

Abstract
A simple, isocratic HPLC method based on HILIC-WAX separation, has been developed for analyzing sulfated disaccharides of glycosaminoglycans (GAGs). To our best knowledge, this is the first successful attempt using this special phase in nano-HPLC-MS analysis. Mass spectrometry was based on negative ionization, improving both sensitivity and specificity. Detection limit for most sulfated disaccharides were approximately 1 fmol; quantitation limits 10fmol. The method was applied for glycosaminoglycan profiling of tissue samples, using surface digestion protocols. This novel combination provides sufficient sensitivity for GAG disaccharide analysis, which was first performed using prostate cancer tissue microarrays. Preliminary results show that GAG analysis may be useful for identifying cancer related changes in small amounts of tissue samples (ca. 10 μg).
Keywords: Glycosaminoglycan, Heparan sulfate, Hydrophilic interaction chromatography, Mass spectrometry, Prostate cancer, Tissue microarray
1. Introduction
Nano-scale liquid chromatography (nano-LC) coupled to mass spectrometry (MS) is an excellent technique used in routine characterization of different types of molecules (e.g. peptides [1,2], glycans [3], etc.), with still growing application areas. The field of proteomics greatly benefited from its constant improvements; however, a similar breakthrough is still waiting to occur in case of glycomics and especially glycosaminoglycan (GAG) analysis. GAGs are an often overlooked class of compounds, characterized by highly sulfated, long, linear polysaccharide chains. Their characterization poses several challenges [4]. The saccharide units can be sulfated at various positions and epimerization may also occur along the chain. Most GAGs are covalently attached to core proteins and form proteoglycans (PGs). They are localized in the extracellular matrix, cell surfaces and intercellular granules and mediate various physiological and pathophysiological processes including coagulation, cancer metastasis, and inflammation [5–8]. Involvement of both GAGs and PGs in cancer progression has been reported [7,9,10]. GAG chains may interact with different effector proteins (e.g. chemokines, cytokines and growth factors) and thus regulate processes such as tumor cell growth, metastasis, and angiogenesis [10]. Interaction between the GAG chains and the effector proteins strongly depends on sulfation motifs within the chain.
The building blocks of GAG chains are repeating disaccharide units of a uronic acid or galactose unit and an amino sugar (often abbreviated as HexA/Gal-HexNAc). GAGs are divided into four different classes: hyaluronan (HA), heparan sulfate (HS)/heparin, chondroitin sulfate/dermatan sulfate (CS/DS) and keratan sulfate (KS). Perhaps the most widely known group is HS, consisting of a hexuronic acid (HexA) and N-acetyl glucosamine (GlcNAc) disaccharide units.
Analytical characterization of GAGs is usually performed after hydrolysis of the polymeric chain into the constituent disaccharide units. Bacterial polysaccharide lyase enzymes can cleave the glycosidic bond between the HexA and HexNAc sugars via an eliminative mechanism and produce Δ4,5-unsaturated disaccharides with varying degrees of sulfation; HS disaccharides may be sulfated by up to three sulfate groups. The structures, masses and nomenclature [11] of HS disaccharides are summarized in Table 1. Determining the ratio of these different structures is important in understanding mechanisms underlying diseases such as cancer.
Table 1.
Structure and nomenclature of the most common unsaturated HS disaccharides obtained following heparin lyases digestion. Note, that D2A0/D0A6 and D2S0/D0S6 are positional isomers and are not distinguished in the present study.
| HS disaccharide structure | Traditional name | Lawrence code [11] | m/z in negative mode |
|---|---|---|---|
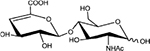 |
ΔHexA-GlcNAc | D0A0 | 378.1 |
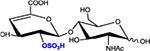 |
ΔHexA2S-GlcNAc | D2A0 | 458.1 |
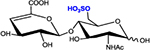 |
ΔHexA-GlcNAc6S | D0A6 | 458.1 |
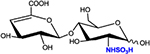 |
ΔHex-GlcNS | D0S0 | 416.1 |
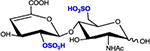 |
ΔHexA2S-GlcNAc6S | D2A6 | 538.1 |
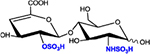 |
ΔHexA2S-GlcNS | D2S0 | 496.1 |
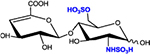 |
ΔHex-GlcNS6S | D0S6 | 496.1 |
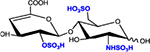 |
ΔHexA2S-GlcNS6S | D2S6 | 576.1 |
Several different chromatographic techniques can be used to analyze Δ4,5-unsaturated, and variously sulfated disaccharide units of HS. These include derivatization followed by reversed-phase chromatography (RP) [12–17]; reversed-phase ion-pairing chromatography (RPIP) [18–20]; size exclusion chromatography (SEC) [21–23]; graphitized carbon [24,25]; amide-HILIC [26,27]; or HILIC-WAX [28] chromatography. These separation methods can be on-line coupled to mass spectrometry (MS) and yield useful information about the involvement of GAGs in various biochemical processes [29,30].These analytical approaches have been reviewed recently [31].The main disadvantage of the above mentioned methods is their relatively low sensitivity, which is generally in the low pmol range [23,28]. Perhaps the currently most sensitive method for GAG disaccharide profiling is fluorescent labelling and HPLC separation [15]. Limit of detection of this method for most HS disaccharides was ca. 0.3 pmol (~0.1 ng); for the trisulfated D2S6 it was ca. 2 pmol (~1 ng). This sensitivity is sufficient to study bulk tissues, but is insufficient to study tissue slices or tissue microarrays (TMAs). In fact, recent examples of GAG analysis as much as 1 g tissue amount were required [16].
The main reason for developing our method was the need to have a technology capable of GAG analysis of small size tissue slices. Our plan is utilizing tissue microarrays (set of 1.5 mm diameter, 5 μm thick tissue slices arranged in array format, ca. 10 μg tissue), which provide an opportunity to study relatively large number of well characterized, well matched human cancer tissue samples. Target sensitivity is the low fmol range (for individual disaccharides), which is the amount foreseen to be present in tissue microarray digests. To achieve this goal, special, self-packed, commercially unavailable capillary columns were designed. A mixed mode resin combining hydrophilic interaction (HILIC) and weak anion exchange (WAX) retention mechanisms was chosen as packing material, as it enables separation of glycans based on charge, size and polarity [28,32]. Use of capillary columns allows the use of on-line nano-UHPLC-MS coupling; needed to achieve the necessary sensitivity. The packing material was obtained by opening a commercially available analytical (2.1 mm diameter) column, and repacked into 100 μm i.d. capillaries.
2. Materials & methods
2.1. Chemicals and reagents
The following standards and enzymes were purchased from Iduron (Cheshire, UK): unsaturated heparan sulfate disaccharides, heparinase I, II and III enzymes. LC-MS grade solvents, ammonium formate solution were purchased from Sigma-Aldrich (Sigma-Aldrich Kft., Budapest, Hungary).
2.2. Capillary column packing
A GlycanPac™ AXH-1 1.9 μm analytical column (2.1 × 100 mm, Thermo Fisher Scientific, Waltham, MA USA) was unpacked and repacked into 100 μm internal diameter capillaries using a pressure injection cell (Next Advance Inc., NY, USA). For this purpose, 20–30 cm 100 μm internal diameter capillaries were cut and fritted as previously reported [33]. Briefly, capillaries were dipped in a solution containing Kasil® 1624, Kasil 1 and formamide in a ratio of 3:1:1. The capillaries were then placed in an oven at 80 °C for 4 h. Capillaries were examined under a microscope and in case of fully porous frit the excess was trimmed to 0.5 cm in order to reduce dead volume. Capillary was then placed in the pressure injection cell and was washed with methanol. A 1 mg/mL suspension was prepared from the GlycanPac™ AXH-1 resin in 75% Acetonitrile - 25% Methanol. The slurry was continuously vortexed using a magnetic stir bar and the column was packed using nitrogen at 2000 psi. After reaching the desired length - generally between 10 and 11 cm - the pressure was carefully released overnight. Due to the pressure drop inhomogeneity may appear, therefore 30 min long compression step (at 5000 psi) was necessary following mounting the column on the nano-HPLC.
2.3. Liquid chromatography-mass spectrometry
The in-house packed capillary column was mounted on a Waters® nanoAcquity UPLC system (Waters, Milford, MA, USA) coupled to a high resolution Waters® Q-Tof Premier™ Mass Spectrometer (Waters, Milford, MA, USA) via nanoelectrospray ionization source. Precut silica tips (360 μm OD, 20 μm ID, 10 μm tip ID, DNU - MS GbR, Berlin, Germany) were used as emitters.
2.3.1. LC parameters
A flow rate of 0.6 μL/min was found to give stable signal and gave reasonable back pressure (2000 psi). Solvent A was 50 mM ammonium formate pH 4.4 (pH adjusted with formic acid) and solvent B was 95% acetonitrile and 5% water, without any buffer. Method optimization was performed on the self-packed columns using commercially available Δ4,5-unsaturated HS disaccharide standards. The following isocratic methods were tested: 90%B, 85%B, 80%B, 75%B and 70% B.
2.3.2. MS parameters
Negative ionization mode was used. Correct tuning of MS parameters is critical when analyzing labile molecules such as sulfated sugars. The instrument was tuned by direct infusing the most labile triply sulfated heparan sulfate disaccharide (D2S6). Decreasing the cone voltage to 15 eV from the generally used 35 eV resulted in minimal (<5%) sulfate loss. Extraction cone voltage was also lowered to 1 V. MS data was acquired in the m/z 180–800 mass range.
2.4. Enzymatic digestion of TMA cores
Prostate cancer tissue microarrays T191a and T196 were obtained from US Biomax, Inc. (Derwood, MD, USA). Tissue sections were processed (dewaxing and antigen retrieval) as previously described [34]. TMA cores corresponding to normal (healthy prostate tissue, male, 33 years), grade 1 (adenocarcinoma, male, 72 years) and grade 2–3 (adenocarcinoma, male, 60 years) cancer were chosen for the current analysis. In case of each patient the TMA contained 3 cores/case allowing to assess the reproducibility of the developed method. Heparinase I-III digestion was performed on the surface of the TMA cores as previously described [34]. Following 5 cycles of enzyme addition (40 min 37 °C incubation/cycle) two additional cycles were carried out by pipetting the enzyme solution without the enzymes to ensure completion of the enzymatic digestion process. The HS disaccharides were extracted from the individual cores using 0.3% ammonium-hydroxide solution, dried down and re-suspended in the LC starting conditions. During the experiments a quality control sample followed by two blank injections was ran before the tissue microarray samples.
2.5. Data analysis
Data acquisition was controlled by MassLynx, quantitative results were evaluated by QuanLynx software (Waters, Milford, MA).
3. Results & discussion
Detection of both neutral and highly acidic disaccharides by mass spectrometry presents a challenge; initial trials showed that negative electrospray ionization provides the best sensitivity. High mass resolution was needed to improve selectivity of disaccharide analysis as extracts of tissue surface digestion contain a large amount of impurities, the target disaccharides being minor components only. Sulfated disaccharides fragment easily in the mass spectrometer ion source under conventional conditions, compromising analysis. In order to avoid this problem ion source conditions have to be specially tuned. The most critical parameter is the cone voltage, which, in our instrument, had to be reduced from the conventionally used 30–35 eV to as low as 15 eV.
Based on the literature [28] we have selected a mixed mode HILIC-WAX stationary phase, which was shown to be efficient for analysis of acidic oligosaccharides [32]. Capillary columns were packed in-house with this packing material. We have used medium ion strength ammonium formate buffer and acetonitrile solvents. Method development was carried out using commercially available standards with the goal of analyzing small amounts of tissue digests (tissue microarrays) containing disaccharides in the low fmol range.
Initial trials using gradient elution showed that mass spectrometry sensitivity significantly changes with solvent composition; as a result, the late-eluting doubly and triply sulfated disaccharides had the lowest sensitivity among those tested. For example, in equimolar mixtures the signal intensity for the trisulfated HS disaccharide was 30 times less than those of the neutral or the monosulfated disaccharides, due to changes in solvent composition. For this reason, we have decided to develop an isocratic elution method.
3.1. Method optimization
First, the required ion strength of the strong eluent was established. It was found that the method is very sensitive to the salt concentration: sulfated (especially highly sulfated) disaccharides do not elute from the column neither at lower (10–40 mM) nor at higher (80 mM) ammonium formate concentration. Medium ion strength (50 mM ammonium formate) resulted in a reasonable chromatogram; and this was used in the following. The next step was the adjustment of the pH of the buffer, using formic acid. We tested pH values of 4.0, 4.4 and 4.6, respectively. At pH 4.0 resolution of monosulfated, at pH 4.6 resolution of disulfated disaccharides were unsatisfactory. The best results were achieved at pH 4.4 for which it was possible to detect and resolve the HS disaccharides according to composition.
Subsequently we evaluated five different isocratic conditions (90% B, 85% B, 80%B, 75%B and 70% B, Fig. 1). Note, Fig. 1 shows the sum of the peak intensities (sum of selected ion chromatograms at high resolution) of the HS disaccharides. When the eluent contained a high percentage of acetonitrile (90 and 85% B) the corresponding peaks were broad and shallow (Fig. 1E and D) for the highly sulfated structures and the triply sulfated disaccharide did not elute within 15 min. In case of 75% and 70% B partial co-elution of various peaks was observed (Fig. 1A and B). The isocratic method at 80% B was optimal (Fig. 1C) because it resolved the HS disaccharides based on composition; peak shapes were symmetric, and all peaks eluted within 15 min. Individual selected ion chromatograms of the HS disaccharides are shown in Fig. 2. This isocratic method ameliorated the problem of low signal intensity for the triply sulfated HS. The main reason was that using gradient separation water and buffer concentration is increased at the elution of the triply sulfated disaccharide. Under such conditions the MS sensitivity decreases, resulting in low sensitivity for the highly sulfated derivatives. Note that D2S6 (the triply sulfated derivative) elutes in a fairly wide peak. This is due to the highly polar nature of this compound, which binds strongly to the HILIC-WAX resin.
Fig. 1.

Sum of selected ion chromatograms of500fmol heparan sulfate disaccharide standards (D0A0, D0S0, D0A6, D2A0, D2A6, D2S0, D0S6and D2S6) on an in-house packed 100 μm i.d. capillary column packed with 1.9 μm particle size GlycanPac™ AXH-1 resin using the following isocratic methods: 70% B (A), 75% B(B), 80% B(C), 85% B(D), 90% B (E). Note, that D2A0/D0A6 and D2S0/D0S6 are positional isomers.
Fig. 2.

Extracted ion chromatograms of 500fmol heparan sulfate disaccharide standards (D0A0, D0S0, D0A6/D2A0, D2A6, D2S0/D0S6 and D2S6) on an in-house packed 100 μm i.d. capillary column packed with 1.9 μm particle size GlycanPac™ AXH-1 resin using isocratic separation (80% B).
We have determined the repeatability of analysis as well (Table 2). For the 500 fmol sample mixture intra-day repeatability of relative peak areas was 5% on average; inter-day repeatability was 10% (relative standard deviation; 5 measurements in a day, measured on 3 different days). Repeatability (relative standard deviation) of retention times was, on average 0.3% within a day, and 1.2% between days.
Table 2.
Repeatability of analysis. Intra-Day and Inter-Day relative standard deviation of peak areas and retention times for the unsaturated HS disaccharides.
| RSD Peak Area |
RSD Retention time |
|||
|---|---|---|---|---|
| HS disaccharide | Intra-Day | Inter-Day | Intra-Day | Inter-Day |
| D0A0 | 6.68% | 7.91% | 0.22% | 0.45% |
| D0S0 | 5.15% | 15.58% | 0.27% | 0.97% |
| D0A6/D2A0 | 3.11% | 3.44% | 0.26% | 0.75% |
| D2S0/D0S6 | 3.67% | 7.61% | 0.48% | 1.69% |
| D2A6 | 4.03% | 7.37% | 0.19% | 1.35% |
| D2S6 | 10.18% | 20.19% | 0.35% | 1.75% |
| Average | 5.47% | 10.35% | 0.30% | 1.16% |
3.2. Linearity studies
We also demonstrated the linearity (Fig. 3 and Supplementary material Fig. S1) of signal intensity with increasing sample amount, and the limits of quantitation. The following concentrations of HS disaccharide mixtures were tested with 80% B isocratic method: 10 fmol, 20 fmol, 50 fmol, 100 fmol, 200 fmol, 500 fmol and 1000 fmol. As 1 μL sample volume was injected always, these sample amounts correspond to 10, 20, 50, 100 200, 500 and 1000pmol/mL concentrations. Each concentration was measured in triplicate. For the mono and disulfated components at 10 fmol injection the signal-to-noise ratio was greater than 20:1 (indicating that the limit of quantitation (LOQ, defined as S/N>10) is slightly better than 10 fmol). We estimate the limit of detection for these components (S/N >3) to be approximately 1 fmol. For the non-sulfated D0A0 disaccharide, the detection limit was ca. 10 fmol (due to interference from an abundant background ion at a similar mass and retention time). The quantitation limit was ca. 20 fmol in this case. The trisulfated compound gave very low signal intensity at low concentration, detection limit was ca. 20 fmol, and quantitation limit was 50 fmol. We evaluated linearity of peak areas over a 10–1000 fmol range that is appropriate for biological samples. Excellent linearity was observed for four disaccharides in the 10–1000 fmol range (Fig. 3B-E), with an R2 value > 0.99 for each. The non-sulfated D0A0 disaccharide was measured in the 20–1000 fmol range (Fig. 3A), the trisulfated HS in the 50–500 fmol range (Fig. 3F), both with R2 > 0.99 linearity. Average standard deviation of the HS disaccharides, as expected, decreased with increasing concentration of the standard mixture (Table 3). Average standard deviations were found to be 4.2% for the 1000 fmol mixture and 14% for the 100fmol mixture. The present experiments do not distinguish the positional isomers (D2A0/D0A6 and D2S0/D0S6). We have checked, that the ionization efficiencies of the two isomer pairs are within 15%; well within the intended accuracy of the present study. For this reason, quantitation will determine the sum of the two positional isomers. Relative sensitivity of the isomers is shown in Supplementary material Table S1.
Fig. 3.

Linearity of the method, calibration curves for the individual HS disaccharide standards: D0A0 (A), D0S0 (B), D0A6/D2A0 (C), D2S0/D0S6 (D), D2A6 (E), and D2S6 (F). Excellent linearity was observed for four disaccharides (B-E) in the 10–1000 fmol range with an R2 value >0.99 for each. The non-sulfated disaccharide D0A0 (A) was measured in the 20–1000fmol range, the trisulfated HS (F) in the 50–500fmol range, both with R2 >0.99 linearity.
Table 3.
Relative standard deviation of peak areas for the unsaturated HS disaccharides at different concentrations measured in triplicates.
| 10fmol | 20 fmol | 50 fmol | 100 fmol | 200 fmol | 500 fmol | 1000 fmol | |
|---|---|---|---|---|---|---|---|
| D0A0 | ND | 18.04% | 10.98% | 5.82% | 4.57% | 4.92% | 6.31% |
| D0S0 | 18.79% | 13.07% | 11.43% | 15.27% | 10.28% | 4.84% | 1.45% |
| D0A6/D2A0 | 13.95% | 18.21% | 20.89% | 14.86% | 14.97% | 10.81% | 10.98% |
| D2S0/D0S6 | 15.84% | 15.88% | 17.11% | 17.28% | 9.94% | 5.53% | 2.86% |
| D2A6 | 8.53% | 11.56% | 12.40% | 13.75% | 7.50% | 1.20% | 1.96% |
| D2S6 | ND | ND | ND | 19.85% | 7.58% | 3.52% | 1.73% |
3.3. Analysis of tissue microarrays
The next phase of method development was establishing, whether the method developed on standards would be suitable for analyzing small amounts of biological material. As test examples we have chosen tissue microarrays (TMA), which are gaining importance in biomedical workflows; the main challenge being the limited sample amount (1.5 mm diameter, 5 μm thin cores). Sample amount in such cores is sufficient for proteomics workflows based on nanoLC-MS methodologies. TMA cores were digested using heparinase enzymes on their surface (see details in the Materials & Methods section), yielding Δ4,5-unsaturated HS disaccharides. The digestion products were pipetted off the cores, and analyzed by the LC-MS method described above. Selected example chromatograms are shown in Fig. 4 for a grade 2–3 prostate cancer TMA cores. Based on the calibration curve obtained using the standards it was possible to estimate the amount of the extracted disaccharides (Fig. 4).
Fig. 4.

Extracted ion chromatograms of heparan sulfate disaccharides extracted from the surface of a grade 2–3 prostate cancer tissue microarray core following on surface heparinase I-III digestion. The amount of the extracted disaccharides were estimated based on the calibration curve obtained using the standards.
In this study TMA sections were analyzed corresponding to prostate tissue biopsy from one healthy individual, one grade 1 and one grade 2–3 patient, respectively. Three serial biopsies were analyzed for each TMA sample. We observed the 4 most abundant disaccharides in these samples. Among these, one doubly sulfated and the triply sulfated disaccharide were not detected. Approximate quantitation based on Fig. 3 indicated that the most abundant component is the non-sulfated D0A0 disaccharide being in the 20–80 fmol range in the various samples. The big peak in the D0A0 ion chromatogram, before the sample at Rt = 3.5–6.5 min is coming from the background. This does not interfere with detecting and quantifying D0A0. The two mono-sulfated components are present in comparable amounts; 10–40 fmol in the case of D0S0 and 5–20 fmol in the case of D0A6/D2A0. Among the higher sulfated components only the disulfated D2S0/D0S6 HS disaccharide was observed in each case, but only in low amounts (less than 10 fmol).
Three biological samples were analyzed in triplicates. The results showed an increased abundance in cancer of the mono-sulfated D0S0 and the D0A6/D2A0 components (Fig. 5).
Fig. 5.

Average amount of various GAG related HS disaccharides in normal, grade 1 and grade 2–3 prostate cancer TMA cores. The error bars represent standard deviation of the method.
4. Conclusion
We demonstrated that sulfated HS disaccharides can be advantageously studied using LC-MS with isocratic nano-chromatography conditions. Isocratic elution is advantageous for mass spectrometry analysis, as ion spray conditions and sensitivity issues do not complicate analysis. It is of particular importance in those cases, when the analyte has unfavorable MS characteristics. Using a combination of nano-HPLC and negative ionization, we detected HS disaccharides in the low fmol range.
We used the technique to analyze HS in tissue slices, using digestion directly on the tissue surface. Both sensitivity and specificity were sufficient to analyze small, ca. 2 mm2 size areas on the tissue surface, opening up the possibility of studying tissue microarrays. We expect that this will allow systematic studies of human disease biospecimens. In the present paper, as an illustrative example, we studied TMA of healthy, grade 1 and grade 2–3 prostate tissue biopsies. These pilot studies demonstrated the ability to differentiate the biospecimens based on HS disaccharide abundances. This is consistent with known roles of HS proteoglycans in dysregulation of receptor tyrosine kinase signaling in cancer phenotypes [35–37]. In the future studies, we will extend to analyze GAGs from a large number of TMA cores, in order to build statistically relevant models correlating with disease progression.
Supplementary Material
Acknowledgments
Lilla Turiák acknowledges support of the National Research Development and Innovation Office (OTKA PD 121187); Lilla Turiák and Ágnes Révész the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/jxhroma.2018.02.034.
References
- [1].Choi YS, Reaching for the deep proteome: recent nano liquid chromatography coupled with tandem mass spectrometry-based studies on the deep proteome, Arch. Pharm. Res 35 (2012) 1861–1870. [DOI] [PubMed] [Google Scholar]
- [2].Wilson SR, Vehus T, Berg HS, Lundanes E, Nano-LC in proteomics: recent advances and approaches, Bioanalysis 7 (2015) 1799–1815. [DOI] [PubMed] [Google Scholar]
- [3].Hua S, Lebrilla C, An HJ, Application of nano-LC-based glycomics towards biomarker discovery, Bioanalysis 3 (2011) 2573–2585. [DOI] [PubMed] [Google Scholar]
- [4].Zaia J, Principles of mass spectrometry of glycosaminoglycans, J. Biomacromol. Mass Spectrom 1 (2005) 3–36. [Google Scholar]
- [5].Berretta S, Extracellular matrix abnormalities in schizophrenia, Neuropharmacology 62 (2012) 1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ariga T, Miyatake T, Yu RK, Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: amyloidogenesis and therapeutic strategies-a review, J. Neurosci. Res 88 (2010)2303–2315. [DOI] [PubMed] [Google Scholar]
- [7].Binder MJ, McCoombe S, Williams ED, McCulloch DR, Ward AC, The extracellular matrix in cancer progression: role of hyalectan proteoglycans and ADAMTS enzymes, Cancer Lett. 385 (2017) 55–64. [DOI] [PubMed] [Google Scholar]
- [8].Gill S, Wight TN, Frevert CW Proteoglycans, Key regulators of pulmonary inflammation and the innate immune response to lung infection, Anat. Rec.-Adv. Integr. Anat. Evol. Biol 293 (2010) 968–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yip GW, Smollich M, Goette M, Therapeutic value ofglycosaminoglycans in cancer, Mol. Cancer Ther 5 (2006) 2139–2148. [DOI] [PubMed] [Google Scholar]
- [10].Sanderson RD, Yang Y, Purushothaman A, Khotskaya YB, Ritchie JP, et al. , Proteoglycans and cancer, in: Zent R, Pozzi A (Eds.), Cell-Extracellular Matrix Interactions in Cancer, Springer-Verlag, New York, 2010, pp. 191–215. [Google Scholar]
- [11].Lawrence R, Lu H, Rosenberg RD, Esko JD, Zhang L, Disaccharide structure code for the easy representation of constituen oligosaccharides from glycosaminoglycans, Nat. Methods 5 (2008) 291–292. [DOI] [PubMed] [Google Scholar]
- [12].Ramsay SL, Meikle PJ, Hopwood JJ, Determination of monosaccharides and disaccharides in mucopolysaccharidoses patients by electrospray ionisation mass spectrometry Mol. Genet. Metab 78 (2003) 193–204. [DOI] [PubMed] [Google Scholar]
- [13].Galeotti F, Volpi N, Online reverse phase-high-performance liquid chromatography-fluorescence detection-electrospray ionization-mass spectrometry separation and characterization of heparan sulfate, heparin, and low-molecular weight-heparin disaccharides derivatized with 2-aminoacridone, Anal. chem 8 (2011)6770–6777. [DOI] [PubMed] [Google Scholar]
- [14].Volpi N, Galeotti F, Yang B, Linhardt RJ, Analysis of glycosaminoglycan-derived, precolumn, 2-aminoacridone-labeled disaccharides with LC-fluorescence and LC-MS detection, Nat. Protoc 9 (2014)541–558. [DOI] [PubMed] [Google Scholar]
- [15].Yang B, Chang Y, Weyers AM, Sterner E, Linhardt RJ, Disaccharide analysis of glycosaminoglycan mixtures by ultra-high-performance liquid chromatography-mass spectrometry, J. Chromatogr. A 1225 (2012) 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shrikanth CB,Sanjana J, Chilkunda ND, One-pot analysis of sulfated glycosaminoglycans, Glycoconj.J (2017), 10.1007/s10719-017-9809-0. [DOI] [PubMed] [Google Scholar]
- [17].Pan Y, Wang P, Zhang F, Yu Y, Zhang X, et al. , Glycosaminoglycans from fish swim bladder: isolation, structural characterization and bioactive potential, Glycoconj.J (2017), 10.1007/s10719-017-9804-5. [DOI] [PubMed] [Google Scholar]
- [18].Yang B, Weyers A,Baik JY, Sterner E, Sharfstein S, et al. , Ultra-performance ion-pairing liquid chromatography with on-line electrospray ion trap mass spectrometry for heparin disaccharide analysis, Anal. Biochem 415 (2011) 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang B, Buhse LF, Al-Hakim A, li MTB, Keire DA, Characterization of currently marketed heparin products: analysis ofheparin digests by RPIP-UHPLC-QTOF-MS, J. Pharm. Biomed 67–68 (2012) 42–50. [DOI] [PubMed] [Google Scholar]
- [20].Zhang ZQ,Xie J, Liu HY, Liu J, Linhardt RJ, Quantification of heparan sulfate disaccharides using ion-pairing reversed-phase microflow high-performance liquid chromatography with electrospray ionization trap mass spectrometry, Anal. Chem 81 (2009) 4349–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hitchcock AM, Costello CE,Zaia J, Glycoform quantification of chondroitin/dermatan sulfate using a liquid chromatography-tandem mass spectrometry platform, Biochemistry 45 (2006) 2350–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shi XF, Zaia J, Organ-specific heparan sulfate structural phenotypes, J. Biol. Chem 284(2009) 11806–11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shao C, Shi X, Phillips JJ, Zaia J, Mass spectral profiling of glycosaminoglycans from histological tissue surfaces, Anal. Chem 85 (2013) 10984–10991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barroso B, Didraga M, Bischoff R, Analysis of proteoglycans derived sulphated disaccharides by liquid chromatography/mass spectrometry, J. Chromatogr. A 1080 (2005) 43–48. [DOI] [PubMed] [Google Scholar]
- [25].Karlsson NG, Schulz BL, Packer NH, Whitelock JM, Use of graphitised carbon negative ion LC-MS to analyse enzymatically digested glycosaminoglycans, J. Chromatogr. B 824 (2005) 139–147. [DOI] [PubMed] [Google Scholar]
- [26].Takegawa Y, Araki K, Fujitani N,Furukawa J, Sugiyama H, et al. , Simultaneous analysis of heparan sulfate, chondroitin/dermatan sulfates, and hyaluronan disaccharides by glycoblotting-assisted sample preparation followed by single-step zwitter-ionic-hydrophilic interaction chromatography, Anal. Chem 83 (2011)9443–9449. [DOI] [PubMed] [Google Scholar]
- [27].Gill VL, Aich U, Rao S, Pohl C,Zaia J, Disaccharide analysis of glycosaminoglycans using hydrophilic interaction chromatography and mass spectrometry, Anal. Chem 85 (2013) 1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen JH, Kawamura T, Sethi MK, Zaia J, Repunte-Canonigo V, et al. , Heparan sulfate: resilience factorand therapeutic target for cocaine abuse, Sci. Rep 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ucakturk E, Akman O, Sun XJ, Baydar DE, Dolgun A, et al. , Changes in composition and sulfation patterns of glycoaminoglycans in renal cell carcinoma, GlycoconjugateJ.33 (2016) 103–112. [DOI] [PubMed] [Google Scholar]
- [30].Bruinsma IB, Riet LT, Gevers T, ten Dam GB, van Kuppevelt TH, et al. , Sulfation of heparan sulfate associated with amyloid-beta plaques in patients with Alzheimer’s disease, Acta Neuropathol. 119 (2010) 211–220. [DOI] [PubMed] [Google Scholar]
- [31].Zaia J, Glycosaminoglycan glycomics using mass spectrometry, Mol. Cell. Prot 12 (2013) 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Aich U, Saba J, Liu X, Pohl C, Structural Analysis of Native N-Glycans Released from Proteins Using a Novel Mixed-Mode Column and a Hybrid Quadrupole-Orbitrap Mass Spectrometer, Thermo Fisher Scientific Application Note 20827, (2016) retrieved from Thermo Fisher Scientific website: https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AN-20827-Structural-Analysis-Native-N-Glycans-Released-Proteins-AN20827-E.pdf.
- [33].Maiolica A, Borsotti D, Rappsilber J, Self-made frits for nanoscale columns in proteomics, Proteomics 5 (2005) 3847–3850. [DOI] [PubMed] [Google Scholar]
- [34].Turiak L, Shao C, Meng L, Khatri K, Leymarie N, et al. , Workflow for combined proteomics and glycomics profiling from histological tissues, Anal. Chem 86 (2014) 9670–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mundhenke C, Meyer K, Drew S, Friedl A, Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 receptor binding in breast carcinomas, Am.J. Pathol 160 (2002) 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kwabi-Addo B, Ozen M, Ittmann M, The role of fibroblast growth factors and their receptors in prostate cancer, Endocr. Relat. Cancer 11 (2004) 709–724. [DOI] [PubMed] [Google Scholar]
- [37].Afratis N, Gialeli C, Nikitovic D, Tsegenidis T, Karousou E, et al. , Glycosaminoglycans: key players in cancer cell biology and treatment, FEBS J. 279(2012)1177–1197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.