

Abstract
Background
Chronic rhinosinusitis (CRS) with nasal polyps (CRSwNP) is a chronic inflammatory disease often accompanied by impairment of sense of smell. This symptom has been somewhat overlooked, and its relationship to inflammatory cytokines, tissue compression, neuronal loss, and neurogenesis is still unclear.
Methods
In order to elucidate potential mechanisms leading to CRS in humans, we have established a type 2/T helper type 2 cell (Th2)‐mediated allergic CRS mouse model, based on house dust mite (HDM) and Staphylococcus aureus enterotoxin B (SEB) sensitization. The inflammatory status of the olfactory epithelium (OE) was assessed using histology, biochemistry, and transcriptomics. The sense of smell was evaluated by studying olfactory behavior and recording electro‐olfactograms (EOGs).
Results
After 22 weeks, a typical type 2/Th2‐mediated inflammatory profile was obtained, as demonstrated by increased interleukin (IL)‐4, IL‐5, and IL‐13 in the OE. The number of mast cells and eosinophils was increased, and infiltration of these cells into the olfactory mucosa was also observed. In parallel, transcriptomic and histology analyses indicated a decreased number of immature olfactory neurons, possibly due to decreased renewal. However, the number of mature sensory neurons was not affected and neither the EOG nor olfactory behavior was impaired.
Conclusion
Our mouse model of CRS displayed an allergic response to HDM + SEB administration, including the type 2/Th2 inflammatory profile characteristic of human eosinophilic CRSwNP. Although the sense of smell did not appear to be altered in these conditions, the data reveal the influence of chronic inflammation on olfactory neurogenesis, suggesting that factors unique to humans may be involved in CRSwNP‐associated anosmia.
Keywords: chronic rhinosinusitis, olfaction, olfactory epithelium, sensory neurogenesis, Type 2/Th2 inflammation
1. INTRODUCTION
Chronic rhinosinusitis (CRS) is a very common disease, reportedly affecting approximately 12% of the general population1 with significant direct medical costs. CRS is defined in adults as a local inflammation of the nasal and paranasal sinus mucosae with symptoms including nasal congestion, loss of smell, rhinorrhea, and facial pain that persists for at least 12 consecutive weeks.1 There are two main phenotypes of CRS: CRS with nasal polyps (CRSwNP), which typically has an eosinophilic component, and CRS without nasal polyps (CRSsNP), which has a neutrophilic component.2 Although the causes of CRSwNP are still unclear, suspected pathogenic mechanisms include an innate barrier defect, primary infections that damage the mucosal barrier and promote its permeability, and dysfunction of several components of immunity.3 Although still a matter of debate,4 fungal colonization has been proposed to promote dysregulation of the sinonasal microbiome. Association of CRSwNP with local colonization by Staphylococcus aureus has been reported,5 and the toxins secreted by S. aureus that act as superantigens have been shown to amplify immune responses and shift inflammation toward the type 2/T helper type 2 cell (Th2) profile, including eosinophil infiltration and activation.6, 7, 8 While systemic atopy is not strongly associated with CRSwNP, elevations of IgE in nasal secretions and tissue indicate a localized allergic condition.3 Levels of IgE antibodies to Staphylococcal enterotoxin superantigens in nasal polyp tissue are thought to be related to disease severity.5, 6, 7
It is generally well accepted that the immune system plays a major role in CRS development.2, 3, 9 Infiltration of innate immune cells and increased levels of pro‐inflammatory chemokines and cytokines have been detected in nasal polyps of CRS patients.3 The injured epithelium releases cytokines interleukin IL‐25 and IL‐33, and thymic stromal lymphopoietin (TSLP), collectively known as alarmins, which direct Th2‐polarized inflammatory response by inducing the production of type 2 cytokines, IL‐5, IL‐4, and IL‐13.2, 9
Patients with CRS generally suffer from many symptoms, including nasal obstruction, congestion, nasal discharge, facial pressure and pain, headache, and reduction or even loss of smell (hyposmia or anosmia, respectively).2 Impairment in sense of smell, one of the most troublesome symptoms in patients with CRSwNP, is correlated with disease severity and may be the first sign of disease reoccurrence.10 This particular symptom has a substantial impact on quality of life and is often overlooked in disease management. The degree of olfactory dysfunction varies across CRS phenotypes. Clinical studies report that olfactory loss is more prevalent in CRSwNP patients than in CRSsNP patients.11 Although impaired olfaction in CRS is associated with conductive and sensorineural components as well as local inflammation,12, 13 its exact mechanisms are still not fully understood. Nasal airflow reduction, edema‐induced compression of olfactory nerves, or hypersecretion of Bowman's glands with altered ion concentrations in the olfactory mucus likely contribute to reducing the sense of smell.12, 14 It has also been suggested that inflammatory mediators could have direct neurotoxic actions on olfactory sensory neurons (OSNs).12, 13, 15, 16 OSNs with abnormal morphologies and potential functional defects,17 as well as higher numbers of immature neurons, suggesting an attempt at regeneration,18 have also been reported in CRS patients.
Because biopsies from CRSwNP patients are somewhat difficult to obtain, several animal models have been developed to study CRS, including allergy models. Ovalbumin (OVA) administration combined with or without low doses of Staphylococcal enterotoxin B (SEB) has been shown to induce eosinophil infiltration, to release type 2 cytokines from the olfactory mucosa, and to form structures that resemble nasal polyps.19 Recently, house dust mite (HDM), a well‐known allergen in humans, was used in combination with SEB in an allergic CRS mouse model to induce nasal inflammation resembling that observed in CRSwNP patients, such as infiltration of eosinophils, mast cells, and polypoid lesions.20 However, these animal studies did not directly measure the sense of smell or the OSN turnover within the olfactory epithelium (OE).
In our present study, we used HDM and SEB as inducers of allergic CRS in mice to investigate the link between the inflammatory state and the OE structure, as well as the impact of type 2/Th2 inflammation on olfactory function. Our data demonstrate an elevation of type 2/Th2 markers, concomitant with changes in other transcriptomic and histology markers, and a decrease in immature olfactory neuron number and renewal without affecting the sense of smell in mice.
2. MATERIALS AND METHODS
Experiments were carried out on 76 BALB/cByJ mice, all 4‐week‐old females (Charles River Laboratories, Saint‐Germain‐Nuelles, France) housed in animal facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). All animal experiments were conducted in accordance with European guidelines for care and use of laboratory animals and were approved by the local ethics committee (Comité d'Ethique pour la Protection de l'Animal de Laboratoire C2EA‐24). The experimental protocol (Figure 1) of an allergic CRS model activated by purified HDM extract from Dermatophagoides pteronyssinus (Der p1; Greer Laboratories, Lenoir, NC, USA) and SEB from S. aureus (List Biological Laboratories, Inc., Campbell, CA, USA) was adapted from Khalmuratova et al20 Statistical analyses were performed by a statistician in collaboration with our biostatistical department and using SAS v9.2 (SAS Institute, Cary, NC, USA).
Figure 1.
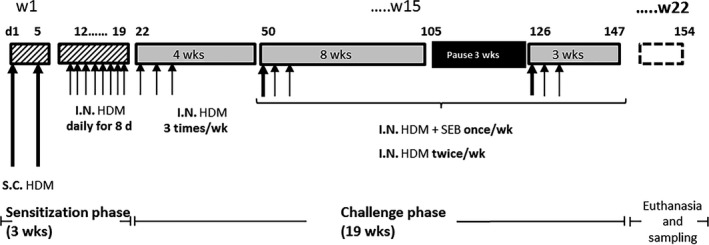
Protocol for allergen HDM sensitization and SEB administration. Two groups of mice of 38 animals were treated either with PBS (control group) or with HDM + SEB (treated group). On Days 1 and 5, S.C. HDM (HDM 100 μg + 200 μg Alum in PBS [100 μL]) was administered, followed by daily I.N. HDM (HDM 20 μg in 40 μL PBS [20 μL/nare]) from Days 12 to 19. I.N. HDM + SEB: 20 μg HDM + SEB 10 ng (20 μL/nare). HDM, house dust mite; I.N., intranasally; PBS, phosphate buffer saline; S.C. subcutaneously; SEB, Staphylococcal enterotoxin type B
Detailed materials and methods are fully described in Supporting Information of this article.
3. RESULTS
3.1. HDM + SEB intranasal treatment induces local type 2/Th2 inflammation
Type 2/Th2 inflammation markers were found to be enhanced in the OE of mice treated with intranasal injections of HDM + SEB as compared to controls.
After 22 weeks of allergen treatment, mice presented a significant (P < 0.001) elevation of IL‐13, IL‐4, and IL‐5 mRNA levels in the OE compared to the control group (51‐fold, 1.8‐fold, and 1.4‐fold, respectively) (Table 1). Expression of the chemokine eotaxin 1 was dramatically and significantly increased (10‐fold; P < 0.001 compared to control group) (Table 1). mRNA analysis of additional inflammatory markers in the HDM + SEB‐treated OE compared to control OE further revealed an upward trend for the expression of alarmins (eg, IL‐25 and TSLP), a significant (P < 0.001) increase in IL‐13 receptor subunits (6.8‐fold in IL‐13Rα2 and 1.3‐fold in IL‐13Rα1, Table 1), and a significant increase in IL‐10 (1.7‐fold; P < 0.001).
Table 1.
Transcriptomic analysis of olfactory epithelium (OE) inflammation and cytoarchitecture
| Function | Gene | FC | Mus musculus gene name |
|---|---|---|---|
| General inflammation | TNF‐α | ×0.6 | Tumor necrosis factor |
| Th2 inflammation | Eotaxin 1 (CCL11) | ×10*** | Chemokine (c‐c motif) ligand 11 |
| IL‐13 | ×51*** | Interleukin 13 | |
| IL‐13Rα1 | ×1.3*** | Interleukin 13 receptor alpha 1 | |
| IL‐13Rα2 | ×6.8*** | Interleukin 13 receptor alpha 2 | |
| IL‐4 | ×1.8*** | Interleukin 4 | |
| IL‐5 | ×1.4 | Interleukin 5 | |
| Innate inflammation (alarmins from epithelial cells) | IL‐25 | ×1.4 | Interleukin 25 |
| IL‐33 | ×1 | Interleukin 33 | |
| IL‐1RL1 | ×7.1*** | Interleukin 1 receptor L1 (ST2 IL‐33 receptor) | |
| CCR4 | ×0.2*** | Chemokine (c‐c motif) receptor 4 | |
| TSLP | ×1.3 | Thymic stromal lymphopoietin | |
| T2R 38 | ×1.4 | Putative taste receptor | |
| Th1 inflammation | IFN‐γ | ×1 | Interferon γ |
| Tolerance | IL‐10 | ×1.7*** | Interleukin 10 |
| Sustentacular cells | EPAS1 | ×1.1 | Endothelial PAS domain protein 1 |
| SIX1 | ×1.1 | Sine oculis‐related homeobox 1 homolog | |
| Polyp | APRIL | ×1.2 | A proliferation‐inducing ligand |
| BAFF | ×0.4*** | B‐cell activating factor | |
| Horizontal basal cells (HBC) | ICAM1 | ×0.9 | Intercellular adhesion molecule 1 |
| RGS1 | ×1.6** | Regulator of G‐protein signaling 1 | |
| TRP63 | ×1.4 | Transformation related protein p63 | |
| Globose basal cells (GBC) | NEUROG1 | ×0.6*** | Neurogenin 1 |
| SOX2 | ×1 | SRY (sex‐determining region Y)‐box 2 | |
| Immature olfactory neurons | GAP43 | ×0.4*** | Growth‐associated protein 43 |
| NHLH2 | ×0.3*** | Nescient helix loop helix 2 | |
| Mature olfactory neurons | NEU2 | ×1.1 | Neuraminidase 2 transcript variant 1 |
| OMP | ×1.1 | Olfactory marker protein |
Data are expressed as a fold change (FC) between the ΔCT of HDM + SEB‐treated mice and ΔCT of vehicle‐treated mice. Statistical differences between groups were assessed using the Wilcoxon‐Mann‐Whitney U test, and P values below P < 0.05 were considered statistically significant.
CT, cycle threshold; HDM, house dust mite; OE, olfactory epithelium; OMP, olfactory marker protein; PAS, Per‐Arnt‐Sim; SEB, Staphylococcal enterotoxin type B; TSLP, thymic stromal lymphopoietin.
P < 0.001 vs controls.
P < 0.01 vs controls.
These data were corroborated by protein titration (Figure 2A), showing a significant increase (P < 0.0001) in IL‐13 and IL‐5 in the HDM + SEB‐treated group vs control group (IL‐13: 6.3 ± 0.5 pg/mg of tissue vs 2.8 ± 0.3 pg/mg; IL‐5: 0.30 ± 0.04 pg/mg vs 0.07 ± 0.01 pg/mg). IL‐4 levels were under the detection threshold in the control group but detectable in HDM + SEB‐treated mice (3.7 ± 0.6 pg/mg). In contrast to the transcriptomic analysis, eotaxin protein levels were not significantly (P = 0.06) different between HDM + SEB‐treated vs control groups (75.3 ± 3.8 pg/mg vs 85.1 ± 3.6 pg/mg). Moreover, total IgE, considered as an allergic index, was significantly (P < 0.0001) increased in the OE (22.4 ± 3.9 ng/mg vs 3.1 ± 0.2 ng/mg for the HDM + SEB‐treated group vs control group). In addition, IgE was the only inflammatory marker found elevated in serum (24,393 ± 1613 ng/mL vs 6989 ± 1783 ng/mL for the HDM + SEB‐treated group vs control group, Figure 2B).
Figure 2.
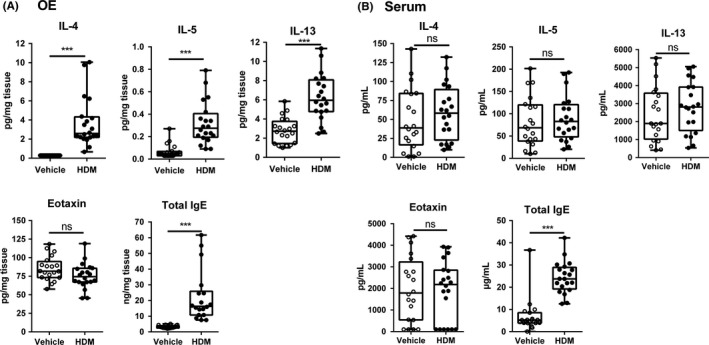
Differential expression of cytokines and IgE in (A) OE and (B) serum. Measurement of cytokines, IL‐4, IL‐5, and IL‐13, and eotaxin (CCL11) by Bio‐Plex multiplex and IgE by ELISA in OE of 20 controls (vehicle) and 20 HDM + SEB‐treated mice (HDM). All individual data are expressed in box‐and‐whisker plots showing the median values with interquartile range and minimum and maximum values. Statistical analysis was performed using a Wilcoxon‐Mann‐Whitney U test, and P values below P < 0.05 were considered statistically significant. ***P < 0.001 vs controls. ELISA, enzyme‐linked immunosorbent assay; HDM, house dust mite; IL, interleukin; ns, not significant; OE, olfactory epithelium
The infiltrations in both olfactory and respiratory mucosa tissues by eosinophils and mast cells were quantified by Sirius red staining and immunohistochemistry (IHC), respectively, and showed significantly (P < 0.001) higher levels in HDM + SEB‐treated mice vs control mice (in olfactory mucosa 0.03 ± 0.01% vs 0.0008 ± 0.0005% for eosinophils and 629 ± 214 cells/mm² vs 3.3 ± 3.3 cells/mm² for mast cells; in respiratory mucosa 0.4 ± 0.1% vs 0.003 ± 0.001% for eosinophils and 4.6 ± 4.6 cells/mm² vs 1655 ± 491 cells/mm² for mast cells; Figure 3A and B). Qualitative histological analysis revealed a differential distribution of infiltrating eosinophils (Figure 3C and D) and mast cells in the allergic OE: Mast cells were located into the epithelium layer, whereas eosinophils were observed within the lamina propria, near to the nerve bundle (Figure 4A).
Figure 3.
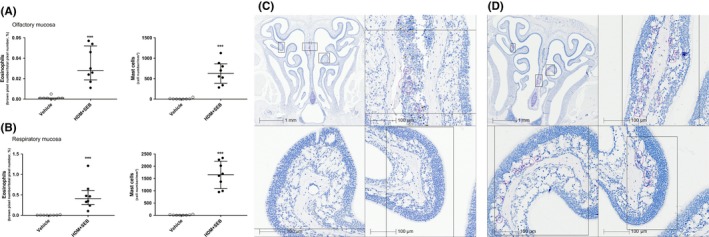
Eosinophils and mast cell counts in (A) olfactory mucosa and in (B) respiratory mucosa. Eosinophil infiltration was evaluated using Sirius red staining and quantified by pixel count (number of red pixels/total number of pixels) and expressed as a percentage in an olfactory or respiratory area previously defined. Mast cells were stained based on IHC using mast cell tryptase antibody. Positive cells were counted as a number of cells per squared millimeter. Graphs represent median with interquartile range of eight mice (plots) for each group. Statistical differences between groups were assessed using the Wilcoxon‐Mann‐Whitney U test, and P values below P < 0.05 were considered statistically significant. (C) Sirius red staining of a vehicle animal at low magnification with higher magnification inset. (D) Sirius red staining of an HDM + SEB animal at low magnification with higher magnification inset. Pink circles indicate areas of eosinophil infiltration. ***P < 0.001 vs controls. HDM, house dust mite; IHC, immunohistochemistry; SEB, Staphylococcal enterotoxin type B
Figure 4.
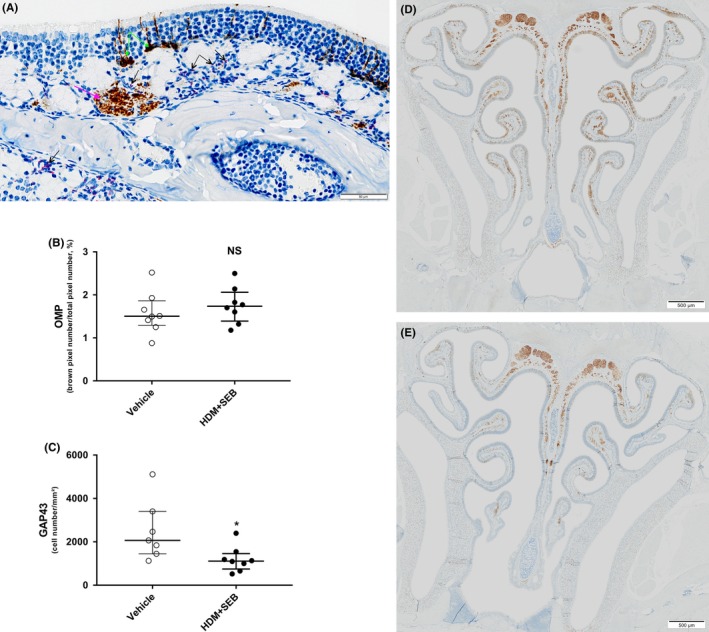
(A) Distribution of GAP43‐positive immature cells and interaction between eosinophils and nerve bundles within olfactory mucosa of HDM + SEB exposed mouse (IHC GAP43 combined with Sirius red staining); black arrows point to eosinophils stained by Sirius red, pink arrow points to a nerve bundle stained with GAP43 antibody, and green arrows point to GAP43‐positive immature olfactory neurons. (B) Number of mature olfactory neurons and (C) immature olfactory neurons in OE. Mature olfactory neurons were stained by IHC using OMP antibody and quantified by pixel count (brown pixels/total pixel number) and expressed as a percentage. Immature olfactory neurons were stained by IHC using GAP43 antibody and counted as a number of cells per squared millimeter. Graphs represent median with interquartile range of 7‐8 mice (plots) for each group. Statistical differences between groups were assessed using the Wilcoxon‐Mann‐Whitney U test, and P values below P < 0.05 were considered statistically significant. (D) GAP43 IHC at low magnification in a vehicle animal. (E) GAP43 IHC at low magnification in an HDM + SEB animal. *P < 0.05 vs controls. GAP43, growth‐associated protein 43; HDM, house dust mite; IHC, immunohistochemistry; NS, not significant; OE, olfactory epithelium; OMP, olfactory marker protein; SEB, Staphylococcal enterotoxin type B
3.2. HDM + SEB treatment affects olfactory mucosa structure
The impact of allergen treatment on the olfactory markers was further analyzed. Expression levels of the olfactory marker protein (OMP), which is uniquely associated with mature OSNs, were not significantly different between groups (fold change [FC] = 1.1). In contrast, a significant decrease in expression levels of growth‐associated protein 43 (GAP43), a marker of immature OSNs, was observed in the HDM + SEB‐treated group (0.4‐fold, P < 0.001) compared to controls (Table 1). Immunolabeling of mature and immature OSNs also supported a differential response of these populations. In HDM + SEB‐treated mice, the number of mature OMP‐positive OSNs was unchanged in the OE (1.7 ± 0.3% for HDM + SEB‐treated group vs 1.5 ± 0.2% for control group), whereas there was a significant (P < 0.05) reduction in the number of immature GAP43‐positive OSNs (1145 ± 271 cells/mm² for HDM + SEB group vs 2067 ± 617 cells/mm² for control group; P < 0.05; Figure 4B‐E). Additional transcriptomic hallmarks indicated a 0.6‐fold reduction in the expression of NEUROG1, a marker of globose basal cells (GBC) in HDM + SEB group compared to control (P < 0.001; Table 1). In contrast, expression of the horizontal basal cell (HBC) markers TRP63 and RGS1 was slightly increased compared to controls (1.4‐fold and 1.6‐fold, respectively).
3.3. HDM + SEB treatment does not induce OSN dysfunction and loss of smell
The heptanal‐evoked electro‐olfactogram (EOG) characteristically displayed a fast depolarization of approximately 1 mV in amplitude followed by a slow and progressive repolarization lasting about 10 seconds (Figure 5A). Electro‐olfactograms amplitudes observed in each group were not significantly different (P = 0.4536) overall heptanal doses tested (Figure 5B).
Figure 5.
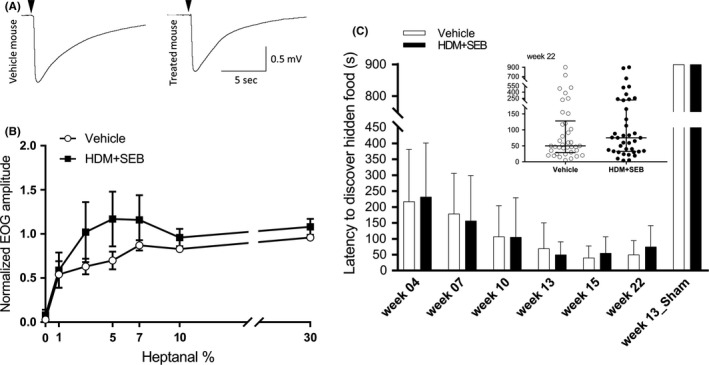
Characterization of sense of smell (A) typical recordings of 10% heptanal‐evoked EOGs of OSNs from vehicle‐treated and HDM + SEB‐treated mice. Black arrows indicate odorant (heptanal 10%) stimulation (100 ms). (B) EOG responses to increasing odorant percentage in vehicle and HDM + SEB groups. Values represent the means of peak amplitudes ± SEM (n = 7‐8/group), normalized to the response to 100% heptanal. (C) Buried food test. Bar graph represents the time course of median latency to discover a hidden food ± MAD in the vehicle and HDM + SEB‐treated groups (n = 38/group). In the Week 13_Sham experiment, mice had to discover a food pellet without odorant. The inset shows the latency distribution in Week 22 for both groups. Graphs represent median with interquartile range of 38 mice (plots) for each group. EOG, electro‐olfactograms; HDM, house dust mite; MAD, median absolute deviation; OSN, olfactory sensory neurons; SEB, Staphylococcal enterotoxin type B
After 22 weeks of allergen treatment, a buried food test was performed and the median latency to discover the hidden food in HDM + SEB‐treated mice group was not significantly different compared to control mice (P = 0.4365) (Figure 5C).
The performance of the mice changed over the course of the study. The latency to discover hidden food declined over time in both HDM + SEB‐treated and control groups (Figure 5C). A sham trial was performed as a negative control at Week 13, with an odor‐free polystyrene pellet; mice were unable to find this odor‐free pellet (latency of 900s ± 0 median absolute deviation) (Figure 5C), suggesting that mice relied on their sense of smell to find buried pellets and that they were not overtrained to burrow and find any object regardless of its odor.
4. DISCUSSION
To our knowledge, this study is the first to evaluate the impact of inflammation on both sense of smell and olfactory neurogenesis in a mouse model of allergic CRS. Mice treated for 22 weeks with a combination of HDM + SEB showed an elevation of several type 2/Th2 markers, as measured by transcriptomic and biochemical analyses. This elevation was concomitant with changes in other transcriptomic and histology markers revealing a decrease of immature olfactory neuron number and renewal, without affecting the sense of smell of mice as measured by EOG and behavioral food test.
In our present study, we combined and adapted the mouse models developed by Kim et al19 and Khalmuratova et al.20 In order to induce a robust type 2/Th2 inflammatory profile, the impact of several factors (eg, mouse strain, type of allergen, anesthesia, sex, age, and stress21) that could modulate allergy development following airway exposure of nonsensitized laboratory rodents to antigen was considered in our experimental procedure. BALB/cByJ female mice were chosen because it has been established that they have a more pronounced type 2/Th2 response in dermatitis or airway allergy inflammation models.22, 23 However, the impact of allergen exposure regimen (dose and duration) on tolerance, a reaction that can occur during a long period of continuous allergen administration, was more difficult to ascertain. To decrease tolerance, we introduced an interruption in the allergen (HDM) administration and monitored IL‐10 mRNA levels, which are known as a tolerogenic marker. However, after the immunization pause, IL‐10 mRNA levels were still up‐regulated, while type 2/Th2 markers remained elevated. These data are in good agreement with those of a long‐term study by Kim et al.,24 where mice treated with OVA + SEB presented increased type 2/Th2 inflammation markers despite a twofold increase in IL‐10 cytokine levels, suggesting an incomplete immune tolerance mechanism. House dust mite is an allergen rarely used to induce allergic CRS in animals, but, as shown by Khalmuratova et al20 its use may represent an improvement of CRS models, as HDM induces a more robust inflammatory profile than OVA, enhances the infiltration of mast cells known to be involved in the development of CRSwNP, and is more relevant to human disease than OVA.2, 25
In their HDM CRS murine model study,20 Khalmuratova et al reported an elevation of total serum IgE, as well as increased eosinophil and mast cell numbers in the nasal mucosa. Further describing this model, our transcriptomic analysis showed an increase in inflammatory markers related to type 2/Th2 inflammation in the OE (Table 1), including IL‐4, IL‐13, and IL‐5 mRNA levels. These results were confirmed at the protein level. In contrast, no change in the level of these three cytokines was observed in the serum. These results strongly suggest that our CRS mice model presents a type 2/Th2 inflammatory profile restricted to the nasal cavity. Focusing on the OE and combining two different stimuli (protease activity with HDM and bacterial protein with SEB), we found a strong increase in IL‐33 receptor (ST2) mRNA and a slight increase in IL‐25 mRNA without affecting TSLP and IL‐33 mRNA levels (Table 1). In our model, the cellular sources of cytokine release have yet to be determined. Recently, in a short‐term (3 months) mouse model where CRSwNP was induced by OVA and SEB, a correlation between the up‐regulation of IL‐25 and TSLP was observed, suggesting that IL‐25 could drive TSLP release, leading to type 2/Th2 inflammation. However, this result was not reported in a long‐term (6 months) model.24 These observations pointed to these innate inflammatory markers as participants in the early stages of the disease that drive inflammation toward type 2/Th2 response. It is now clearly established in the literature that innate inflammation markers (eg, alarmin molecules such as IL‐25, TSLP, and IL‐33) are involved in the generation of CRS.9, 26
In our present study, the structure of the OE was similar when comparing HDM + SEB‐treated and control groups, while previous studies have reported differences.20, 24 The thickness of the OE was unchanged, in contrast to the data shown by Khalmuratova et al.20 This discrepancy could possibly be due to the fact that we measured the thickness of the mature olfactory neuron layer, whereas Khalmuratova et al20 investigated the subepithelial layer. Further, nasal polypoid lesions described by Kim et al24 were not observed in our HDM + SEB‐treated group, and this was corroborated by qPCR analysis, showing low B‐cell activating factor (BAFF) mRNA levels and no modification of A proliferation‐inducing ligand (APRIL) mRNA, both markers of nasal polyps in humans and mice.27
Histological differences were observed between control and HDM + SEB‐treated groups. Olfactory mucosa from HDM + SEB‐treated animals displayed a significant reduction in the number of immature OSNs. This observation was further correlated with the decreased expression of GAP43 mRNA, which encodes a protein believed to be involved in the growth and development of immature neurons.28 Reduced expression of GAP43 has been associated with impaired development and maturation of olfactory neurons in severe neurotoxic or inflammatory conditions.16, 29
Elevations in TNF release are known to induce apoptosis of immature and mature OSNs.29, 30 However, in our present study, we did not observe any change in TNF mRNA (Table 1) or protein levels (data not shown), which may be explained by differences in protocols for inducing inflammation: nasal administration of Aspergillus fumigatus 16 or genetically induced local pro‐inflammatory cytokine release30 vs nasal administration of combined allergens HDM + SEB in our model. These different induction strategies might result in differential cytokine profiles (T helper 1 [Th1], Th2, Th1/Th2‐mixed, or T helper 17 [Th17]) that could differently impact OSN survival or renewal. For example, a study by Mori et al31 suggests that IL‐4 and IL‐13 cytokines could contribute to pathological mechanisms leading to the loss of dopaminergic neurons in Parkinson's disease. Interestingly, IL‐13Rα1 and IL‐4Rα mRNAs are detected in mature OSNs as well as in HBCs and GBCs,32 which are involved in OSN renewal. Moreover, the expression of IL‐13Rα2, a decoy IL‐13 receptor that may inhibit IL‐4 and IL‐13‐dependent pathways, was dramatically increased in the HDM + SEB‐treated group. Further studies are required to decipher the exact roles of neural IL‐4 and IL‐13 pathways and their possible regulatory impacts on neurogenesis and homeostasis of olfactory neurons.
In our model, the induced inflammation of the OE may have affected the neurogenesis cascade without impairing the survival of mature OSNs. Indeed, in addition to GAP43, NEUROG1 transcripts were reduced, suggesting that GBC‐derived neurogenesis may be impaired, as NEUROG1‐expressing GBCs are immediate neuronal precursors that can give rise to a small number of OSNs in a few days.33 Given that adult mouse olfactory neurons display a half‐life of 90 days and are continuously renewed,34 it is possible that the duration of our protocol was not long enough to observe a significant and measurable reduction in the total number of OMP‐expressing mature OSNs. Horizontal basal cells are another class of stem cell in the OE that are activated by severe epithelial damage.35 TRP63, a master negative regulator of the multipotent progenitor HBC activation,33, 35, 36 was found to be up‐regulated in our model. Recently, a study using methimazole‐induced olfactory lesions in mice demonstrated the importance of a transient inflammation (Th1) in promoting repair, especially through the proliferation of TRP63‐expressing HBCs prior to their differentiation.37 In our model, the increased expression of RGS1, another HBC‐specific gene,32 together with TRP63, may further suggest that sustained, rather than transient, inflammation could maintain the proliferation of undifferentiated HBCs.
The sense of smell and EOG recordings were not affected by the development of inflammation in our allergic CRS model, possibly due to the preservation of the mature olfactory layer. In an allergic rhinitis (AR) mouse model, Ozaki et al38 demonstrated an impact of inflammation and olfactory defect by choosing a more discriminatory behavioral model to detect olfaction and by electrophysiological records of the olfactory bulb. Unfortunately, this study did not mention the state of the mature OSNs. Moreover, it has been shown previously that sense of smell in mice was unaffected by a dramatic depletion of up to 90% of carnosine, a peptide marker for mature OSNs, in the OE.39 Nevertheless, the link between OSN number and sense of smell has not been clearly determined in the literature.
Furthermore, it has been proposed that eosinophil infiltrations are involved in sense of smell deficiency. AR mouse models have evaluated the differences in eosinophil infiltration between respiratory epithelium and OE.16, 38, 40, 41 These models used OVA or A. fumigatus as allergens and presented that eosinophils infiltrated or did not infiltrate the OE, or were limited to the respiratory‐olfactory epithelia transition. These discrepancies can be explained by various factors, including the amount of allergen, immunization time course, and induction of an allergic response coupled or not with innate inflammation. In CRS animal models, the use of different allergens (eg, OVA/HDM with or without SEB) and sensitization protocol19, 20 focused on nasal cavities without any distinction between olfactory and respiratory epithelia.
In our present work, eosinophils in the HDM + SEB‐treated group were more present in the respiratory than in the olfactory area; this may result from a greater number of epithelial cells releasing alarmins and eosinophil‐attracting chemokines in that region. Interestingly, eosinophils in OE were mainly observed in the subepithelial compartment (lamina propria), at times near to nerve bundles which was also observed by Carr et al.40 Given that eosinophil granules are neurotoxic,42 this could contribute to OSN dysfunction and particularly to impaired signal transmission. The EOG recordings in our study assessed normal events related to the cilia and olfactory receptor‐mediated transduction pathway. However, this technique does not provide any information on signal propagation through the OSN axons, which may be damaged by inflammation. A recent clinical study by Lavin et al43 demonstrated a correlation between elevated eosinophil markers in the superior turbinate of CRSwNP patients and poor performance in an olfactory test. Although human and murine olfactory mucosa cytoarchitectures are similar, their relative surfaces and cell subpopulation ratios display some differences.44 For example, the total number of olfactory neurons is much higher in mice than in humans45 and could explain why olfactory function in rodents appears to be more resistant to inflammatory challenges.
In conclusion, our study presents a mouse model of allergic CRS describing for the first time a decrease in immature olfactory neurons associated with a type 2/Th2 response within the olfactory area. Our findings strengthen the emerging view that olfactory loss may not only result from physical features, such as mechanical nasal obstruction, but also involve an inflammatory component. In agreement with and reinforcing previously published data on the potential impact of inflammatory cytokines on OSN regeneration, these results also raise the question of a potential toxicity of type 2/Th2 inflammatory cells on olfactory signal transmission, potentially opening new perspectives to explain human olfactory dysfunction in CRSwNP.
CONFLICT OF INTEREST
Biton B, Bock M‐D, Classe M, Clément M, Didier M, Fourgous V, Françon D, Gorski R, Guillemot J‐C, Haddad E‐B, Le‐Guern J, Leonetti M, Mikol V, Orsini C, Paul P, Ponsolles C, Remaury A, Roche S, Rocheteau‐Beaujouan L, Rouyar A are Sanofi employees and may hold stock and/or stock options in the company.
AUTHOR CONTRIBUTIONS
E.‐B.H., B.B., M.D., J.‐C.G., V.M., C.O., and M.L. conceived and designed the experiments and interpreted the results. R.G., C.P., J.L.‐G., S.R., V.F., and M.‐D.B. performed the ex vivo experiments and analyzed and interpreted the data. M. Classe, A. Remaury, P.P., and M.‐D.B. performed the ex vivo experiment, analyzed and interpreted the data, and wrote the article. M. Clément, L.R.‐B., and D.F. performed the in vivo experiment. A. Rouyar and M.L. performed the in vivo and ex vivo experiments, analyzed and interpreted the data, and wrote the article. A. Rouyar, B.B., M.D., and M.L. wrote the article. All the authors revised and contributed to the manuscript.
Supporting information
ACKNOWLEDGMENTS
The authors would like to thank Isabel Lefevre for a critical reading of the manuscript and Valérie Martin for her statistical assistance. The research was sponsored by Sanofi and Regeneron Pharmaceuticals, Inc. Editorial assistance was provided by Ravi Subramanian, PhD, of Excerpta Medica and funded by Sanofi Genzyme and Regeneron Pharmaceuticals, Inc.
Rouyar A, Classe M, Gorski R, et al. Type 2/Th2‐driven inflammation impairs olfactory sensory neurogenesis in mouse chronic rhinosinusitis model. Allergy. 2019;74:549–559. 10.1111/all.13559
REFERENCES
- 1. Hastan D, Fokkens WJ, Bachert C, et al. Chronic rhinosinusitis in Europe–an underestimated disease. A GA(2)LEN study. Allergy. 2011;66:1216‐1223. [DOI] [PubMed] [Google Scholar]
- 2. Schleimer RP. Immunopathogenesis of chronic rhinosinusitis and nasal polyposis. Annu Rev Pathol. 2017;12:331‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hulse KE, Stevens WW, Tan BK, Schleimer RP. Pathogenesis of nasal polyposis. Clin Exp Allergy. 2015;45:328‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fokkens WJ, van Drunen C, Georgalas C, Ebbens F. Role of fungi in pathogenesis of chronic rhinosinusitis: the hypothesis rejected. Curr Opin Otolaryngol Head Neck Surg. 2012;20:19‐23. [DOI] [PubMed] [Google Scholar]
- 5. Bachert C, Claeys SEM, Tomassen P, van Zele T, Zhang N. Rhinosinusitis and asthma: a link for asthma severity. Curr Allergy Asthma Rep. 2010;10:194‐201. [DOI] [PubMed] [Google Scholar]
- 6. Gevaert P. Co‐morbidities of allergic rhinitis: nasal polyposis In: Akdis CA, Hellings PW, Agache I, eds. Global Atlas of Allergic Rhinitis and Chronic Rhinosinusitis. European Academy of Allergy and Clinical Immunology, San Diego, CA: European Academy of Allergy and Clinical Immunology; 2015:124‐126. [Google Scholar]
- 7. Bachert C, Zhang N. The role of superantigens in allergic rhinitis, asthma and chronic rhinosinusitis In: Akdis CA, Hellings PW, Agache I, eds. Global Atlas of Allergic Rhinitis and Chronic Rhinosinusitis. European Academy of Allergy and Clinical Immunology, San Diego, CA: European Academy of Allergy and Clinical Immunology; 2015; 289‐291. [Google Scholar]
- 8. Seiberling KA, Conley DB, Tripathi A, et al. Superantigens and chronic rhinosinusitis: detection of staphylococcal exotoxins in nasal polyps. Laryngoscope. 2005;115:1580‐1585. [DOI] [PubMed] [Google Scholar]
- 9. Kato A. Immunopathology of chronic rhinosinusitis. Allergol Int. 2015;64:121‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simmen DB, Jones NS. Olfaction and nasal polyposis In: Önerci T, Ferguson B, eds. Nasal Polyposis. Berlin, Heidelberg: Springer; 2010:163‐173. [Google Scholar]
- 11. Kohli P, Naik AN, Harruff EE, Nguyen SA, Schlosser RJ, Soler ZM. The prevalence of olfactory dysfunction in chronic rhinosinusitis. Laryngoscope. 2017;127:309‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rombaux P, Huart C, Levie P, Cingi C, Hummel T. Olfaction in chronic rhinosinusitis. Curr Allergy Asthma Rep. 2016;16:41. [DOI] [PubMed] [Google Scholar]
- 13. Kern RC. Chronic sinusitis and anosmia: pathologic changes in the olfactory mucosa. Laryngoscope. 2000;110:1071‐1077. [DOI] [PubMed] [Google Scholar]
- 14. Huart C, Franceschi D, Rombaux P. Chronic rhinosinusitis and olfactory dysfunction In: Marseglia GL, ed. Peculiar Aspects of Rhinosinusitis. Rijeka, Croatia: InTech; 2011:39‐52. [Google Scholar]
- 15. Imamura F, Hasegawa‐Ishii S. Environmental toxicants‐induced immune responses in the olfactory mucosa. Front Immunol. 2016;7:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Epstein VA, Bryce PJ, Conley DB, Kern RC, Robinson AM. Intranasal Aspergillus fumigatus exposure induces eosinophilic inflammation and olfactory sensory neuron cell death in mice. Otolaryngol Head Neck Surg. 2008;138:334‐339. [DOI] [PubMed] [Google Scholar]
- 17. Yee KK, Pribitkin EA, Cowart BJ, Rosen D, Feng P, Rawson NE. Analysis of the olfactory mucosa in chronic rhinosinusitis. Ann NY Acad Sci. 2009;1170:590‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holbrook EH, Leopold DA, Schwob JE. Abnormalities of axon growth in human olfactory mucosa. Laryngoscope. 2005;115:2144‐2154. [DOI] [PubMed] [Google Scholar]
- 19. Kim DW, Khalmuratova R, Hur DG, et al. Staphylococcus aureus enterotoxin B contributes to induction of nasal polypoid lesions in an allergic rhinosinusitis murine model. Am J Rhinol Allergy. 2011;25:e255‐e261. [DOI] [PubMed] [Google Scholar]
- 20. Khalmuratova R, Lee M, Kim DW, Park JW, Shin HW. Induction of nasal polyps using house dust mite and Staphylococcal enterotoxin B in C57BL/6 mice. Allergol Immunopathol (Madr). 2016;44:66‐75. [DOI] [PubMed] [Google Scholar]
- 21. Guibas GV, Makris M, Spandou E, Priftis KN. Exposure of immunologically naive laboratory rodents to antigen via the airways. Where does tolerance stop and sensitization begin? Clin Exp Allergy. 2012;42:1552‐1565. [DOI] [PubMed] [Google Scholar]
- 22. Melgert BN, Postma DS, Kuipers I, et al. Female mice are more susceptible to the development of allergic airway inflammation than male mice. Clin Exp Allergy. 2005;35:1496‐1503. [DOI] [PubMed] [Google Scholar]
- 23. Luckett‐Chastain L, Calhoun K, Kemp J, Gallucci R. Immunological difference in Th1 and Th2 dominant mouse strains in an ICD model. (IRM15P.601). J Immunol. 2015;194(199):113.25452562 [Google Scholar]
- 24. Kim DW, Eun KM, Jin HR, Cho SH, Kim DK. Prolonged allergen exposure is associated with increased thymic stromal lymphopoietin expression and Th2‐skewing in mouse models of chronic rhinosinusitis. Laryngoscope. 2016;126:E265‐E272. [DOI] [PubMed] [Google Scholar]
- 25. London NR, Lane AP. Innate immunity and chronic rhinosinusitis: what we have learned from animal models. Laryngoscope Investig Otolaryngol. 2016;1:49‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hamilos DL. Drivers of chronic rhinosinusitis: inflammation versus infection. J Allergy Clin Immunol. 2015;136:1454‐1459. [DOI] [PubMed] [Google Scholar]
- 27. Kim DY, Lee SH, Carter RG, Kato A, Schleimer RP, Cho SH. A recently established murine model of nasal polyps demonstrates activation of B cells, as occurs in human nasal polyps. Am J Respir Cell Mol Biol. 2016;55:170‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Turner JH, Liang KL, May L, Lane AP. Tumor necrosis factor alpha inhibits olfactory regeneration in a transgenic model of chronic rhinosinusitis‐associated olfactory loss. Am J Rhinol Allergy. 2010;24:336‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Turner JH, May L, Reed RR, Lane AP. Reversible loss of neuronal marker protein expression in a transgenic mouse model for sinusitis‐associated olfactory dysfunction. Am J Rhinol Allergy. 2010;24:192‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lane AP, Turner J, May L, Reed R. A genetic model of chronic rhinosinusitis‐associated olfactory inflammation reveals reversible functional impairment and dramatic neuroepithelial reorganization. J Neurosci. 2010;30:2324‐2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mori S, Maher P, Conti B. Neuroimmunology of the interleukins 13 and 4. Brain Sci. 2016;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Colquitt BM, Allen WE, Barnea G, Lomvardas S. Alteration of genic 5‐hydroxymethylcytosine patterning in olfactory neurons correlates with changes in gene expression and cell identity. Proc Natl Acad Sci USA. 2013;110:14682‐14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwob JE, Jang W, Holbrook EH, et al. Stem and progenitor cells of the mammalian olfactory epithelium: taking poietic license. J Comp Neurol. 2017;525:1034‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gogos JA, Osborne J, Nemes A, Mendelsohn M, Axel R. Genetic ablation and restoration of the olfactory topographic map. Cell. 2000;103:609‐620. [DOI] [PubMed] [Google Scholar]
- 35. Duggan CD, Ngai J. Scent of a stem cell. Nat Neurosci. 2007;10:673‐674. [DOI] [PubMed] [Google Scholar]
- 36. Im S, Moon C. Transcriptional regulatory network during development in the olfactory epithelium. BMB Rep. 2015;48:599‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen M, Reed RR, Lane AP. Acute inflammation regulates neuroregeneration through the NF‐κB pathway in olfactory epithelium. Proc Natl Acad Sci USA. 2017;114:8089‐8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ozaki S, Toida K, Suzuki M, et al. Impaired olfactory function in mice with allergic rhinitis. Auris Nasus Larynx. 2010;37:575‐583. [DOI] [PubMed] [Google Scholar]
- 39. Harding JW, Getchell TV, Margolis FL. Denervation of the primary olfactory pathway in mice. V. Long‐term effect of intranasal ZnSO4 irrigation on behavior, biochemistry and morphology. Brain Res. 1978;140:271‐285. [DOI] [PubMed] [Google Scholar]
- 40. Carr MV, Robinson AM, Kern RC. Tissue‐specific effects of allergic rhinitis in mouse nasal epithelia. Chem Senses. 2012;37:655‐668. [DOI] [PubMed] [Google Scholar]
- 41. Sousa G, Chen M, Smith A, Lazarini PR, Lane AP. Role of the type I tumor necrosis factor receptor in inflammation‐associated olfactory dysfunction. Int Forum Allergy Rhinol. 2017;7:160‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bandeira‐Melo C, Weller PF. Mechanisms of eosinophil cytokine release. Mem Inst Oswaldo Cruz. 2005;100:73‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lavin J, Min JY, Lidder AK, et al. Superior turbinate eosinophilia correlates with olfactory deficit in chronic rhinosinusitis patients. Laryngoscope. 2017;127:2210‐2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Borgmann‐Winter K, Willard SL, Sinclair D, et al. Translational potential of olfactory mucosa for the study of neuropsychiatric illness. Transl Psychiatry. 2015;5:e527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harkema JR, Carey SA, Wagner JG, Dintzis SM, Liggitt D. Nose, sinus, pharynx, and larynx In: Piper M, Treuting SD, Liggitt D, Frevert CW, eds. Comparative Anatomy and Histology A Mouse and Human Atlas. London, Waltham, San Diego: Elsevier; 2012:71‐94. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials