

Abstract
Although many large mammal species went extinct at the end of the Pleistocene epoch, their DNA may persist due to past episodes of interspecies admixture. However, direct empirical evidence of the persistence of ancient alleles remains scarce. Here, we present multifold coverage genomic data from four Late Pleistocene cave bears (Ursus spelaeus complex) and show that cave bears hybridized with brown bears (Ursus arctos) during the Pleistocene. We develop an approach to assess both the directionality and relative timing of gene flow. We find that segments of cave bear DNA still persist in the genomes of living brown bears, with cave bears contributing 0.9 to 2.4% of the genomes of all brown bears investigated. Our results show that even though extinction is typically considered as absolute, following admixture, fragments of the gene pool of extinct species can survive for tens of thousands of years in the genomes of extant recipient species.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
It is increasingly apparent that admixture among closely related mammalian species may have occurred frequently over the course of their evolution1. Many extant Holarctic mammals existed in widespread sympatry with now-extinct megafauna species during the Pleistocene, providing the opportunity for admixture. Palaeogenomic studies have shown evidence of gene flow from two archaic hominins into modern humans2,3, but it remains debated whether these represent distinct species, or early archaic populations within the broader human radiation4. Thus, a genetic contribution of ecologically and morphologically divergent Pleistocene megafauna to living mammal populations represents a plausible hypothesis that is largely untested by empirical evidence.
Cave bears are an iconic component of the Pleistocene megafauna. Cave bears went extinct around 25,000 years ago5, following a protracted period of population decline, with interactions with humans being a likely contributing factor6,7. Admixture between brown bears (Ursus arctos) and polar bears (Ursus maritimus), which form the sister clade to cave bears8,9, is well documented10–12, and recent studies suggest that interspecies admixture may be widespread among representatives of the Ursidae13. However, the genetic contribution, if any, of extinct bear species to their living congeners is largely unknown. Specifically, it is unknown whether admixture occurred between brown bears and cave bears, which coexisted in widespread sympatry and local syntopy in Eurasia for hundreds of thousands of years14,15 before extinction of the cave bear.
Results
Sampling of bear genomes.
To investigate whether brown and cave bears admixed during the Pleistocene, we extracted and sequenced nuclear genomic DNA from the petrous bones of four Late Pleistocene cave bears.
These samples were assigned to recognized cave bear taxa based on morphology and geographic location, and subsequently verified by analysis of mitochondrial sequences16. Although we refrain from forming any taxonomic conclusions based on our genomic datasets, we retain these assigned names for consistency with the published cave bear literature. Three of the cave bear samples are from Europe: an individual from the Gamssulzen cave, Austria, which is assigned to the taxon ingressus and has been 14C dated to 35,062 ± 966 yr BP7; an individual from the Eirós cave, Spain, which is assigned to the taxon spelaeus and has been 14C dated to 34,806 ± 931 yr BP7; and a third individual from the Windischkopf cave, Austria, assigned to the taxon eremus, which has been 14C dated to >49,000 yr BP and dated by phylogenetic tip dating analysis to 71,992 yr BP (95% credibility interval 54,640–91,860 yr BP)7. The fourth cave bear is from the Hovk-1 cave in the southern Caucasus (Armenia), and is assigned to the taxon kudarensis. The Caucasus cave bear is 14C dated to >49,000 yr BP17 and stratigraphic unit 6, from which the bone was excavated, is optically stimulated luminescence dated to 54,600 ± 5,700 yr BP18. We also sequenced DNA extracted from the petrous bone of a Late Pleistocene brown bear from the Winden cave, Austria, which has been 14C dated to 41,201 ± 895 yr BP7. This individual was contemporaneous with cave bears and is located in geographic proximity to the two sequenced Austrian cave bears, ingressus and eremus. We additionally sequenced brown bears from modern Georgian, Slovenian, Russian and Spanish populations, which, when combined with published datasets, provides representative sampling of the complete Holarctic distribution of the brown bear. Moreover, our sampling design provides approximate geographic pairing of each sequenced cave bear with a modern brown bear individual. Our analyses also included published datasets of three polar bears, an American black bear (Ursus americanus)12, an Asiatic black bear (Ursus thibetanus), a spectacled bear (Tremarctos ornatus)13 and the genome assembly of the giant panda (Ailuropoda melanoleuca)19. Full details of samples and datasets are provided in Supplementary Tables 1–3.
Cave bear relationships.
Phylogenetic analysis of aligned genomic sequences supported the monophyly of cave bears and their position as sister to the brown and polar bear clade (Fig. 1a). Within cave bears, the Caucasus cave bear kudarensis is sister to a clade containing the European cave bears eremus, ingressus and spelaeus, with the latter two forming sister taxa within that clade. This result is consistent with relationships inferred from morphology20 but contradicts those inferred using mitochondrial DNA, which instead indicate spelaeus and eremus as sister clades7,16 (Supplementary Fig. l). This incongruence may reflect either incomplete sorting of mitochondrial lineages in the population ancestral to ingressus and spelaeus, or transfer and fixation of introgressed mitochondrial lineages as a result of admixture between spelaeus and eremus.
Fig. 1 |. Phylogenetic relationships of the sequenced brown, polar and cave bear genomes.
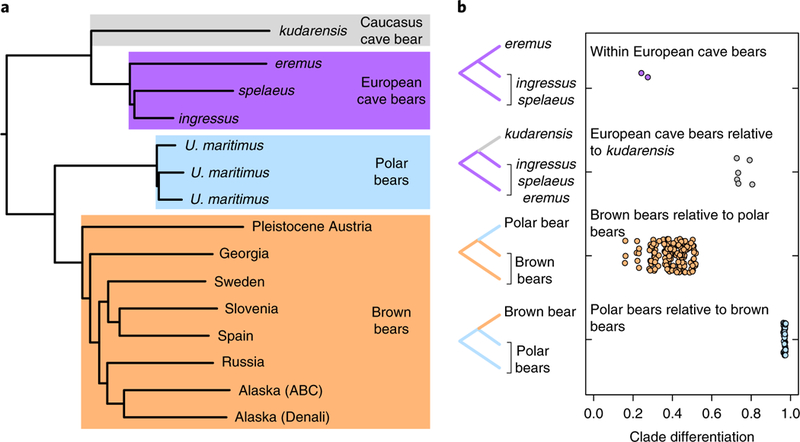
a, The maximum-likelihood phylogeny of individuals used in this study based on whole-genome transversion differences, rooted using the American black bear outgroup. b, Measures of clade differentiation based on D statistic tests inconsistent with the species tree. The tested topologies are shown on the left, with points showing the calculated clade differentiation (D values) for all possible combinations of the sampled individuals. This measure of clade differentiation scales between 0 (indicating a phylogenetic trifurcation) and 1 (indicating complete lineage sorting and absence of post-divergence gene flow). Measures of differentiation for the brown bear clade relative to polar bears, and for the polar bear clade relative to brown bears, are shown for comparison with values obtained for cave bears.
We then examined the extent to which cave bear clades are differentiated from one another by calculating the proportion of derived alleles an individual shares with members of the same clade relative to those shared with their sister clade, expressed as D statistics2,21 for topologies that are inconsistent with the species tree (Fig. 1b). This measure of clade differentiation scales between 0 and l, with low values indicating much incomplete lineage sorting and/or admixture among clades, and high values indicating that lineage sorting is approaching completion. This analysis revealed relatively low differentiation of the European cave bears ingressus and spelaeus clade from their sister eremus. In contrast, the European cave bear clade as a whole is highly differentiated from the Caucasus cave bear kudarensis lineage, suggesting deep and temporally sustained structuring among these respective populations.
Admixture between cave bears and brown bears.
We investigated interspecies admixture using D statistics2,21. All brown bears exhibited a significant excess of shared derived alleles with cave bears relative to polar bears (Supplementary Fig. 2), supporting admixture between cave bears and brown bears. We then used the related f̂ statistic21 to estimate the genomic proportion shared between brown bears and cave bears as a consequence of admixture following the divergence of brown and polar bears. This admixture proportion corresponds to the fraction of the cave bear genome that has introgressed into the brown bear genome assuming unidirectional gene flow. Values of f̂ were variable among brown bears, and highest in the Late Pleistocene Austrian brown bear (at least 2.4%, averaged across all comparisons), which was contemporaneous with cave bears. Among modern brown bears, the cave bear admixture proportion is highest in the Georgian brown bear (at least 1.8%), intermediate in Western European brown bears (at least 1.3 to l.4%), and lowest in the Russian and American brown bears (at least 0.9 to 1.0%). Since American brown bears exist outside of the Pleistocene distribution of cave bears, the variation in admixture proportions among brown bears shows some correspondence with their geographic and temporal proximity to the sampled Late Pleistocene cave bears. However, we did not find any obvious localized pattern of increased admixture between geographically paired brown bears and cave bears (Supplementary Fig. 2). Variable post-admixture diffusion of cave bear alleles via gene flow among brown bear populations may therefore be responsible for the observed patterns at a more geographically localized scale.
We also investigated variability in the proportion of brown bear admixture among pairs of cave bears. All comparisons between European cave bears fell below the Z > 3 threshold for strong significance, probably because these individuals were admixed with brown bears in similar proportions. However, comparisons involving the Caucasus cave bear kudarensis and European cave bears suggested an increased proportion of admixture with brown bears (around 1%) for European cave bears, with moderate statistical support (Z > 2) for many comparisons (Fig. 2). This suggests that some admixture with brown bears may have postdated the basal divergence of the sampled Caucasian and European cave bear lineages.
Fig. 2 |.
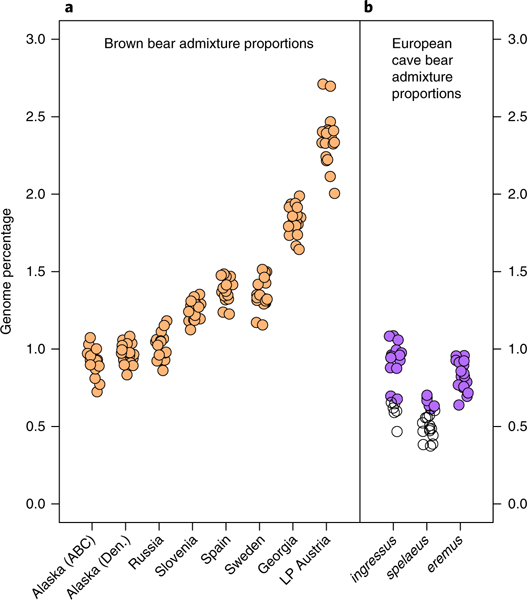
a, The proportion of the genomes of individual brown bears derived from admixture with cave bears, following the divergence of brown bears and polar bears (f̂results). Eighteen admixture estimates per brown bear represent all possible combinations of European cave bear introgressor, relative to three polar bears. Almost all values are strongly significant (Z > 3). A small number of comparisons involving the Russian and American bears were more moderately supported (Z > 2.4). ABC, Admiralty, Baranof and Chichagof Islands; Den., Denali; LP, Late Pleistocene. b, The proportion of the genomes of individual European cave bears derived from admixture with brown bears, following the divergence of the European cave bear clade and the Caucasus cave bear kudarensis (f̂results). Twenty dots per cave bear sample represent all possible combinations of modern Eurasian brown bear introgressors. All comparisons involving eremus are at least moderately supported (Z > 2), but 30% and 70% of comparisons involving ingressus and spelaeus, respectively, fell below this threshold (indicated by open circles).
Models of unidirectional gene flow do not explain the observed patterns.
The D statistic does not provide explicit information on the direction of gene flow. To explore this, we first investigated the possibility of unidirectional gene flow from cave bears to brown bears, and vice versa. For both scenarios, we calculated the expected D statistic values under a variety of demographic scenarios. Using a previously described approach21, the expected value of D was calculated for a given underlying demographic model, assuming a range of parameter values for the phylogeny, divergence times, population sizes, gene flow direction, the time and strength of gene flow, and generation times. After obtaining a single value of D expected for each combination of parameter values, we then used approximate Bayesian computation to compare the expected D with the value of D we observed from the sampled genomes. The closer the observed and expected D values are (based on the Euclidean distance), the higher the chance of the particular underlying model(s) generating those D values being close to the true demographic model. However, comparisons did not yield conclusive evidence for gene flow in either direction or for a specific range of demographic parameter values of the underlying model, because the parameter combinations that generated the closest D value covered a range of models that was too wide to point to a specific conclusive subset of scenarios (see Methods). Failure to identify demographic models consistent with the observed patterns may result from several confounding factors, including the accumulation of errors in parameter value estimates, population structure and other model misspecifications (such as bidirectional gene flow) that our model does not take into account.
A phylogenetic test of directional admixture.
To further investigate the direction of gene flow, we implemented a test based on the distribution of rooted tree topologies along a non-overlapping sliding genomic window. This approach is similar in principle to recently developed DFOIL statistics22, but involves comparisons of phylogenetic trees rather than diagnostic nucleotide positions. Our analyses used five individuals: a polar bear, a brown bear, the least admixed Caucasus cave bear (kudarensis), a more admixed European cave bear (spelaeus), and an Asiatic black bear as an outgroup. The ingroup species tree has a symmetrical topology: ((polar, brown),(least admixed cave, more admixed cave)). We divided the aligned haploidized genome sequences into ~50,000 non-overlapping blocks of 25 kb length, and computed the rooted maximum-likelihood phylogeny of each block. Conversion of diploid genome data into a single pseudohaploid sequence complicates phylogenetic analysis of regions where an individual is heterozygous for both admixed and unadmixed alleles. To overcome this problem (see Methods and Supplementary Fig. 3), we grouped each of the twelve possible non-symmetrical topologies into one of four topology classes: class 1, polar bear basal; class 2, brown bear basal; class 3, least admixed cave bear basal; and class 4, more admixed cave bear basal (Fig. 3a). The null hypothesis of no admixture would result in equal representation of classes 1 and 2, and of classes 3 and 4. In the absence of gene flow, the absolute numbers of blocks in each of these sets would reflect the degree of incomplete lineage sorting, which is dependent on effective population size and divergence times within each clade. In contrast, gene flow from cave bears into brown bears would result in an overrepresentation of class 1 relative to class 2. Gene flow in the opposite direction, from the brown/polar bear lineage into the more admixed cave bear, would result in an overrepresentation of class 3 relative to class 4. An overrepresentation in both cases is indicative of bidirectional gene flow.
Fig. 3 |. Test of gene flow direction based on the distribution of rooted tree topologies along a non-overlapping 25 kb sliding window.
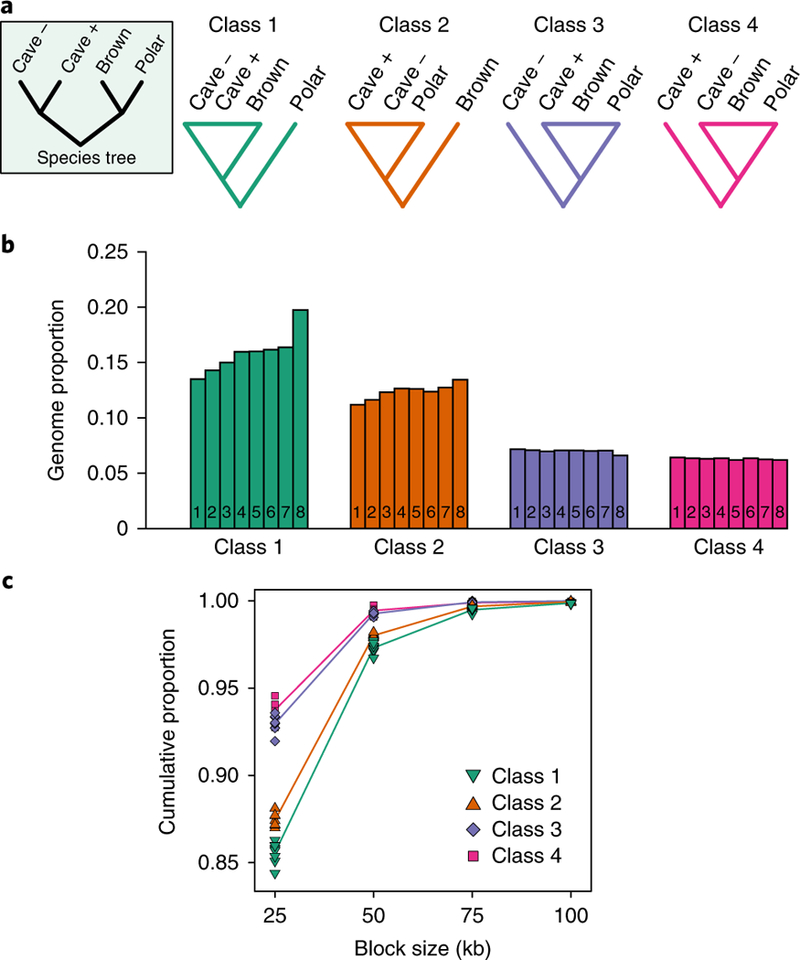
Five individuals are used in the test: a brown bear, a polar bear, the least admixed Caucasus cave bear (kudarensis, ‘cave –’), a more admixed European cave bear (spelaeus, ‘cave +’) and the Asiatic black bear outgroup. a, The symmetrical rooted ingroup species tree and the four alternative classes of topology that are informative on gene flow directionality. b, The proportion of 25 kb genomic blocks returning each topology class for eight brown bears, each represented by individual numbered bars: 1, Alaska (ABC Islands), North America; 2, Alaska (Denali), North America; 3, Russia; 4, Slovenia; 5, Sweden; 6, Georgia; 7, Spain; 8, Late Pleistocene Austria. The results support bidirectional gene flow among cave bears and brown bears: overrepresentation of topology class 1 relative to class 2 for all brown bears investigated indicates gene flow from cave bears into brown bears, and overrepresentation of topology class 3 relative to class 4 indicates gene flow in the opposite direction, from the brown/polar bear lineage into the more admixed cave bear. c, The cumulative size distributions of regions returning each topology class, determined by counting the number of consecutive 25 kb blocks returning the same topology. Absolute counts are provided in Supplementary Table 5. Individual points represent the cumulative proportions for individual brown bears. For the purpose of visualization, average cumulative proportions for each topology class are shown as coloured lines centred on the mean proportion observed for each block size and linked by linear interpolation.
We first evaluated the ability of our approach to detect directional admixture using a well characterized system: recent gene flow from polar bears into the brown bear populations inhabiting the Alaskan Admiralty, Baranof and Chichagof10,12 (ABC) Islands. We divided the aligned pseudohaploid genome sequences of two polar bears, an ABC islands brown bear, a European brown bear and the American black bear outgroup into non-overlapping genomic blocks and computed the maximum-likelihood phylogeny based on transversion sites. After the species tree, the next most abundant topology class comprised non-symmetrical trees in which the ABC Islands brown bear and polar bear formed a clade and the European brown bear occupied the basal ingroup position, which is consistent with gene flow from polar bears into the ABC Islands brown bear population (see Methods and Supplementary Table 4). Based on this result, we concluded that the distribution of rooted tree topologies along a non-overlapping sliding window is a reliable method to test for directional admixture.
Applying this test to the newly generated cave bear and brown bear genome data revealed clear and consistent evidence of gene flow from cave bears into brown bears. Topology class 1 outnumbered class 2 by a ratio of 1.2–1.5 for all brown bears investigated (Fig. 3b), representing an absolute difference of 1,226–3,052 blocks (Supplementary Fig. 4). The overrepresentation was greatest in the Late Pleistocene Austrian brown bear, with a rank order of diminishing levels of cave bear introgression broadly consistent with that inferred from D statistic analyses (Supplementary Fig. 2). Assuming introgressed blocks exist in a heterozygous state, the total genomic overrepresentation of topology class 1 suggests that between 1.2 to 3.1% of the genomes of the brown bears investigated is derived from cave bear introgression (Supplementary Fig. 4), which exceeds f̂estimates. This proportion may still be an underestimate, because some introgressed blocks are likely to exist in a homozygous state. We also found evidence of gene flow from the brown/polar bear lineage into the more admixed cave bear, with topology class 3 outnumbering class 4 for all brown bears by a ratio of approximately 1.1, representing an absolute difference of 196–448 blocks (Fig. 3b and Supplementary Fig. 4).
The length distribution of introgressed genomic blocks in admixed individuals reflects the number of intervening generations (recombination events) separating them from their F1 hybrid ancestor, and can therefore be used to infer the relative timings of different admixture events23. Of the four topology classes, the largest block sizes were associated with class 1 (13.7 to 15.6% of occurrences spanning more than one 25 kb block), which suggests that the most recent episodes of admixture involved gene flow from cave bears into brown bears (Fig. 3c and Supplementary Table 5). Length distributions are remarkably similar among modern and ancient brown bears, suggesting that all introgressed blocks derive from a similar time period or from the same admixed population. Thus, the reduced admixture proportions observed in modern brown bears relative to the Late Pleistocene Austrian individual probably result from diffusion into less admixed populations, rather than from an additional gene flow event into the ancient population. We also found a marginally larger length distribution for topology class 3 relative to class 4 (6.4 to 8.0% versus 5.4 to 7.2% of occurrences spanning more than one 25 kb block, respectively; see Fig. 3c and Supplementary Table 5). This is consistent with the low-level gene flow from the brown/polar bear lineage into cave bears inferred from the relative abundance of topology classes. Interestingly, we found the length distribution of topology class 2 (11.9 to 13.0% of occurrences spanning more than one 25 kb block; see Fig. 3c and Supplementary Table 5) to be intermediate between class 1 and classes 3 and 4, suggesting that class 2 fragments are not solely the result of lineage sorting, but may reflect earlier gene flow from cave bears into either the common ancestor of polar and brown bears or the population ancestral to polar bears after their divergence from the lineage leading to modern brown bears.
Introgressed cave bear genes potentially under selection in brown bears.
Introgressed cave bear genes in brown bears may increase fitness and undergo positive selection, as suggested for Neanderthal and Denisovan alleles in modern humans24–26. Identifying such genes is, however, challenging, given our low coverage unphased genomic datasets and fragmented reference genome assembly. With this in mind, we undertook a search for candidate introgressed genes by identifying 25 kb genomic blocks returning a topology consistent with being homozygous for introgressed cave bear alleles (brown bear nested within the cave bear clade) for all eight investigated brown bears. This uncovered 34 blocks which were compared against all UniProt vertebrate sequences in the Swissprot division. Five blocks were found to contain notable matches to UniProt sequences: matches to zinc finger protein PLAGL1 gene in pig, human and chicken; matches to the Titin gene in human; matches to the Tigger transposable element-derived protein 1 gene in human; and two separate blocks with matches to a LINE-1 reverse transcriptase homologue in slow loris and LINE-1 retrotransposable element ORF2 protein in human and mouse. These observations may result from selection on introgressed cave bear genes in brown bears, incomplete lineage sorting or selection on ancestral polymorphism. Although our analysis does not allow rigorous testing of these alternative explanations, it does provide pertinent evolutionary hypotheses for future investigation.
Discussion
Our results provide conclusive evidence of admixture between brown bears and extinct cave bears. This admixture involved gene flow in both directions, but at least the most recent episode of admixture transferred cave bear alleles into recipient brown bear populations. The evolutionary consequences of such admixture are difficult to assess. Introgression of novel alleles may facilitate adaptation when exposed to certain selection regimes24–27. Equally, such alleles may prove maladaptive, providing short-term fitness reductions and possibly leading to expulsion from the gene pool28. Brown bears are a widespread and successful species that has been recipient of alleles from two ecologically and morphologically divergent lineages in their recent evolutionary history: cave bears and polar bears10–12. Brown bears thus represent an excellent opportunity to study the biological implications of admixture in a dynamic multispecies system, with the five potentially introgressed loci identified by our functional analysis providing a logical starting point. Substantial progress in Ursid genetics will be required before this opportunity can be exploited fully.
Determining the direction of gene flow is of key importance in the study of admixture since the evolutionary implications of admixture processes strongly depend on the donor–recipient relationship. Previous inferences have generally relied on either high coverage, phased genomic sequence data2,29, or allele frequency estimates30,31, requiring either high genomic coverage or prior knowledge of the site frequency spectrum. Inference based on D statistics has been possible when one of the admixing species has extremely low genetic diversity12, but such approaches cannot be generalized to all species. Thus, none of these approaches are particularly well suited for studies of extinct Pleistocene megafauna, where genomic coverage and the number of individuals sampled are likely to be low. Recently developed DFOIL statistics22—which, similarly to the D statistic, summarize allelic patterns as continuously distributed statistics—may provide a suitable approach in such cases. However, identification of specific introgressed regions of the genomes of recipient species may be more straightforward using the phylogenetic approach described here, which instead relies on a finite number of diagnostic tree topologies. Furthermore, the phylogenetic approach has the potential to make use of a broad array of existing nucleotide substitution, molecular clock and coalescent-based population models, allowing further analytical refinement and development.
Here, we have shown that studying the distribution of rooted tree topologies along a non-overlapping sliding genomic window is a simple but powerful approach to determine gene flow directionality, which is robust to low genomic coverage and requires data from as few as four individuals. Using this method, we have shown that a fraction of the cave bear gene pool survives in the genomes of living brown bears, mirroring the Neanderthal ancestry found in non-African humans2, but at much deeper phylogenetic divergence. This result forces a reevaluation of the very concept of species extinction. At the genetic level, species may survive and participate in the theatre of evolution for tens of thousands of years after their disappearance from the fossil record.
Methods
Samples.
Ancient DNA data was obtained from cave bear and brown bear petrous bones. Data from modern brown bears was obtained from either skin or soft tissue samples. Complete details of all samples are provided in Supplementary Tables 1 and 2. No sample randomization or investigator blinding was undertaken.
Laboratory methods for ancient samples.
All laboratory work preceding library amplification was carried out in dedicated ancient DNA facilities at the University of York or the University of Potsdam, following established guidelines32. The ancient data were generated over a series of experiments, with the aim of optimizing DNA extraction and library preparation. The outcome of these experiments has been described previously17. The contribution of sequence data from each experiment is reported in Supplementary Table 6. For the majority of data, DNA was obtained from 50 mg of bone, sampled from the densest regions of the petrous bones, then ground to a fine powder using ceramic mortar and pestles, and digested overnight in 1 ml EDTA/proteinase K extraction buffer33. DNA was then isolated using a published method based on silica columns33, and eluted in 25 μl TET buffer. 20 μl of each DNA extract was used for preparation of Illumina sequencing libraries. First, DNA extracts were treated with uracil-DNA glycosylase and endonuclease VIII to remove uracil residues resulting from cytosine deamination, which are typically prevalent in ancient DNA fragments34, and then converted into libraries using a published protocol based on singlestranded DNA35. A unique eight base-pair index sequence was incorporated within the P7 adapter sequence of each library during amplification to facilitate data demultiplexing, with the optimal number of PCR cycles applied to each library determined in advance using a qPCR approach35. Amplified libraries were purified using commercial silica spin columns (Qiagen MinElute) and quantified using a Qubit 2.0 fluorometer (ThermoFisher Scientific) and either the Agilent 2100 Bioanalyzer or the 2200 TapeStation Instrument, before paired-end sequencing on Illumina platforms. Full details of alternative laboratory methods and the data obtained are provided in Supplementary Table 6.
The authenticity of the sequences obtained from the ancient samples was verified by mapping sequences to the polar bear reference genome assembly36 and then calculating fragment length distributions and checking for an excess of C to T substitutions at the fragment ends, which is indicative of ancient DNA damage (Supplementary Fig. 5), using the program mapDamage37.
Laboratory methods for modern samples.
DNA was extracted from modern tissue samples using a commercial kit (Qiagen DNeasy). Extract concentrations were measured using Qubit and DNA quality assessed using TapeStation genomic DNA assay. 500 ng of DNA in a volume of 50 μl was then sheared by sonication to an average fragment length of 500 bp using a Covaris S220 System. Sheared DNA was converted into Illumina sequencing libraries using a published protocol based on double-stranded DNA38, with modifications39. Library amplification and indexing was carried out as described previously for single stranded libraries. Library molecules corresponding to insert sizes <300 bp and >1,000 bp were removed prior to sequencing using a PippinPrep instrument (Sage Science). Modern libraries were sequenced on an Illumina NextSeq 500 platform generating approximately 80–200 million 150 bp read pairs for each library.
Processing of sequencing data.
We merged overlapping paired end reads with SeqPrep40 and mapped either both merged and unmerged reads (modern samples), or merged reads only (ancient samples), to the reference genome assembly of the giant panda19 with bwa aln v0.7.741. To account for the evolutionary divergence between the reference genome and the samples, we increased the allowed mismatch rate by setting the −n flag to 0.01 rather than the default of 0.04. We excluded reads with a MapQuality score less than 30 and removed duplicate reads with samtools v0.1.1942.
We selected the giant panda reference genome rather than the less evolutionarily distant and more contiguous polar bear reference genome36 for evolutionary analyses. As an ingroup to the group of samples being studied, the polar bear reference genome might introduce bias into our mappings that would disproportionately impact admixture inference. Cave bear reads from regions of the cave bear genome potentially introgressed from the polar/brown bear lineage would have a greater probability of mapping to the polar bear genome than reads from other parts of the genome because of their lower divergence. This biased assembly would produce the artefact of an inflated frequency of shared derived alleles between polar bears and cave bears in the cave bear assemblies. By contrast, mapping to the outgroup giant panda reference would have no bias for or against mapping introgressed regions making it a more suitable reference genome for this study.
For analysis, we generated haploidized sequences for each individual by randomly selecting a single high quality base call (BaseQuality ≥30, read MapQuality ≥30) at each site in the panda reference genome. This method better represents non-reference alleles for low coverage samples than genotype calling, which tends to be biased toward the reference allele, potentially confounding downstream analyses2. To avoid inclusion of repetitive or duplicated genomic elements, we masked sites where an individual’s coverage was above the 95th percentile of genome wide coverage.
Phylogenetic analysis of aligned nuclear and mitochondrial sequences.
The aligned pseudohaploid nuclear sequences were recoded into binary characters to only score transversions (Rs: 0, Ys: 1) and all aligned columns containing missing data were removed. The maximum-likelihood phylogeny was then computed under the BINGAMMA model with the American black bear as outgroup using RaxML43.
The mitochondrial sequences of the investigated European cave bears have been published previously7. A consensus mitochondrial sequence for the Caucasus cave bear kudarensis was generated from the shotgun sequencing data using an iterative mapping approach. First, merged reads were mapped to a published spelaeus mitogenome (Genbank EU327344) using the ‘mem’ algorithm in bwa v0.7.841. Reads with MapQuality score less than 30 were removed with samtools, and duplicate reads removed using MarkReadsByStartEnd.jar (https://github.com/dariober/Java-cafe/tree/master/MarkDupsByStartEnd). From this alignment, an alternative reference sequence was created using FastaAlternateReferenceMaker from the GenomeAnalysisToolKit v3.144 based on the variants detected using GATK’s UnifiedGenotyper. Then, merged reads were again mapped to this newly generated reference with the more stringent ‘aln’ algorithm in bwa, and reads with low mapping quality and duplicates removed as described previously. A final consensus was generated from this alignment in Geneious v7.0, using a minimum sequence depth of 3× and a 90% majority rule for base calling.
Mitochondrial sequences were aligned using MUSCLE45 and a problematic section of the control region removed, as described previously7. The final alignment contained 16,361 aligned nucleotide positions. Phylogenetic analysis was conducted using maximum-likelihood under the GTR-GAMMA model using RAxML-HPC2 8.2.343 on the CIPRES Portal46 using the American black bear as outgroup. Clade support was assessed by 500 bootstrap replicates using the GTR-CAT substitution model.
D statistic tests of lineage differentiation and admixture.
We tested for admixture between cave bears and their nearest extant relatives, polar bears and brown bears, with the D statistic (ABBA, BABA test)2,21. All D statistics calculated during this study are reported in the Supplementary information (Supplementary Data 1–4). To avoid bias resulting from ancient DNA cytosine deamination (C- > T error) damage, we restricted our analysis to transversion sites. To test for significance, we applied the weighted block jackknife2. Because of the low contiguity of the giant panda genome, we used 1 Mb non-overlapping blocks, rather than the 5 Mb non-overlapping blocks used in previous studies2,10,12, which would exclude most of the panda scaffolds (N50 = 1,281,781)19. Despite their smaller size, 1 Mb non-overlapping bins are adequate for testing cave bear introgression into brown bears because they are substantially longer than the longest estimated length of introgressed blocks of cave bear ancestry which is 175 kb (Supplementary Table 5). We consider results more than three standard errors different from zero (Z > 3) as strong evidence of admixture, and more than two standard errors different from zero (Z > 2) as providing moderate evidence of admixture.
We investigated four alternative outgroups for admixture tests. These were, in order of increasing phylogenetic distance from the focal clade13: American black bear, Asiatic black bear, spectacled bear and giant panda. All outgroups resulted in a significant signal of admixture between all cave bears and all modern brown bears, following the divergence of polar bears and brown bears (Supplementary Fig. 6). In contrast, tests involving the Late Pleistocene brown bear similarly supported admixture between brown bears and cave bears when the less divergent outgroups were used, but the two most divergent outgroups resulted in a reversal of the admixture signal (Supplementary Fig. 6). We attribute this effect to accumulated errors in the ancient pseudohaploid sequences as a result of both sequencing error and spurious read mapping, both of which tend to occur at higher rates in ancient compared to modern DNA datasets. Specifically, at sites where the outgroup has a private allele, accumulated errors in an ancient sample occupying the P2 position would convert a proportion of these BBBA sites to BABA sites, causing that individual to appear unadmixed relative to P1. This effect is amplified with more divergent outgroups since they will posses more private alleles relative to the ingroup. For the American black bear outgroup, we also observed consistently elevated D values relative to those generated using other outgroups, suggesting differential allele sharing between American black bear and P1 and P2 ingroup lineages (polar and brown bear, in this case). We calculated D statistics to assess this imbalance using the giant panda as outgroup, and found a significant excess of derived alleles shared between American black bear and polar bears, relative to brown bears, in almost all comparisons (Supplementary Fig. 7). This may reflect admixture between American black bear and polar bear, admixture with a ghost lineage, ancestral population structure, or the transfer of archaic alleles into brown bears by a cave bear vector, but we did not investigate these alternative explanations further. All other outgroups did not show any clear or consistent imbalance in allele sharing with either ingroup lineage. Overall, we selected Asiatic black bear as the most appropriate outgroup, being sufficiently closely related to the ingroup to avoid artefacts associated with ancient datasets (Supplementary Fig. 6), and showing no consistent pattern of differential allele sharing with either polar bears or brown bears (Supplementary Fig. 7). Asiatic black bear was thus used as outgroup for all subsequent admixture tests.
To quantify the amount of admixture, we used the f̂statistic21, which is the excess of shared derived alleles between the admixed individual and candidate introgressor standardized by the maximum excess of shared derived alleles expected in an entirely (100%) introgressed individual. All f̂values calculated during this study are reported in the Supplementary Data 5 and 6. The f̂expected value is best calculated by using individuals that we hypothesize best approximate the diversity within the introgressing populations. We considered the European cave bears to best represent the diversity within a potentially introgressing cave bear lineage. For brown bear introgressors, we selected Eurasian brown bears as best representing diversity in a potential brown bear introgressor. As with the D statistic, we determined significance based on weighted block jackknife with 1 Mb blocks.
Inferring the direction of gene flow based on observed values of D.
In order to obtain more insights into the underlying demographic model, we followed the methodology of a previous study21 by calculating the expected counts of ABBA and BABA generated under the model of instantaneous unidirectional admixture (IUA). Populations P1, P2 and P3 represent polar, brown and cave bear, respectively. We assumed two versions of this model, a single admixture event from P3 into P2, and the same model but with the direction of gene flow from P2 into P3. The range of parameter values tested were as follows (following the notation described elsewhere21): a constant population size of Ne 10,000 to 30,000 in steps of 5,000 in all populations; a generation time of 13 years; tP2 800 ka (thousand years ago) to 1.4 Ma (million years ago); tP3 1.5 Ma to 2 Ma, both in steps of 50 ka; f̂ 0.01 to 0.1 in steps of 0.01 and tgf 150 ka to tP2 – 50 ka in steps of 50 ka. For each of the 135.850 combinations of parameter values, we then calculated the expected counts of ABBA and BABA.
We then applied a procedure based on approximate Bayesian computation (described in detail previously47) to identify combinations of parameter values that generate expected D values closely matching the observed D. We used the grid of parameter values as discrete uniform prior distributions and the expected D as summary statistics. We kept the closest 1% parameter combinations based on the Euclidean distance between observed and expected D. However, this approach proved inconclusive, because too many different parameter values generated D values that closely matched the observed D. We could not identify a specific conclusive subset of parameter values that would support specific demographic models. The distributions with the kept parameter values were broadly uniform and, therefore, uninformative. Based on these observations, we did not investigate this approach in more detail. In summary, we did not obtain any clear evidence to identify the direction of gene flow or a specific range of demographic parameter values from the IUA model.
Phylogenetic test of directional admixture.
This test is based on the assertion that if an admixed individual of a recipient species possesses an introgressed allele at a given locus, then phylogenetic analysis of that locus will cluster the introgressed allele within the diversity of the donor species and not within the diversity of the recipient species. Such patterns can also result from incomplete lineage sorting; however, using the same principles upon which the D statistic is based21, this can be differentiated from admixture by looking for an imbalance in the frequencies of specific topologies across a large number of loci. We devised an implementation of this test using a sample of four individuals plus a suitable outgroup. There are fifteen possible rooted tree topologies. Three of these rooted trees are symmetrical, and we selected individuals such that one of these symmetrical trees represented the species tree. As such, rooted trees that may reflect the possession of introgressed alleles in one individual are non-symmetrical.
Precise phylogenetic placement of putatively introgressed alleles is confounded by the use of unphased pseudohaploid sequences when individuals are heterozygous for both introgressed and non-introgressed alleles (here, allele refers to a linked haplotype sequence spanning a particular region of the genome). Random selection of nucleotides from both introgressed and non-introgressed alleles from such a heterozygous individual would be likely to result in a phylogeny with branch lengths representing an average between the symmetrical species tree and the non-symmetrical admixed tree (Supplementary Fig. 3). In this averaged tree, the position of the admixed individual within the donor species clade may shift, but the overall topology will remain non-symmetrical. For this reason, we grouped the twelve possible non-symmetrical rooted topologies into four topology classes (three topologies each) according to the individual occupying the basal ingroup position, which formed the basis of the directional admixture test (Fig. 3a). This procedure accounts for the variable positioning of introgressed individuals within the donor clade resulting from heterozygosity by exploiting the preservation of tree non-symmetry.
A further consideration for implementing the test is the selection of an appropriate block size. Phylogenetic analysis of whole-genomic alignments returns the reciprocal monophyly of cave bears, brown bears and polar bears (Fig. 1a). Thus, it is likely that phylogenetic analysis of very large genomic blocks would, in general, also return this species tree. A suitable block size is therefore sufficiently small to be able to detect regions resulting from admixture and incomplete lineage sorting, while also being of sufficient size for accurate phylogenetic estimation. We therefore investigated a range of block sizes to identify an appropriate size providing good sampling of topological variation along the aligned genome sequences.
Evaluation of the method using the example of recent gene flow from polar bears into the brown bear populations inhabiting the ABC Islands involved genomic data from two polar bears, an ABC Islands brown bear, a European brown bear, and the American black bear outgroup. Details of read processing, mapping and pseudohaploidisation were as described above. Introgressed polar bear alleles in the ABC Islands brown bear should be detectable as a relative increase in the frequency of genomic blocks returning the ABC islands brown bear and polar bears as monophyletic relative to the number returning the European brown bear and the polar bears as monophyletic. To test this hypothesis, we divided aligned pseudohaploid sequences into non-overlapping blocks of equal sizes and computed the maximum-likelihood phylogeny based on transversion sites as described previously. Blocks that contained >50% missing data for any of the individuals were recorded as having insufficient data and not used for phylogenetic analysis. The topology of each phylogeny was evaluated using a custom Perl script that made use of the ETE3 software48. Analysis of both 0.25 Mb and 0.1 Mb block sizes produced topology frequencies consistent with the hypothesized pattern: following the species tree, the next most abundant topology class comprised non-symmetrical trees in which the ABC Islands brown bear and polar bears formed a clade, with the European brown bear occupying the basal ingroup position, which is consistent with gene flow from polar bears into the ABC Islands brown bear population (Supplementary Table 4). Based on this result, we concluded that the distribution of rooted tree topologies along a non-overlapping sliding window is a reliable method to test for directional admixture.
We then applied the test to investigate directional gene flow between cave bears and brown bears. We tested a range of block sizes and found that a block size of 25 kb provided good sampling of topological variation along the aligned sequences. The estimated abundance of topology classes were also consistent using larger block sizes (Supplementary Table 7). At these block sizes, ingroup lineages may not have sorted with respect to the outgroup, leading to erroneous outgroup rooted tree topologies. In the four taxon framework of the admixture test, if the assumed outgroup is allowed to occupy all positions within the tree, there are 105 possible rooted topologies representing three alternative tree shapes. We considered the effect of rooting each of these tree shapes when the assumed outgroup occupies an internal position within the tree (Supplementary Fig. 8). We identified four possible outcomes of this outgroup misspecification. In some cases, both incomplete and complete lineage sorting of ingroup taxa with respect to their sister clade was correctly identified, but with incorrect outgroup positioning. In other cases, we predicted artefactual grouping of taxa into both admixture-informative and admixture-uninformative topologies, but importantly in all cases equilibrium of topology class frequencies are maintained. Thus, although outgroup misspecification may mislead absolute measures of lineage sorting, we predict no systematic bias that may lead to erroneous inference of admixture. We empirically tested this hypothesis by replicating the analysis with the addition of the more distantly related panda, which served as outgroup for the rooting of phylogenetic trees. Aligned sequences from the panda genome assembly were included in each 25 kb block containing the four ingroup taxa and the assumed Asiatic black bear outgroup. A rooted maximum-likelihood phylogenetic tree was computed as described previously for each block, using the panda as outgroup. These were then filtered for only those trees where the ingroup were monophyletic with respect to the Asiatic black bear, using BioPerl49 functions. Phylogenies for each of these blocks were then recomputed using the Asiatic black bear as outgroup, and the abundance of alternative topologies counted as described previously. Although the absolute abundance of topology classes was reduced in comparison to the complete dataset, differences in abundance between topology classes were consistent and congruent with admixture patterns inferred from the complete dataset (Supplementary Fig. 9).
Finally, we investigated the potential of biases in the estimation of topology frequencies introduced by missing data by comparing the distribution of missing data among blocks returning each topology (Supplementary Fig. 10). The occurrence of missing data was found to be highly consistent among topologies, indicating that the presence or absence of missing data is not influencing our frequency estimates.
The lengths of potentially admixed genomic regions were estimated from the complete dataset by counting the number of consecutive blocks returning the respective tree topology. Due to the presence of blocks with insufficient data, these measurements are likely to be underestimates. Length comparisons were made by calculating empirical cumulative densities and plotted in R50, with all other parameters set at default.
Identification of introgressed genes potentially under selection.
We identified genomic blocks where all eight investigated brown bears had sufficient data for phylogenetic reconstruction, and where phylogenetic analysis indicated that each was nested within the cave bear clade (topologies 5 and 6, Supplementary Table 7). For each block identified, the corresponding sequence of the panda was retrieved and compared against all UniProt vertebrate sequences in the swissprot division using blastx, requiring an E value of 10−40 or better, and requiring that at least 50% of the UniProt sequence was covered in one or more of the resulting alignments.
Supplementary Material
Acknowledgements
This work was funded by European Research Council (ERC) consolidator grant ‘gene flow’ 310763 to M.H. G.G.F. and R.P. were supported by ERC starting grant 263441 to R.P. A.G.-d’A. and A.G.-V were supported by research project CGL2014-57209-P of the Spanish Ministry of Economy and Competitiveness to A.G.-d’A. J.A.C. and B.S. were supported by a grant from the Gordon and Betty Moore Foundation (GBMF-3804) and NSF ARC-1417036 to B.S. U.S. was supported by grant IUT20-32 from the Estonian Ministry of Education and Research, and PA. by the Estonian Science Foundation DoRa programme. We thank the regional governments of Asturias and Castilla y León, in Spain, for providing tissue samples of Cantabrian bears. The authors would like to acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure (NGI), Sweden, the Knut and Alice Wallenberg Foundation and UPPMAX for providing assistance in massively parallel DNA sequencing and computational infrastructure.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41559-018-0654-8.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Code availability. Scripts for generation of pseudohaploid sequences and calculation for D and f̂statistics are available from https://github.com/jacahill/Admixture for public non-commercial use. Scripts used for the phylogenetic test of admixture are available from the corresponding author upon request.
Data availability. Raw nucleotide sequence data generated in this study have been deposited in the European Nucleotide Archive (ENA) with the run accession codes ERR2678614-ERR2678640. The haploidized fasta sequences have been deposited in the Dryad Digital Repository: https://doi.org/10.5061/dryad.cr1496b. The kudarensis (sample HV74) consensus mitochondrial sequence has been deposited in GenBank with the accession code MH605139.
References
- 1.Shurtliff QR Mammalian hybrid zones: a review. Mamm. Rev. 43, 1–21 (2013). [Google Scholar]
- 2.Green RE et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer M et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reich D et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468, 1053–1060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baca M et al. Retreat and extinction of the Late Pleistocene cave bear (Ursus spelaeus sensu lato). Naturwissenschaften 103, 11–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stiller M et al. Withering away—25,000 years of genetic decline preceded cave bear extinction. Mol. Biol. Evol. 27, 975–978 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Fortes GG et al. Ancient DNA reveals differences in behaviour and sociality between brown bears and extinct cave bears. Mol. Ecol. 25, 4907–4918 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Noonan JP et al. Genomic sequencing of Pleistocene cave bears. Science 309, 597–599 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Krause J et al. Mitochondrial genomes reveal an explosive radiation of extinct and extant bears near the Miocene-Pliocene boundary. Evol. Biol. 8, 220 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cahill JA et al. Genomic evidence of geographically widespread effect of gene flow from polar bears into brown bears. Mol. Ecol. 24, 1205–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cahill JA et al. Genomic evidence of globally widespread admixture from polar bears into brown bears during the last ice age. Mol. Biol. Evol. 35, 1120–1129 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Cahill JA et al. Genomic evidence for island population conversion resolves conflicting theories of polar bear evolution. PLoS Genet. 9, e1003345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar V et al. The evolutionary history of bears is shaped by gene flow across species. Sci. Rep. 7, 46487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurtén B Pleistocene Mammals of Europe (Weidenfeld & Nicholson, London, 1968). [Google Scholar]
- 15.Kurtén B The Cave Bear Story: Life and Death of a Vanished Animal (Columbia Univ. Press, New York, NY, 1976). [Google Scholar]
- 16.Stiller M et al. Mitochondrial DNA diversity and evolution of the Pleistocene cave bear complex. Quat. Int. 339, 224–231 (2014). [Google Scholar]
- 17.Barlow A et al. Massive influence of DNA isolation and library preparation approaches on palaeogenomic sequencing data. Preprint at 10.1101/075911 (2016). [DOI]
- 18.Pinhasi R et al. Middle Palaeolithic human occupation of the high altitude region of Hovk-1, Armenia. Quat. Sci. Rev. 30, 3846–3857 (2011). [Google Scholar]
- 19.Li R et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baryshnikov GF & Puzachenko AY Craniometrical variability in the cave bears (Carnivora, Ursidae): multivariate comparative analysis. Quat. Int. 245, 350–368 (2011). [Google Scholar]
- 21.Durand EY, Patterson N, Reich D & Slatkin M Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28, 2239–2252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pease JB & Hahn MW Detection and polarization of introgression in a five-taxon phylogeny. Syst. Biol. 64, 651–662 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Węcek K et al. Complex admixture preceded and followed the extinction of wisent in the wild. Mol. Biol. Evol. 34, 598–612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dannemann M, Andrés AM & Kelso J Introgression of Neandertal- and Denisovan-like haplotypes contributes to adaptive variation in human toll-like receptors. Am. J. Hum. Genet. 98, 22–33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Racimo F, Sankararaman S, Nielsen R & Huerta-Sánchez E Evidence for archaic adaptive introgression in humans. Nat. Rev. Genet. 16, 359–371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huerta-Sánchez E et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miao B, Wang Z & Li Y Genomic analysis reveals hypoxia adaptation in the Tibetan mastiff by introgression of the gray wolf from the Tibetan plateau. Mol. Biol. Evol. 34, 734–743 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Sankararaman S, Mallick S, Patterson N & Reich D The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr. Biol. 26, 1241–1247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohse K & Frantz LAF Neandertal admixture in Eurasia confirmed by maximum-likelihood analysis of three genomes. Genetics 196, 1241–1251 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutenkunst RN, Hernandez RD, Williamson SH & Bustamante CD Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5, e1000695 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pickrell JK & Pritchard JK Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fulton TL in Ancient DNA: Methods and Protocols Vol. 840 (eds. Shapiro B & Hofreiter M) 1–11 (Springer, New York, NY, 2012). [Google Scholar]
- 33.Dabney J et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl Acad. Sci. USA 110, 15758–15763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dabney J, Meyer M & Pääbo S Ancient DNA damage. Cold Spring Harb. Perspect. Biol. 5, a012567 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gansauge M-T & Meyer M Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 8, 737–748 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Li B, Zhang G, Willerslev E, Wang J & Wang J Genomic data from the polar bear (Ursus maritimus). GigaScience http://gigadb.org/dataset/100008 (2011). [Google Scholar]
- 37.Ginolhac A, Rasmussen M, Gilbert MTP, Willerslev E & Orlando L mapDamage: testing for damage patterns in ancient DNA sequences. Bioinformatics 27, 2153–2155 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Meyer M & Kircher M Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb.prot5448 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Fortes GG & Paijmans JLA in Whole Genome Amplification (ed. Kroneis T) 179–195 (Humana, New York, NY, 2015). [Google Scholar]
- 40.St John J SeqPrepv1.1 (2013); https://github.com/jstjon/SeqPrep
- 41.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stamatakis A RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenna A et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller MA, Pfeiffer W & Schwartz T in 2010 Gateway Computing Environments Workshop (GCE) 1–8 (IEEE, 2010); 10.1109/GCE.2010.5676129 [DOI] [Google Scholar]
- 47.Beaumont MA, Zhang W & Balding DJ Approximate Bayesian computation in population genetics. Genetics 162, 2025–2035 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huerta-Cepas J, Serra F & Bork P ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stajich JE et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 12, 1611–1618 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.R Core Team. R: a language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, 2014); http://www.r-project.org/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.