/F. In a previous dedicated drug‐drug interaction study conducted in healthy subjects, it was observed that subjects on a strong CYP3A inhibitor (ketoconazole 200 mg a day) had an 83% decrease in CL/F and a 3.5‐ and 5.4‐fold increase in peak plasma concentration (Cmax) and area under the concentration‐time curve, respectively, as compared with subjects not taking ketoconazole.24 Based on the current analysis, CYP3A inhibitor coadministration (Inh) with tolvaptan reduced CL/F by only 23.2%; this relatively small effect occurred because very few strong CYP3A inhibitors were used during the open‐label trial, as the protocol indicated that “strong CYP3A inhibitors were to be avoided.” Dose reductions to 30 mg or 15 mg once daily, depending on tolerated dose, are recommended when tolvaptan is to be administered with a strong CYP3A inhibitor.7
In a previous healthy‐subject study evaluating the effect of a strong CYP3A4 inducer on tolvaptan concentrations, it was observed that tolvaptan Cmax and area under the concentration‐time curve ratios for subjects on rifampin and tolvaptan over tolvaptan alone were 0.17 and 0.13, respectively.24 However, fractional change of CL/F due to CYP3A4 inducer coadministration was excluded from the forward analysis because fewer than 1% of the observations were reported to follow coadministration with CYP3A4 inducers (Table 2).
The final Pop PK model with inclusion of covariates adequately described the PK of tolvaptan; however, due to sparse sampling and the limited number of observations in the absorption phase, higher interindividual variability was observed for the duration of absorption.The VPC plots showed acceptable model predictive performance across all dose regimens, and few peak tolvaptan plasma concentrations were outside the 90%CI. One of the reasons we speculated that some of these observations may have been captured by the model if dosing in the fasted or fed state had been able to be incorporated as a covariate was that as tolvaptan dose was increased from 30 mg25 to 90 mg (noncompartmental analysis, data on file), mean Cmax values were increased 15% to 96% following a high‐fat meal.
Conclusions
This is the first published article describing a Pop PK analysis of tolvaptan in ADPKD subjects. Tolvaptan PK was well described by a 1‐compartmental model with eGFR, Inh, and WT as significant covariates on CL/F and WT as a significant covariate on Vd/F. Covariate models have improved our understanding of tolvaptan PK. Bootstrap analysis shown that the Pop PK model is stable and robust. Internal and external validation using VPC confirmed the ability of the model to describe the data optimally.
Declaration of Conflicting Interests
All authors were employees of OPDC at the time this article was submitted. These trials were supported by OPDC.
Supporting information
Table S1. Pharmacokinetic Sampling Plans
Table S2. Population Pharmacokinetic Model‐Building Summary
Figure S1. Tolvaptan diagnostic plots for the final PK model.
Figure S2. Internal validation.
Acknowledgments
The authors would like to thank Timothy Goggin for help with the analysis, Frank Czerwiec, Arash Raoufinia, and Olga Sergeyeva for their review of this manuscript, and all subjects who participated in our tolvaptan clinical trials.
Data Sharing
Otsuka Pharmaceutical Development & Commercialization (OPDC) will not share the population pharmacokinetic data used in this report.
Funding
Otsuka Pharmaceutical Development & Commercialization, Inc, Princeton, NJ. provided all funding for the analysis.
References
- 1. Matsuzaki M, Hori M, Izumi T, Asanoi H, Tsutamoto T. Effects of tolvaptan on volume overload in Japanese patients with heart failure: results of a phase II, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study. Cardiovasc Drugs Ther. 2011;25(Suppl 1):S19–S31. [DOI] [PubMed] [Google Scholar]
- 2. Fukunami M, Matsuzaki M, Hori M, Izumi T. Efficacy and safety of tolvaptan in heart failure patients with sustained volume overload despite the use of conventional diuretics: a phase III open‐label study. Cardiovasc Drugs Ther. 2011;25(Suppl 1):S47–S56. [DOI] [PubMed] [Google Scholar]
- 3. Sakaida I. Tolvaptan for the treatment of liver cirrhosis oedema. Expert Rev Gastroenterol Hepatol. 2014;8(5):461–470. [DOI] [PubMed] [Google Scholar]
- 4. European Medical Agency . Samsca: summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000980/WC500048716.pdf Accessed May 15, 2018.
- 5. US Food and Drug Administration . Samsca prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/022275s009lbl.pdf Accessed June 12, 2018.
- 6. Hori M. Tolvaptan for the treatment of hyponatremia and hypervolemia in patients with congestive heart failure. Future Cardiol. 2013;9(2):163–176. [DOI] [PubMed] [Google Scholar]
- 7. European Medicines Agency . Jinarc: summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002788/WC500187921.pdf Accessed June 12, 2018.
- 8. US Food andDrug Administration . Jynarque: NDA approval letter. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/204441Orig1s000ltr.pdf Accessed June 12, 2018.
- 9. Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25(1):18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shoaf SE, Chapman AB, Torres VE, Ouyang J, Czerwiec FS. Pharmacokinetics and pharmacodynamics of tolvaptan in autosomal dominant polycystic kidney disease: phase 2 trials for dose selection in the pivotal phase 3 trial. J Clin Pharmacol. 2017;57(7):906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Torres VE, Devuyst O, Chapman AB, et al. Rationale and design of a clinical trial investigating tolvaptan safety and efficacy in autosomal dominant polycystic kidney disease. Am J Nephrol. 2017;45(3):257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. US Food and Drug Administration . Drug interactions & labeling. https://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm. Accessed November 5, 2018.
- 14. Shoaf SE, Wang Z, Bricmont P, Mallikaarjun S. Pharmacokinetics, pharmacodynamics, and safety of tolvaptan, a nonpeptide AVP antagonist, during ascending single‐dose studies in healthy subjects. J Clin Pharmacol. 2007;47(12):1498–1507. [DOI] [PubMed] [Google Scholar]
- 15. Bauer RJ. NONMEM Users Guide. Introduction to NONMEM 7.3. ICON Deployment Solutions: Hanover, MD, 2013. [Google Scholar]
- 16. Joerger M. Covariate pharmacokinetic model building in oncology and its potential clinical relevance. AAPS J. 2012;14(1):119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lindbom L, Pihlgren P, Jonsson EN. PsN‐Toolkit‐a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Meth Prog Biomed. 2005;79(3):241–257. [DOI] [PubMed] [Google Scholar]
- 19. Mentre F, Escolano S. Prediction discrepancies for the evaluation of nonlinear mixed‐effects models. J Pharmacokinet Pharmacodyn. 2006;33(3):345–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Magar DD, Magar MD, Sakarkar DM, Jaiswal SB. Design and evaluation of solid dispersion based tolvaptan immediate release tablets. Indo Am J Pharm Res. 2014;4(3):1333–1343. [Google Scholar]
- 21. Dave RA, Morris ME. Novel high/low solubility classification methods for new molecular entities. Int J Pharm. 2016;511(1):111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Y, Zhang L, Abraham S, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clin Pharmacol Ther. 2009;85(3):305–311. [DOI] [PubMed] [Google Scholar]
- 23. Rowland Yeo K, Aarabi M, Jamei M, Rostami‐Hodjegan A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev Clin Pharmacol. 2011;4(2):261–274. [DOI] [PubMed] [Google Scholar]
- 24. Shoaf SE, Bricmont P, Mallikaarjun S. Effects of CYP3A4 inhibition and induction on the pharmacokinetics and pharmacodynamics of tolvaptan, a non‐peptide AVP antagonist in healthy subjects. Br J Clin Pharmacol. 2012;73(4):579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shoaf SE, Kim SR, Bricmont P, Mallikaarjun S. Pharmacokinetics and pharmacodynamics of single‐dose oral tolvaptan in fasted and non‐fasted states in healthy Caucasian and Japanese male subjects. Eur J Clin Pharmacol. 2012;68(12):1595–1603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Pharmacokinetic Sampling Plans
Table S2. Population Pharmacokinetic Model‐Building Summary
Figure S1. Tolvaptan diagnostic plots for the final PK model.
Figure S2. Internal validation.