

Abstract
Small alterations during early stages of innate immune response can drive large changes in how adaptive immune cells develop and function during protective immunity or disease. Controlling these events creates exciting potential in development of immune engineered vaccines and therapeutics. This progress report discusses recent biomaterial technologies exploiting innate immunity to dissect immune function and to design new vaccines and immunotherapies for infectious diseases, cancer, and autoimmunity. Across these examples, an important idea is the possibility to co-opt innate immune mechanisms to enhance immunity during infection and cancer. During inflammatory or autoimmune disease, some of these same innate immune mechanisms can be manipulated in different ways to control excess inflammation by promotion of immunological tolerance.
Keywords: innate immunity, nanoparticles and microparticles, autoimmunity and tolerance, biomaterials and nanotechnology, antigen-presenting cells
Graphical Abstract
Control of innate immunity is the basis of immune engineered vaccines and therapeutics. Biomaterial technologies provide modular platforms to control display and delivery of combinations of innate immune signals alongside molecular fragments termed antigens. These antigens are processed and presented by antigen-presenting cells (APCs) to adaptive immune cells. This biomaterial-controlled delivery can co-opt innate mechanisms to direct immunity and tolerance during infection, cancer, or autoimmunity.
1. Introduction
The innate immune system provides a rapid, but non-specific, defense against invading pathogens. This activity is enabled in part by recognition of generalized molecular patterns that are characteristic of invading pathogens (e.g., viruses, bacteria), but uncommon in humans. While these innate cells play an important role in directly preventing pathogens from infecting a host, they play an equally important role in activating the highly specific adaptive immune system for infections that do occur. In particular, a small difference in the ability of innate immune cells to recognize a pathogen and communicate the information to adaptive immune cells significantly impacts long-term immunity and the ultimate disease course. This non-linear behavior in immunity draws parallels with the mathematical concept of chaos and the butterfly effect, where small changes at early stages result in large differences at later stages.1 Thus, controlling the initial events that dictate signals between the innate and adaptive immune system is crucial in developing engineered approaches to direct immune function.
Biomaterials are materials designed to interact with biological systems. These materials may incorporate synthetics – including polymers, metals, and ceramics, or naturally occurring components such as proteins or living cells. Biomaterials are useful for their ability to incorporate multiple signaling molecules. Likewise, biomaterials can be modified to protect and control the release of biological signals. These properties make biomaterials very attractive for the design of technologies that control innate immunity. This progress report discusses recent biomaterial technologies exploiting innate immunity to dissect immune function and to design new vaccines and immunotherapies for infectious diseases, cancer, and autoimmunity.
2. Innate Immunity Helps Direct Reactivity and Specificity of Adaptive Immunity
2.1. Adaptive Immunity is a Dynamic Balance Between Activation and Immune Tolerance
Adaptive immunity is regulated by T and B cells that have a highly diverse set of cell surface receptors for recognizing specific molecular fragments of a given pathogen, termed “antigens”. To regulate the reactivity of T cells, the innate immune system has evolved a set of three signals to direct the attack against pathogens or maintain tolerance against healthy, self-tissue (Figure 1).2 First, the innate immune cell processes and presents an antigen from a pathogen to a naïve T cell specific for that antigen (Signal 1). Second, the innate immune cell binds to the naïve T cell through “costimulatory” receptors (Signal 2), which are upregulated in innate cells upon recognition of warning signals present on pathogens. These warning signals are not present in healthy, self-tissue. Third, following pathogen detection, the innate immune cell secretes signaling proteins – termed cytokines – that help activate and condition the T cell (Signal 3). Integration of all three of these signals by the T cell results in activation and proliferation of effector T cells (TEFF) specific for the presented antigen; these cells then seek out and eliminate pathogens, executing the process known as immunity. If the costimulatory signals are absent, the T cell remains inactive. Likewise, if secreted cytokines are regulatory or anti-inflammatory, T cells can develop alternate phenotypes, such as regulatory T cells (TREG). TREGS suppress the function of TEFFS and help drive immune tolerance that restrains or suppresses immunity in peripheral tissue. The interactions between innate immune cells and naïve T cells that lead to immunity or tolerance occur in specialized immune tissues: spleen and lymph nodes (LNs). At these sites, antigen-presenting cells, activated T cells, and soluble antigen may also drive activation of a second adaptive immune cell type: B cells. This process involves B cell proliferation, maturation, and secretion of antibodies that exert adaptive immunity by tagging extracellular pathogens for elimination and neutralizing other toxins or dangerous molecules. This reductionist view of the balance between immunity and tolerance portrays the general relationship between innate and adaptive immune cells (Figure 1). Immune signals that control this balance are highly diverse, as exemplified by variants in antigen structures that lead to changes in antigen affinity, stability, and recognition.3 Other reviews have highlighted the potential of manipulating a single signal, such as guides synthesis of a novel antigens based on computational biology.4 The control of antigen structure, valency, and stability through biomaterial platforms has also been recently reviewed.5,6 Consideration of individual signal complexity, as well as combination of multiple signals, is vital to controlling the balance between immunity and tolerance. The emerging field of innate immune engineering aims to manipulate this balance through biomaterial and nanotechnology platforms to exert precise control over features of an immune response.
Figure 1. Biomaterial platforms promote immunity and tolerance through delivery of cargo to innate immune cells.
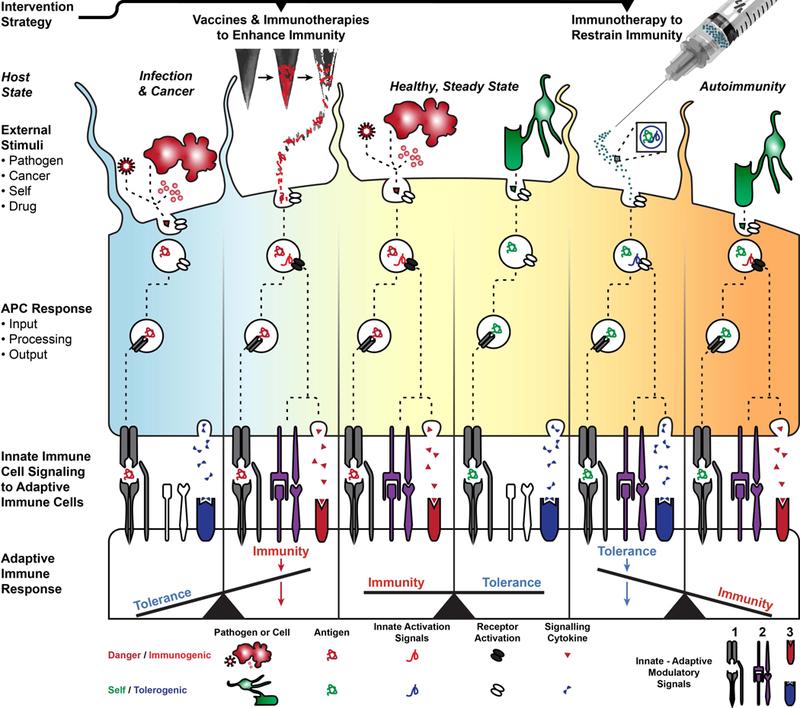
In a healthy, steady state, innate immune cells like DCs become activated against dangerous external stimuli like pathogens or tumor cells and do not become activated against self tissue. This difference in response is communicated to the adaptive immune sytem by three generalized signals at the innate-adaptive cell interface, which include (1) antigen-presentation, (2) costimulation, and (3) cytokine conditioning. These signals generate a balance of immune response during healthy, steady state. In an infectious disease or during cancer, the immune system is unable to mount an appropriate response to counter the pathogen or tumor growth. To address this deficit in immunity, biomaterial platforms like degradable microneedles may be loaded or coated with antigen and immunogenic cargo to activate inflammatory receptors and induce therapeutic immunity. In autoimmunity, the overstimulation of the immune system leads to loss of tolerance and production of self-reactive immunity against host cells like oligodendrocytes that maintain myelin in the central nervous system. Biomaterials like NPs may be loaded with self antigen and a tolerogenic, innate immune cue that suppresses inflammatory receptor activity and facilitates the production of immune tolerance. Collectively, biomaterials facilitate the modular loading of cargo to treat a spectrum of immunological diseases.
2.2. Innate Immune Cells and Signaling have Multiple Connections to Adaptive Immunity
2.2.1. Antigen-Presenting Cells are Diverse and Specialized
Antigen-presenting cells (APCs) are specialized innate immune cells that provide the signaling cues outlined in the previous section to adaptive immune cells (e.g., T and B cells). These cues regulate the balance between the immunity needed to combat pathogens and tumors and the immune tolerance needed to prevent host tissue from mistaken attack. Antigen presentation is the process by which innate immune cells uptake pathogens or cellular debris, degrade the proteins into presentable peptide fragments, and then localize these peptide antigens on their surface for recognition by adaptive immune cells that are molecularly specific to the antigen. Thus, this step is the connection between the pathogen causing an infection, the antigen displayed, and the reactivity of a T or B cells specific for the antigen. Antigen binding drives either immunity (turning on the immune system) or tolerance (restraining the immune system); this is dependent on the other signals presented by the APC alongside the antigen. Figure 1 illustrates the immune cue inputs, processing, and signaling outputs that drive APC function during disease and biomaterial delivery of immune cues that shift the balance of immunity and tolerance from diseased states toward a healthy, steady state. In particular, during infection and cancer, pro-immune responses are desirable, while during autoimmune and inflammatory diseases, immunological tolerance or suppression is desired. The most common APCs are dendritic cells (DCs) and macrophages. Monocytes and B cells possess lower antigen-presenting functionality that is overshadowed by their effector and antibody-production functions, respectively. Understanding of different APC subsets and their paths for interacting with adaptive immune cells have informed immune engineered materials that target specific cell types and deliver therapeutics to important immunological sites, like the LN.
Dendritic Cells – The Antigen-Presenter:
DCs are a specialized APC that reside in host tissues where they survey for antigens that they present to adaptive immune cells. DCs are classified by a star-like shape, rapid uptake of antigen, and high expression of the cell-surface protein complexes that antigen is presented in. Once a DC detects an antigen, the DC becomes activated, engulfs and processes the antigen, and migrates to the LNs or spleen where T cells are localized. This migration and the close proximity with T cells facilitate the activation of an adaptive response if the appropriate signals (i.e., Signals 1–2-3 discussed above) are presented by the DC to naïve T cells that are specific for the antigen.7 APC antigen uptake, antigen trafficking, and presentation of signals to T cells maintain the balance between immunity and tolerance. Thus, these pathways may be co-opted by biomaterials for new vaccines and therapeutics.
Different tissues have unique systems in place for immune surveillance.8 For example, skin has a high density of resident immune cells, particularly DCs, to combat the constant exposure to antigens from external environments and the bacteria that reside on the skin of a host. In skin there are also unique DC subtypes, like Langerhans cells, which have been identified as mediators of immune tolerance.9 Interestingly, similar DC phenotypes serve opposing functions in different tissues, which may be important for identifying the appropriate sites for introducing a vaccine or therapeutic agent.10 Biomaterials are able to target and exploit specific DC subsets that can control the highly specific, and potentially protective or therapeutic, aspects of adaptive immunity.
Macrophages – The Phagocyte:
Macrophages are an innate cell that reside in tissue as sentinels, prepared to quickly and non-specifically respond to invading pathogens or damaged tissue. Macrophages are associated with their function as a phagocyte, where they clear pathogens, dead cell debris, and damaged tissue to aid in tissue homeostasis.11 Macrophages have a broad phenotypic range that stems from their highly upregulated sensing and effector functions. The specialized phagocytic function of these cells is enabled by surface-bound scavenger receptors, yet they are also capable of antigen presentation. For example, within LNs, macrophages occupy an interface where they are exposed to components from lymph and blood. These specialized, subcapsular sinus macrophages are positioned to capture antigen or pathogens that enter the LN and readily process and present antigen to B cells to stimulate the generation of protective antigen-specific antibodies.12 Co-opting the phagocytic function of macrophages to deliver immune cues is one route that biomaterials might use to alter the course of immunity.
Monocytes – The Circulating Sentinel:
Monocytes are associated with surveillance in circulation, migration into tissue, and differentiation into macrophages. While a large portion of monocytes is confined to circulatory surveillance, other monocyte subtypes can migrate into tissue without activation. Once in tissue, migratory monocytes may become activated, uptake antigen, and drain to LNs to present antigen, similar to DCs. They may also adopt properties of macrophages, their maturation and phenotype being dependent on the inflammatory state of the invaded tissue.13 Identifying the targetability of monocyte subsets could be advantageous for localizing a therapeutic to sites of inflammation or altering surveillance in circulation.
2.2.2. Innate Cell Subsets Contribute to Clearance and Immunity
As discussed above, adaptive immunity involves molecularly specific attack of pathogens that stems from non-specific recognition by APCs. Natural killer (NK) cells are a specialized innate immune cell that straddles the innate-adaptive spectrum as they exhibit an innate recognition of pathogens and attack functions similar to adaptive immunity. NK cells were originally named for their ability to kill tumor cells but are now known to play a key role in protection against viral infections. NK cells function by detecting cells that lack expression of inhibitory signals, like those that identify a cell as ‘self’.14 Healthy cells constitutively express these signals, but these signals are often downregulated in tumor and virus-infected cells.15 Thus, when NK cells encounter these diseased cells, they identify them as ‘non-self’ and attack with a more specific reaction than that of macrophages or DCs, but less specific than adaptive immune mechanisms recognizing specific antigens. Design of therapeutic strategies for both cancer and viruses have sought to exploit NK cells ‘self’ versus ‘non-self’ discriminatory mechanisms which highlights the importance of cell-surface receptors in initiating immunity or tolerance.
2.2.3. Pattern Recognition Receptors Drive Potent Innate Immune Responses
The innate immune system has evolved many different pattern recognition receptors (PRRs) to recognize signals from a diverse set of pathogens and tissue damage. Two of the main classes of molecules that these receptors recognize are pathogen associated molecular patterns (PAMPs) – molecules that are present in pathogens but not in mammalian cells – and danger associated molecular patterns (DAMPs), endogenous signals that arise from tissue damage and elicit a noninfectious immune response.16 One of the best characterized families of PRRs is the toll-like receptors (TLRs).17 TLRs are spatially restricted within cells to align with the routes by which different pathogens may be encountered. For example, plasma membrane-associated TLRs recognize components found on the outer surfaces of pathogens, such as lipopolysaccharides (LPS) on the surface of gram-negative bacteria, while endosomal TLRs recognize intracellular components exposed after degradation of a pathogen, such as viral RNA or DNA. A PAMP binding to a TLR initiates a signal transduction cascade resulting in activation of downstream transcriptions factors (e.g., Nf-κB, AP-1). The activated transcription factors then translocate from the cytoplasm into the nucleus and initiate transcription of type I interferons (IFN-α/β) and inflammatory cytokines, such as tumor necrosis factor (TNF), IL-1, IL-6, IL-8 and IL-12. Aside from the TLR family, other common PRRs include NOD-like receptors (NLRs), RIG-like receptors (RLRs), and AIM-2 like receptors (ALRs).16 These PRRs recognize a variety of PAMPS within the cytosol of the cell and initiate signal transduction pathways that lead to secretion of type I interferons and inflammatory cytokines IL-1 and IL-18. Overall, the PRRs allow the innate immune system to recognize a broad class of pathogenic molecules and drive a potent and rapid first line of defense. By generating inflammatory responses, PRRs also influence how antigens from the pathogens are presented by APCs, and thereby have important function in initiating the more specific adaptive response.
2.2.4. APC Signals Promote T Cell Mediated Immunity and Tolerance
The purpose of this manuscript is to discuss the use of biomaterials to harness innate immunity for engineering therapeutics. To provide context for the targeting of innate mechanisms, the following examples illustrate the control of specific adaptive responses that are mediated by innate immune cells.
Targeting APCs to Activate Immunity:
An important cell type in tumors is tumor-associated macrophages (TAMs), which have a phenotype that promotes tumor cell growth and the lethal spread of tumor cells to distal organs (i.e., metastasis). TAMs suppress the anti-tumor function of T and NK cells, which has motivated therapeutics aimed at eliminating TAMs or their suppressive functions using antibodies and small molecule inhibitors.18 Approaches to eliminate or suppress TAMs could counter their pro-tumor phenotype, yet may also eliminate beneficial aspects of immunity.19 An alternative to eliminating TAMs is shifting the phenotype from pro-tumor to anti-tumor using stimulatory cues that promote inflammatory characteristics and activate antigen-presenting function that support adaptive immune clearance of tumor cells (Figure 1).20–22 Shifting the inflammatory balance within tumors is an important target for biomaterials, which can localize stimulatory immune cues that promote the transition of TAMs to an anti-tumor state.
Targeting APCs to Suppress Immunity:
In a healthy host the immune system does not attack ‘self’ tissue in part because it lacks danger signals like those found on pathogens. This tolerance of ‘self’ can break down during autoimmune disease, which results in inflammation and attack of host tissue. Current autoimmune therapies can broadly suppress these detrimental functions, but also hinder healthy aspects of immunity, leaving patient susceptible to infection. Retaining the beneficial aspects of immunity while correcting only the defects is possible through induction of immune tolerance that suppresses the attack of ‘self’ tissue as discussed in Section 2.1 (Figure 1). Of note, tolerance promoting cells like TREGS may be inducible or controllable by APCs. For example, APCs orchestrate the enrichment of organ- and antigen-specific TREGS in a healthy host to maintain immunological tolerance of organs.23,24 Controlling APC signaling is an opportunity for biomaterial platforms that regulate uptake of antigen while suppressing inflammation to present antigen to adaptive immune cells in a tolerogenic manner.
2.3. Innate Immune Functionality Diverges from Steady State During Disease
2.3.1. Immunity is Ill-Prepared to Eliminate Some Infectious Diseases
Infectious diseases are caused by bacteria, viruses, fungi, or parasites passed between individuals directly, through an insect or animal bite, or present in food. The immune system effectively combats many of these organisms, but some can evade or overpower the immune system and lead to infection. Vaccines are an important tool to prevent spread of infection, but for diseases like malaria and human immunodeficiency virus (HIV), the development of vaccines has been challenging. Malaria, for example, has a high mortality rate in specific regions of the world. Vaccines that prevent malaria transmission have been challenging to produce because the adaptive response and production of parasite-targeting antibodies is insufficient.25 One hurdle is that the functional state of APCs are altered in children that contract malaria, which can make vaccine efficacy unpredictable.26 This challenge suggests an opportunity for immune engineered biomaterials that sequentially deliver factors that first establish a baseline state in APCs, then present antigen and signals to direct adaptive immunity. For HIV vaccines, clinical trials have demonstrated a lack of or limited efficacy.27 However, innate immune activation has recently been associated with improved protection following initial vaccine administration in HIV models.27,28 Studies in these and other challenging infectious diseases indicate advantageous correlations between innate immune states and vaccine efficacy, motivating immune engineered biomaterials that exploit innate immunity to enhance vaccine efficacy.
2.3.2. Transformative Cancer Therapies Control Signals Between Innate and Adaptive Immunity
As another clinical example of the link between innate and adaptive immunity, checkpoint blockade is an important recent advance in cancer immunotherapy. Tumors suppress normal innate immune functions to evade immune recognition and support tumor growth. To reestablish normal immune functions and tumor clearance, checkpoint blockade uses antibodies to modulate the communication between innate and adaptive immunity. Antibodies that block cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) expression on T cells reestablish the costimulatory signaling between APCs and T cells that lead to an adaptive response. Another common checkpoint blockade therapy targets programmed cell death protein 1 (PD-1) and the paired ligand (PD-L1) to improve T cell engagement and elimination of tumor cells.29 Checkpoint blockade therapies are a monumental achievement, yet these powerful approaches only work in subsets of patients. In patients with cancers that resist checkpoint blockade therapy, it will be beneficial to engineer complementary approaches to improve the efficacy and specificity of this approach. For example, solid tumors have been challenging in general for immunotherapies, due in part to the accumulation of immunosuppressive cells, like myeloid-derived suppressor cells (MDSCs), that inhibit T cell effector function in the tumor. Therapeutic targeting of MDSCs has been attempted in combination with checkpoint blockade and MDSC accumulation has been reported as a possible predictor of anti-CTLA-4 therapeutic efficacy.30,31 TAMs also contribute to immunosuppression in tumors, but recent evidence indicates that different phagocytic pathways can drive pro-tumor or anti-tumor response.32 Collectively, the control of innate cell populations or specific mechanisms within these populations that accumulate in tumors could help overcome obstacles that hinder treatment of solid tumors.
2.3.3. Autoimmunity Occurs due to Aberrant Maintenance of Self-Reactive Adaptive Immune Cells
In autoimmunity, loss of tolerance, as discussed in Section 2.2.4, is driven by adaptive immune cells that mistakenly recognize antigens originating from host cells as foreign and drive an inflammatory response against the cells expressing these self-antigens. Normally, self-reactive adaptive immune cells are kept at bay through a variety of tolerance mechanisms utilized by the body to delete the cells or render the cells anergic (i.e., functionally unresponsive to antigen). For example, T cells that bind antigen presented by APCs in the absence of the proper co-stimulatory signals (e.g., CD80, CD86) can become inactivated and/or undergo apoptosis. Autoimmune disease occurs due to a lack of elimination and/or control of self-reactive adaptive immune cells. It is generally hypothesized that autoimmunity is triggered due to a combination of genetic mutations that impair innate regulatory mechanisms and environmental stimuli that initiate activation of self-reactive adaptive immune cells.33 Increasing evidence confirms aberrant activation of innate pathways, such as TLR signaling, have an important role in development of autoimmune diseases such as multiple sclerosis (MS), type 1 diabetes (T1D) and systemic lupus erythematous (SLE).34,35 Given the critical role of innate immunity in shaping adaptive, antigen-specific responses, it is also reasonable to assume that self-reactivity during autoimmune disease is influenced by innate immune cells and signaling. Biomaterial-based therapies that reprogram how APCs process and present self-antigen to T and B cells, and thus reprogram these cells away from inflammatory function and toward tolerogenic phenotypes, show promising pre-clinical results that could ultimately be part of the key to permanently reversing autoimmune disease.
3. Biomaterial-Based Treatments Function Through Modulating Innate Immunity
Biomaterials offer unique physicochemical features and tunability that have catalyzed there use in a multitude or pre-clinical studies, and increasingly, clinical trials.36–38 These properties also make biomaterials very attractive to study and control innate immunity. In this section we focus exclusively on the emerging opportunities in which biomaterial- and nanotechnology-based strategies are being applied to understand and manipulate innate immunity to address challenges facing infectious disease, cancer, and autoimmunity.
3.1. Biomaterials Improve Vaccine Capabilities for Infectious Diseases
Synthetically engineered and biomaterial strategies have been employed in pre-clinical studies and clinical trials to improve the efficacy and dissect the mechanism of treatments and vaccines against infections originating from various bacteria, viruses, and parasites. This section highlights materials-based strategies for combating infectious disease by engaging and controlling innate immunity with biomaterials.
3.1.1. Bacterial Membrane Coatings Activate Innate Immunity
Bacterial infections involve proliferation and colonization of extracellular pathogens. As a result, the immune system often relies on generation of antibodies to tag or neutralize the bacteria. Historically, vaccines to protect against these pathogens consist of bacterial components and immune agonists to stimulate antibody production; however, biomaterials offer unique features to control activation of innate immunity that stimulate adaptive cellular responses to aid clearance of infections.39
One interesting strategy is to preserve the natural cell membrane from bacteria and coat it onto synthetic nanoparticles (NPs). This approach preserves the natural cellular surface, which contains a robust set of PAMPs. This is useful for stimulating innate immune cells through multiple innate pathways developed to recognize bacteria. Coating these bacterial membranes on a NP core allows for co-delivery of immune modifying cargo. Using this idea, Gao et al. designed vaccines mimicking bacterial infections by isolating the outer membrane vesicles from E. coli bacteria, which were subsequently coated on the surface of gold NPs (AuNPs) to create bacterial membrane coated NPs (BM-AuNPs, Figure 2A). These coated NPs drove a modest increase in the ability to activate DCs in comparison to outer membrane vesicles alone (Figure 2B–C).40,41 DC activation was defined as the upregulation of costimulatory surface receptors CD40 and CD80, which are Signal 2 in the innate-adaptive signaling (as described in Section 2.1). An open question remains as to the tunability of the core material, which in these experiments utilized a gold NP that did not contain any additional immune modifying factors. Additional studies are using cell membranes from neutrophils – an innate immune cell with potent effector functions – to coat polymer NPs, which can be loaded with immune stimulating factors.42 It will be exciting to see if polymer NPs loaded with immune modifying cues and coated with bacterial membranes are able to induce more efficient adaptive cellular response to clear bacterial infections.
Figure 2. Biomaterial platforms for vaccination and engineering immunity against infectious diseases.
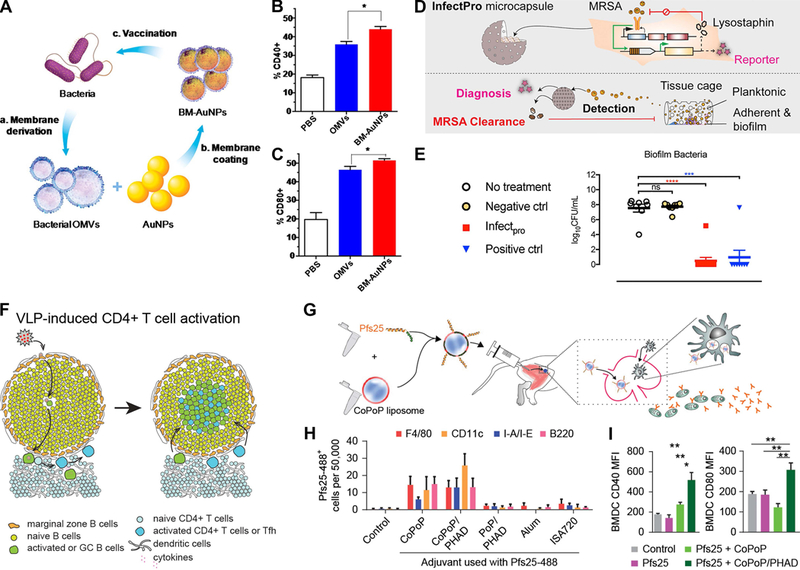
A) Bacterial outer membrane vesicle (OMV) coated gold NPs (BM-AuNPs) increase DC activation markers B) CD40 and C) CD80 compared to bacterial OMVs alone.41 D) To control MRSA at implant interfaces, encapsulated cells were genetically engineered to elute anti-bacterial lysotaphin in response to TLR stimulation, which E) prevented MRSA biofilm development.43 F) Virus-like particles (VLPs) induce antigen-presentation through B cells in a model of HIV to promote an immunogenic germinal center (GC).44 G) Admixed formation of malaria antigen-presenting NPs with a TLR agonist embeded in the liposome was delivered intramuscularly and produced long-lived plasma cells that produce antibodies against malaria antigen Pfs25. H) This was associated with innate immune uptake of liposomes containing the TLR agonist and I) activation of bone-marrow derived cells (BMDCs) that includes APCs.51 The uptake by innate immune cells was highest for liposomes that contained cobalt porphyrin-phospholipid (CoPoP) when compared against combinations of porphyrin-phospholipid (PoP) and synthetic monophosphoryl lipid A (PHAD), or alum and montanide (ISA 720) adjuvants alone. Panels adapted with permission from the indicated references.
3.1.2. Engineered Antimicrobial Cells are Triggered by Innate Pathways to Inhibit Bacterial Infections
Improved bacterial vaccines are needed but would be limited in clearance of bacterial biofilms. Biofilms develop when bacteria aggregate together and develop a microenvironment that protects them from antibiotics and innate immunity. The surface of medical implants are likely sites for bacterial biofilm formation, and the inability to treat biofilms on implant surfaces often necessitates implant removal. Thus, there is great interest in design of biomaterial strategies to prevent the development of biofilms. One team recently tackled this challenge by engineering cells to secrete an antimicrobial enzyme, lysostaphin (Figure 2D).43 Production and secretion of lysostaphin in these cells were triggered by activated TLRs that detect bacteria. These engineered cells – denoted as InfectPro – were encapsulated in a porous Teflon scaffold and then implanted to test their inhibition of biofilms. To induce a biofilm and challenge the function of the engineered cells, methicillin-resistant staphylococcus aureus (MRSA) were injected at the implant site. The inclusion of lysostaphin secreting cells in the implant prevented MRSA biofilm at the implant interface (Figure 2E).43 This work demonstrated that genetic engineering innate immune mechanisms to control cell function can combat an emerging drug-resistant bacterial health threat. It will be exciting to see how this strategy might be used to probe other innate immune interactions and to develop new anti-effectives.
3.1.3. B Cells Act as APCs to Activate Cellular Response Against Viral Vaccines
B cells that recognize specific antigens may function as an APC and activate T cells. This innate function and the antibody production of B cells make them an interesting target for modulating immunity. In one example, Hong et al. constructed bacterial phage Qb-derived virus-like particles (Qb-VLPs), which are NPs assembled from viral coat proteins.44 VLPs are strongly immunogenic due to their encapsulation of CpG-containing nucleic acids that activate inflammation through TLR signaling. Using transgenic mice as a source of antigen-specific B cells and as a means to selectively deplete DCs, the authors confirmed VLPs can activate T cells in the absence of DCs (Figure 2F). To demonstrate robustness the authors used the same model lacking DCs and administered an influenza virus, which again showed that B cells act as APCs and activate T cells. While B cells are often associated with adaptive response, this work reinforced the potential of exploiting mechanisms most often associated with innate immune function for eliciting an effective adaptive response.
3.1.4. Biomaterials Improve Gene Delivery for Vaccines Against HIV
HIV continues to be a serious global health concern. Vaccines for this virus have been limited by difficulties in generating strong responses again protective HIV antigens. Thus, an important area that biomaterials can contribute to is the control of innate immune activation to enhance vaccine efficacy.45 To improve efficacy, one solution is to deliver genes that encode HIV antigens; biomaterials have been extensively exploited for gene delivery.46,47 Biomaterial platforms are also being investigated for delivery of HIV antigens. In particular, biomaterials allow for delivery to specialized innate immune cells in the skin that can efficiently induce immunity. To target this immunological niche, DeMuth et al. utilized a degradable polymer microneedle skin patch to deliver an HIV antigen juxtaposed with a TLR3 agonist, poly(I:C), in polyelectrolyte multilayer films.48 Delivery of these cues through microneedles resulted in a skin-localized innate immune response – measured by inflammatory enzyme production – that lead to generation of systemic, antigen-specific T cells. This highlights the potential of biomaterials for controlling a local innate immune response alongside delivery of antigen to generate a systemic adaptive response.
3.1.5. Malaria Vaccine Enhanced by Co-Delivery of Antigens and Innate Immune Stimuli
The malaria lifecycle involves mosquitos infected with a parasite that are able to infect many individuals because each bite leads to transfer of the malaria parasite to the victim. A malaria vaccine has been challenging to develop because the parasite replicates rapidly and exhibits different features during different stages of infection, complicating targeting of specific critical antigens by a vaccine. However, recent clinical evidence suggests malaria vaccine protection may correlate with innate immune signatures such as DC activation.49 Thus, biomaterials that control innate mechanisms during antigen delivery may improve vaccine efficacy. One approach from Moon et al. used interbilayer-crosslinked multilamellar vesicles to entrap antigen and deliver a payload to the draining LN.50 Using this platform they showed that administered multilamellar vesicles were localized within LN resident macrophages and enhanced neutralizing antibody production when benchmarked against standard adjuvants like alum. This approach focused on the delivery of a single antigen, yet unique features of malaria over the parasite lifecycle requires approaches that integrate multiple antigens.
Huang et al. reported a malaria vaccine based on self-assembling NPs.51 These NPs readily incorporate multiple antigens, which is particularly relevant for addressing the multiple stages of malaria. These NPs are formed by a process called ‘spontaneous nanoliposome-antigen particleization’ (SNAP). SNAP NPs with malaria antigens induced long-lasting functional antibodies against malaria in mice and rabbits (Figure 2G).51 The liposome core of the antigen-presenting NPs integrated a synthetic monophosphoryl lipid A (PHAD), which is a TLR4 agonist. To investigate the impact of liposomes containing PHAD and a malaria antigen (Pfs25) on innate immunity, the draining LNs were analyzed by flow cytometry 48 hours following intramuscular administration. SNAP liposomes increased accumulation of macrophages and infiltrating monocytes in the draining LN. Further, malaria antigen presented on the SNAP liposomes was taken up more efficiently by DCs, macrophages, and B cells relative to other adjuvant formulations (e.g., alum+Psf25, Figure 2H). Activation of DCs and expression of costimulatory factors (i.e., CD40, CD80) on their cell surface also increased when formulations included PHAD (Figure 2I). Lastly, these alterations in APC trafficking and uptake in the presence of SNAP liposomes correlated with an increase in germinal centers and long-lived plasma cells. Germinal centers are domains formed in LNs that help promote long-lived plasma cells that produce antibodies to block malaria transmission. This study illustrates the key theme of integrating innate immune agonists to achieve sustained modification of immunity. The examples in this section also highlight the important role enhanced innate function has on a range of immune responses, connections that are also being exploiting in cancer immunotherapy.
3.2. Biomaterials Enhance Cancer Immunotherapy by Promoting Immunogenic APCs
Biomaterials have already been applied in a range of approaches for pre-clinical cancer therapies. Several clinical trials using biomaterial particles have completed early stages and the first cancer immunotherapy using biomaterial implants is also now in progress.52 This section discusses several of the biomaterial- and nanotechnology strategies exploiting innate immunity to probe the mechanism of tumor development and to develop immunotherapeutic platforms. We highlight strategies spanning DC vaccines – where APCs are activated against tumors – and emerging strategies that target immune-cargos to APCs in tumors.
3.2.1. DC Vaccines and Artificial APCs Program Antigen Presentation Ex Vivo
DC vaccines are a promising avenue for cancer therapeutics based on the isolation, expansion, and conditioning of DCs in controlled ex vivo conditions. DC conditioning is the process of providing immune modifying cues and tumor antigens to immature innate immune cells to promote their maturation towards an activated, antigen-presenting phenotype. These cells are then administered to patients to efficiently activate patient T cells against cancerous cells. Despite substantial investments, DC vaccines have only transiently enhanced anti-tumor responses, and had modest clinical impact.53,54 These vaccines have been limited by a lack of understanding of how signals and suboptimal antigens are integrated to condition DC function. Thus, biomaterials that control innate immune signaling may provide beneficial avenues to improve DC vaccines.
Achieving the full potential of DC-based vaccines will require studies to determine how to effectively select optimal antigens that are strongly or uniquely expressed on tumors and how best to present these antigens to drive effective immune responses. (Figure 3A).53 Additionally, the dosing and delivery of DCs or APCs for these cell vaccines is an important question. All of these questions center on innate immune cells and functions as APCs. Biomaterials offer unique properties to elucidate answers to these questions by defining design parameters for APC-based vaccines. For example, functionalized biomaterials can display tumor antigens at specific densities. Biomaterials may also serve a manufacturing role in these cancer vaccines by controlling which antigens are engulfed and presented during priming of APCs before administration to patient or in controlling the other signals co-delivered to APCs.
Figure 3. Targeting and mimicry of APCs to promote immunity against cancer.
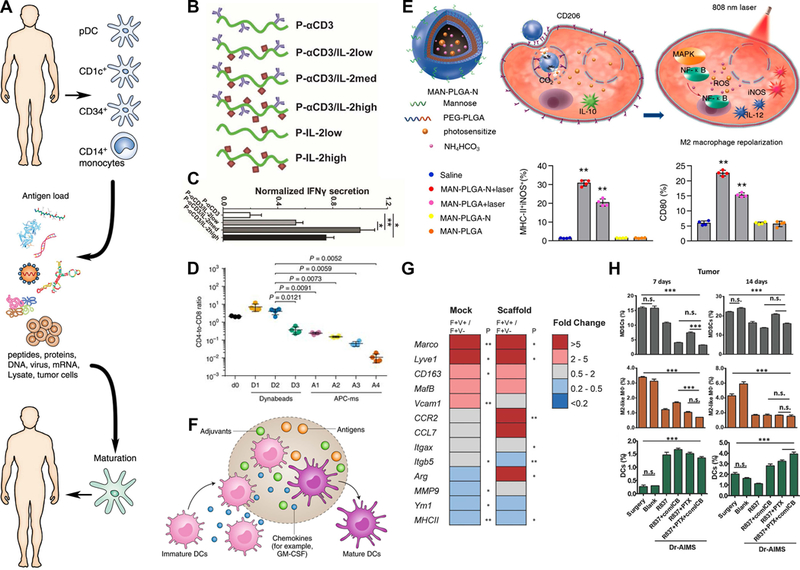
A) Conventional paradigm for DC-based vaccines where innate immune populations are removed from a patient, conditioned ex vivo with factors like tumor lysate to promote maturation, the reintroduced to provide signals to the patient’s adaptive immune cells.53. B-C) Artificial APCs that control the presentation of cytokines to T cells and D) incorporate T cell costimulatory factors to generate cytotoxic T cells.55,56 This incorporation of T cell costimulatory factors in artificial APC scaffolds (APC-ms) at a range of loadings (A1-A4) was benchmarked against T cell activating Dynabeads administered at a range of doses (D1-D3). E) Combinatorial approach to switch TAMs toward immunogenic phenotype by including mannose binding sites on PLGA NPs that were loaded with drug and photosensitizers to control macrophage inflammation at specific sites.63 F) Implanted biomaterials scaffolds are able to mature DC phenotypes in situ through recruitment and exposure to antigens, adjuvants, and chemokines.52. G) Presence of an implanted scaffold alone impacts the phenotype of TAMs, as demonstrated through gene expression analysis of TAM populations following a mock surgery (left column) or scaffold implantation (right column).68 H) Implants for release of different drug combinations at tumor resection site were compared at Days 7 and 14 for the impact on MDSCs, TAMs (M2-like macrophages), and DCs.70 Panels adapted with permission from the indicated references.
In addition to enabling APC vaccines, biomaterials have a new emerging role as artificial APCs that could entirely replace the need for actual cells. These artificial APC strategies have a similar goal to DC vaccines but rely on NPs and engineered protein constructs to control antigen and costimulatory molecule presentation with the appropriate orientation and density needed for T cell activation. Within the NP core there is also the potential to load cytokines such as IL-2 that induce proliferation of T cells. To mimic the APC presentation of cytokines to T cells, Eggermont et al. recently utilized a polyisocyanopeptide (PIC) backbone appended with controlled densities and ratios of an antibody that bind to a T cell receptor (cluster of differentiation 3, CD3); these constructs also integrated IL-2 or IFNα to stimulate T cell proliferation (Figure 3B).55 One desired element of this modular platform is the flexibility that supports dynamic rearrangement of the signaling cues during T cell interactions. Formulations that included either IL-2 or IFNα demonstrated an ability to induce proliferation and activation of T cells (Figure 3C).55 Another study integrated IL-2 alongside tumor antigen and T cell costimulatory factors in a composite scaffold and benchmarked their results against commercial microbeads that promote T cell proliferation when cultures are supplemented with IL-2. This mimicry of APC signaling from the scaffold drove a significant increase in the proliferation of cytotoxic T cells, which could then be injected to attack tumor cells. (Figure 3D).56 This control over the density of innate-adaptive modulatory signals on artificial APCs is a strength of biomaterials and nanotechnology that can be leveraged further through incorporation of more complex signaling cues, optimized display densities, or display of multiple antigens.
Another approach to activate innate immunity in tumors is targeting tumor-associated DCs. Targeting DCs in tumors may shift tumors from immunosuppressed states to activation of anti-tumor immune function. In one strategy, small interfering RNAs (siRNAs) that block pro-tumor cell functions were loaded into polyethyleneimine (PEI) NPs. These siRNA-PEI NPs were preferentially engulfed by DCs at ovarian cancer sites. The uptake activated TLR signaling and shifted the DC phenotypes towards an antigen-presenting, immunostimulatory phenotype. Interestingly, TLR5 was activated by blank PEI NPs while TLR3 and TLR7 were only activated by siRNA loaded NPs.57 In another study focusing on the cationic nature of PEI and dextran, the experiments revealed intra-tumor administration of cationic polymers induced inflammatory phenotypes in TAMs, including an increase of IL-12. Antibody inhibition of TLR4 or TAMs from TLR4 knockout mice exhibited significantly reduced reactivity to cationic polymers. This result suggests that polymers of a specific charge can modulate innate immune responses through TLR activation. During mouse challenge studies, this enhanced immunogenicity of TAMs by cationic polymer stimulation increased survival and accumulation of TEFF and NK cells in the tumor.58 Thus, the use of biomaterials to promote innate immune activation provided a way to improve the treatment of tumors.
3.2.2. Biomaterials can Deliver Payload in Response to Unique Features of Tumor Microenvironment
The inflammatory response to biomaterials is different in the tumor microenvironment as compared to healthy tissue.59 In particular, the tumor-secreted factors modulate the activation ability of the innate immune cells that respond to and aggregate around implants. It would be beneficial to utilize this difference in tissue states as a control for therapeutic release where stimuli-responsive NPs activate release of a payload within tumors. Most stimuli-responsive NPs rely on hypoxic or acidic dysregulation within tumors. This payload can specifically target various facets of the tumor, including the innate immune functions that help maintain suppression of T cells, for example, from MDSCs.60 There are many approaches that have leveraged pH-regulated degradation in tumors as a means to release a therapeutic, like TNFα.61 Alongside therapeutic release, stimuli-responsive polymer NPs have been developed for real-time imaging of tumor pH.62
A combinatorial approach was recently investigated by Shi et al. with mannose functionalized PEGylated PLGA NPs that encapsulated ammonium bicarbonate to disrupt lysosomal/endosomal compartments in TAMs; TAMs express a high density of mannose receptors to facilitate uptake (Figure 3E). Following uptake and during NP degradation, photosensitizers were released into phagocytic cells, including TAMs. Illumination of the released photosensitizers with the necessary wavelength of light generated inflammatory reactive oxygen species and shifted the TAMs towards an inflammatory and immunogenic phenotype. Last, tumor associated antigen was incorporated into the NPs to be released during the tumor transition toward an immunogenic phenotype. This system was shown to suppress melanoma tumor growth and significantly improve survival in a preclinical mouse model. To test the role of TAMs in this efficacy, clodronate liposomes were administered to deplete macrophages. This study revealed a complete loss of therapeutic efficacy.63 These data emphasize the utility of targeting a biological mechanism of innate immune cells in the primary tumor.
To integrate immunotherapy with APC stimulating agents, Wang et al. generated a reactive oxygen species (ROS) responsive hydrogel that degraded in the tumor microenvironment and released a chemotherapeutic (gemcitabine) with anti-PD-L1 blocking antibody. This approach led to innate activation and increased expression of PD-L1 in tumor-resident APCs.64 This increase in PD-L1 on APCs makes the cells responsive to checkpoint blockade therapy, where antibodies blocking PD-L1 reestablish the immune attack on tumor cells. In clinical studies, elevated PD-L1 expression in patient tumor-resident APCs is correlated with improved therapeutic response. Thus, a link exists between efficacy of checkpoint blockade and the host’s innate immune response.65
3.2.3. Biomaterial Scaffolds Instruct APCs Towards an Anti-Tumor Phenotype
Most DC vaccines are conditioned outside of a host, as discussed in Section 3.2.1, to maximize the exposure of DCs to immune modifying cues and tumor antigens. One innovative approach is to load a biomaterial scaffold with the immune cues necessary to recruit and condition DC populations in situ (Figure 3F).52 This concept is currently being tested in a phase I clinical trial (ClinicalTrials.gov # NCT01753089) using a degradable PLGA scaffold. This implanted scaffold integrates antigens from the lysate of skin cancer cells and innate immune growth factors (GM-CSF) and agonists (CpG). This exciting use of biomaterials as a platform highlights the potential translation of engineering innate immunity in cancer patients.
The preceding example used scaffolds loaded with antigen and stimulatory cues, yet the presence of an implanted polymer scaffold alone alters the phenotype of innate immune cells in the primary tumor. Microporous polymer implants designed to facilitate tissue ingrowth and capture of metastatic tumor cells for early detection have recently been studied for their impact on the gene expression of innate immune cells in the primary tumor.66–68 In mice receiving either a polymer scaffold or mock surgery, TAMs from primary tumors were separated based on their expression of Vcam1, which is indicative of blood-borne macrophages, as opposed to tissue-resident macrophages. In mice with implants, blood-borne macrophages (Vcam1+) expressed altered gene expression (Figure 3G). Thus, the implantation of a biomaterial scaffold altered the phenotype of TAMs, indicating a systemic role of implants in altering disease state. Data from implanted blank scaffolds, scaffolds loaded with antigen and agonists, and different surgical parameters provides a flexible system for modulating APCs in cancer towards an anti-tumor phenotype.69
As discussed in Section 2.2.4., the concept of reprogramming APCs in tumors from a pro-tumor to an anti-tumor phenotype could aid rejection of tumors. Ren et al. focused on this concept to address tumor regrowth after an incomplete therapeutic excision in a pre-clinical model, where residual tumor cells will result in disease recurrence.70 A biomaterials scaffold containing both structural and sacrificial components was fabricated to release a payload that contained paclitaxel to deplete tumor cells and TAMs, imiquimod to activate APCs, and a combination of immune checkpoint blockade antibody anti-CTLA-4 and anti-OX40. This diverse range of cargo decreased tumor regrowth and improved survival against individual components loaded in scaffolds, indicating additive or synergistic benefit. When compared with soluble systemic administration at a 10-fold higher dose (which also improved survival), the loaded scaffolds prolonged survival (42.5 d vs. 21 d). Immune cell profiling indicated that all scaffolds releasing therapeutics resulted in an increase in DCs and a decrease in TAMs and MDSCs at the site of regrowth (Figure 3H). This work demonstrates the utility of biomaterials for controlled drug release that shift APCs toward an immunogenic phenotype to prevent primary tumor recurrence, yet extending these principles to distant metastatic recurrence is needed to improve patient survival.
3.3. Engineered Antigen Delivery to APCs Induces Tolerance during Autoimmunity
Treatment strategies for autoimmune diseases have come a long way in effectively mitigating symptoms, making living with devastating diseases like MS and type 1 diabetes more manageable for patients and the families. However, the most effective therapeutic approaches tend to also be non-specific immunosuppressants that are non-curative, require life-long treatment, and lack specificity. Even monoclonal antibody-based drugs that are molecularly-specific do not distinguish between healthy and self-reactive immune cells expressing a targeting ligand. The challenge for new strategies is then to induce sustained, antigen-specific immune tolerance without hindering normal immune function. Given that adaptive immune responses are inherently antigen-specific and are mediated by APCs of the innate immune system, targeting innate pathways to induce antigen-specific tolerance is a promising approach currently being explored. The unique features of biomaterials, including co-delivery of multiple immune signals, have been exploited in a variety of contexts to control delivery and presentation of antigen to specific innate populations or signaling mechanisms. The general approach involves delivering self-antigens attacked during an autoimmune disease to APCs either alone or along with an immunomodulatory signal. These strategies seek to control the processing and presentation of antigen and regulatory cues to foster interactions with the adaptive immune system in a way that programs selective, antigen-specific tolerance.
3.3.1. Coupling Antigen to Apoptotic Cells Targets Self-Antigen to Innate Clearance Pathways
One promising strategy that has emerged in recent years to induce antigen specific tolerance is targeting disease relevant antigen to apoptotic cell clearance pathways.71 Phagocytic cells routinely clear apoptotic debris without eliciting an adaptive immune response. This implies that the antigens from apoptotic debris are processed and presented through innate mechanisms that allow the immune system to recognize these antigens as “self”. To harness this naturally occurring tolerogenic pathway, several groups have conjugated disease relevant antigens to apoptotic splenocytes or red blood cells (RBCs) as a treatment strategy in mouse models of autoimmunity.72–79 The hypothesis is that because these cells are normally recycled in the spleen at high rates, the antigens will be processed through the same tolerogenic mechanisms and presented by APCs to effector cells in a way that promotes selective tolerance and reverses disease. Getts et al. have shown that antigen coupled splenocytes (Ag-SP) effectively induced tolerance in a mouse model of MS (EAE). The strategy was not effective in inducing tolerance in splenectomized mice or when administered with intraperitoneal or subcutaneous injection, indicating that the apoptotic clearance pathways in the spleen are necessary for inducing tolerance with this strategy.75 Upon injection, Ag-SP upregulated expression of scavenger receptor SRBII and co-localized with macrophages, but not DCs, in the marginal zone of the spleen (Figure 4A–D).75 Tolerogenic processing of the disease relevant antigen led to subsequent production of antigen specific TREGS and was dependent on regulatory cytokine IL-10 secretion and PD-L1 surface expression by APCs. More recently, Pishesha et al. showed that when antigen coupled RBCs were employed in a therapeutic setting, treatment was also able to protect mice from EAE.74 Protection from disease was associated with T cells producing less of inflammatory cytokines IFN-γ and IL-17. These data indicate that this treatment strategy leads to tolerogenic processing and presentation of antigen to T cells, resulting in polarization towards regulatory T cell phenotypes that mitigate disease. Although this strategy has shown great promise and has already been attempted in a clinical setting,76 further understanding of how tolerance is generated is needed so it can be optimized for clinical translation.
Figure 4. Engineering tolerance in autoimmunity.
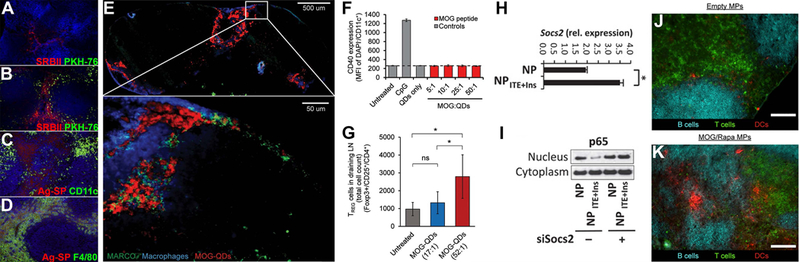
A) Untreated mice did not express high levels of scavenger receptor SRBII in the spleen. B) However, upon intravenous infusion of Ag-SP (PKH-76), SRBII was upregulated and some co-localization of SRBII and Ag-Sp was observed. C-D) Ag-SP co-localized with macrophages (F4/80), not DCs (CD11c), in the spleen.75 E) Subcutaneous treatment of mice with antigen coupled QDs (MOG-QDs) resulted in drainage of QDs to the LN and co-localization with scavenger receptor MARCO expressing macrophages. F) Treating DCs with QDs coupled to varying doses of peptide did not elicit any inflammatory activation, measured by CD40 expression, in vitro. G) Treating EAE-induced mice with QDs coupled to a higher dose of peptide (52:1) resulted in a greater reduction in disease severity compared to mice treated with QDs coupled to a lower dose of peptide (17:1). Improvement was correlated with increased expansion of TREGS in the LN.80 H) Ag-ITE NPs increased expression of Socs2 in DCs in culture. I) Ag-ITE NPs inhibited translocation of Nf-ΚB into the nucleus. When treated with an inhibitor to Socs2 (siSocs2), translocation of Nf-ΚB into the nucleus was restored, indicating that Ag-ITE NP inhibited Nf-ΚB signaling via upregulation of Socs2.91 J-K) Intra-LN injection of Ag/Rapa-MPs resulted in reorganization of the LN while treatment with empty MPs did not cause any changes.105 Panels adapted with permission from the indicated references.
3.3.2. Delivery of Antigen in Particulate Form Promotes Antigen-Specific Tolerance
Interestingly, several labs have shown that encapsulating or displaying self-antigens on NPs or microparticles (MPs) has similar therapeutic effects in mouse models of autoimmune disease.80–87 Upon administration, these antigen bearing particles co-localized with scavenger receptor expressing macrophages,80,81 indicating that delivering antigen in particulate form to target them to innate clearance pathways could be a potential strategy to induce selective tolerance. For example, a recent study from Hess et al. showed that CdSe/ZnS core/shell quantum dots (QDs) coupled with self-antigen localized with macrophages expressing scavenger receptor MARCO in the LN (Figure 4E).80 These QDs did not elicit inflammatory signaling when exposed to DCs in culture (Figure 4F). Tolerogenic processing is further supported by the fact that therapeutic efficacy is associated with increased TREGS and upregulation of regulatory markers PD-L1 and CTLA-4 by APCs.84–86
Being able to harness apoptotic cell clearance pathways without the need to use live cells could provide great benefit towards understanding of how tolerance is generated. As mentioned above, one of the limitations of coupling antigen to apoptotic cells was that efficacy was only achieved when administered intravenously so that cells reached the spleen. Studies using antigen coupled particles have already demonstrated efficacy in mouse models using a variety of administration strategies, including subcutaneous injection to target draining LNs,80 oral administration to target DCs found in the gut,85 and intravenous injection to target APCs in the liver84 as well as the spleen.81,83 Biomaterials, in particular, offer unique features to understand the “design rules” that could enable these strategies targeting innate clearance pathways.80,88 For example, studies by Hess et al. revealed that the dose and density at which self-antigen is delivered on QDs correlates with therapeutic efficacy.80 Mice induced with EAE responded to treatment more efficiently when the dose of peptide was higher; efficacy was associated with enhanced expansion of TREGS (Figure 4G). Intriguingly, however, efficacy was inversely correlated with the density at which the peptide was delivered on the QDs. Disease severity was reduced when mice were treated with a higher number of particles displaying fewer peptides per particle, compared to treatment with a lower number of particles displaying more peptides per particle. This outcome was interesting because in all cases, the total dose of peptide remained fixed. These results indicate that elucidating the design features that promote tolerogenic processing of antigen by APCs will be a crucial aspect to understanding how innate pathways can be harnessed to reverse autoimmunity.
3.3.3. Employing Polymeric Carriers to Co-Deliver Antigen and Tolerogenic Cues Promotes Tolerance
Biomaterials have also been exploited to co-deliver disease relevant antigen with regulatory immune signals to APCs.88–107 The hypothesis for this type of strategy is that inhibiting inflammatory signaling or promoting regulatory signaling while antigen is being processed and presented by APCs will promote presentation of antigen in a manner that polarizes effector cells towards tolerance. Yeste et al. have shown that delivery of gold NPs loaded with antigen and 2-(1H-Indol-3-ylcarbonyl)-4-thiazolecarboxylic acid methyl ester (ITE), an aryl hydrocarbon receptor (AhR) agonist, reduced incidence of disease in a mouse model of T1D.90,91 DCs isolated from mice and treated with these particles after stimulation with LPS in culture expressed lower levels of inflammatory cytokines IL-6 and IL-12, increased regulatory cytokine IL-10, and induced less proliferation and inflammatory cytokine secretion by antigen-specific mouse T cells. The tolerogenic particles increased expression of a protein (SOCS2) that inhibited an important transcription factor (Nf-κB) of the TLR signaling cascade (Figure 4H–I).91 When these particles were tested in cells from human patients, particle treated DCs similarly expressed lower levels of activation markers and reduced polarization of T cells towards inflammation. Other studies have employed polymeric carriers, such as PLGA and dextran, to co-deliver disease relevant antigen with known regulatory signals such as rapamycin,92,96,97,101,105 IL-10,89 dexamethasone,93 and TGF-β.94 A common theme underlying therapeutic efficacy in these studies is reduced expression of activation markers (e.g., CD80, CD86) by particle treated DCs, reduced inflammatory cytokine signaling, and increased numbers of antigen-specific TREGS.
Recently, the Jewell lab employed a novel strategy of injecting PLGA MPs loaded with antigen and rapamycin directly into the LN to treat EAE.105 This strategy directly targeted the site where innate immune cells converge to program adaptive responses and provided an opportunity to study tolerance induction with respect to the local structure and function of the LN microenvironment. Treatment with these particles via intra-LN injection led to local LN reorganization (Figure 4J–K), systemic expansion of TREGS, and a systemic, antigen-specific tolerance response that permanently reversed paralysis in mice with EAE. An alternative strategy that has been attempted is to establish an artificial microenvironment that attracts APCs to the site and promotes tolerogenic processing of antigen.95,98,99,108 This strategy involves the use of chemical signals (e.g., GM-CSF) to recruit local APCs to the site and regulatory immune signals to facilitate processing of antigen in a tolerogenic manner. Interestingly, data have shown that DCs recruited to the sites are polarized to tolerogenic phenotypes, expansion of TREGS is promoted, and disease severity is improved in mouse models after treatment.
3.3.4. Carrier-Free Nanotechnologies Juxtaposing Self-Antigen and a Regulatory Signal Promote Tolerogenic Processing by APCs
One potential limitation for translation of strategies involving polymeric carriers is the intrinsic immunogenicity of commonly used polymeric particles such as PLGA.109 Considering that added inflammation could exacerbate autoimmune disease, several strategies are being developed based on self-assembled, carrier free materials.110 One approach involves electrostatic self-assembly of carrier free structures composed entirely of immune signals to eliminate any “carrier-effects”. Tostanoski et al. engineered capsules composed of antigen and a TLR antagonist assembled into immune polyelectrolyte multilayer (iPEM) capsules. Since TLR signaling is over active in many human autoimmune diseases (see Section 2.3.3), the goal was to test if inhibition of TLR signaling while delivering self-antigen to DCs could induce tolerance during EAE.106 Treatment with capsules completely prevented paralysis in mice and reduced antigen-specific inflammatory responses in samples from human MS patients. A separate study by Hess et al. employed electrostatic interactions to self-assemble the self-antigen and TLR antagonist into polyplex-like nanostructures also designed to blunt TLR signaling during antigen presentation. Polyplex-treated DCs reduced antigen-specific T cell proliferation in culture and reduced severity and incidence of disease in mice induced with EAE. These self-assembled particles not only provide a potential strategy for treating autoimmune disease, but their simple design and modularity could also be employed as a tool to gain more mechanistic insight into how combinations of signals are processed and the relative role of specific components on tolerance induction. The impact of intrinsic design features, such as size and charge, on processing by APCs can also be studied to determine how this and other strategies can be optimized for cargo delivery.
4. Improving Design and Delivery of Therapeutics Remain as Challenges in Programming Innate Immunity
The preceding sections have highlighted the disease-centric studies, yet the mechanistic interactions during the synthetic presentation of immune signals often elicits unforeseen responses or variable magnitudes of response. This is due in part to a lack of understanding of the fundamental interactions between synthetic and natural biomaterials with the biological response. This section focuses on insights gained from recent studies that systematically evaluate how different immune cues and biomaterial properties affect innate immune response. Also highlighted are new methods being developed to improve delivery of therapeutics to innate immune cells.
4.1. Both the Biomaterial Carrier and Loaded Immune Cues Modulate the Innate Immune Response
4.1.1. Varying Modulatory Cues Changes Innate Response to Therapeutics
For vaccine platforms, a fundamental element is the inclusion of an agonist (i.e., adjuvant) with the antigen to ensure stimulation of an immune response. Many of these agonists exploit the potent inflammatory response mounted by TLR signaling. However, the innate system has evolved to detect pathogens with a multitude of PRRs, so use of a single agonist, while controlled, may underutilize the potential of APCs. Madan-Lala utilized a PLGA MP as a platform for testing the innate immune response to different combinations of clinically relevant TLR agonists including: Pam3CSK4 (TLR2 agonist), MPLA (TLR4 agonist), R837 (TLR7 agonist) and CpG (TLR9 agonist). Ten different combinations were compared by in vitro assessment of DC IL12p70 and IL10 secretion, which impact T cell phenotypes during antigen presentation. Results of these studies show that different TLR agonist combinations result in differential modulation of DC cytokine output, which could aid control of adaptive immune responses during antigen-presentation.111
Building off the comparison of multiple agonists, another study recently compared the transcriptional profile of DCs exposed to tolerogenic agents. Vitamin D3, dexamethasone, and rapamycin treated DCs were compared against the transcriptional profile of mature and immature DCs (Figure 5A). These studies indicated drastically different profiles with rapamycin treatment compared to those observed when treated with vitamin D3 and dexamethasone. While this work did not focus on the release of modulatory agents from biomaterials, it does point towards the use of deep transcriptional analysis of multiple factors that control immune responses, with benchmarks against DCs at different stages of activation.112 A study from Gammon et al. extended this mechanistic data by integration of rapamycin into biodegradable MPs for the control of T cell phenotypes. Administration of these rapamycin-loaded MPs to DCs decreased their activation state and modulated secretion of inflammatory cytokines in a non-linear manner, demonstrating the possibility of tuning immune cell phenotype (Figure 5B–D).113
Figure 5. Biomaterial integration of regulatory cues, carrier propulsion, and multiple immune cues for targeted delivery to skin.
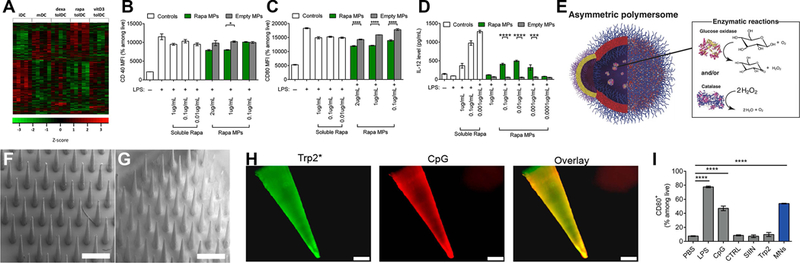
A) Systematic investigation of tolerogenic cues dexamethasone, rapamycin, and vitamin D3 benchmarked against immature and mature DCs serves as a biological basis for the design of loaded NPs.112 B-C) Biodegradable, rapamycin loaded MPs decrease the activation state of APCs and D) modulate the secretion of inflammatory cytokines as a function of dose.113 E) Asymmetric polymersome are able to mobilize towards chemotactic gradients based on polar surface chemistry.118 F) Scanning electron microscopy of uncoated polymer microneedles compared to G) microneedles with polyelectrolyte multilayer coatings containing H) antigen against tumor cells and a TLR9 agonist, CpG, for delivery of cancer vaccine mediate cancer immunity through I) activation of APCs.121 Panels adapted with permission from the indicated references.
4.1.2. Chemical Properties of Biomaterials Dictate Interactions with Innate Immune Cells
There are a variety of biomaterial approaches used to integrate different immune modulatory cues alongside antigens. More so, there are a range of studies dedicated to design of tunable biomaterial properties like surface charge, degradation, surface functionalization versus encapsulation, and novel biomaterial formulations. Given that all biomaterials introduced in vivo elicit a foreign body response that is directed by innate immune cells, understanding how intrinsic properties of biomaterials affect these responses will be necessary to design clinically translatable therapeutics.
Surface charge of the carrier is a fundamental characterization for NPs and has been shown to modulate uptake by APCs. In a study delivering anionic and cationic NPs to the lung, positively charged NPs were preferentially engulfed by DCs. Interestingly, tissue-resident macrophages engulfed a higher percentage of negatively charged NPs.114 In addition to charge, the biomaterial composition plays a role in the activation of differential innate immune populations. Comparisons between polystyrene and PLGA have demonstrated recruitment of different cell types and differential activation of inflammasomes, which operate through the NLR pathway to trigger innate activation.115
One study used a systematic combination of alginate chemistries to build a library of alginate analogs (n=634), which were then evaluated in vivo for immune activation and fibrous encapsulation. The results of this study indicate that specific compositions based on triazole elicited a reduced inflammatory profile. This decrease in inflammation was associated with a decrease in the fibrous encapsulation component of foreign body response. The goal of this study was to identify biomaterials that could mitigate the foreign body response, which is largely dictated by innate immunity, to improve long-term performance of implants. While this line of investigation is more typical for biocompatibility and implant/device development, benchmarking a family of polymers can also provide novel insight into the relationship between synthetic biomaterials and innate immunity for vaccine and immunotherapeutic applications.116
4.2. Improved Delivery of Therapeutics Aids Control Over Innate Immune Responses
4.2.1. Carrier Propulsion Systems Can Improve Targeted Particle Delivery
One parameter important in targeting specific immune cell populations is the orientation of targeting moieties on the surface of a carrier. Active targeting has been studied and reviewed to great extent for cancer.117 For example, a breast cancer binding site oriented on the surface of a liposome or polymer NP loaded with Doxorubicin has been used to direct delivery of chemotherapy. An example of this concept for innate immunity was already detailed above (Figure 3E) involving the NP surface conjugated with mannose to promote uptake by macrophages (which express a high density of mannose receptors). However, the concept of active targeting is fundamentally challenging because the binding of a moiety on a carrier surface to a target cell requires the carrier to be in close proximity with the target cell. Additionally, uptake of particles by innate immune cells that are not being targeted can deplete the administered dose and cause off target effects (i.e., immunodeficiency). An exciting concept for actively directing guidance is the inclusion of propulsion systems within nanocarriers. These carriers respond to chemical gradients in a similar manner to the mechanism of cellular chemotaxis, where a chemical gradient orients cellular migration. A recent study by Joseph et al. established a framework for self-propelled NPs that respond to glucose levels. These researchers observed the ability of this propulsion to aid in blood-brain barrier crossing (Figure 5E).118 Collectively, while active targeting is challenging, the rapid advance of synthetic nanotechnologies may soon enable carriers to not only co-opt cellular pathways of inflammation, but also chemical gradients for targeting.
4.2.2. Microneedles Allow for Localized Delivery of Cargo to Skin-Resident APCs
The immune cells found in skin are uniquely positioned to mediate immunity, as illustrated earlier in the discussion on skin-resident DCs. A biomaterials platform that takes advantage of the skin immunological niche is microneedle patches, which contain hundreds of microneedles to deliver an immunological payload. This payload is delivered either through delamination of coatings from solid microneedles, diffusion of encapsulated agents from dissolvable microneedles, or injection of solutions from hollow microneedles. Microneedle patches are also pain-free and easy to apply, which can aid patient compliance. These technologies have recently been investigated in humans for administration of an influenza vaccine.119 Of note, delivery of the influenza vaccine through microneedles generated similar immune responses when compared against the standard intramuscular injection.120
Our group has leveraged microneedles as a delivery platform for a melanoma cancer vaccine. Zeng et al. coated polylactic acid microneedles with a melanoma antigen and a TLR9 agonist (CpG) using an automated layer-by-layer coating process that resulted in self-assembled polyelectrolyte multilayers (Figure 5F–H). Following insertion into skin, the coating is delivered and can be engulfed and processed by APCs. Introduction of the coating in co-cultures of DCs and T cells resulted in DC activation (Figure 5I), supporting in vivo measurements of increased tumor-specific CD8+ T cells in peripheral blood.121 Another recent study demonstrated that within a single microneedle insertion site, it was possible to administer antigen- and adjuvant-laden NPs from a microneedle coating. Following injection, immune cells and soluble factors that migrate or diffuse into the microneedle coating can be collected for diagnostics. Microneedles are likely to find an increasingly useful tool for targeting and assessing the skin immunological niche.122 When considered alongside approaches that directly engineer the LN, as discussed in Section 3.3.3, the choice of delivery site is an important experimental parameter for engineering innate immunity.37
5. Conclusion
Control of innate mechanisms is increasingly important in immune engineering strategies to induce immunity against infectious disease and cancer or to promote tolerance against self-reactivity in autoimmune disease. A common theme in engineering innate immunity is the co-opting of innate mechanisms like antigen-presentation and co-stimulation to control adaptive immune responses. Biomaterial-based platforms have a tunable and modular nature that makes these technologies well suited for probing established and newly discovered innate immune mechanisms. Leveraging the precision of biomaterials to load cargo that directs innate immunity provides a technical foundation for future studies to develop specific and durable therapeutics.
Acknowledegements
This work was supported in part by the United States Department of Veterans Affairs (Award # 1I01BX003690), the National Institutes of Health (Award # R01EB026896), and NSF CAREER Award # 1351688. E. F. is a Clark Doctoral Fellow supported in part by Clark School of Engineering.
Author Biographies
 Robert S. Oakes is a Postdoctoral Research Associate in the Fischell Department of Bioengineering at the University of Maryland, College Park. Dr. Oakes’ research focuses on immune engineering platforms and molecular regulation of the biomaterial-tissue interface. He earned his PhD in Bioengineering from the University of Utah, then worked as a Postdoctoral Research Fellow at the University of Michigan in the field of cancer diagnostics. He is completing his postdoctoral training at the University of Maryland, College Park where his work focuses on engineered therapeutics for autoimmunity and cancer.
Robert S. Oakes is a Postdoctoral Research Associate in the Fischell Department of Bioengineering at the University of Maryland, College Park. Dr. Oakes’ research focuses on immune engineering platforms and molecular regulation of the biomaterial-tissue interface. He earned his PhD in Bioengineering from the University of Utah, then worked as a Postdoctoral Research Fellow at the University of Michigan in the field of cancer diagnostics. He is completing his postdoctoral training at the University of Maryland, College Park where his work focuses on engineered therapeutics for autoimmunity and cancer.
 Eugene Froimchuk is a graduate student in the Fischell Department of Bioengineering at the University of Maryland, College Park. His research focuses on studying how biomaterial properties and design parameters of therapeutics impact immune function in order to develop targeted vaccines for autoimmunity. He earned his B.S. in Biochemistry and Biological Sciences from the University of Maryland, College Park, and then worked at the National Institutes of Health as an Intramural Research Training Award (IRTA) Fellow for a year before beginning his graduate training.
Eugene Froimchuk is a graduate student in the Fischell Department of Bioengineering at the University of Maryland, College Park. His research focuses on studying how biomaterial properties and design parameters of therapeutics impact immune function in order to develop targeted vaccines for autoimmunity. He earned his B.S. in Biochemistry and Biological Sciences from the University of Maryland, College Park, and then worked at the National Institutes of Health as an Intramural Research Training Award (IRTA) Fellow for a year before beginning his graduate training.
 Christopher M. Jewell is an Associate Professor and Associate Chair in the Fischell Department of Bioengineering at the University of Maryland. Dr. Jewell also holds an appointment in the United States Department of Veterans Affairs at the VA Maryland Health Care System. Dr. Jewell’s research spans engineering and immunology to study and manipulate immune function for therapeutic vaccines targeting autoimmunity and cancer. He earned his PhD in Chemical Engineering from the University of Wisconsin, then worked as a consultant at the Boston Consulting Group in the Healthcare Practice. Dr. Jewell completed his postdoctoral training at MIT and Harvard, launching his own lab 2012.
Christopher M. Jewell is an Associate Professor and Associate Chair in the Fischell Department of Bioengineering at the University of Maryland. Dr. Jewell also holds an appointment in the United States Department of Veterans Affairs at the VA Maryland Health Care System. Dr. Jewell’s research spans engineering and immunology to study and manipulate immune function for therapeutic vaccines targeting autoimmunity and cancer. He earned his PhD in Chemical Engineering from the University of Wisconsin, then worked as a consultant at the Boston Consulting Group in the Healthcare Practice. Dr. Jewell completed his postdoctoral training at MIT and Harvard, launching his own lab 2012.
Footnotes
Disclosures
C.M.J. is an employee of VA Maryland Health Care System. The views reported in this paper do not reflect the views of the Department of Veterans Affairs or the United States Government. C.M.J. has an equity position in Cellth Systems, LLC.
References
- [1].Callard R, George AJ & Stark J, Immunity 1999, 11, 507–513. [DOI] [PubMed] [Google Scholar]
- [2].Jain A & Pasare C, J Immunol 2017, 198, 3791–3800, doi: 10.4049/jimmunol.1602000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dormitzer PR, Ulmer JB & Rappuoli R, Trends Biotechnol 2008, 26, 659–667, doi: 10.1016/j.tibtech.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rappuoli R, Bottomley MJ, D’Oro U, Finco O & De Gregorio E, J Exp Med 2016, 213, 469–481, doi: 10.1084/jem.20151960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moyer TJ, Zmolek AC & Irvine DJ, J Clin Invest 2016, 126, 799–808, doi: 10.1172/JCI81083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hartwell BL, Antunez L, Sullivan BP, Thati S, Sestak JO & Berkland C, J Pharm Sci 2015, 104, 346–361, doi: 10.1002/jps.24273. [DOI] [PubMed] [Google Scholar]
- [7].Worbs T, Hammerschmidt SI & Forster R, Nat Rev Immunol 2017, 17, 30–48, doi: 10.1038/nri.2016.116. [DOI] [PubMed] [Google Scholar]
- [8].Kushwah R & Hu J, Immunology 2011, 133, 409–419, doi: 10.1111/j.1365-2567.2011.03457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ali N, Zirak B, Truong HA, Maurano MM, Gratz IK, Abbas AK & Rosenblum MD, J Immunol 2018, 200, 3100–3108, doi: 10.4049/jimmunol.1701206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Audiger C, Rahman MJ, Yun TJ, Tarbell KV & Lesage S, J Immunol 2017, 198, 2223–2231, doi: 10.4049/jimmunol.1601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lavin Y, Mortha A, Rahman A & Merad M, Nat Rev Immunol 2015, 15, 731–744, doi: 10.1038/nri3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gordon S, Pluddemann A & Mukhopadhyay S, Cold Spring Harb Perspect Biol 2014, 7, a016378, doi: 10.1101/cshperspect.a016378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jakubzick CV, Randolph GJ & Henson PM, Nat Rev Immunol 2017, 17, 349–362, doi: 10.1038/nri.2017.28. [DOI] [PubMed] [Google Scholar]
- [14].Hammer Q, Ruckert T & Romagnani C, Nat Immunol 2018, 19, 800–808, doi: 10.1038/s41590-018-0163-6. [DOI] [PubMed] [Google Scholar]
- [15].Molgora M, Bonavita E, Ponzetta A, Riva F, Barbagallo M, Jaillon S, Popovic B, Bernardini G, Magrini E, Gianni F, Zelenay S, Jonjic S, Santoni A, Garlanda C & Mantovani A, Nature 2017, 551, 110–114, doi: 10.1038/nature24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tang D, Kang R, Coyne CB, Zeh HJ & Lotze MT, Immunol Rev 2012, 249, 158–175, doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kawasaki T & Kawai T, Front Immunol 2014, 5, 461, doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Noy R & Pollard JW, Immunity 2014, 41, 49–61, doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Komohara Y, Fujiwara Y, Ohnishi K & Takeya M, Adv Drug Deliv Rev 2016, 99, 180–185, doi: 10.1016/j.addr.2015.11.009. [DOI] [PubMed] [Google Scholar]
- [20].Peng J, Tsang JY, Li D, Niu N, Ho DH, Lau KF, Lui VC, Lamb JR, Chen Y & Tam PK, Cancer Lett 2013, 331, 239–249, doi: 10.1016/j.canlet.2013.01.001. [DOI] [PubMed] [Google Scholar]
- [21].Kang SW, Lee SC, Park SH, Kim J, Kim HH, Lee HW, Seo SK, Kwon BS, Cho HR & Kwon B, Cancer Res 2017, 77, 5989–6000, doi: 10.1158/0008-5472.CAN-17-0610. [DOI] [PubMed] [Google Scholar]
- [22].Haabeth OAW, Fauskanger M, Manzke M, Lundin KU, Corthay A, Bogen B & Tveita AA, Cancer Res 2018, 78, 4573–4585, doi: 10.1158/0008-5472.CAN-17-2426. [DOI] [PubMed] [Google Scholar]
- [23].Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, Amit AS, Kang C, Geddes JE, Allison JP, Socci ND & Savage PA, Science 2013, 339, 1219–1224, doi: 10.1126/science.1233913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Leventhal DS, Gilmore DC, Berger JM, Nishi S, Lee V, Malchow S, Kline DE, Kline J, Vander Griend DJ, Huang H, Socci ND & Savage PA, Immunity 2016, 44, 847–859, doi: 10.1016/j.immuni.2016.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Murray CJL, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R & Lopez AD, Lancet 2012, 379, 413–431, doi:Doi 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- [26].de Oca Montes M., Good MF, McCarthy JS & Engwerda CR, J Immunol 2016, 197, 4518–4526, doi: 10.4049/jimmunol.1600619. [DOI] [PubMed] [Google Scholar]
- [27].Vaccari M, Fourati S, Gordon SN, Brown DR, Bissa M, Schifanella L, de Castro Silva I., Doster MN, Galli V, Omsland M, Fujikawa D, Gorini G, Liyanage NPM, Trinh HV, McKinnon KM, Foulds KE, Keele BF, Roederer M, Koup RA, Shen X, Tomaras GD, Wong MP, Munoz KJ, Gach JS, Forthal DN, Montefiori DC, Venzon DJ, Felber BK, Rosati M, Pavlakis GN, Rao M, Sekaly RP & Franchini G, Nat Med 2018, 24, 847–856, doi: 10.1038/s41591-018-0025-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, Zueva E, Maurin M, Nadalin F, Knott GJ, Zhao B, Du F, Rio M, Amiel J, Fox AH, Li P, Etienne L, Bond CS, Colleaux L & Manel N, Cell 2018, 175, 488–501 e422, doi: 10.1016/j.cell.2018.08.062. [DOI] [PubMed] [Google Scholar]
- [29].Ribas A & Wolchok JD, Science 2018, 359, 1350–1355, doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, Lepage P, Boneca IG, Chamaillard M, Kroemer G & Zitvogel L, Immunity 2016, 44, 1255–1269, doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- [31].Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, Merghoub T & Wolchok JD, Cell Rep 2015, 13, 412–424, doi: 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, Quarato G, Boada-Romero E, Kalkavan H, Johnson MDL, Natarajan S, Turnis ME, Finkelstein D, Opferman JT, Gawad C & Green DR, Cell 2018, 175, 429–441 e416, doi: 10.1016/j.cell.2018.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rosenblum MD, Remedios KA & Abbas AK, J Clin Invest 2015, 125, 2228–2233, doi: 10.1172/jci78088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Waldner H, Autoimmun Rev 2009, 8, 400–404, doi: 10.1016/j.autrev.2008.12.019. [DOI] [PubMed] [Google Scholar]
- [35].Mohammad Hosseini A., Majidi J, Baradaran B & Yousefi M, Adv Pharm Bull 2015, 5, 605–614, doi: 10.15171/apb.2015.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bookstaver ML, Tsai SJ, Bromberg JS & Jewell CM, Trends Immunol 2018, 39, 135–150, doi: 10.1016/j.it.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gosselin EA, Eppler HB, Bromberg JS & Jewell CM, Nat Mater 2018, 17, 484–498, doi: 10.1038/s41563-018-0077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Irvine DJ, Swartz MA & Szeto GL, Nat Mater 2013, 12, 978–990, doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lin LC, Chattopadhyay S, Lin JC & Hu CJ, Adv Healthc Mater 2018, 7, e1701395, doi: 10.1002/adhm.201701395. [DOI] [PubMed] [Google Scholar]
- [40].Angsantikul P, Thamphiwatana S, Gao W & Zhang L, Vaccines (Basel) 2015, 3, 814–828, doi: 10.3390/vaccines3040814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gao W, Fang RH, Thamphiwatana S, Luk BT, Li J, Angsantikul P, Zhang Q, Hu CM & Zhang L, Nano Lett 2015, 15, 1403–1409, doi: 10.1021/nl504798g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang Q, Dehaini D, Zhang Y, Zhou J, Chen X, Zhang L, Fang RH, Gao W & Zhang L, Nat Nanotechnol 2018, doi: 10.1038/s41565-018-0254-4. [DOI] [PubMed] [Google Scholar]
- [43].Liu Y, Bai P, Woischnig AK, Charpin-El Hamri G., Ye H, Folcher M, Xie M, Khanna N & Fussenegger M, Cell 2018, 174, 259–270 e211, doi: 10.1016/j.cell.2018.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hong S, Zhang Z, Liu H, Tian M, Zhu X, Zhang Z, Wang W, Zhou X, Zhang F, Ge Q, Zhu B, Tang H, Hua Z & Hou B, Immunity 2018, 49, 695–708 e694, doi: 10.1016/j.immuni.2018.08.012. [DOI] [PubMed] [Google Scholar]
- [45].Barouch DH, Nature 2008, 455, 613–619, doi: 10.1038/nature07352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Youngblood RL, Truong NF, Segura T & Shea LD, Mol. Ther. 2018, 26, 2087–2106, doi: 10.1016/j.ymthe.2018.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Skoumal M, Seidlits S, Shin SJ & Shea L, Biotechnol. Bioeng. 2016, 113, 2033–2040, doi: 10.1002/bit.25961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].DeMuth PC, Min Y, Huang B, Kramer JA, Miller AD, Barouch DH, Hammond PT & Irvine DJ, Nat Mater 2013, 12, 367–376, doi: 10.1038/nmat3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kazmin D, Nakaya HI, Lee EK, Johnson MJ, van der Most R, van den Berg RA, Ballou WR, Jongert E, Wille-Reece U, Ockenhouse C, Aderem A, Zak DE, Sadoff J, Hendriks J, Wrammert J, Ahmed R & Pulendran B, Proc Natl Acad Sci U S A 2017, 114, 2425–2430, doi: 10.1073/pnas.1621489114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A & Irvine DJ, Proc Natl Acad Sci U S A 2012, 109, 1080–1085, doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huang WC, Deng B, Lin C, Carter KA, Geng J, Razi A, He X, Chitgupi U, Federizon J, Sun B, Long CA, Ortega J, Dutta S, King CR, Miura K, Lee SM & Lovell JF, Nat Nanotechnol 2018, doi: 10.1038/s41565-018-0271-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang H & Mooney DJ, Nat Mater 2018, 17, 761–772, doi: 10.1038/s41563-018-0147-9. [DOI] [PubMed] [Google Scholar]
- [53].Santos PM & Butterfield LH, J Immunol 2018, 200, 443–449, doi: 10.4049/jimmunol.1701024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, Congdon KL, Reap EA, Archer GE, Desjardins A, Friedman AH, Friedman HS, Herndon JE 2nd, Coan A, McLendon RE, Reardon DA, Vredenburgh JJ, Bigner DD & Sampson JH, Nature 2015, 519, 366–369, doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Eggermont LJ, Hammink R, Blank KG, Rowan AE, Tel J & Figdor CG, Advanced Therapeutics 2018, 1, 1800021, doi: 10.1002/adtp.201800021. [DOI] [Google Scholar]
- [56].Cheung AS, Zhang DKY, Koshy ST & Mooney DJ, Nat Biotechnol 2018, 36, 160–169, doi: 10.1038/nbt.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cubillos-Ruiz JR, Engle X, Scarlett UK, Martinez D, Barber A, Elgueta R, Wang L, Nesbeth Y, Durant Y, Gewirtz AT, Sentman CL, Kedl R & Conejo-Garcia JR, J Clin Invest 2009, 119, 2231–2244, doi: 10.1172/JCI37716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Huang Z, Yang Y, Jiang Y, Shao J, Sun X, Chen J, Dong L & Zhang J, Biomaterials 2013, 34, 746–755, doi: 10.1016/j.biomaterials.2012.09.062. [DOI] [PubMed] [Google Scholar]
- [59].Oliva N, Carcole M, Beckerman M, Seliktar S, Hayward A, Stanley J, Parry NM, Edelman ER & Artzi N, Sci Transl Med 2015, 7, 272ra211, doi: 10.1126/scitranslmed.aaa1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dai Y, Xu C, Sun X & Chen X, Chem Soc Rev 2017, 46, 3830–3852, doi: 10.1039/c6cs00592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kienzle A, Kurch S, Schloder J, Berges C, Ose R, Schupp J, Tuettenberg A, Weiss H, Schultze J, Winzen S, Schinnerer M, Koynov K, Mezger M, Haass NK, Tremel W & Jonuleit H, Adv Healthc Mater 2017, 6, doi: 10.1002/adhm.201700012. [DOI] [PubMed] [Google Scholar]
- [62].Wang Y, Zhou K, Huang G, Hensley C, Huang X, Ma X, Zhao T, Sumer BD, DeBerardinis RJ & Gao J, Nat Mater 2014, 13, 204–212, doi: 10.1038/nmat3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shi C, Liu T, Guo Z, Zhuang R, Zhang X & Chen X, Nano Lett 2018, 18, 7330–7342, doi: 10.1021/acs.nanolett.8b03568. [DOI] [PubMed] [Google Scholar]
- [64].Wang C, Wang J, Zhang X, Yu S, Wen D, Hu Q, Ye Y, Bomba H, Hu X, Liu Z, Dotti G & Gu Z, Sci Transl Med 2018, 10, doi: 10.1126/scitranslmed.aan3682. [DOI] [PubMed] [Google Scholar]
- [65].Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, Vats P, Chinnaiyan AM, Lao CD, Lawrence TS, Wicha M, Hamanishi J, Mandai M, Kryczek I & Zou W, J Clin Invest 2018, 128, 805–815, doi: 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Azarin SM, Yi J, Gower RM, Aguado BA, Sullivan ME, Goodman AG, Jiang EJ, Rao SS, Ren Y, Tucker SL, Backman V, Jeruss JS & Shea LD, Nat Commun 2015, 6, 8094, doi: 10.1038/ncomms9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rao SS, Bushnell GG, Azarin SM, Spicer G, Aguado BA, Stoehr JR, Jiang EJ, Backman V, Shea LD & Jeruss JS, Cancer Res 2016, 76, 5209–5218, doi: 10.1158/0008-5472.CAN-15-2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Aguado BA, Hartfield RM, Bushnell GG, Decker JT, Azarin SM, Nanavati D, Schipma MJ, Rao SS, Oakes RS, Zhang Y, Jeruss JS & Shea LD, Adv Healthc Mater 2018, 7, e1700903, doi: 10.1002/adhm.201700903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, Dougan M, Lambert AW, Bierie B, Ploegh HL, Dougan SK & Weinberg RA, Sci Transl Med 2018, 10, doi: 10.1126/scitranslmed.aan3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ren L & Lim YT, Adv Funct Mater 2018, 28, 1804490, doi: 10.1002/adfm.201804490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Getts DR, McCarthy DP & Miller SD, J Immunol 2013, 191, 5341–5346, doi: 10.4049/jimmunol.1302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Smith CE & Miller SD, J Autoimmun 2006, 27, 218–231, doi: 10.1016/j.jaut.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Turley DM & Miller SD, J Immunol 2007, 178, 2212–2220. [DOI] [PubMed] [Google Scholar]
- [74].Pishesha N, Bilate AM, Wibowo MC, Huang NJ, Li Z, Deshycka R, Bousbaine D, Li H, Patterson HC, Dougan SK, Maruyama T, Lodish HF & Ploegh HL, Proc Natl Acad Sci U S A 2017, 114, 3157–3162, doi: 10.1073/pnas.1701746114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo X, Terry RL, King NJ & Miller SD, J Immunol 2011, 187, 2405–2417, doi: 10.4049/jimmunol.1004175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, Schippling S, Miller SD, Sospedra M & Martin R, Sci Transl Med 2013, 5, 188ra175, doi: 10.1126/scitranslmed.3006168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Prasad S, Kohm AP, McMahon JS, Luo X & Miller SD, J Autoimmun 2012, 39, 347–353, doi: 10.1016/j.jaut.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Grimm AJ, Kontos S, Diaceri G, Quaglia-Thermes X & Hubbell JA, Sci Rep 2015, 5, doi: 10.1038/srep15907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kontos S, Kourtis IC, Dane KY & Hubbell JA, Proc Natl Acad Sci U S A 2013, 110, E60–68, doi: 10.1073/pnas.1216353110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hess KL, Oh E, Tostanoski LH, Andorko JI, Susumu K, Deschamps JR, Medintz IL & Jewell CM, Adv Funct Mater 2017, 27, doi: 10.1002/adfm.201700290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD & Miller SD, Nat Biotechnol 2012, 30, 1217–1224, doi: 10.1038/nbt.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kuo R, Saito E, Miller SD & Shea LD, Molecular Therapy 2017, 25, 1676–1685, doi: 10.1016/j.ymthe.2017.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD & Miller SD, ACS Nano 2014, 8, 2148–2160, doi: 10.1021/nn405033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].McCarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, Miller SD & Shea LD, Nanomedicine 2017, 13, 191–200, doi: 10.1016/j.nano.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chen Y, Wu J, Wang J, Zhang W, Xu B, Xu X & Zong L, Diabetologia 2018, 61, 1384–1396, doi: 10.1007/s00125-018-4593-3. [DOI] [PubMed] [Google Scholar]
- [86].Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD & Miller SD, J Autoimmun 2018, 89, 112–124, doi: 10.1016/j.jaut.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Pearson RM, Casey LM, Hughes KR, Wang LZ, North MG, Getts DR, Miller SD & Shea LD, Mol Ther 2017, 25, 1655–1664, doi: 10.1016/j.ymthe.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Roberts RA, Eitas TK, Byrne JD, Johnson BM, Short PJ, McKinnon KP, Reisdorf S, Luft JC, DeSimone JM & Ting JP, Biomaterials 2015, 72, 1–10, doi: 10.1016/j.biomaterials.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, Gigliotti CL, Boggio E, Clemente N, Comi C, Dianzani C, Boldorini R, Chiocchetti A, Reno F & Dianzani U, Vaccine 2014, 32, 5681–5689, doi: 10.1016/j.vaccine.2014.08.016. [DOI] [PubMed] [Google Scholar]
- [90].Yeste A, Nadeau M, Burns EJ, Weiner HL & Quintana FJ, Proc Natl Acad Sci U S A 2012, 109, 11270–11275, doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, Tukpah AM, Babon JA, DeNicola M, Kent SC, Pozo D & Quintana FJ, Sci Signal 2016, 9, ra61, doi: 10.1126/scisignal.aad0612. [DOI] [PubMed] [Google Scholar]
- [92].Chen N, Kroger CJ, Tisch RM, Bachelder EM & Ainslie KM, Adv Healthc Mater 2018, 7, e1800341, doi: 10.1002/adhm.201800341. [DOI] [PubMed] [Google Scholar]
- [93].Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, Peng H, Yang Y, Vangundy Z, Papenfuss TL, Lovett-Racke AE, Bachelder EM & Ainslie KM, Mol Pharm 2014, 11, 828–835, doi: 10.1021/mp4005172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Casey LM, Pearson RM, Hughes KR, Liu JMH, Rose JA, North MG, Wang LZ, Lei M, Miller SD & Shea LD, Bioconjug Chem 2018, 29, 813–823, doi: 10.1021/acs.bioconjchem.7b00624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Verbeke CS, Gordo S, Schubert DA, Lewin SA, Desai RM, Dobbins J, Wucherpfennig KW & Mooney DJ, Adv Healthc Mater 2017, 6, doi: 10.1002/adhm.201600773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].LaMothe RA, Kolte PN, Vo T, Ferrari JD, Gelsinger TC, Wong J, Chan VT, Ahmed S, Srinivasan A, Deitemeyer P, Maldonado RA & Kishimoto TK, Front Immunol 2018, 9, 281, doi: 10.3389/fimmu.2018.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O’Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH & Kishimoto TK, Proc Natl Acad Sci U S A 2015, 112, E156–165, doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Cho JJ, Stewart JM, Drashansky TT, Brusko MA, Zuniga AN, Lorentsen KJ, Keselowsky BG & Avram D, Biomaterials 2017, 143, 79–92, doi: 10.1016/j.biomaterials.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Yoon YM, Lewis JS, Carstens MR, Campbell-Thompson M, Wasserfall CH, Atkinson MA & Keselowsky BG, Sci Rep 2015, 5, 13155, doi: 10.1038/srep13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lewis JS, Roche C, Zhang Y, Brusko TM, Wasserfall CH, Atkinson M, Clare-Salzler MJ & Keselowsky BG, J Mater Chem B Mater Biol Med 2014, 2, 2562–2574, doi: 10.1039/c3tb21460e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Gammon JM, Tostanoski LH, Adapa AR, Chiu YC & Jewell CM, J Control Release 2015, 210, 169–178, doi: 10.1016/j.jconrel.2015.05.277. [DOI] [PubMed] [Google Scholar]
- [102].Chittasupho C, Sestak J, Shannon L, Siahaan TJ, Vines CM & Berkland C, Mol Pharm 2014, 11, 367–373, doi: 10.1021/mp4003909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Hartwell BL, Smalter Hall A., Swafford D, Sullivan BP, Garza A, Sestak JO, Northrup L & Berkland C, Mol Pharm 2016, 13, 330–343, doi: 10.1021/acs.molpharmaceut.5b00825. [DOI] [PubMed] [Google Scholar]
- [104].Northrup L, Sestak JO, Sullivan BP, Thati S, Hartwell BL, Siahaan TJ, Vines CM & Berkland C, Aaps j 2014, 16, 1204–1213, doi: 10.1208/s12248-014-9671-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS & Jewell CM, Cell Rep 2016, 16, 2940–2952, doi: 10.1016/j.celrep.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Tostanoski LH, Chiu YC, Andorko JI, Guo M, Zeng X, Zhang P, Royal W 3rd & Jewell CM, ACS Nano 2016, doi: 10.1021/acsnano.6b04001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Hess KL, Andorko JI, Tostanoski LH & Jewell CM, Biomaterials 2017, 118, 51–62, doi: 10.1016/j.biomaterials.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ & Keselowsky BG, Clin Immunol 2015, 160, 90–102, doi: 10.1016/j.clim.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Andorko JI, Hess KL, Pineault KG & Jewell CM, Acta Biomater 2016, 32, 24–34, doi: 10.1016/j.actbio.2015.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Tostanoski LH & Jewell CM, Adv Drug Deliv Rev 2017, 114, 60–78, doi: 10.1016/j.addr.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Madan-Lala R, Pradhan P & Roy K, Sci Rep 2017, 7, 2530, doi: 10.1038/s41598-017-02804-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Navarro-Barriuso J, Mansilla MJ, Naranjo-Gomez M, Sanchez-Pla A, Quirant-Sanchez B, Teniente-Serra A, Ramo-Tello C & Martinez-Caceres EM, Sci Rep 2018, 8, 14985, doi: 10.1038/s41598-018-33248-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Gammon JM, Gosselin EA, Tostanoski LH, Chiu YC, Zeng X, Zeng Q & Jewell CM, J Control Release 2017, 263, 151–161, doi: 10.1016/j.jconrel.2017.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Fromen CA, Rahhal TB, Robbins GR, Kai MP, Shen TW, Luft JC & DeSimone JM, Nanomedicine 2016, 12, 677–687, doi: 10.1016/j.nano.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, Singh M, O’Hagan DT, Petrilli V, Tschopp J, O’Neill LA & Lavelle EC, Proc Natl Acad Sci U S A 2009, 106, 870–875, doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Vegas AJ, Veiseh O, Doloff JC, Ma M, Tam HH, Bratlie K, Li J, Bader AR, Langan E, Olejnik K, Fenton P, Kang JW, Hollister-Locke J, Bochenek MA, Chiu A, Siebert S, Tang K, Jhunjhunwala S, Aresta-Dasilva S, Dholakia N, Thakrar R, Vietti T, Chen M, Cohen J, Siniakowicz K, Qi M, McGarrigle J, Graham AC, Lyle S, Harlan DM, Greiner DL, Oberholzer J, Weir GC, Langer R & Anderson DG, Nat Biotechnol 2016, 34, 345–352, doi: 10.1038/nbt.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Bertrand N, Wu J, Xu X, Kamaly N & Farokhzad OC, Adv Drug Deliv Rev 2014, 66, 2–25, doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Joseph A, Contini C, Cecchin D, Nyberg S, Ruiz-Perez L, Gaitzsch J, Fullstone G, Tian X, Azizi J, Preston J, Volpe G & Battaglia G, Sci Adv 2017, 3, e1700362, doi: 10.1126/sciadv.1700362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Arya J, Henry S, Kalluri H, McAllister DV, Pewin WP & Prausnitz MR, Biomaterials 2017, 128, 1–7, doi: 10.1016/j.biomaterials.2017.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Rouphael NG, Paine M, Mosley R, Henry S, McAllister DV, Kalluri H, Pewin W, Frew PM, Yu T, Thornburg NJ, Kabbani S, Lai L, Vassilieva EV, Skountzou I, Compans RW, Mulligan MJ, Prausnitz MR & Group T-MS, Lancet 2017, 390, 649–658, doi: 10.1016/S0140-6736(17)30575-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zeng Q, Gammon JM, Tostanoski LH, Chiu YC & Jewell CM, ACS Biomater Sci Eng 2017, 3, 195–205, doi: 10.1021/acsbiomaterials.6b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Mandal A, Boopathy AV, Lam LKW, Moynihan KD, Welch ME, Bennett NR, Turvey ME, Thai N, Van JH, Love JC, Hammond PT & Irvine DJ, Sci Transl Med 2018, 10, doi: 10.1126/scitranslmed.aar2227. [DOI] [PubMed] [Google Scholar]