

Abstract
Stearoyl‐coenzyme A desaturase 1 (SCD‐1) in sebaceous glands is a key enzyme in the synthesis of monounsaturated fatty acids essential for acne development. GSK1940029 gel, a novel SCD‐1 inhibitor, is being developed as a potential treatment for acne. To assess the irritation potential, pharmacokinetics (PK), and safety of topical GSK1940029 to the skin of healthy adults, two interdependent studies were conducted in parallel. Study 1 (n = 54) investigated the irritation potential of GSK1940029 (0.3% and 1%, occluded application) to allow for its application to larger surface areas in study 2 (n = 39), which investigated the safety, tolerability, and PK of GSK1940029 after single and repeat doses as occluded and nonoccluded applications. GSK1940029 was not a primary or cumulative irritant after 2 and 21 days of dosing in study 1. In study 2, single and repeat applications of GSK1940029 (0.1% to 1%) doses were well tolerated with little or no influence on AUC and Cmax under occluded or unoccluded conditions. Systemic exposure increased proportionally with surface area and was higher in occluded conditions. Design of these interdependent studies allowed for the assessment of the irritation potential for topical GSK1940029 in parallel with the investigation of PK and safety profiles.
Keywords: acne, irritation potential, pharmacokinetics, stearoyl‐coenzyme A (CoA) desaturase 1 (SCD‐1) inhibitor, topical
Development of topical therapeutics includes early understanding of the potential irritation liabilities and systemic exposure. Exact methods to assess these vary based on the characteristics and needs of the investigational or drug product being developed, and should be validated.1 Here we present the early phase 1 irritation potential and pharmacokinetic (PK) and safety evaluation of GSK1940029 gel being developed for acne (Figure 1).
Figure 1.
Structure of GSK1940029.
Acne vulgaris is a chronic inflammatory condition of the pilosebaceous follicles, characterized by lesions that are noninflammatory (comedones) and/or inflammatory (papules, pustules, nodules).2 There are 4 key factors responsible for the development of acne lesions: (1) excess sebum production, (2) Propionibacterium acnes infection, (3) inflammation, and (4) follicular epidermal hyperproliferation and hyperkeratinization. Prescription medications used to treat acne include both oral and topical agents and, with the exception of oral retinoids, only target 1 or more of the last 3 key acne development factors.2, 3
Currently, no topical drugs, including topical retinoids, treat excess sebum production.4 The main drug that induces a reduction in sebum production is oral (systemic) 13‐cis retinoic acid (isotretinoin), which carries severe adverse events, such as teratogenicity. Oral isotretinoin is only used for the treatment of severe acne under highly controlled environments with severe restrictions.5, 6 Thus, an unmet need exists for a topical drug that will reduce or eliminate excess sebum production with good dermal tolerability and no systemic adverse events.
Stearoyl‐coenzyme A desaturase 1 (SCD‐1) is a key enzyme involved with the synthesis of monounsaturated fatty acids from saturated fatty acids.7 Inhibitors of SCD‐1 have been developed to target the pathway in several diseases including metabolic syndrome, nonalcoholic steatohepatitis, hepatitis C virus, cancer, and skin disorders including acne.8 Production of fatty acids and lipids in sebaceous glands is essential for the development of acne as follows. Excessive production of sebum (seborrhea) promotes the growth of P. acnes and, in turn, contributes to inflammation, keratinocyte proliferation, and papule formation. An SCD‐1 inhibitor has the potential to decrease or eliminate sebum production, halting this cycle.
GSK1940029 is an SCD‐1 inhibitor that caused atrophy of sebaceous glands in mice. The effect of GSK1940029 gel on sebaceous glands was evaluated through twice‐daily topical applications to Crl:NMRI(Han) mice. Minimal to moderate sebaceous gland atrophy was observed in mice given both 0.3% and 2% GSK1940029 gel. Evaluation of irritation and sensitization potential in rabbits and mice, respectively, found that GSK1940029 gel (up to 2% concentration) and gel vehicle alone did not produce dermal irritation and were not contact sensitizers. These effects may have a beneficial impact on stopping or reversing the development of acne lesions.
This is the first report on the application of the GSK1940029 gel formulation on human skin. The purpose of this study was to provide information on the irritation potential, PK, and safety of topical applications of GSK1940029 to intact skin of healthy adult subjects.
Methods
Study Design and Population
Two interdependent studies were conducted in parallel, as illustrated in Figure 2. Both study 1 and study 2 enrolled healthy male or female subjects aged 18–65 years, inclusive. Study 1 investigated the irritation potential of GSK1940029 to allow for its application to larger surface areas in study 2, which was designed to investigate the safety, tolerability, and preliminary pharmacokinetics (PK) of topical application of GSK1940029 after single and repeat doses.
Figure 2.
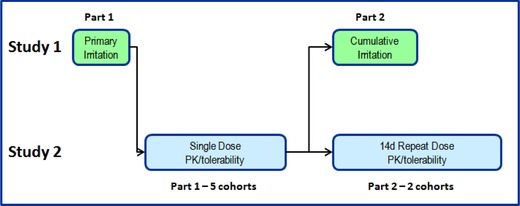
Study design schematics.
Both study protocols were funded by GSK and were conducted at CMAX Clinical Research Party Limited, Adelaide, South Australia, Australia. The protocols were reviewed and approved by the Human Research Ethics Committee, Bellberry Limited, Eastwood South Australia, Australia. Written informed consent was obtained from all subjects at screening, and the study was performed in compliance with International Conference on Harmonization/Good Clinical Practice (ICH/GCP) guidelines and the Declaration of Helsinki.
Irritation Potential Study (Study 1)
This study (NCT01984801; GSK protocol 117225) was designed as a randomized, single‐blind, placebo‐controlled trial in 2 parts. In part 1 primary irritation was examined after 2 days of dosing. In part 2 cumulative irritation was examined after 21 days of dosing. Both parts were randomized (with respect to location of treatments on the body), single‐blind, vehicle‐, positive‐, negative‐, and patch‐controlled. In each part, subjects were randomized to receive treatment to 1 of 6 designated locations on either the upper arm or other locations such as the lower or upper back. Doses of GSK1940029 included 0.3% and 1% gel (w/w). The negative irritant control was sterile distilled water for injection. The primary and cumulative irritation positive controls were 0.5% and 0.1% sodium lauryl sulfate in sterile distilled water for injection, respectively. Administration of the study medication was via occluded applications using patches.
Single and Repeat Doses (Study 2)
Study 2 (NCT01938482; GSK protocol 117226) was a randomized, single‐blind, vehicle‐controlled dose‐rising study comprising 2 parts. Part 1 of study 2 was a single‐dose, concentration and surface area‐rising (dose applied‐rising), randomized, single‐blind, and vehicle‐controlled. Subjects in part 1 received 0.3% or 1% GSK1940029 (or matching vehicle), as a single ∼24‐hour (22.5‐hour) occluded or unoccluded application over 400 and 1600 cm2 in cohorts 1 −5. In part 2 of study 2, subjects received 0.1% or 0.3% GSK1940029 (or matching vehicle) in cohorts 1 and 2, as a daily ∼24‐hour (22.5‐hour) unoccluded application to a 400‐cm2 surface area for 14 days in a randomized, single‐blind fashion.
Safety end points in study 2 included assessment of adverse events (AEs), clinical laboratory evaluations, vital signs, and 12‐lead electrocardiograms (ECGs).
Study Interdependencies
Part 1 of study 1 (primary irritation) of all GSK1940029 concentrations and controls was initiated first. Part 1 of study 2 began in the absence of significant primary irritation signal in part 1 of study 1, for the first of 3 single‐dose dose‐rising cohorts. Safety, tolerability, and preliminary PK data were assessed in making dose‐escalation decisions within study 2. Higher‐formulation concentrations were applied to larger surface areas to provide dose escalation. Extensive subject monitoring, including safety, tolerability, and PK observations, was conducted prior to dose‐escalation decisions.
Once safety, tolerability, and exposure information were determined in part 1 of study 2, part 2 of study 1 (21‐day cumulative irritation part) was initiated using compound amounts equal to those used for the primary irritation. Simultaneously, part 2 of cohort 1 of study 2, the first of 2 repeat‐dose dose‐rising cohorts was initiated with extensive subject monitoring (Figure 2).
Pharmacokinetic Assessments and Analysis
PK samples were only collected in study 2. Blood samples of part 1 (single dose, approximately 126 mL) and part 2 (repeat dose, approximately 185 mL) were collected predose and 1, 2, 4, 6, 8, 12, 16, 22.5, and 24 hours postdose. In part 1, an additional sample was collected 36 hours postdose.
Plasma PK analyses were performed by GlaxoSmithKline (Department of Drug Metabolism and Pharmacokinetics, King of Prussia, Pennsylvania). Concentrations of GSK1940029 in plasma samples were determined with validated analytical methods over the range of 10 to 5000 pg/mL (part 1) and 1 to 500 pg/mL (part 2) by ultra‐high‐performance liquid chromatography‐high‐resolution mass spectroscopy (UHPLC‐HRMS). GSK1940029 was extracted from 250 μL of human plasma with an isotopically labeled internal standard ([13C7]‐GSK1940029) by double liquid‐liquid extraction using methyl tert‐butyl ether. Extracts were injected (5 μL for part 1, 25 μL for part 2) onto an Acquity UPLC HSS T3 column (50 × 2.1 mm, 1.8 μm; Waters Corporation, Milford, Massachusetts), maintained at 65°C and eluted with a binary mobile‐phase gradient using 0.1% ammonium hydroxide in water (A) and 10% methanol in acetonitrile (B), with a constant flow rate of 0.9 mL/min.
The initial mobile‐phase condition of 55:45, A:B, was held until 0.5 minutes. From 0.5 to 1.0 minutes, the mobile phase changed to 20:80, A:B, and was held at this composition until 1.1 minutes, when the composition changed to 5:95, A:B, from 1.2 until 1.7 minutes and reverted to the initial conditions, 55:45, A:B, at 1.75 minutes. Detection was performed by positive ion HRMS/MS using TurboIonSpray interface set to 650°C on a (part 1) Waters Xevo TQ‐S mass spectrometer or on a (part 2) Waters Synapt G2‐S, Q‐TOF (Waters Corporation, Milford, Massachusetts) with multiple reaction monitoring (m/z 391→m/z 159 for GSK1940029 and m/z 398→m/z 159 for the internal standard).
For part 1, the maximum within‐run and between‐run precision values observed were 13.9% and 4.5%, respectively. Accuracy ranged from −12.7% to 5.5% bias. For part 2, the maximum within‐run precision observed was 21.0% at the lower limit of quantification (LLQ, 1 pg/mL) and 16.9% at the other levels. Maximum between‐run precision observed was 5.7%. For part 2, the acceptance criteria were extended to ±20% (±25% at the LLQ). Accuracy ranged from −11.2% to 13.2% bias.
PK parameters were derived using standard noncompartmental methods using Phoenix WinNonlin version 6.4 (Pharsight Corporation, St. Louis, Missouri). Calculations of the PK parameters for GSK1940029 were based on the actual sampling and collection times relative to the start of the last topical application recorded during the study (ie, day 1 postdose was relative to day 1 dosing, day 14 postdose was relative to day 14 dosing).
From the plasma concentration‐time data, the following parameters were determined: maximum observed plasma concentration (Cmax), time to Cmax (tmax), area under the plasma concentration‐time curve (AUC0‐24, and AUC0‐∞), and apparent terminal‐phase half‐life (t1/2), as data permitted. To estimate the extent of accumulation after repeat dosing in part 2, the observed accumulation ratio (day 14 vs day 1) for AUC (Ro) and Cmax (Rcmax) were determined.
Data Analyses
Irritation Potential Study (Study 1)
No sample‐size calculations were performed for study 1. For both parts of the study, sample size was based on feasibility and the generally accepted cohort sizes for studies of this type.
All subjects who received at least 1 application of the study medication were used in the evaluation of safety and tolerability and for study population displays. Primary and cumulative irritations were assessed by a study review team at the end of parts 1 and 2, respectively. The primary and cumulative irritation grading Scales were based on scales developed by Hill Top Research, Inc. and cited by the United States Food and Drug Administration.9
Primary irritation was collected in the form of a numeric grade (0, 0.5, 1, 2, 3 from none to greatest visible reaction) and when applicable a letter grade for additional specified adverse reactions (B, E, I, J, P, S, V, W, C, D, F, G, H, Y). For the purpose of summarization, the letter grades were assigned a corresponding number: each C, D, F, G, H, Y, or J = 0.5 and each B, E, I, P, S, V, or W = 1. The primary irritation converted score was calculated as the sum of the numeric grade and the converted letter grade.
Cumulative irritation was collected in the form of a numeric score (0, 1, 2, 3, 4, 5, 6, 7 from none to greatest visible reaction) and when applicable a letter score for effects on superficial layers of the skin (A, B, C, F, G, H). For the purpose of calculating further scores, the letter scores were assigned a corresponding number: A = 0.5; B = 1; C = 2; F, G, or H = 3.
Single and Repeat Doses (Study 2)
There was no formal calculation of power or sample size for study 2. A sample size of 6 subjects per active dose group was considered a feasible sample size to provide sufficient safety and PK data for a topical formulation study. The sample size for the vehicle at each dose level was set at 2 to avoid subject bias in safety assessment.
All data were descriptively analyzed. All subjects who received at least 1 application of the study medication were used in the evaluation of safety and tolerability and for study population displays. All subjects who received at least 1 dose of the study medication and for whom a PK sample was obtained and analyzed were included in the PK population.
Results
Demographic Characteristics and Disposition
Irritation Potential Study (Study 1)
A total of 54 subjects participated in study 1 including 15 subjects in part 1 (primary irritation) and 39 subjects in part 2 (cumulative irritation). Subjects were well matched between the 2 parts. The majority of subjects were white men with a mean age of approximately 34 years (range, 18‐65 years). All 15 enrolled subjects in part 1 received GSK1940029 ge1 0.3% and 1.0% for 2 days. A total of 39 subjects enrolled in part 2. Of these, 36 subjects received the study medication for 21 days and were included in the cumulative irritation analysis. Of the 3 withdrawn subjects in part 2, 1 was for an AE of unrelated folliculitis, and 2 were because of failing assessment of eligibility criteria.
Single and Repeat Doses (Study 2)
All 39 enrolled subjects in the single‐dosing portion of the study (part 1) completed the study. Of the 16 subjects enrolled in the repeat‐dosing portion (part 2), 1 subject withdrew consent after 12 days. Subjects were well matched across all dosing regimens. The majority of subjects were white men with an average age of approximately 29 years (range, 18‐62 years) in both parts of the study.
Safety
Irritation Potential Study (Study 1)
Topical occluded administration of GSK1940029 gel at 0.3% and 1% was well tolerated for up to 21 days. After 2 days of administration, the most common AE in part 1 was headache (4 of 15 subjects). All AEs were mild to moderate in intensity. After 21 days of dosing in part 2, the majority of the 39 subjects (>97%) reported AEs that were mild in intensity and considered drug related (59% of subjects) by the investigator. There were no clinically significant changes in clinical laboratory values, vital signs, or ECGs during this study.
Single and Repeat Doses (Study 2)
The number and types of reported AEs were generally similar across all GSK1940029 doses and the vehicle after single and repeat dosing, and all were mild to moderate in intensity (Table 1 and Table 2).
Table 1.
Summary of All Reported Adverse Events in ≥2 Subjects and All Drug‐Related Adverse Events—Study 2, Single Dosing
| GSK1940029 | |||||||
|---|---|---|---|---|---|---|---|
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | |||
| 0.3% | 1% | 1% | 0.3% | 1% | |||
| 400 cm2 | 400 cm2 | 1600 cm2 | 1600 cm2 | 1600 cm2 | Vehicle | Total | |
| Preferred Term | (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 5) | (N = 10) | (N = 39) |
| Subjects with ≥1 AE | 3 (50.0) | 2 (33.3) | 4 (66.7) | 4 (66.7) | 5 (100.0) | 8 (80.0) | 26 (66.7) |
| Subjects with ≥1 drug‐related AE | 1 (16.7) | 1 (16.7) | 3 (50.0) | 0 | 2 (40.0) | 3 (30.0) | 10 (25.6) |
| All AEs reported by ≥2 subjects | |||||||
| Headache | 1 (16.7) | 1 (16.7) | 1 (16.7) | 1 (20.0) | 1 (10.0) | 5 (12.8) | |
| Injection‐site bruising | 2 (33.3) | 1 (20.0) | 1 (10.0) | 4 (10.3) | |||
| Nausea | 3 (30.0) | 3 (7.7) | |||||
| Erythema | 1 (16.7) | 1 (20.0) | 1 (10.0) | 3 (7.7) | |||
| Application‐site erythema | 2 (33.3) | 1 (16.7) | 3 (7.7) | ||||
| Ocular discomfort | 2 (40.0) | 2 (5.1) | |||||
| Contusion | 2 (33.3) | 2 (5.1) | |||||
| Dermatitis contact | 2 (33.3) | 2 (5.1) | |||||
| Rash erythematous | 1 (16.7) | 1 (16.7) | 2 (5.1) | ||||
| Drug‐related AEs reported by ≥1 subject | |||||||
| Headache | 1 (16.7) | 1 (16.7) | 1 (20.0) | 1 (10.0) | 4 (10.3) | ||
| Nausea | 2 (20.0) | 2 (5.1) | |||||
| Ocular discomfort | 2 (40.0) | 2 (5.1) | |||||
| Rash erythematous | 1 (16.7) | 1 (16.7) | 2 (5.1) | ||||
| Photophobia | 1 (10.0) | 1 (2.6) | |||||
| Abdominal discomfort | 1 (20.0) | 1 (2.6) | |||||
| Diarrhea | 1 (20.0) | 1 (2.6) | |||||
| Acne | 1 (16.7) | 1 (2.6) | |||||
| Application‐site erythema | 1 (16.7) | 1 (2.6) | |||||
| Application‐site pruritus | 1 (16.7) | 1 (2.6) | |||||
| Tear breakup time decreased | 1 (16.7) | 1 (2.6) | |||||
Table 2.
Summary of All Reported Adverse Events in ≥2 Subjects and All Drug‐Related Adverse Events—Study 2, Repeat Dosing
| GSK1940029 | ||||
|---|---|---|---|---|
| Cohort 1 | Cohort 2 | |||
| 0.1% | 0.3% | |||
| 400 cm2 | 400 cm2 | Vehicle | Total | |
| Preferred Term | (N = 6) | (N = 6) | (N = 4) | (N = 16) |
| Subjects with ≥1 AE | 6 (100.0) | 4 (66.7) | 3 (75.0) | 13 (81.3) |
| Subjects with ≥1 drug‐related AE | 3 (50.0) | 4 (66.7) | 1 (25.0) | 8 (50.0) |
| All AEs reported by ≥2 subjects | ||||
| Dry skin | 3 (50.0) | 1 (25.0) | 4 (25.0) | |
| Injection‐site bruising | 2 (33.3) | 1 (25.0) | 3 (18.8) | |
| Pruritus | 2 (33.3) | 2 (12.5) | ||
| Headache | 2 (33.3) | 2 (12.5) | ||
| Tear breakup time decreased | 1 (16.7) | 1 (25.0) | 2 (12.5) | |
| Dermatitis contact | 1 (16.7) | 1 (25.0) | 2 (12.5) | |
| Injection‐site pain | 1 (16.7) | 1 (25.0) | 2 (12.5) | |
| Drug‐related AEs reported by ≥1 subject | ||||
| Dry skin | 3 (50.0) | 1 (25.0) | 4 (25.0) | |
| Pruritus | 2 (33.3) | 2 (12.5) | ||
| Erythema | 1 (16.7) | 1 (6.3) | ||
| Mouth ulceration | 1 (16.7) | 1 (6.3) | ||
| Injection‐site rash | 1 (16.7) | 1 (6.3) | ||
| Dysgeusia | 1 (16.7) | 1 (6.3) | ||
| Headache | 1 (16.7) | 1 (6.3) | ||
| Rash | 1 (16.7) | 1 (6.3) | ||
After single dosing, a total of 10 of 39 subjects (26%) were reported with investigator‐assessed drug‐related AEs with no apparent dose relationship (Table 1). After repeat dosing, 8 of 16 subjects (50%) had drug‐related AEs without a dose relationship (Table 2).
There were no clinically significant findings in vital signs, clinical laboratory tests, and ECGs after single‐ or repeat‐dose applications of GSK1940029 0.1% or 0.3% (occluded and nonoccluded). In addition, there were no clinically significant findings in ocular evaluations and renal function and liver function tests. There were no serious AEs or withdrawals because of AEs.
Irritation Potential Results
The primary and cumulative irritation scores for GSK1940029 gel (0.3% and 1%) and the vehicle were similar to each other, and all were less than the positive control, as illustrated in Figure 3 and listed in Table 3 and Table 4. The data indicate that the positive, negative, and patch controls performed as expected (Table 3 and Table 4).
Figure 3.
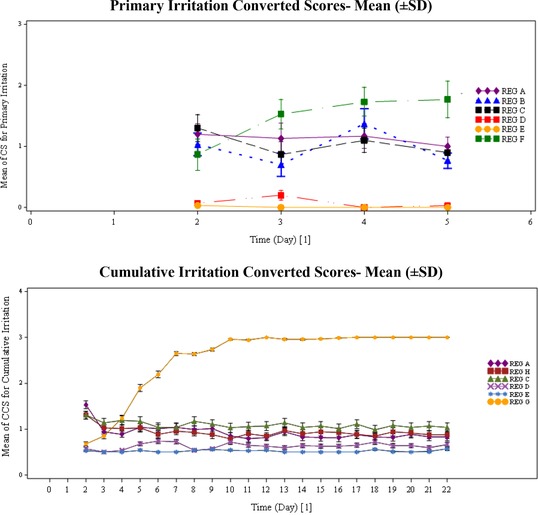
Irritation scores—study 1.
Table 3.
Summary of Average and Maximum Values for Primary Irritation Converted Scores—Study 1, Part 1
| Mean (n = 15) | 95%CI Lower | 95%CI Upper | SD | Median | Min | Max | |
|---|---|---|---|---|---|---|---|
| A‐ 0.3 % GSK1940029 | |||||||
| Average converted score | 1.13 | 0.81 | 1.44 | 0.573 | 1.00 | 0.4 | 2.4 |
| Maximum converted score | 1.77 | 1.41 | 2.13 | 0.651 | 1.50 | 1.0 | 3.0 |
| B‐ 1 % GSK1940029 | |||||||
| Average converted score | 0.97 | 0.68 | 1.25 | 0.519 | 1.00 | 0.0 | 1.8 |
| Maximum converted score | 1.67 | 1.18 | 2.15 | 0.880 | 1.50 | 0.0 | 3.5 |
| C‐Vehicle control | |||||||
| Average converted score | 1.04 | 0.74 | 1.34 | 0.540 | 1.00 | 0.4 | 2.3 |
| Maximum converted score | 1.70 | 1.31 | 2.09 | 0.702 | 1.50 | 0.5 | 3.0 |
| D‐Negative control | |||||||
| Average converted score | 0.08 | 0.02 | 0.13 | 0.104 | 0.00 | 0.0 | 0.3 |
| Maximum converted score | 0.23 | 0.06 | 0.41 | 0.320 | 0.00 | 0.0 | 1.0 |
| E‐Patch control | |||||||
| Average converted score | 0.01 | −0.01 | 0.03 | 0.032 | 0.00 | 0.0 | 0.1 |
| Maximum converted score | 0.03 | −0.04 | 0.10 | 0.129 | 0.00 | 0.0 | 0.5 |
| F‐Positive control | |||||||
| Average converted score | 1.48 | 1.07 | 1.88 | 0.737 | 1.50 | 0.0 | 2.8 |
| Maximum converted score | 2.33 | 1.77 | 2.89 | 1.012 | 2.50 | 0.0 | 4.0 |
Converted score, calculated as the sum of the numeric score and converted letter score.
Table 4.
Summary of Actual Values for Cumulative Irritation Total Score and TS10 (n = 36)—Study 1, Part 2
| Regimen | Total Score | TS10 | Cumulative Irritation Classa |
|---|---|---|---|
| A‐ 0.3 % GSK1940029 | 698.5 | 194.0 | Class 2 |
| H‐ 0.1 % GSK1940029 | 708 | 196.7 | Class 2 |
| C‐ Vehicle control | 825 | 229.2 | Class 3 |
| D‐ Negative control | 477.5 | 132.6 | Class 2 |
| E‐ Patch control | 393.5 | 109.3 | Class 2 |
| G‐ Positive control | 1932 | 536.7 | Class 4 |
TS10, total scores standardized to 10 subjects (only those subjects who completed all 21 days of patch application per protocol).
Score categories: 0 to 49, class 1, mild material, no experimental irritation (essentially no evidence of cumulative irritation under conditions of the test); 50 to 199, class 2, probably mild in normal use (evidence of a slight potential for very mild cumulative irritation under conditions of the test); 200 to 449, class 3, possibly mild in normal use (evidence of a moderate potential for mild cumulative irritation under normal conditions of the test); 450 to 580, class 4, experimental cumulative irritation (evidence of a strong potential for mild to moderate cumulative irritation under conditions of the test); 581 to 630, class 5, experimental primary irritant (evidence of potential for primary irritation under conditions of the test).
Pharmacokinetic Results
Pharmacokinetic parameters after single and repeat dosing are summarized in Table 5 and Table 6, respectively. The mean GSK1940029 plasma concentration versus time after single or repeat dosing is illustrated in Figure 4. The gel strength of GSK1940029 (ranging from 0.1% to 1%) exerted little or no influence on systemic exposure (AUC and Cmax) under occluded or nonoccluded conditions following a single dose and under nonoccluded conditions following repeated doses.
Table 5.
Summary of Plasma GSK1940029 Pharmacokinetic Parameters After Single Topical Application of GSK1940029 (Day 1) — Study 2
| Cohorta | Dose/Area | Application Occlusion | Load (mg/cm2) | AUC0‐24 (ng·h/mL) Mean (SD) | AUC0‐∞ (ng·h/mL) Mean (SD) | Cmax (ng/mL)Mean (SD) | AUC0‐24 (ng·h/mL) Geo Mean (SD)b | AUC0‐∞ (ng·h/mL) Geo Mean (SD)b | Cmax (ng/mL) Geo Mean (SD)b | tmax (h) Median (Min‐Max) | t1/2 (h) Mean (SD) | t1/2 (h) Geo Mean (SD)b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Part 1—single dosing | ||||||||||||
| 1 | 0.3%/400 cm2 | Yes | 10 | 1.93 (1.93) | N/A | 0.14 (0.129) | 1.26 (1.04) | N/A | 0.10 (0.939) | 16.0 (16.0–22.5) | N/A | N/A |
| 2 | 1%/400 cm2 | Yes | 10 | 2.66 (1.97) | N/A | 0.21 (0.111) | 2.02 (0.846) | N/A | 0.19 (0.540) | 19.3 (8.00–24.0) | N/A | N/A |
| 3 | 1%/1600 cm2 | Yes | 10 | 9.75 (4.16) | 11.6 (4.08) | 0.73 (0.334) | 9.03 (0.429) | 11.0 (0.353) | 0.67 (0.463) | 16.0 (8.00–16.0) | 4.53 (1.61) | 4.31 (0.337) |
| 4 | 0.3%/1600 cm2 | No | 1 | 1.57 (1.17) | N/A | 0.16 (0.171) | 1.26 (0.722) | N/A | 0.11 (0.801) | 7.00 (4.00–22.5) | N/A | N/A |
| 5 | 1%/1600 cm2 | No | 1 | 1.68 (1.82) | N/A | 0.14 (0.141) | 1.02 (1.14) | N/A | 0.09 (1.073) | 4.02 (4.00–22.6) | N/A | N/A |
| Part 2—day 1 of repeat dosing | ||||||||||||
| 1 | 0.1%/400 cm2 | No | 1 | 0.22 (0.142) | N/A | 0.03 (0.014) | 0.19 (0.641) | N/A | 0.02 (0.645) | 4.00 (4.00–16.0) | N/A | N/A |
| 2 | 0.3%/400 cm2 | No | 1 | 0.20 (0.124) | N/A | 0.02 (0.016) | 0.17 (0.756) | N/A | 0.02 (0.867) | 4.00 (4.00–23.7) | N/A | N/A |
n = 6 except for part 1 of cohort 5, where n = 5.
Geometric mean (SD of ln[geometric mean]).
Table 6.
Summary of Plasma GSK1940029 Pharmacokinetic Parameters After Repeat Dose Application of GSK1940029—Study 2, Part 2, Day 14
| Cohorta | Dose/Area | Application Occlusion | Load (mg/cm2) | AUC0‐24 (ng·h/mL) Mean (SD) | Cmax (ng/mL) Mean (SD) | AUC0‐24 (ng·h/mL) Geo Mean (SD)b | Cmax (ng/mL) Geo Mean (SD)b | tmax (h) Median (Min‐Max) | Ro Mean (SD) | Rcmax Mean (SD) | Ro Geo Mean (SD)b | Rcmax Geo Mean (SD)b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.1%/400 cm2 | No | 1 | 0.22 (0.045) | 0.03 (0.018) | 0.21 (0.210) | 0.03 (0.627) | 3.00 (1.00– 6.00) | 1.34 (0.855) | 1.50 (0.784) | 1.14 (0.621) | 1.33 (0.537) |
| 2 | 0.3%/400 cm2 | No | 1 | 0.24 (0.100) | 0.02 (0.010) | 0.23 (0.476) | 0.02 (0.477) | 4.00 (2.00– 6.00) | 1.69 (0.584) | 1.74 (0.637) | 1.61 (0.330) | 1.66 (0.348) |
n = 6.
Geometric mean (SD of ln[geometric mean]).
Figure 4.
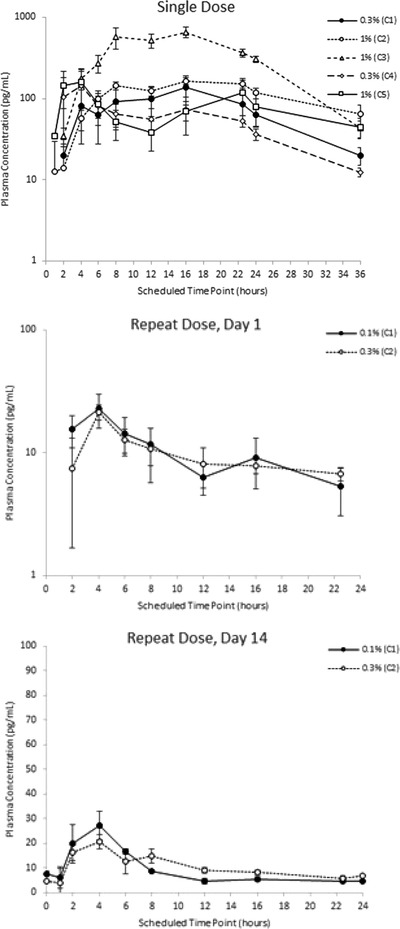
Mean plasma GSK1940029 concentration (SE) versus time – study 2, single and repeat dosing.
As illustrated in Figure 5, there was a relationship between occlusion condition, application area, and single‐dose exposure parameters of GSK1940029 on day 1. There was no relationship between gel strength (ranging from 0.1% to 1%) and single‐dose exposure.
Figure 5.
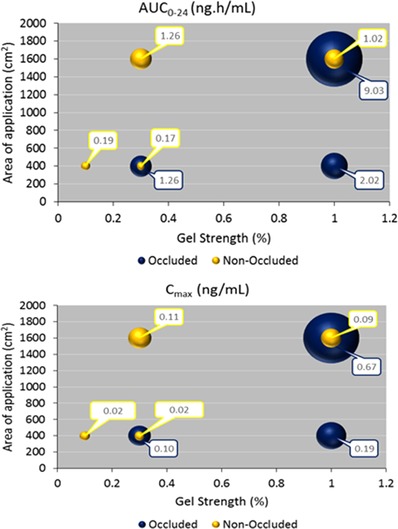
Influence of gel strength and area of application on select plasma GSK1940029 pharmacokinetic parameters after single dose of GSK1940029 (day 1).
Discussion
GSK1940029 gel is a novel SCD‐1 inhibitor for topical administration being developed as a potential treatment for acne. The data from the first application of the GSK1940029 gel formulation on human skin are presented.
This investigation was designed as 2 interdependent studies conducted in parallel. The irritation potential of different GSK1940029 gels was studied first after single and multiple doses. In the absence of irritation, the safety and pharmacokinetics of increasing gel strengths applied to successively larger surface areas were investigated in the second study after single and multiple doses, respectively.
The results of the first study indicated that GSK1940029 gel (0.3% and 1%) and the vehicle were not primary or cumulative irritants with 2 and 21 days of dosing, respectively, under the conditions of this study (Figure 1, Table 3, and Table 4). Overall, the irritation scores were lower than the positive controls and within normal limits of use (Figure 1, Table 3, and Table 4).
The results of the first part of study 1 allowed for progress into study 2, in which GSK1940029 gel strengths of 0.3% and 1% were administered as occluded applications at a higher load (10‐mg formulation/cm2) over 400 and 1600 cm2 and as nonoccluded applications at a lower load (1‐mg formulation/cm2) over 1600 cm2. In addition topical nonoccluded applications of GSK1940029 at a lower load (1‐mg formulation/cm2) evaluated 0.1% and 0.3% gel over a smaller surface area of 400 cm2.
Single and repeat administration of GSK1940029 at all doses was well tolerated during this study. There were no clinically significant findings in vital signs, clinical laboratory tests, and ECGs during either part of this study. In addition, there were no clinically significant findings in ocular evaluations and renal function and liver function tests. There were no SAEs or withdrawals because of AEs.
After single topical application of GSK1940029 gel (day 1), the 6 dose cohorts together involved changes in the load (either 1‐ or 10‐mg formulation/cm2), gel strength (0.1%, 0.3%, or 1%), surface area of application (either 400 or 1600 cm2), either under occlusion or nonocclusion. This diversity of tested conditions provided an opportunity to explore how these factors could have influenced the systemic exposure to GSK1940029.
The potential influence of gel strength on GSK1940029 exposure was documented by the side‐by‐side comparison of the following cohorts: part 1 of cohort 2 versus part 1 of cohort 1 (common 400 cm2 application area, same load of 10‐mg formulation/cm2, both in occluded conditions), part 1 of cohort 5 versus part 1 of cohort 4 (common 1600 cm2 application area, same load of 1‐mg formulation/cm2, both in nonoccluded conditions), and part 2 of cohort 2 versus part 2 of cohort 1 (common 400 cm2 application area, same load of 1‐mg formulation/cm2, both in nonoccluded conditions). These data analyses revealed that the gel strength of GSK1940029 (ranging from 0.1% to 1%) exerted little or no influence on systemic exposure (AUC and Cmax) under occluded or nonoccluded conditions following a single dose and under non‐occluded conditions following repeated doses.
The side‐by‐side comparison of part 1 of cohort 3 versus part 1 of cohort 2 (common 1% gel strength, same load of 10‐mg formulation/cm2, both in occluded conditions) and of part 1 of cohort 4 versus part 2 of cohort 2 (common 0.3% gel strength, same load of 1‐mg formulation/cm2, both in nonoccluded conditions) provided a basis for exploring the influence of the area of surface application.
When part 1 of cohort 3 (1% gel, 1600 cm2) was compared with part 1 of cohort 2 (1% gel, 400 cm2), the geometric mean Cmax and AUC0‐24 were increased 3.6‐ and 4.4‐fold, respectively, for a 4‐fold increase in the application area (from 400 to 1600 cm2). This apparent dose (via surface area) proportionality in systemic exposure was confirmed by the power model analysis of plasma GSK1940029 pharmacokinetic parameters of cohort 3 versus cohort 2. When comparing exposure in part 1 of cohort 4 (0.3% gel, 1600 cm2) versus part 2 of cohort 2 (0.3% gel, 400 cm2), geometric mean Cmax and AUC0‐24 increased 5.5‐ and 7.4‐fold, respectively, for a 4‐fold increase in the application area (from 400 to 1600 cm2), in general still in agreement with proportionality. These observations illustrate that the area of surface application of the GSK1940029 gel is a major determinant of GSK1940029 systemic exposure.
The potential influence of occluding the application area is documented by the side‐by‐side comparison of the following cohorts: part 2 of cohort 2 versus part 1 of cohort 1 (common 0.3% gel strength, same application area of 400 cm2, however with different loads) and part 1 of cohort 5 versus part 1 of cohort 3 (common 1% gel strength, same application area of 1600 cm2, however with different loads). Similar to gel strength, it is believed that load did not play a role in systemic absorption/exposure. It has been observed that the majority of the applied dose was retained on the surface of the skin from in vitro experiments (data not shown); therefore, regardless of whether the dose was increased via gel strength or load increase, the majority of the applied dose did not get absorbed to affect systemic exposure. Hence, the exposure difference from this comparison could be viewed as solely because of occlusion versus nonocclusion rather than load difference.
In line with the known positive effect of occlusion on drug permeability through human skin, application of the GSK1940029 gel under occluded conditions resulted in a 7.4‐ to 8.9‐fold increase in geometric mean AUC0‐24 relative to nonoccluded conditions, assuming load (similar to gel strength) did not play a role in systemic absorption/exposure. The geometric mean Cmax increased to a slightly lesser extent, 5.0‐ to 7.4‐fold. Occlusion has been shown to increase percutaneous penetration of caffeine ex vivo because of the increase in relative humidity in the occluded condition combined with less evaporation of ethanol from skin surface compared with the open condition.10
The data obtained in this study were used to develop a plasma/dermis population pharmacokinetic model.11 The model predicted a free dermis concentration 1.6‐fold the human IC50, suggesting a different formulation with a higher flux would be needed for efficacy in clinical trials.
The design of these 2 interdependent studies allowed for the adequate assessment of irritation potential for topical GSK1940029 in parallel with the investigation of its PK and safety profiles. The design allows for the rapid evaluation of several formulation concentrations and allows direct progression into patient studies.
Acknowledgments
We thank Robert Noble for help with the statistical analyses. Editorial support was provided by GD Scientific & Medical Writing, LLC (Wynnewood, Pennsylvania), funded by GSK, for the following services: development of manuscript first draft, editorial suggestion to draft versions, assembling tables and figures, collating author comments, and referencing.
Declaration of Conflicting Interests
R.A.B., J.Z., A.A.M., and B.A.R. are employees of GSK and hold company stock. S.S. was the study investigator and was paid by GSK.
Funding
This study was sponsored and funded by GlaxoSmithKline (GSK).
Author Contributions
All listed authors met the criteria for authorship set forth by the International Committee for Medical Journal Editors. All authors were involved with study design, review of analyzed data and manuscript. Each author contributed important intellectual content during manuscript drafting or revision and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved. R.A.B. takes responsibility that this study has been reported honestly, accurately, and transparently, that no important aspects of the study have been omitted, and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
References
- 1. Mugglestone CJ, Mariz S, Lane ME. The development and registration of topical pharmaceuticals. Int J Pharm. 2012;435:22–26. [DOI] [PubMed] [Google Scholar]
- 2. Bellew S, Thiboutot D, Del Rosso JQ. Pathogenesis of acne vulgaris: What's new, what's interesting and what may be clinically relevant. J Drugs Dermatol. 2011;10:582–585. [PubMed] [Google Scholar]
- 3. Thiboutot D, Gollnick H. New insights into the management of acne: An update from the Global Alliance to Improve Outcomes in Acne Group. J Am Acad Dermatol. 2009;60:S1–S50. [DOI] [PubMed] [Google Scholar]
- 4. Janiczek‐Dolphin N, Cook J, Thiboutot D, et al. Can sebum reduction predict acne outcome? Br J Dermatol. 2010;163:683–688. [DOI] [PubMed] [Google Scholar]
- 5. Fain K, Alexander GC. Are Food and Drug Administration prescription drug safety plans working? A case study of isotretinoin. Pharmacoepidemiol. Drug Saf. 2013;22:1258–1262. [DOI] [PubMed] [Google Scholar]
- 6. Leyden JJ, Del Rosso JQ, Baum EW. The use of isotretinoin in the treatment of acne vulgaris clinical considerations and future directions. J Clin Aesthet Dermatol. 2014;7(2 suppl.):S3–S21. [PMC free article] [PubMed] [Google Scholar]
- 7. Guillou H, Zadravec D, Martin PGP, Jacobsson A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Prog Lipid Res. 2010;49:186–199. [DOI] [PubMed] [Google Scholar]
- 8. Uto Y. Recent progress in the discovery and development of stearoyl CoA desaturase inhibitors. Chem Phys Lipids. 2016;197:3–12. [DOI] [PubMed] [Google Scholar]
- 9. Food and Drug Administration . Draft Guidance for Industry: Skin Irritation and Sensitization Testing of Generic Transdermal Drug Products. Rockville, MD, December 1999. [Google Scholar]
- 10. Lboutounne Y, Silva J, Pazart L, et al. Microclimate next to the skin: influence on percutaneous absorption of caffeine (ex‐vivo study). Skin Res Techno. 2014;20:293–298. [DOI] [PubMed] [Google Scholar]
- 11. Zhu J, Wilde T, Brigandi R. Development of a plasma/dermis population pharmacokinetic model for GSK1940029. J Pharmacokinet Pharmacodyn. 2015;42:S99. [Google Scholar]