

Abstract
Background
The authors evaluated mocetinostat (a class I/IV histone deacetylase inhibitor) in patients with urothelial carcinoma harboring inactivating mutations or deletions in CREB binding protein [CREBBP] and/or E1A binding protein p300 [EP300] histone acetyltransferase genes in a single‐arm, open‐label phase 2 study.
Methods
Eligible patients with platinum‐treated, advanced/metastatic disease received oral mocetinostat (at a dose of 70 mg 3 times per week [TIW] escalating to 90 mg TIW) in 28‐day cycles in a 3‐stage study (ClinicalTrials.gov identifier NCT02236195). The primary endpoint was the objective response rate.
Results
Genomic testing was feasible in 155 of 175 patients (89%). Qualifying tumor mutations were CREBBP (15%), EP300 (8%), and both CREBBP and EP300 (1%). A total of 17 patients were enrolled into stage 1 (the intent‐to‐treat population); no patients were enrolled in subsequent stages. One partial response was observed (11% [1 of 9 patients; the population that was evaluable for efficacy comprised 9 of the 15 planned patients]); activity was deemed insufficient to progress to stage 2 (null hypothesis: objective response rate of ≤15%). All patients experienced ≥1 adverse event, most commonly nausea (13 of 17 patients; 77%) and fatigue (12 of 17 patients; 71%). The median duration of treatment was 46 days; treatment interruptions (14 of 17 patients; 82%) and dose reductions (5 of 17 patients; 29%) were common. Mocetinostat exposure was lower than anticipated (dose‐normalized maximum serum concentration [Cmax] after TIW dosing of 0.2 ng/mL/mg).
Conclusions
To the authors’ knowledge, the current study represents the first clinical trial using genomic‐based selection to identify patients with urothelial cancer who are likely to benefit from selective histone deacetylase inhibition. Mocetinostat was associated with significant toxicities that impacted drug exposure and may have contributed to modest clinical activity in these pretreated patients. The efficacy observed was considered insufficient to warrant further investigation of mocetinostat as a single agent in this setting.
Keywords: CREB binding protein (CREBBP), E1A binding protein p300 (EP300), histone deacetylase, mocetinostat, urothelial carcinoma
Short abstract
After the genomic‐based selection of patients with urothelial cancer with inactivating mutations/deletions in the histone acetyltransferase genes CREBBP and/or EP300, single‐agent mocetinostat appears to be associated with significant toxicities that limit drug exposure. This may have contributed to the limited activity noted in the current phase 2 study (response rate of 11%) among heavily pretreated patients with platinum‐refractory disease.
Introduction
Worldwide, urothelial carcinoma of the upper urinary tract and bladder results in 165,000 deaths annually.1 The majority of patients with metastatic disease experience disease progression despite platinum‐based chemotherapy, and salvage chemotherapy is reported to have only modest efficacy.2, 3 Recently, 5 immune checkpoint inhibitors were approved for patients with platinum‐refractory urothelial carcinoma and, although the anti–programmed cell death protein 1 (PD‐1) agent pembrolizumab has improved overall survival (OS) versus chemotherapy in this setting, many patients do not benefit from such therapy.4 Consequently, new treatment options are needed.
Dysregulated histone acetylation is implicated in the pathogenesis of several cancers, including urothelial carcinoma. Acetylation of chromatin by histone acetyltransferases (HATs) generally is associated with elevated transcription, whereas deacetylation, mediated by histone deacetylases (HDACs), is associated with repressed transcription.5, 6 Histone acetylation can become dysregulated through the upregulation of HDACs and/or genetic inactivation of HATs, resulting in the silencing of tumor supressor and other genes.5, 6 Inhibition of HDAC1 and HDAC2 resulted in antitumor activity in urothelial carcinoma in vitro, whereas elevated HDAC1 is linked with poor prognosis in patients with urothelial carcinoma.7, 8 HDAC inhibitors have demonstrated promise in clinical trials across a range of tumor types, and several have been approved by the US Food and Drug Administration, including vorinostat for patients with cutaneous T‐cell lymphoma, romidepsin for patients with cutaneous T‐cell lymphoma and peripheral T‐cell lymphoma, belinostat for patients with peripheral T‐cell lymphoma, and panobinostat for patients with multiple myeloma.9
Mocetinostat is an investigational HDAC inhibitor that targets class I and class IV HDACs (isoforms 1, 2, 3, and 11),10 and has demonstrated antitumor activity in patients with hematologic malignancies.11, 12, 13 In vivo, mocetinostat induces cell cycle arrest and apoptosis and inhibits tumor growth.10 Furthermore, a HAT inactivation signature associated with muscle‐invasive bladder cancer was found to be inversely influenced by mocetinostat in breast cancer cells.14 Mocetinostat also demonstrated preferential activity in CREB binding protein (CREBBP)–mutated and/or E1A binding protein p300 (EP300)–mutated (HAT genes) xenograft models and solid tumor cell lines, including urothelial cell carcinoma (see Supporting Tables S1 and S2 and Supporting Fig. S1). Thus, we hypothesized that treating patients with urothelial carcinoma harboring inactivating mutations in CREBBP and EP300 with selective HDAC inhibitors may restore the expression of tumor suppressor genes, resulting in antitumor responses.
This phase 2 study investigated single‐agent mocetinostat in patients with locally advanced or metastatic urothelial carcinoma who previously were treated with platinum‐based chemotherapy and inactivating tumor mutations or deletions in CREBBP and/or EP300.
Materials and Methods
Patients and Study Design
The current phase 2, open‐label, single‐arm, 3‐stage, multicenter study was conducted between November 2014 and July 2016 (ClinicalTrials.gov identifier NCT02236195). Patients with histologically confirmed, locally advanced, unresectable or metastatic urothelial (transitional cell) carcinoma who developed disease progression after receipt of platinum‐based chemotherapy were recruited. Eligible patients had adequate bone marrow, hepatic, and renal function and an inactivating mutation or deletion (homozygous or hemizygous) in CREBBP and/or EP300 (see Supporting Materials). Genomic prescreening of tumor tissue (primary or metastatic; archival tissue was permitted if a fresh biopsy was not available) was performed centrally using next‐generation sequencing (Foundation Medicine, Cambridge, Massachusetts) or a sponsor‐approved, local sequencing platform (FoundationOne [Foundation Medicine] and MSK‐IMPACT [Memorial Sloan Kettering Cancer Center, New York, New York]) or next‐generation sequencing (Oncopanel; Center for Advanced Molecular Diagnostics, Brigham and Women’s Hospital, Boston, Massachusetts) capturing the full coding regions for CREBBP and EP300. Key exclusion criteria included prior or current treatment with an HDAC inhibitor and symptomatic or uncontrolled brain metastases.
Oral mocetinostat (Mirati Therapeutics Inc, San Diego, California) was administered in continuous 28‐day cycles at a starting dose of 70 mg 3 times per week (TIW) for stage 1 of the study. Escalation to 90 mg TIW on day 1 of cycle 2 was planned for patients without treatment‐related grade ≥3 adverse events (AEs), and 90 mg TIW was the planned starting dose for the cohorts in stage 2 and stage 3 of the study. Mocetinostat was continued until disease progression or unacceptable AEs occurred.
The protocol was approved by the institutional review boards at each institution, and the study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines. All patients provided written informed consent.
Study Endpoints and Assessments
The primary endpoint was the objective response rate (ORR; complete response and partial response [PR] as per Response Evaluation Criteria in Solid Tumors [RECIST], version 1.1 ). Secondary endpoints included duration of response, progression‐free survival (PFS; overall and at month 4), OS, 1‐year survival rate, safety, and pharmacokinetics.
Computed tomography scans for tumor evaluation were performed at baseline and at 8‐week intervals for the first 12 months and at 12‐week intervals thereafter. AEs were graded as per National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03).
Plasma concentrations of mocetinostat were determined using high‐performance liquid chromatography and tandem mass spectrometry during stage 1 (before the dose and 1 hour after the dose on day 1 of cycles 1 and 2) with more timepoints planned for stage 2.
Tumor total mutation burden was estimated retrospectively in the 322 target genes included in FoundationOne for patients with central testing (see Supporting Materials).
Statistical Analyses
The primary endpoint, ORR, was assessed using an exact test for a single proportion (2‐sided α=0.05; ORR ≤15% [H0] vs >15% [H1]) in a 3‐stage study design to include 15 patients, 18 patients, and 67 patients, respectively, in the population that was evaluable for efficacy (patients meeting the entry criteria who received mocetinostat and had at least baseline and one on‐study disease assessment) (see Supporting Materials). Safety was assessed in patients receiving ≥1 dose of mocetinostat. Pharmacokinetics were evaluated in all patients with sufficient data. Time‐to‐event efficacy endpoints were estimated using Kaplan‐Meier methodology (see Supporting Materials).
Results
Patient Disposition and Baseline Disease Characteristics
Of the 175 patients who consented to undergo genomic screening, testing was feasible for 155 (89%; sample quantity/quality was insufficient for 20 patients). Frequently altered genes included TP53, AT‐rich interaction domain 1A [ARID1A], MLL2 (KMT2D), KDM6A, MLL3 (KMT2C), retinoblastoma protein (RB1), and cyclin‐dependent kinase inhibitor 2A/B (CDKN2A/B) (Fig. 1).
Figure 1.
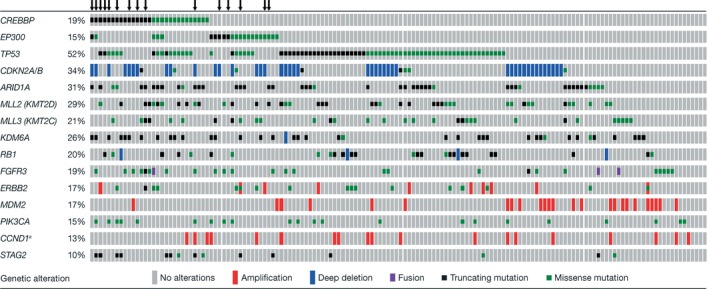
OncoPrint of genetic alterations in 150 of the 155 patients in whom genetic profiling of the tumor was feasible. Alterations included truncating mutations, gene amplifications, homozygous deletions, annotated recurrent missense mutations, and missense variants of uncertain significance (variants of unknown significance were excluded from the main study analysis) that were present in ≥10% of the population. The 150 patients included 144 patients who were tested centrally at Foundation Medicine and 6 patients who were tested at local institutions. An arrow (↓) denotes a patient enrolled in the clinical trial (reports from 5 patients tested locally were not available, including 4 patients who were prescreened using FoundationOne testing and including 2 enrolled patients). aIn cases of CCND1 amplification, this co‐occurred with fibroblast growth factor 3 (FGF3), FGF4, or FGF19 amplification in >80% of cases. In addition, a significant correlation for the co‐occurrence of retinoblastoma protein (RB1) and TP53 mutations and CREB binding protein (CREBBP) and STAG2 mutations and the mutual exclusivity of cyclin‐dependent kinase inhibitor 2A (CDKN2A) homozygous deletion and TP53 mutation or mouse double minute 2 homolog (MDM2) amplification and TP53 mutation was observed. ARID1A indicates AT‐rich interaction domain 1A; CDKN2A/B, cyclin‐dependent kinase inhibitor 2A/B; EP300, E1A binding protein p300; PIK3CA, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit alpha.
Thirty‐three patients (21%) had ≥1 of the 40 qualifying tumor mutations in CREBBP or EP300 identified: 27 CREBBP mutations were identified among 23 patients (15%), 13 EP300 mutations were identified among 12 patients (8%), and mutations in both genes were identified in 2 patients (1%). Each qualifying mutation was observed only once within the study. Qualifying CREBBP alterations were most commonly nonsense (5% [8 patients]), frameshift (5% [7 patients]), or missense (3% [5 patients]) mutations. EP300 mutations were most commonly missense mutations (3% [5 patients]). Nonqualifying mutations in CREBBP and EP300 (putative passenger mutations) were detected in 18 patients (12%) (see Supporting Table S3).
Seventeen of 33 patients with qualifying mutations were enrolled into stage 1 (Fig. 2); baseline demographic and disease characteristics of the enrolled patients are shown in Table 1. Twenty‐two qualifying mutations were identified in these 17 patients: 14 CREBBP mutations in 12 patients and 8 EP300 mutations in 7 patients, and 2 patients had qualifying mutations of both CREBBP and EP300 (see Supporting Table S3). Sixteen patients with qualifying mutations were not enrolled, most often because they were receiving an earlier line of therapy (Fig. 2). The patients received a median of 2 prior systemic therapies (range, 1‐5 prior systemic therapies) (Table 1) and all had discontinued mocetinostat at the time of analysis, most due to disease progression (53%) or AEs (24%) (Fig. 2). Based on the decision of the sponsor, the current study was closed after the enrollment of 17 patients, including 9 patients in the population that was evaluable for efficacy (8 patients withdrew from mocetinostat treatment prior to the on‐study disease assessment [4 due to AEs and 3 due to symptomatic deterioration, and 1 patient withdrew consent]); patients were not recruited for stages 2 and 3 of the current study.
Figure 2.
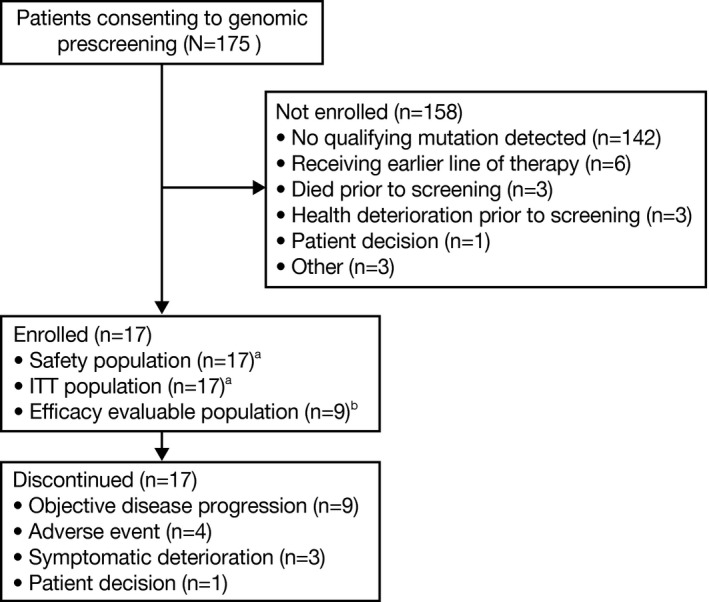
Patient disposition. aThe safety population and intent‐to‐treat (ITT) population included all patients who received at least 1 dose of the study medication. bThe population evaluable for efficacy included all patients in the ITT population who met prespecified entry criteria and had at least a baseline and 1 on‐study disease assessment that were adequate for evaluation using Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1).
Table 1.
Patient Demographics and Disease Characteristics in the ITT Population
| Patient Characteristics | Mocetinostat |
|---|---|
| N=17 | |
| Median age (range), y | 67 (35‐83) |
| Male sex, no. (%) | 15 (88) |
| Race, no. (%) | |
| White | 15 (88) |
| Asian | 1 (6) |
| Black | 1 (6) |
| Smoking history, no. (%) | |
| Past smoker | 8 (47) |
| Never smoker | 7 (41) |
| Current smoker | 2 (12) |
| AJCC/UICC TNM stage of disease, no. (%)a | |
| IVA | 1 (6) |
| IVB | 16 (94) |
| ECOG PS, no. (%) | |
| 0 | 5 (29) |
| 1 | 10 (59) |
| 2 | 2 (12) |
| Scores according to Bellmunt et al, no. (%)b | |
| 0 | 5 (29) |
| 1 | 7 (41) |
| 2 | 5 (29) |
| Median baseline albumin (range), g/dL | 4.1 (3.1‐4.7) |
| Median baseline hemoglobin (range), g/dL | 12.5 (9.0‐14.5) |
| Time from diagnosis of urothelial carcinoma (range), mo | 26.4 (4.3‐95.5) |
| Location of disease, no. (%)c | |
| Lung | 13 (77) |
| Liver | 6 (35) |
| Lymph noded | 15 (88) |
| Bladder | 3 (18) |
| Bone | 4 (24) |
| Other | 8 (47) |
| Prior systemic therapy, no. (%) | 17 (100) |
| Median no. of prior regimens (range) | 2 (1‐5) |
| Patients with prior neoadjuvant/adjuvant regimens, no. (%) | 10 (59) |
| Patients with prior advanced disease regimens, no. (%) | 12 (71) |
| Patients who completed prior systemic therapy ≤3 mo before initiating study treatment, no. (%) | 7 (41) |
| Prior RT, no. (%) | 6 (35) |
| Prior surgery, no. (%)c | 15 (88) |
| Cystectomy | 10 (59) |
| Transurethral resection of bladder tumor | 9 (53) |
| Urethrectomy | 4 (24) |
| Other | 4 (24) |
Abbreviations: AJCC/UICC TNM, American Joint Committee on Cancer/Union for International Cancer Control (T) Tumor‐(N) Lymph Node‐(M) Metastasis; ECOG PS, Eastern Cooperative Oncology Group performance status; ITT; intent to treat (all patients receiving study medication); RT, radiotherapy.
Disease subsite (bladder, ureter, or renal pelvis) and disease stage were not specifically collected in the current study; disease stage using definitions for bladder cancer was assessed retrospectively.
Scores according to Bellmunt et al were assessed retrospectively.15
Patients may have >1 disease location or surgery.
Baseline disease was confined only to the lymph nodes in 2 patients.
Efficacy
One objective response was observed in the population of patients who were evaluable for efficacy. This PR lasted 3.9 months and occurred in a 67‐year‐old man with disease restricted to the lymph nodes. His primary tumor contained 2 qualifying EP300 missense mutations (G1347E and P925T) and other mutations (truncating mutations in ARID1A, MLL2 [KMT2D], and CHEK2; a missense mutation in ATM; and amplification of TERC and PRKCI). The ORR of 11% (95% confidence interval [95% CI], 0.3%‐48%) was not statistically significant (null hypothesis of ≤15% could not be rejected; P=1.00). Two patients (22%) were found to have achieved stable disease lasting 3.5 months and 3.8 months, respectively, and progressive disease was reported in 6 patients (67%) (see Supporting Fig. S2). The median PFS was 57 days (95% CI, 23‐117 days) in the population of patients who were evaluable for efficacy. The estimated PFS at 4 months was 10% (95% CI, 0%‐40%); the PFS at 1 year could not be estimated. The median OS was 3.5 months (95% CI, 2.1‐15.7 months) and the 1‐year survival rate was 30% (95% CI, 10%‐60%) in the intent‐to‐treat (ITT) population (all patients who received the study medication). Similar efficacy results were observed in the population that was evaluable for efficacy and the ITT population.
Safety
The median duration of mocetinostat therapy was 46 days (range, 8‐225 days), and the cumulative median dose administered was 930 mg (range, 280‐7730 mg). The median relative dose intensity was 99% (range, 37%‐117%) during cycle 1 and was 84% (range, 14%‐117%) in subsequent cycles. Eleven of the 17 enrolled patients initiated ≥2 treatment cycles. The dose of mocetinostat was escalated from 70 mg TIW to 90 mg TIW in 9 patients (4 of whom received ≤1 full cycle of mocetinostat at a dose of 90 mg TIW). Five patients (29%) underwent dose reductions due to AEs (3 patients; 18%) or other reasons (2 patients; 12%), and 14 patients (82%) had at least 1 dose interruption, most commonly due to AEs (11 patients; 65%).
All patients experienced ≥1 treatment‐emergent (all causality) AE, and the majority of patients (14 patients; 82%) experienced ≥1 treatment‐related AE. The most frequent treatment‐emergent AEs were nausea (13 patients; 77%), fatigue (12 patients; 71%), decreased appetite (8 patients; 47%), and diarrhea (8 patients; 47%) (Table 2); these events also were the most frequent treatment‐related AEs. Grade ≥3 treatment‐related AEs were fatigue and hyponatremia (in 2 patients each, respectively; 12%). A total of 21 treatment‐emergent serious AEs were reported in 10 patients (59%), including vomiting, lower gastrointestinal hemorrhage, abdominal pain, and pericardial effusion (2 patients each; 12%). One serious AE of pericardial effusion was assessed as being related to mocetinostat (both pericardial effusion events resolved). Ten patients died during the study, all due to their underlying disease.
Table 2.
Treatment‐Emergent (All Causality) Adverse Events Occurring in at Least 3 Patients (Safety Population)
| MedDRA Preferred Term | All Gradesa | Grade 3/4a |
|---|---|---|
| No. (%) | N=17 | N=17 |
| Nausea | 13 (77) | 1 (6) |
| Fatigue | 12 (71) | 3 (18) |
| Decreased appetite | 8 (47) | NR |
| Diarrhea | 8 (47) | NR |
| Hyponatremia | 6 (35) | 3 (18) |
| Vomiting | 6 (35) | 1 (6) |
| Abdominal pain | 5 (29) | 2 (12) |
| Anemia | 5 (29) | 2 (12) |
| Back pain | 5 (29) | NR |
| Constipation | 5 (29) | NR |
| Hypoalbuminemia | 5 (29) | NR |
| Hematuria | 4 (24) | NR |
| Muscular weakness | 4 (24) | NR |
| Alkaline phosphatase increased | 3 (18) | NR |
| Chills | 3 (18) | NR |
| Cough | 3 (18) | NR |
| Creatinine increased | 3 (18) | NR |
| Dehydration | 3 (18) | 1 (6) |
| Dizziness | 3 (18) | NR |
| Dysgeusia | 3 (18) | NR |
| Hypocalcemia | 3 (18) | NR |
| Lymphocyte count decreased | 3 (18) | 1 (6) |
| Pain | 3 (18) | 1 (6) |
| Urinary tract infections | 3 (18) | NR |
Abbreviations: MedDRA, Medical Dictionary for Regulatory Activities; NR, not reported.
Adverse events were graded as per National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03).
Pharmacokinetics
Due to the limited blood sampling schedule for stage 1 of the current study, the 1‐hour post‐dose sample was considered representative of the maximum serum concentration (Cmax) based on data from prior studies (see Supporting Information), and pharmacokinetic analyses were restricted to Cmax and time to Cmax.
After a single 70‐mg dose of mocetinostat, the mean Cmax was found to be 105 ng/mL. The mean dose‐normalized Cmax was 1.2 ng/mL/mg and intersubject variability (coefficient of variation, geometric mean) was 90%. After multiple TIW doses of mocetinostat of 50 mg and 90 mg, respectively, the mean Cmax was 41 ng/mL and 39 ng/mL (see Supporting Table S4). The mean dose‐normalized Cmax was 0.2 ng/mL/mg and the intersubject variability was 423%.
Discussion
Inactivating alterations of CREBBP and EP300 are relatively frequent (approximately 13% and approximately 15%, respectively) in patients with urothelial carcinoma14, 16, 17 and are implicated in the dysregulation of key acetylation pathways and oncogenesis.18, 19 Based on promising findings in urothelial carcinoma cell lines and tumor models (see Supporting Tables S1 and S2 and Supporting Fig. S1), we postulated that patients with urothelial carcinoma and inactivating alterations in CREBBP and/or EP300 could be treated by class I HDAC inhibition via a mechanism of increased histone acetylation leading to an open chromatin state with decreased transcriptional repression of tumor suppressor genes. Although the maximum tolerated dose of mocetinostat as a single agent was determined to be 110 mg TIW in other tumor settings, a lower recommended dose of 90 TIW was considered for the current study based on prior observations of pericardial infusion and balancing pharmacodynamic and clinical data as well as regulatory guidance.20 However, single‐agent mocetinostat at doses up to 90 mg TIW demonstrated only modest activity in this cohort of heavily pretreated patients with factors indicative of a poor prognosis. The ORR of 11% and the finding that of 9 evaluable patients, only 1 patient (with lymph node–only disease and multiple genomic alterations) was alive and free of disease progression for 4 months was not consistent with meaningful clinical activity. Although mocetinostat‐related AEs, including gastrointestinal events and fatigue, were consistent with the safety profiles reported in other settings,11, 12, 21 frequent dose interruptions and reductions were required. Mocetinostat exposure (mean dose‐normalized Cmax of 0.2 ng/mL/mg) was lower than in prior trials of mocetinostat TIW (range, 0.8‐1.6 ng/mL/mg). It is feasible that underlying disease and prior treatments may have contributed to limited functional reserve, resulting in poor tolerability. These findings underscore the limitations of preclinical models in predicting clinical activity and toxicity issues related to anticancer treatments. Further evaluation of mocetinostat at lower doses may be useful for guiding dose reduction in future study protocols to maximize each patient’s exposure to treatment.
Studies of other HDAC inhibitors in patients with urothelial carcinoma reported mixed results, with responses noted with single‐agent vorinostat but not when vorinostat was combined with doxorubicin or docetaxel.22, 23, 24 An ORR of 20% was reported in a small study of belinostat or panobinostat, and prolonged stable disease was noted in 1 of 3 patients with urothelial carcinoma who were treated with entinostat plus 13‐cis‐retinoic acid.25, 26 These data suggest that HDAC inhibitors can be active in patients with urothelial carcinoma, but predictive biomarkers are needed for patient selection. To our knowledge, data regarding genomic predictors of response to HDAC inhibitors are limited. In a phase 2 study of panobinostat in patients with recurrent diffuse large B‐cell lymphoma, mutations in MEF2B were found to be associated with response, whereas 14 genes, including TOX4, PSMD13, and CCNK, were associated with resistance to vorinostat based on a study of human colon cancer cell lines.27, 28 To our knowledge, this is the first clinical trial using genomic‐based selection to identify patients with urothelial carcinoma for treatment with selective HDAC inhibition. The results of the current study demonstrate the feasibility of this approach while also providing genomic tumor characterization for this population of patients.
There was considerable genomic variation in CREBBP and EP300, with each qualifying mutation observed only once in the current study. It is interesting to note that the patient with a confirmed PR harbored 2 EP300 mutations in trans, P925T, and G1347E, suggesting that biallelic loss of function in this pathway could be therapeutically significant; however, this patient had lymph node–only metastasis, which is a favorable prognostic factor. It is feasible that mocetinostat activity might be greater as an earlier line of therapy when a longer duration of treatment may be feasible and potentially confer meaningful disease‐modifying activity. Furthermore, we hypothesized a mechanism of action of mocetinostat to reactivate the transcription of tumor suppressor genes, but a relatively high frequency of inactivating alterations in the tumor suppressor genes TP53, CDKN2A/B, and RB1 may have limited the potential of epigenetic modulation by mocetinostat to induce tumor response. Potential future treatment strategies could include combining mocetinostat with an inhibitor of PD‐1/programmed death–ligand 1 (PD‐L1) to take advantage of the former’s potential immunomodulatory effects. Indeed, mocetinostat has been shown to increase the expression of PD‐L1 and augment PD‐1/PD‐L1 checkpoint blockade immunotherapy.29 Other combination partners could be considered within the appropriate molecular context.
In summary, single‐agent mocetinostat was found to be associated with significant toxicities and limited activity in heavily pretreated patients with advanced/metastatic urothelial carcinoma and poor prognostic factors. Few patients received the intended dose of 90 mg TIW, which may have compromised efficacy. Nevertheless, the clinical activity observed does not warrant the further investigation of mocetinostat as a single agent in this setting. Mocetinostat currently is being investigated in other tumors and in combination with immunotherapy.
Funding Support
Supported by Mirati Therapeutics Inc.
Conflict of Interest Disclosures
Petros Grivas has served as consultant for Genentech/Roche, Bristol‐Myers Squibb, Merck and Company, AstraZeneca, EMD Serono, Clovis Oncology, Foundation Medicine, Driver Inc, Seattle Genetics, Dendreon, Bayer, Pfizer, Exelixis, QED Therapeutics, and Biocept and received research funding from Genentech/Roche, Bayer, Merck and Company, Mirati Therapeutics Inc, OncoGenex, Pfizer, Bristol‐Myers Squibb, AstraZeneca, and Clovis Oncology for work performed outside of the current study; his institution received research funding from Mirati Therapeutics for work performed as part of the current study. Amir Mortazavi has served as a consultant and advisory board member for Genentech/Roche, received honoraria from Motive Medical Intelligence, and his institution has received research funding from Acerta Pharma, Merck, Genentech/Roche, Novartis, Seattle Genetics, and Bristol‐Myers Squibb for work performed outside of the current study and from Mirati Therapeutics for work performed as part of the current study. Joel Picus's institution has received research funding from Mirati Therapeutics for work performed as part of the current study. Noah M. Hahn has served as a consultant for OncoGenex, AstraZeneca, Merck, Bristol‐Myers Squibb, Genentech, Inovio, Principia Biopharma, Ferring, TARIS, Eli Lilly, Advanced Health, Seattle Genetics, Rexahn Pharmaceuticals, and Pieris Pharmaceuticals, received honoraria from the Bladder Cancer Academy and his institution has received research funding from OncoGenex, Seattle Genetics, Merck, Genentech, Bristol‐Myers Squibb, AstraZeneca, Principia Biopharma, Pieris Pharmaceuticals, and Inovio for work performed outside of the current study and from Mirati Therapeutics for work performed as part of the current study. Matthew I. Milowsky has served as a consultant for BioClin Therapeutics and his institution has received research funding from Acerta Pharma, Astellas, Seattle Genetics, Bristol‐Myers Squibb, Incyte, Merck, Pfizer, and Roche/Genentech for work performed outside of the current study and from Mirati Therapeutics for work performed as part of the current study. Lowell L. Hart's institution has received research funding from Mirati Therapeutics for work performed as part of the current study. Ajjai Alva's institution has received research funding from Mirati Therapeutics for work performed as part of the current study. Joaquim Bellmunt has served as a consultant, member of the advisory board, and provided lectures for Merck; acted as a member of the advisory board and provided lectures for Roche and Pfizer; acted as a consultant and member of the advisory board for AstraZeneca and Pierre Fabre; and acted as a member of the advisory board for Bristol‐Myers Squibb for work performed outside of the current study; and his institution received research funding from Mirati Therapeutics for work performed as part of the current study. Sumanta K. Pal has served as a member of the advisory board for Exelixis, Bristol‐Myers Squibb, Astellas, GlaxoSmithKline, Novartis, and Pfizer and has acted as a member of the advisory board and received honoraria from Genentech for work performed outside of the current study; her institution received research funding from Mirati Therapeutics for work performed as part of the current study. Peter H. O'Donnell owns stock in Allergan; has received honoraria from Algeta ASA, American Medical Forum, Astellas, AstraZeneca/MedImmune, Genentech/Roche, Harrison Consulting, Inovio Pharmaceuticals, Janssen Biotech, Kantar Health, Merck, Novartis, Parexel, Quintiles, Seattle Genetics, and Xcenda; has received travel expenses from Merck; has provided expert testimony for Trinity Health and Temple Health; is a coinventor of a genomic prescribing system (patent pending); and his institution has received research funding from Genentech/Roche, Merck, Boehringer Ingelheim, Acerta Pharma, Seattle Genetics, Janssen Biotech, and AstraZeneca/MedImmune for work performed outside of the current study and from Mirati Therapeutics for work performed as part of the current study. Sumati Gupta's institution received research support from Bristol‐Myers Squibb, Clovis Oncology, Five Prime Therapeutics, Hoosier Oncology Group, Incyte, LSK BioPharma, Merck, Novartis, Pfizer, and Rexahn Pharmaceuticals for work performed outside of the current study and from Mirati Therapeutics for work performed as part of the current study. Elizabeth A. Guancial has served as a consultant for TARIS and as a member of the Speakers' Bureau for Genentech for work performed outside of the current study, and her institution received research funding from Mirati Therapeutics for work performed as part of the current study. Guru P. Sonpavde has served as a consultant for Bristol‐Myers Squibb, Exelixis, Bayer, Sanofi, Pfizer, Novartis, Eisai, Janssen, Amgen, AstraZeneca, Merck, Genentech, Pfizer, Biotheranostics, the National Comprehensive Cancer Network, and Astellas/Agensys; acted as a speaker for Clinical Care Options, Physicians Education Resource, Research to Practice, and OncLive; received author royalties from UpToDate; acted as a member of the Data Safety Monitoring Board for AstraZeneca and his institution has received research/clinical trial support from Celgene, Bayer, Onyx‐Amgen, Boehringer‐Ingelheim, Merck, and Pfizer for work performed outside of the current study. Demiana Faltaos is an employee of Mirati Therapeutics Inc. Diane Potvin has acted as a paid consultant for Mirati Therapeutics Inc. James G. Christensen and Richard C. Chao are employees and shareholders of Mirati Therapeutics Inc. Jonathan E. Rosenberg has received trial funding from Novartis and Genentech/Roche; has served as a consultant for Genentech/Roche, Bristol‐Myers Squibb, Merck, AstraZeneca, Inovio, Bayer, Seattle Genetics, Mirati Therapeutics Inc, BioClin, EMD Serono, QED Therapeutics, and Astellas; has received travel funding from Genentech/Roche; and his institution has received research funding from AstraZeneca, Genentech/Roche, Astellas, and Seattle Genetics for work performed outside of the current study and from Mirati Therapeutics for work performed as part of the current study.
Author Contributions
Petros Grivas: Methodology, resources, investigation, writing–original draft, and writing–review and editing. Amir Mortazavi, Joel Picus, Noah M. Hahn, Matthew I. Milowsky, Lowell L. Hart, Ajjai Alva, Joaquim Bellmunt, Sumanta K. Pal, Richard M. Bambury, Peter H. O’Donnell, Sumati Gupta, Elizabeth A. Guancial, and Guru P. Sonpavde: Resources, investigation, and writing–review and editing. Demiana Faltaos and Diane Potvin: Data curation, formal analysis, and writing–review and editing. James G. Christensen: Conceptualization, methodology, formal analysis, writing–review and editing, visualization, and project administration. Richard C. Chao: Methodology, formal analysis, writing–review and editing, visualization, and project administration. Jonathan E. Rosenberg: Conceptualization, methodology, resources, investigation, writing–original draft, and writing–review and editing.
Supporting information
We thank the patients and their families who participated in this study, Josée Morin (Excelsus Statistics Inc, Montreal, Quebec, Canada) for her review of the article, and Mary Collier (Mirati Therapeutics Inc) for coordinating the conduct of the trial. This work was supported in part by National Cancer Institute Cancer Center Support grant P30 CA008748. Medical writing services were provided by Siân Marshall (Siantifix Limited, Cambridge, United Kingdom) and funded by Mirati Therapeutics Inc.
References
- 1. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol. 2017;71:96‐108. [DOI] [PubMed] [Google Scholar]
- 2. Bellmunt J, Theodore C, Demkov T, et al. Phase III trial of vinflunine plus best supportive care compared with best supportive care alone after a platinum‐containing regimen in patients with advanced transitional cell carcinoma of the urothelial tract. J Clin Oncol. 2009;27:4454‐4461. [DOI] [PubMed] [Google Scholar]
- 3. Choueiri TK, Ross RW, Jacobus S, et al. Double‐blind, randomized trial of docetaxel plus vandetanib versus docetaxel plus placebo in platinum‐pretreated metastatic urothelial cancer. J Clin Oncol. 2012;30:507‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bellmunt J, de Wit R, Vaughn DJ, et al. KEYNOTE‐045 Investigators. Pembrolizumab as second‐line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898‐3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roy DM, Walsh LA, Chan TA. Driver mutations of cancer epigenomes. Protein Cell. 2014;5:265‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Poyet C, Jentsch B, Hermanns T, et al. Expression of histone deacetylases 1, 2 and 3 in urothelial bladder cancer. BMC Clin Pathol. 2014;14:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pinkerneil M, Hoffmann MJ, Deenen R, et al. Inhibition of class I histone deacetylases 1 and 2 promotes urothelial carcinoma cell death by various mechanisms. Mol Cancer Ther. 2016;15:299‐312. [DOI] [PubMed] [Google Scholar]
- 9. Suraweera A, O’Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Oncol. 2018;8:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou N, Moradei O, Raeppel S, et al. Discovery of N‐(2‐aminophenyl)‐4‐[(4‐pyridin‐3‐ylpyrimidin‐2‐ylamino)methyl]benzamide (MGCD0103), an orally active histone deacetylase inhibitor. J Med Chem. 2008;51:4072‐4075. [DOI] [PubMed] [Google Scholar]
- 11. Batlevi CL, Crump M, Andreadis C, et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br J Haematol. 2017;28:2047‐2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blum KA, Advani A, Fernandez L, et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br J Haematol. 2009;147:507‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Younes A, Oki Y, Bociek RG, et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: an open‐label, single‐arm, phase 2 trial. Lancet Oncol. 2011;12:1222‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duex JE, Swain KE, Dancik GM, et al. Functional impact of chromatin remodeling gene mutations and predictive signature for therapeutic response in bladder cancer. Mol Cancer Res. 2018;16:69‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bellmunt J, Choueiri TK, Fougeray R, et al. Prognostic factors in patients with advanced transitional cell carcinoma of the urothelial tract experiencing treatment failure with platinum‐containing regimens. J Clin Oncol. 2010;28:1850‐1855. [DOI] [PubMed] [Google Scholar]
- 16. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robertson AG, Kim J, Al‐Ahmadie H, et al. Comprehensive molecular characterization of muscle‐invasive bladder cancer. Cell. 2017;171:540‐556.e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225‐4231. [DOI] [PubMed] [Google Scholar]
- 19. Wang F, Marshall CB, Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol Life Sci. 2013;70:3989‐4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boumber Y, Younes A, Garcia‐Manero G. Mocetinostat (MGCD0103): a review of an isotype‐specific histone deacetylase inhibitor. Expert Opin Investig Drugs. 2011;20:823‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chan E, Chiorean EG, O’Dwyer PJ, et al. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother Pharmacol. 2018;81:355‐364. [DOI] [PubMed] [Google Scholar]
- 22. Schneider BJ, Kalemkerian GP, Bradley D, et al. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Invest New Drugs. 2012;30:249‐257. [DOI] [PubMed] [Google Scholar]
- 23. Munster PN, Marchion D, Thomas S, et al. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer. 2009;101:1044‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheung EM, Quinn DI, Tsao‐Wei DD, et al. Phase II study of vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced transitional cell urothelial cancer after platinum‐based therapy. J Clin Oncol. 2008;26(suppl 15):16058. [Google Scholar]
- 25. Pili R, Salumbides B, Zhao M, et al. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13‐cis retinoic acid in patients with solid tumours. Br J Cancer. 2012;106:77‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gupta S, Albertson DJ, Parnell T, et al. Clinical and translational investigation of pan‐HDAC inhibition in advanced urothelial carcinoma. J Clin Oncol. 2017;35(suppl 6):379.28095275 [Google Scholar]
- 27. Assouline SE, Nielsen TH, Yu S, et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B‐cell lymphoma. Blood. 2016;128:185‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Falkenberg KJ, Gould CM, Johnstone RW, Simpson KJ. Genome‐wide functional genomic and transcriptomic analyses for genes regulating sensitivity to vorinostat. Sci Data. 2014;1:140017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD‐1 ligands in melanoma and augments immunotherapy with PD‐1 blockade. Cancer Immunol Res. 2015;3:1375‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials