

Abstract
Hepatitis B virus (HBV) alters the expression of host cellular genes to support its replication and survival and to promote the liver cell injury. However, the underlying mechanism remained incompletely understood. In this study, we investigated HBV-induced epigenetic changes in HepG2 cells by profiling the landscapes of the active histone modification mark H3K4me3 and repressive mark H3K27me3 using chromatin immunoprecipitation-sequencing. HBV caused the altered histone modifications at thousands of genomic loci, which are critically involved in HBV entry, inflammation, fibrosis and carcinogenesis of host cells. Interestingly, treatment of the HBV-transformed HepG2 cells with the anti-HBV drug Telbivudine substantially restored the H3K4me3 level to that of untransformed HepG2 cells. More importantly, our analysis of liver samples from control and chronic hepatitis B patients revealed that treatment of the patients with Telbivudine not only corrected the target gene expression but also the epigenetic modification of critical genes. In addition, the expression of the histone methyltransferases SMYD3 and EZH2 that regulate histone H3-specific methylation showed no difference in HepG2 cell with or without HBV existence. Thus, our data suggest that abnormal histone modifications might critically involved in HBV-mediated liver pathogenesis and Telbivudine therapy might benefit patients with HBV-related chronic infection, liver cirrhosis and even hepatic carcinoma.
Summary: Telbivudine substantially restores in vitro and in vivo HBV-caused abnormal expressions and histone H3K4me3 and H3K27me3 modifications at thousands of genomic loci that are involved in the pathogenesis of liver cells, revealing a novel mechanism for HBV-mediated liver damage.
Introduction
Chronic hepatitis B (CHB) infection is strongly associated with subsequent development of liver cirrhosis and hepatocellular carcinoma. Despite extensive research, many aspects of the molecular pathogenic mechanisms of the hepatitis B virus (HBV) remain to be fully elucidated. Activation of host immune responses is generally recognized as a significant feature of HBV pathogenesis, but HBV-mediated intracellular effects, such as those involving transcription and signaling in hepatocytes, also play important roles. In recent years, a number of comprehensive studies have provided evidence of HBV causing aberrant expression, both up- and downregulation, of target host genes with functions in virtually every aspect of the molecular processes underlying liver injury, including positive feedback of hepatic HBV endocytosis [e.g. upregulated asialoglycoprotein receptors (ASGR) (1,2)], inflammation [e.g. upregulated interleukin (IL)-8 (3,4) and downregulated CD59 (5)], liver fibrosis [e.g. upregulated fibronectin 1 (FN1) (2,6,7)] and carcinogenesis [e.g. downregulated glutathione-S-transferase P1 (GSTP1) (8,9)]. However, the mechanism by which altered host gene expression is established by HBV and maintained in the host cells is currently unknown.
Eukaryotic gene regulation can be achieved through several mechanisms, including structural and chemical modification of chromatin. The open chromatin structure facilitates gene expression, whereas the condensed heterochromatin structure often promotes gene silencing (10). Covalent modifications of the histone N-terminal tails, by acetylation, methylation or phosphorylation, also regulate the chromatin state (11). Among different histone modifications, methylation displays a more complex relationship with chromatin states (12,13). Methylation of histone H3 lysines 4, 36 and 79 (H3K4me, H3K36me and H3K79me) is often associated with transcription activation, whereas methylation of H3K9, H3K27 and H4K20 is associated with transcription repression (12,13). Furthermore, the histone tail residues may be methylated by mono-, di- or trimethyl groups, each of which produce different chromatin structures, and possibly different effects on the transcriptional activity of the associated gene (12,14). To date, however, the role of histone methylation in the regulation of HBV-induced differential gene expression has not been examined.
Currently, the research on the mechanisms underlying HBV-mediated alteration of host gene expression has been hindered by the fact that the human HBV virion is unable to directly infect the immortalized cell lines available for study (15). To overcome this technical limitation, the HepG2.2.15 cell line, which constitutively expresses HBV, was generated from the HepG2 human hepatoblastoma cell line by integrating a 2-fold version of the HBV genome (16). Chimpanzees intravenously inoculated with supernatant medium from HepG2.2.15 culture manifested typical symptoms of human hepatitis (17), indicating that this transformed cell line might represent a useful model for studying the effects of HBV in host cells, despite the fact that it only partially recapitulates the natural HBV replication cycle. Since its development, the HepG2.2.15 cell line has been widely used to study various molecular features of CHB pathogenesis.
To investigate whether HBV itself may regulate host gene expression in liver cells by inducing modifications to the epigenetic landscape, we detected the genome-wide histone methylation patterns of HepG2 and HepG2.2.15 cells by chromatin immunoprecipitation-sequencing (ChIP-Seq) and constructed the digital gene expression (DGE) profiles by RNA-sequencing (RNA-Seq). Analysis of the HBV-related differential profiles indicated that the virus was associated with not only changes in expression of key genes related to HBV entry into host cells, inflammation, fibrosis and carcinogenesis but also alteration in histone methylation patterns at their specific genetic loci. Interestingly, the HBV-related changes in genes’ expressions and corresponding epigenetic modifications were not detected in HepG2.2.15 cells treated with the well-established anti-HBV drug Telbivudine. The same effect was also observed in clinical liver samples from CHB patients who underwent Telbivudine treatment. As such, CHB pathogenesis may involve, at least partly, virus-mediated changes to the histone methylation profile in hepatocytes and the consequent abnormal expressions of many key genes related to host cell susceptibility to virus entry and activation of signaling mechanisms with potential to injure the organ’s structural and functional integrity. Collectively, our results suggest a novel role for histone methylation as a potential target of epigenetic-based molecular therapy for HBV infection.
Materials and methods
Patients
Liver biopsy specimens from CHB patients who were treatment naive (n = 31) or who had been treated with a 2 year course of Telbivudine (600mg orally per day) (n = 8) and individuals confirmed as hepatitis virus negative (n = 7, for use as healthy controls) were collected for analysis. CHB diagnosis was based on the Chinese Management Scheme of Diagnostic and Therapy Criteria of Viral Hepatitis (18,19). Exclusion criteria were the following: positive test results for hepatitis C virus, hepatitis D virus or human immunodeficiency virus; any concomitant illness and detection of serological markers of autoimmune disease. All study participants with CHB were adults, hospitalized and followed up in the Department of Infectious Diseases, Southwest Hospital, Third Military Medical University (Chongqing, China) between December 2007 and December 2010. The clinical characteristics of all study participants are shown in Supplementary Table S1, available at Carcinogenesis Online. Written, informed consent was obtained from all study participants prior to enrollment. This study was approved by the Ethics Committee of the Third Military Medical University (Chongqing, China).
Cell culture, quantitation of HBV covalently closed circular DNA and representative gene expression analysis
Cell culture, quantitation of HBV covalently closed circular DNA (cccDNA) and representative gene expression analysis are described in Supplementary DataSupplementary DataSupplementary Data, available at Carcinogenesis Online. The oligonucleotide sequences of primers and probes used in this study are listed in Supplementary Table S2, available at Carcinogenesis Online.
ChIP-Seq and DGE
ChIP-Seq experiments were performed as described previously (20,21). DGE experiments were performed with total RNA isolated from both the HepG2 and HepG2.2.15 cells. The analysis of ChIP-Seq data and DGE data were performed as described in Supplementary DataSupplementary DataSupplementary Data, available at Carcinogenesis Online.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software 5.0 (GraphPad Software). The two-tailed Student’s t-test was used to analyze the differences of messenger RNA (mRNA) expression and histone methylation amount in the two cell types. Statistical significance was defined by a P value ≤0.05.
Results
HBV induces genome-wide alteration of chromatin histone methylation in host cells
It has been reported that H3K4me3 modification is associated with target gene activation, whereas H3K27me3 modification is associated with gene silencing (20). To determine the HBV-related global histone methylation status and its contribution to differential gene expression, we detected the genome-wide distribution profile of H3K4me3 and H3K27me3 in the HepG2 and HBV-transformed HepG2.2.15 cells by ChIP-Seq. The chromatin DNA fragments (200–300bp) immunoprecipitated with H3K4me3 and H3K27me3 antibodies were sequenced by the Illumina HiSeq 2000 system, as previously described (12,22). A total of ~40 million sequence reads were generated for H3K4me3 (17 626 114 for HepG2 cells and 22 508 720 for HepG2.2.15 cells) and ~21 million reads were generated for H3K27me3 (12 184 634 for HepG2 cells and 8 932 974 for HepG2.2.15 cells) and mapped to the human genome (Supplementary Table S3, available at Carcinogenesis Online). Prior to and postsequencing, the ChIP samples were confirmed for the target sites in both cell types by routine ChIP–quantitative PCR with the indicated primers (Supplementary Table S2 and Supplementary Figure S1, available at Carcinogenesis Online). In addition, to obtain the overall pictures of the H3K4me3 and H3K27me3 distributions in HepG2 and HepG2.2.15 cells, we divided the entire human genome into four distinct regions, according to the annotated ‘known genes’ from the UCSC Genome Browser: proximal promoters (1kb upstream and downstream of the transcription starting site), exons, introns and intergenic regions. Results showed that about 52 and 26% of H3K4me3 peaks were located in proximal promoter regions for HepG2 cells and HepG2.2.15 cells, respectively (Supplementary Table S4, available at Carcinogenesis Online). However, the majority of H3K27me3 peaks were found in intergenic regions (Supplementary Table S4, available at Carcinogenesis Online).
Differential expression of host genes is induced by HBV and is closely associated with locus-specific histone methylations
Global gene expression profiles were determined for both the HepG2 and HepG2.2.15 cells by RNA-Seq. Significantly different gene expression between the two cell types was defined as ≥2-fold intensity changes and ≤0.001 false discovery rate (23). A total of 4021 genes were identified as significantly differentially expressed in HepG2.2.15 cells, as compared to HepG2 cells by DGE analysis; among those genes, 2100 were upregulated and 1921 genes were downregulated. Most of the differentially expressed host genes were within the ±2-fold intensity threshold and characterized as poised (Figure 1A). The main functions of the differentially expressed genes (DEGs) with obvious relevance to HBV pathogenesis are summarized in Table I.
Fig. 1.
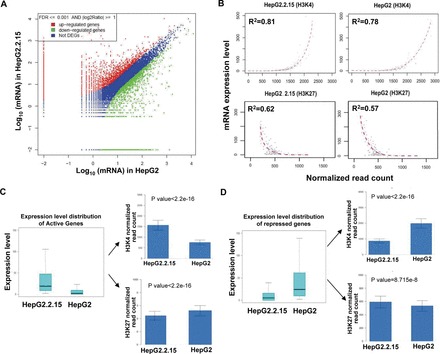
DEGs and comparison of the differential histone methylation patterns between HepG2 and HepG2.2.15 cells. (A) DEGs between the HepG2 and HepG2.2.15 cells. (B) Global correlation between gene expression and histone methylation in HepG2 and HepG2.2.15 cells. The amount of H3K4me3 modification positively correlates with gene expressions, and the amount of H3K27me3 negatively correlates with gene expressions. The normalized read counts of H3K4me3 or H3K27me3 were plotted against the gene expression of all annotated genes. Each dot in the plots represents the mean value of 100 genes that were grouped according to their rank of mRNA expression, from the highest to the lowest. (C and D) Comparison of H3K4me3 or H3K27me3 amounts in active gene loci between the HepG2 and HepG2.2.15 cells. The normalized H3K4me3 or H3K27me3 counts of active genes are plotted as mean ± standard error of the mean, with significant differences determined by Student’s t-test.
Table I.
DEGs between HepG2 and HepG2.2.15 cells
| Biological process | HepG2 | Fold change (HepG2/HepG2.2.15) | HepG2.2.15 | Fold change (HepG2.2.15/HepG2) |
|---|---|---|---|---|
| Transport | ALB | 4553.66 | ||
| AP2M1 | 3.12 | APOH | 41892.00 | |
| CAV2 | 68.59 | ASGR | 12153.00 | |
| NTCP | 18.70 | |||
| Complement activation | CD46 | 2.03 | C2 | 260.53 |
| CD55 | 154.27 | C3 | 124.79 | |
| CD59 | 4.59 | C5 | 40.53 | |
| Regulation of cytokine production | IL18 | 2.79 | IGF2BP2 | 2.43 |
| Inflammatory response | IL8 | 70.17 | ||
| Acute-phase response | STAT3 | 2.42 | FN1 | 494.19 |
| AHSG | 17068.00 | |||
| SERPINA1 | 3140.17 | |||
| Regulation of apoptosis | GSTP1 | 15 112.00 | ||
| Chromatin modification | SMARCA4 | 2.42 | DNMT3A | 8.52 |
| DNMT3B | 4.39 | |||
| HDAC1 | 2.64 | |||
| Defense response to virus | BNIP3L | 2.25 |
The total 11 021 annotated genes including 2100 active genes, 1921 repressed genes and 7000 poised genes were selected for further analysis. For each, the corresponding quantitated mRNA expression level was compared to the amount of H3K4me3 and H3K27me3 detected in the gene body, as determined by the following equation and expressed as normalized read counts: [(count of sequence reads from 1kb upstream of the transcription starting site to the end of the gene/total number of sequence reads for each modification) × the larger number of total sequence reads between the two cell types for each modification]. This approach revealed that substantial differences exist between the annotated genes’ expressions and histone methylations of the HepG2 cells and the HepG2.2.15 cells. Furthermore, both cell types showed a general positive correlation between the amount of H3K4me3 and mRNA expression and a general negative correlation between the amount of H3K27me3 and mRNA expression (Figure 1B). These results suggest that HBV itself might alter host cell gene expression by regulating locus-specific histone modifications, particularly the H3K4me3 and H3K27me3 modifications.
The 2100 active genes in HepG2.2.15 showed significantly higher amounts of H3K4me3 and significantly lower amounts of H3K27me3 in their gene loci, as compared to that detected in HepG2 cells (Figure 1C). In contrast, the 1921 repressed genes showed significantly lower amounts of H3K4me3 and significantly higher amounts of H3K27me3, which was in agreement with their repressed status (Figure 1D). The comprehensive set of annotated DEGs of the HepG2.2.15 cells represent a diverse set of functions, including control of HBV life cycle, hepatic inflammation and injury, acute-phase response, complement activation and chromatin modification (summarized in Table I).
Telbivudine restores the HBV-related altered expressions of genes involved in virus entry into host cells
Upon gaining entry into the host cell, HBV can manipulate cellular processes to enhance susceptibility to further viral entry and promote proliferation. For example, interaction between HBV particles and the host-encoded apolipoprotein H (APOH) protein has been reported to facilitate entry of the virus into hepatocytes (24). The current analysis found that the mRNA expression level of the APOH gene was >40 000-fold higher in the HBV-transformed HepG2.2.15 cells than in the untransformed HepG2 cells (Table I). This increased expression of APOH might be attributed to locus histone modifications because the amount of H3K4me3 detected in HepG2.2.15 cells was also significantly higher, whereas the amount of H3K27me3 was significantly lower, than in HepG2 cells (Supplementary Figure S2A, available at Carcinogenesis Online). In addition, the host-encoded albumin (ALB) and ASGR proteins have also been shown in previous studies to bind specifically to HBV particles, thereby mediating hepatic endocytosis (25,26). The current analysis also found the expressions of these two genes to be remarkably elevated, by >12 000-fold and >4500-fold, respectively, in the HBV-transformed HepG2.2.15 cells (Table I).
A well-studied member of the host-encoded solute carrier protein family that facilitates membrane transport, the sodium taurocholate cotransporting polypeptide (NTCP), has been recently characterized as a functional receptor for human hepatitis B and D viruses (27). Although the current analysis only detected minimal NTCP gene expression in the untransformed HepG2 cells, >10-fold higher expression was detected in the HBV-transformed HepG2.2.15 cells. This upregulated expression pattern was accompanied by corresponding H3K4me3 modifications that were much higher than those observed in the HepG2 cells; however, the amounts of H3K27me3 modifications in the NTCP gene locus were not appreciably different between the two cell types (Supplementary Figure S2B, available at Carcinogenesis Online).
IL-18 has been demonstrated to directly inhibit HBV replication in HepG2.2.15 cells (28). The current DGE analysis indicated, and quantitative PCR verified, that IL-18 expression was downregulated by ~3-fold in the HBV-transformed HepG2.2.15 cells (Table I). In addition, the amount of H3K4me3 modification in the IL-18 gene locus was markedly decreased in the HepG2.2.15 cells, as compared to that detected in the HepG2 cells (Supplementary Figure S3, available at Carcinogenesis Online). These results suggest an advantageous condition for HBV, in which replication may occur in host cells with less impediment.
To address the question of whether HBV itself is the causative agent of the altered expression and locus-specific histone methylation detected for the genes with functions related to HBV pathogenesis, the effects of inhibiting HBV replication with Telbivudine treatment were assessed both in vitro and in vivo. Here, the differential expression of ASGR was selected for this focused analysis. Telbivudine treatment of the cultured HepG2.2.15 cells led to significant decrease in HBV copy number, and this therapeutic effect was accompanied by decreases in both ASGR gene expression and amount of H3K4me3 modification in the gene locus; however, the total amount of H3K27me3 modification in the gene locus in HepG2 cells did not significantly differ from that in HepG2.2.15 cells (Figure 2A and B). To investigate the clinical relevance of this finding, the ASGR gene expression and histone H3 methylations were evaluated in liver tissues from CHB patients before and after Telbivudine therapy. The in vivo results agreed the in vitro results (Figure 2C). Moreover, correlation analysis of the in vivo data indicated that a positive association exists between HBV copy number and both ASGR mRNA level and H3K4me3 amount. However, but no significant correlation was detected between the H3K27me3 amount and ASGR mRNA or HBV cccDNA (Figure 2D).
Fig. 2.
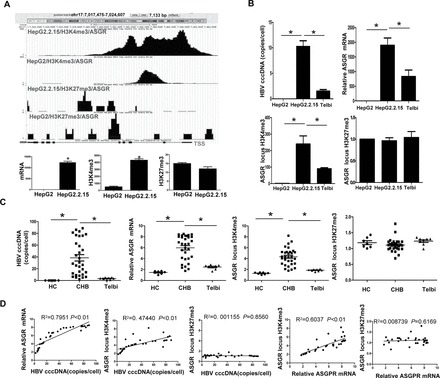
Expression and histone modifications of the ASGR gene. (A) ASGR gene locus and corresponding methylation states are illustrated at the top of each panel. Quantitation of H3K4me3, H3K27me3 and gene mRNA is presented at the bottom of each panel. *P < 0.01. (B and C) Comparison of HBV cccDNA, ASGR mRNA and H3K4me3 or H3K27me3 amounts in the ASGR gene locus between HepG2.2.15 and HepG2 or Telbivudine-treated HepG2.2.15 cells and between CHB liver and healthy liver or CHB liver with Telbivudine treatment. (D) Correlation between HBV cccDNA and ASGR mRNA expression, H3K4me3 amount or H3K27me3 amount in the ASGR locus. HC, healthy controls; Telbi, Telbivudine treatment; TSS, transcription starting site. *P < 0.01.
Telbivudine restores HBV-altered expression of inflammation-related genes in host cells
Complement components have long been known to promote inflammatory injury of host cells in many disease types (29). However, hepatocytes have evolved an effective resistance mechanism to complement-related injury through of a series of regulatory proteins, including CD46, CD55 and CD59 (30). In the current study, the HBV-transformed H2.2.15 cells showed upregulation, between 40- and 260-fold, of the expressions of complement factors C2, C3 C5, but downregulation, between 2- and 150-fold, of the expressions of CD46, CD55 and CD59 (Table I). Furthermore, the H3K4me3 modifications in the representative C3 gene locus were markedly higher in the HepG2.2.15 cells in the HepG2 cells, whereas the inhibitive H3K27me3 modifications were much lower (Supplementary Figure S4A, available at Carcinogenesis Online). The amount of H3K4me3 in the CD46, CD55 and CD59 gene loci was also significantly lower in the HepG2.2.15 cells than that in the HepG2 cells (Supplementary Figure S4B–Supplementary Data, available at Carcinogenesis Online); however, the amounts of H3K27me3 detected in the four gene loci was comparable between the transformed and untransformed cell types (Supplementary Figure S4B–Supplementary Data, available at Carcinogenesis Online).
As CD59 has been studied extensively and its involvement in various types of inflammation is well documented, this complement regulatory protein was selected as a representative gene for focused analysis to determine whether the observed decreases in mRNA expression and modifications in histone methylation are ‘bona fide’ consequences of HBV infection. Treatment of the cultured HepG2.2.15 cells with the anti-HBV drug Telbivudine effectively decreased HBV copy number; this effect was accompanied by a decrease in CD59 gene expression and H3K4me3 modification in the gene locus, but with no observable affect on the locus H3K27me3 modification (Supplementary Figure S5A, available at Carcinogenesis Online). Comparative analysis of liver tissue biopsy specimens from CHB patients with or without Telbivudine treatment, and compared to tissues from healthy controls, confirmed the in vitro results (Supplementary Figure S5B, available at Carcinogenesis Online). Furthermore, the clinical liver samples showed a direct statistical correlation of HBV cccDNA with CD59 mRNA (negative correlation) and with H3K4me3 amount (negative correlation), but no significant correlation was found between H3K27me3 amount and CD59 mRNA or HBV cccDNA (Supplementary Figure S5C, available at Carcinogenesis Online). These findings suggest that HBV-induced downregulation of host inflammation-inhibitive genes’ expression, such as CD59, might be due to HBV-related epigenetic modifications, such as decreased H3K4me3 amount in this gene locus.
In addition to the host complement system, we also investigated whether HBV was able to exert effects on other inflammation-related host genes’ expression. IL-8 was selected for this analysis, based upon its well-characterized activities as a leukocyte chemotactic and proinflammatory cytokine (4). HBV infection is generally known to stimulate expression of IL-8, and, in this study, the HepG2.2.15 cells showed 70-fold higher IL-8 mRNA than the HepG2 cells (Table I). In addition, enhanced serum level of the hepatocyte-synthesized alpha-2-HS glycoprotein (AHSG) is a frequent clinical finding of patients with hepatitis B and non-B/type C hepatitis in the initial stage, making it a useful marker for estimating the severity of acute hepatitis (31). In the current study, the HepG2.2.15 cells showed 17 068-fold higher AHSG mRNA than the HepG2 cells (Table I). Moreover, locus histone modification analyses showed that the trend in upregulation was accompanied by higher H3K4me3 amount in the HepG2.2.15 cells for both IL-8 and AHSG (Figure 3A; Supplementary Figure S6, available at Carcinogenesis Online).
Fig. 3.
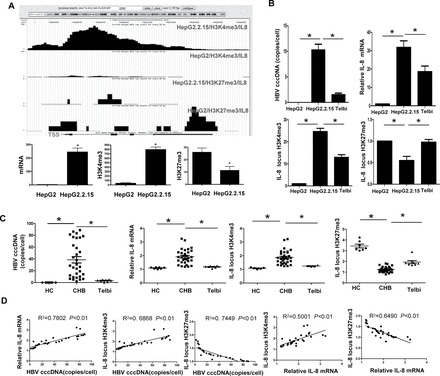
Expression and histone modifications of the IL-8 gene. (A) IL-8 gene locus and corresponding methylation states are illustrated at the top of each panel. Quantitation of H3K4me3, H3K27me3 and gene mRNA is presented at the bottom of each panel. *P < 0.01. (B and C) Comparison of HBV cccDNA, IL-8 mRNA and H3K4me3 or H3K27me3 amounts in the IL-8 gene locus between HepG2.2.15 and HepG2 or Telbivudine-treated HepG2.2.15 cells and between CHB liver and healthy liver or CHB liver with Telbivudine treatment. (D) Correlation between HBV cccDNA and IL-8 mRNA expression, H3K4me3 amount or H3K27me3 amount in the IL-8 locus. HC, healthy controls; Telbi, Telbivudine treatment; TSS, transcription starting site. *P < 0.01.
IL-8 was selected for further investigation to determine whether the increased mRNA expression and the altered histone methylation pattern were bona fide effects of HBV. Similar to the results for CD59, the Telbivudine-induced decrease in HBV copy number was accompanied by significant decreases in both IL-8 gene expression and H3K4me3 modification in the HepG2.2.15 cells, but increased H3K27me3 modification was also observed in the IL-8 gene locus (Figure 3B). Furthermore, the clinical relevance of these findings was borne out in the comparative analysis of liver tissue biopsy specimens (with versus without Telbivudine treatment versus healthy controls; Figure 3C). Correlation analysis of the in vivo data indicated that a positive association exists between HBV cccDNA and IL-8 mRNA or H3K4me3 amount and between IL-8 expression and H3K4me3 modification, whereas a negative correlation was found between H3K27me3 and HBV cccDNA or IL-8 mRNA (Figure 3D).
Telbivudine alleviates HBV-mediated host cell fibrosis and carcinogenesis
The HepG2.2.15 cells showed ~500-fold higher expression of FN1, which encodes a host glycoprotein that is critically involved in hepatic fibrosis (6,7), than the HepG2 cells (Table I), which corresponded with higher H3K4me3 and lower H3K27me3 modifications in the gene promoter (Figure 4A). Again, Telbivudine treatment of HepG2.2.15 was used to investigate whether the observed increase in mRNA expression and alterations in the histone methylation status of the FN1 gene were bona fide consequences of HBV. The Telbivudine-induced decrease in HBV copy number was accompanied by a remarkable decrease in FN1 gene expression and H3K4me3 modification, but with an increase in H3K27me3 modification (Figure 4B). Clinical relevance of these in vitro results was established upon observing similar trends of HBV copy number, FN1 gene expression and histone H3 methylations in liver tissues from CHB patients with or without Telbivudine treatment (Figure 4C). Furthermore, correlation analysis of the in vivo data indicated that a positive association exists between HBV cccDNA and FN1 mRNA or H3K4me3 amount and between FN1 mRNA and H3K4me3 modification, whereas a negative correlation was found between H3K27me3 and HBV cccDNA or FN1 mRNA (Figure 4D).
Fig. 4.
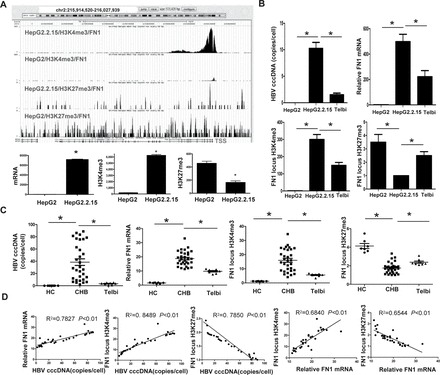
Expression and histone modifications of the FN1 gene. (A) FN1 gene locus and corresponding methylation states are illustrated at the top of each panel. Quantitation of H3K4me3, H3K27me3 and gene mRNA is presented at the bottom of each panel. *P < 0.01. (B and C) Comparison of HBV cccDNA, FN1 mRNA and H3K4me3 or H3K27me3 amounts in the FN1 gene locus between HepG2.2.15 and HepG2 or Telbivudine-treated HepG2.2.15 cells and between CHB liver and healthy liver or CHB liver with Telbivudine treatment. (D) Correlation between HBV cccDNA and FN1 mRNA expression, H3K4me3 amount or H3K27me3 amount in the FN1 locus. HC, healthy controls; Telbi, Telbivudine treatment; TSS, transcription starting site. *P < 0.01.
HBV is also known to promote the expression of some host genes related to carcinogenesis. One such gene is GSTP1, which encodes a detoxification enzyme that acts to protect cells against electrophilic- or oxidant carcinogen-mediated damage, and which has been characterized as a tumor suppressor gene based upon its repressed or silenced state that has been frequently observed in clinical samples of several types of cancers (9). Another key gene of carcinogenesis is alpha-1-antitrypsin (SERPINA1) which is considered a significant risk factor for cirrhosis and hepatocellular carcinoma, and which is used as a diagnostic and prognostic marker of hepatocellular carcinoma (32,33). In this study, the tumor suppressor gene GSTP1 was decreased in the HepG2.2.15 cells by ~15 000-fold and the tumor promoter gene SERPINA1 was increased by ~3000-fold as compared to that in the HepG2 cells (Table I). Similarly, H3K4me3 modification in the GSTP1 gene promoter was much lower in the HepG2.2.15 cells and H3K4me3 modification in the SERPINA1 gene was much higher as compared to that in the HepG2 cells (Figure 5A; Supplementary Figure S7, available at Carcinogenesis Online).
Fig. 5.
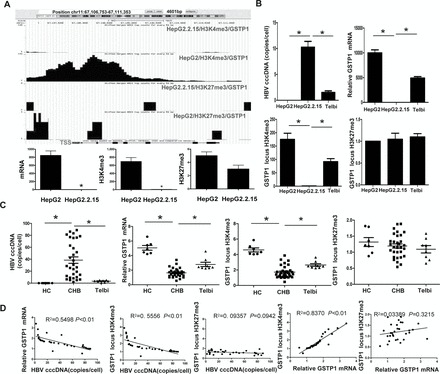
Expression and histone modifications of the GSTP1 gene. (A) GSTP1 gene locus and corresponding methylation states are illustrated at the top of each panel. Quantitation of H3K4me3, H3K27me3 and gene mRNA is presented at the bottom of each panel. *P < 0.01. (B and C) Comparison of HBV cccDNA, GSTP1 mRNA and H3K4me3 or H3K27me3 amounts in the GSTP1 gene locus between HepG2.2.15 and HepG2 or Telbivudine-treated HepG2.2.15 cells and between CHB liver and healthy liver or CHB liver with Telbivudine treatment. (D) Correlation between HBV cccDNA and GSTP1 mRNA expression, H3K4me3 amount or H3K27me3 amount in the GSTP1 locus. HC, healthy controls; Telbi, Telbivudine treatment; TSS, transcription starting site. *P < 0.01.
GSTP1 was selected for further investigation to determine whether the decreased mRNA and altered histone methylation pattern were bona fide effects of HBV. The Telbivudine-induced decrease in HBV copy number in HepG2.2.15 cells was found to be accompanied by significant increases in GSTP1 gene expression and H3K4me3 modification, but the locus H3K27me3 modification appeared to be unaffected (Figure 5B). The clinical relevance of these in vitro results was established upon observing similar trends in liver tissues from CHB patients with or without Telbivudine treatment (Figure 5C). Furthermore, correlation analysis of the in vivo data indicated that a direct correlation exists between HBV cccDNA and GSTP1 mRNA (negative correlation) or H3K4me3 amount (negative correlation), but no significant correlation was found between H3K27me3 amount and GSTP1 mRNA or HBV cccDNA (Figure 5D).
HBV-induced histone 3 methylations were not due to affects on expression of the histone methyltransferases SET and MYND domain-containing protein 3 and enhancer of zeste homolog 2 (Drosophila)
The histone methyltransferases SET and MYND domain-containing protein 3 (SMYD3) and enhancer of zeste homolog 2 (EZH2) regulate histone H3-specific methylation of lysine residues 4 and 27, respectively (34,35). Considering the vast differential patterns of H3K4me3 and H3K27me3 modification events that were detected between the HepG2.2.15 and HepG2 cells, the expression levels of SMYD3 and EZH2 methyltransferases were evaluated by RNA-Seq assay. The differential expression of neither SMYD3 nor EZH2 reached statistical significance as determined by DGE and quantitative PCR, respectively (Supplementary Figure S8, available at Carcinogenesis Online), indicating that HBV affects the histone H3 modification through mechanisms other than influencing the expression of these two particular methyltransferases.
Discussion
Although several studies have begun to elucidate the fundamental transcription changes (activation or repression) that occur in host cells following HBV infection (1,2), the genome-wide relationship between gene expression changes and chromatin states has yet to be addressed. The findings from our current study reveal that HBV-associated modifications in the histone methylation profile correspond to differential gene expression. These in vitro and in vivo data from HepG2.2.15 cells and clinical liver specimens, with or without Telbivudine treatment, suggest that histone methylation may be another level of host gene regulation affected by HBV to support its life cycle or by which subsequent hepatic injury occurs.
Although thousands of HBV-associated DEGs were identified, representing a more comprehensive dataset than obtained by previous studies (1,2), our reliance on a cultured cell system with integrated HBV DNA limits our findings to elements related to the active stage of HBV infection. However, these data reflect the effects of long-term HBV presence on histone methylation and gene expression in host cells, which may be more similar to the chronic HBV infection status of CHB patients. The direct effects of HBV itself were supported by the results obtained in the cell culture system following effective reduction of HBV copy number by the antiviral Telbivudine. More importantly, the clinical relevance of HBV cccDNA associations with host mRNA expression and host epigenetic modifications was revealed by an in vivo evaluation of representative DEGs in biopsied liver tissues of CHB patients (versus that of healthy controls), and in response to a standard Telbivudine drug regimen. Thus, this study provided strong evidence to support the novel finding that HBV-induced changes in host mRNA expression are related to effects on the host epigenetic regulatory mechanism.
In this study, we defined the HBV-associated differential expression patterns of host genes, many of which have functions related to the HBV life cycle, hepatic inflammatory response, fibrosis and carcinogenesis; moreover, these DEGs were strongly associated with HBV-associated modifications of the histone methylation patterns. Based upon these genes’ known roles, we are able to speculate on the functional effects of HBV-associated methylation changes observed in our current study. Firstly, by changing histone modifications in the host cell, the HBV might exert a positive feedback regulation to modulate the functional expression of its life cycle-related genes, thereby maintaining its survival in the liver cells. For example, HBV-induced increase in H3K4me3 and/or decrease in H3K27me3 at some specific gene loci, such as ASGR, APOH or NTCP, may facilitate virus entry into hepatocytes. Furthermore, the HBV-induced decrease in H3K4me3 in the IL-18 gene locus may reduce or eliminate its inhibitory effects on HBV promoters, ultimately promoting HBV replication.
Secondly, by changing histone modifications, HBV could cause the inflammatory reactions in host cells. For example, HBV-induced increase in H3K4me3 and decrease in H3K27me3 in the C3 gene locus may promote inflammation injury of host cells, and HBV-induced decrease in H3K4me3 at the gene loci for CD44, CD46 and CD59 may further sensitize hepatocytes to complement-dependent cytotoxicity. In addition, HBV-induced increase in H3K4me3 and/or decrease in H3K27me3 in other inflammation-related gene loci, such as IL-8 and AHSG, may also promote inflammation injury. Although we detected a substantial amount of differentially expressed inflammation-related genes in host cells harboring HBV, compared to control cells, we did not detect any differences in secretion of alanine transaminase or aspartate transaminase between the HepG2.2.15 and the parental cell line (data not shown). These results might reflect a limitation of the cell line model used in this study, which was isolated from the intrahepatic microenvironment where the host immune system exerts strong proinflammatory effects in the CHB condition (19,36,37). In the complex and dynamically interacting host system, after HBV enters liver cells, it can induce a host signaling mechanism leading to secretion of proinflammatory factors, which serve to prime immune cells that will subsequently target hepatocytes. In addition, HBV and its proteins are also taken up by antigen presenting cells, which subsequently elicit a strong antivirus immune response and destruction of the HBV-infected liver cells. Therefore, although the isolated HBV-infected liver cells did not show apparent injurious effects on alanine transaminase and aspartate transaminase release in vitro, when they are detached from the host immune system, HBV might make the liver cells sensitized to be attacked by immune response in vivo.
Thirdly, by changing histone modifications, HBV itself might promote fibrotic and carcinogenic processes in the affected host tissue. For example, HBV-induced increase in H3K4me3 and decrease in H3K27me3 at the FN1 gene locus may promote hepatic fibrosis through the various and well-characterized activities of FN1. Hepatic fibrosis is a wound healing process involving accumulation of extracellular matrix (ECM) proteins, especially collagen types I and III, as well as an increase in other ECM constituents, such as fibronectin and laminin, in response to liver injury (6). In this pathogenic process, the chronic tissue damage condition stimulates the activation of stellate cells to produce ECM proteins (6). However, in this study, we found that the liver cells themselves could also produce large amounts of the ECM protein FN1, indicating that HBV may be able to induce both hepatocytes and stellate cells to produce ECM proteins, which would further contribute to liver fibrosis (a hallmark of CHB). In addition, HBV-induced increase in H3K4me3 and decrease in H3K27me3 in the tumor-promoting SERPINA1 gene might promote host cell carcinogenesis. Moreover, HBV-induced decrease in H3K4me3 of the tumor suppressor GSTP1 gene might help to further promote an overall profile of gene expression that is amenable to carcinogenesis. The correlation between HBV and carcinogenesis is well established, and these results suggest that the underlying mechanism may involve HBV-mediated regulation of the host’s epigenome.
Histone methylations are catalyzed by respective histone methyltransferases. As such, the altered histone modifications observed in our study may reflect changes in the expression or recruitment of related methyltransferases. The SMYD3 and EZH2 methyltransferases are responsible for specific methylation of H3K4me3 and H3K27me3, respectively (34,35). However, in this study, we did not find any significant differences among the total SMYD3 and EZH2 genes’ expression in HepG2 and HepG2.2.15 cells. Two possible explanations for such observations are: the HBV-associated changes in specific histone methylation observed in this study, increase for certain genes and decrease for other genes, may, on a global scale, amount to a negligible change in the total methyltransferase amount; HBV’s regulation of host gene expression many involve manipulating the recruitment of histone methyltransferase to specific genes’ loci, rather than altering their expression.
In conclusion, we have demonstrated that HBV transformation of HepG2 cells results in changes in histone modifications at thousands of genomic loci, many of which encode proteins that are known to play critical roles in HBV entry, inflammation, fibrosis and carcinogenesis. Treatment of the HBV-transformed HepG2 cells and the CHB patients with Telbivudine substantially restored the H3K4me3 level to almost normal level. Collectively, our data suggest that epigenetic mechanisms may be critically involved in HBV-mediated liver pathogenesis and that epigenetic processes may represent potential targets for HBV infection-induced diseases. Results also suggest that Telbivudine therapy might benefit patients with HBV-related chronic infection, liver cirrhosis and even hepatic carcinoma by correcting the HBV-induced abnormal epigenetic modifications and consequent perturbed expression of key genes essential for normal liver homeostasis.
Supplementary material
Supplementary Methods, Tables S1–Supplementary Data and Supplementary Data–Supplementary Data can be found at Supplementary Data
Funding
Major State Basic Research Development Program of China (2013CB531503); Major Project of NSFC (30930086); Major Collaborative Project of NSFC (81220108024); General Program of NSFC (31070798 and 31200668); Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT 10521); Chongqing Science & Technology Commission (CSTC 2009BB5145).
Conflict of Interest Statement: None declared.
Supplementary Material
Acknowledgements
We would like to thank Dr Lixin Zheng (National Institute of Allergy and Infectious Diseases, Bethesda, MD, USA) for assistance in writing this manuscript.
Glossary
Abbreviations:
- AHSG
alpha-2-HS glycoprotein
- APOH
apolipoprotein H
- ASGR
asialoglycoprotein receptors
- cccDNA
covalently closed circular DNA
- CHB
chronic hepatitis B
- ChIP-Seq
chromatin immunoprecipitation-sequencing
- DEG
differentially expressed gene
- DGE
differential gene expression
- ECM
extracellular matrix
- EZH2
enhancer of zeste homolog
- FN1
fibronectin 1
- GSTP1
glutathione-S-transferase P1
- HBV
hepatitis B virus
- IL
interleukin
- mRNA
messenger RNA
- NTCP
sodium taurocholate cotransporting polypeptide
- SMYD3
SET and MYND domain-containing protein 3.
References
- 1. Otsuka M., et al. (2003). Differential cellular gene expression induced by hepatitis B and C viruses. Biochem. Biophys. Res. Commun., 300, 443–447. [DOI] [PubMed] [Google Scholar]
- 2. Yang J., et al. (2005). Differentially expressed cellular genes following HBV: potential targets of anti-HBV drugs? J. Viral Hepat., 12, 357–363. [DOI] [PubMed] [Google Scholar]
- 3. Mahé Y., et al. (1991). Hepatitis B virus X protein transactivates human interleukin-8 gene through acting on nuclear factor kB and CCAAT/enhancer-binding protein-like cis-elements. J. Biol. Chem., 266, 13759–13763. [PubMed] [Google Scholar]
- 4. Matsushima K., et al. (1989). Interleukin 8 and MCAF: novel inflammatory cytokines inducible by IL 1 and TNF. Cytokine, 1, 2–13. [DOI] [PubMed] [Google Scholar]
- 5. Qu Z., et al. (2009). Hepatitis B virus sensitizes hepatocytes to complement-dependent cytotoxicity through downregulating CD59. Mol. Immunol., 47, 283–289. [DOI] [PubMed] [Google Scholar]
- 6. Kershenobich Stalnikowitz D., et al. (2003). Liver fibrosis and inflammation. A review. Ann. Hepatol., 2, 159–163. [PubMed] [Google Scholar]
- 7. Budkowska A., et al. (1995). Fibronectin of human liver sinusoids binds hepatitis B virus: identification by an anti-idiotypic antibody bearing the internal image of the pre-S2 domain. J. Virol., 69, 840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Niu D., et al. (2009). HBx genotype D represses GSTP1 expression and increases the oxidative level and apoptosis in HepG2 cells. Mol. Oncol., 3, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tischoff I., et al. (2008). DNA methylation in hepatocellular carcinoma. World J. Gastroenterol., 14, 1741–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li B., et al. (2007). The role of chromatin during transcription. Cell, 128, 707–719. [DOI] [PubMed] [Google Scholar]
- 11. Kouzarides T. (2007). Chromatin modifications and their function. Cell, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- 12. Barski A., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell, 129, 823–837. [DOI] [PubMed] [Google Scholar]
- 13. Guenther M.G., et al. (2007). A chromatin landmark and transcription initiation at most promoters in human cells. Cell, 130, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Klose R.J., et al. (2007). Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol., 8, 307–318. [DOI] [PubMed] [Google Scholar]
- 15. Paran N., et al. (2001). HBV infection of cell culture: evidence for multivalent and cooperative attachment. EMBO J., 20, 4443–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sells M.A., et al. (1987). Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl Acad. Sci. USA, 84, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Acs G., et al. (1987). Hepatitis B virus produced by transfected Hep G2 cells causes hepatitis in chimpanzees. Proc. Natl Acad. Sci. USA, 84, 4641–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Z., et al. (2008). Severe dendritic cell perturbation is actively involved in the pathogenesis of acute-on-chronic hepatitis B liver failure. J. Hepatol., 49, 396–406. [DOI] [PubMed] [Google Scholar]
- 19. Zhang J.Y., et al. (2010). Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology, 51, 81–91. [DOI] [PubMed] [Google Scholar]
- 20. Araki Y., et al. (2009). Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity, 30, 912–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tian Y., et al. (2011). Global mapping of H3K4me1 and H3K4me3 reveals the chromatin state-based cell type-specific gene regulation in human Treg cells. PLoS One, 6, e27770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Z., et al. (2008). Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet., 40, 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Audic S., et al. (1997). The significance of digital gene expression profiles. Genome Res., 7, 986–995. [DOI] [PubMed] [Google Scholar]
- 24. Mehdi H., et al. (1994). Hepatitis B virus surface antigen binds to apolipoprotein H. J. Virol., 68, 2415–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Imai M., et al. (1979). A receptor for polymerized human and chimpanzee albumins on hepatitis B virus particles co-occurring with HBeAg. Gastroenterology, 76, 242–247. [PubMed] [Google Scholar]
- 26. Treichel U., et al. (1994). The asialoglycoprotein receptor mediates hepatic binding and uptake of natural hepatitis B virus particles derived from viraemic carriers. J. Gen. Virol., 75 (Pt 11), 3021–3029. [DOI] [PubMed] [Google Scholar]
- 27. Yan H., et al. (2012). Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife, 1, e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Y., et al. (2011). Dual effects of interleukin-18: inhibiting hepatitis B virus replication in HepG2.2.15 cells and promoting hepatoma cells metastasis. Am. J. Physiol. Gastrointest. Liver Physiol., 301, G565–G573. [DOI] [PubMed] [Google Scholar]
- 29. Burkholder P.M., et al. (1971). Immunobiology of complement. Pathobiol. Annu., 1, 215–239. [PubMed] [Google Scholar]
- 30. Halme J., et al. (2009). Primary human hepatocytes are protected against complement by multiple regulators. Mol. Immunol., 46, 2284–2289. [DOI] [PubMed] [Google Scholar]
- 31. Imanishi T. (1981). Clinical and experimental studies on the profiles of serum proteins in acute hepatic injury. Gastroenterol. Jpn., 16, 493–505. [DOI] [PubMed] [Google Scholar]
- 32. Tan X.F., et al. (2011). Alpha-1 antitrypsin is a potential biomarker for hepatitis B. Virol. J., 8, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Topic A., et al. (2012). Alpha-1-antitrypsin in pathogenesis of hepatocellular carcinoma. Hepat. Mon., 12(10 HCC), e7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamamoto R., et al. (2004). SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol., 6, 731–740. [DOI] [PubMed] [Google Scholar]
- 35. Sarma K., et al. (2008). Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol. Cell. Biol., 28, 2718–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Q., et al. (2011). Activated IL-23/IL-17 pathway closely correlates with increased Foxp3 expression in livers of chronic hepatitis B patients. BMC Immunol., 12, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Y., et al. (2011). A proinflammatory role for interleukin-22 in the immune response to hepatitis B virus. Gastroenterology, 141, 1897–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.