

Abstract
Objectives
Infusion of reconstituted HDL (rHDL) leads to changes in HDL metabolism as well as to an increased capacity of plasma to support cholesterol efflux providing an opportunity to investigate mechanisms linking cholesterol efflux to changes in plasma HDL.
Methods and results
Patient plasmas after infusion of rHDL were tested ex vivo for their capacity to stimulate cholesterol efflux. Reconstituted HDL enhanced mobilization of cholesterol from tissues in vivo as shown by rising HDL cholesterol concentrations over the infusion period. Infusion of rHDL in vivo led to increased cholesterol efflux ex vivo; surprisingly, removing apoB-containing lipoproteins while preserving all HDL subfractions eliminated this increase. Infusion of rHDL led to the remodelling of plasma HDL; however, the capacity of plasma to support cholesterol efflux did not correlate with changes in the concentrations of any of HDL subfractions. Unmodified rHDL accounted for only a proportion of the increment in cholesterol efflux capacity. Furthermore, studies using HeLa and BHK cells overexpressing ABCA1, ABCG1, and SR-B1 showed that the contribution of these cellular mediators of cholesterol efflux to the enhanced capacity of plasma for the efflux was minimal.
Conclusion
Enhanced cholesterol efflux from tissues requires the presence of apoB-containing lipoproteins and may involve enhanced flow of cholesterol through multiple components of the reverse cholesterol transport pathway rather than being determined by a specific HDL subfraction.
Keywords: Lipids, Lipoproteins, Cholesterol
Introduction
Drugs affecting lipid metabolism have revolutionized treatment of atherosclerosis reducing the risk of cardiovascular diseases by 30–40%. There is, however, an urgent need for further reduction of the unacceptably high remaining risk of cardiovascular disease. A promising direction is complementing reduction in levels of the pro-atherogenic lipoproteins by increasing the level of the anti-atherogenic lipoprotein, high-density lipoprotein (HDL), an ‘HDL therapy’. One type of HDL therapy, infusion of reconstituted HDL (rHDL),1,2 has shown promising results.
An unresolved issue in understanding the mechanism of the anti-atherogenic action of HDL is that the saturating concentrations of HDL in various in vitro functional assays range from 30 to 100 µg/ml of apolipoprotein A-I (apoA-I). This is well below its normal level in human plasma (1–1.5 mg/ml) making it difficult to envisage how even large variations in HDL levels could affect HDL functions. A number of studies show that there is no direct relationship between plasma level of HDL and capacity of plasma to induce cholesterol efflux ex vivo.3,4 It has been suggested that concentration of HDL in extravascular spaces may be different to that in plasma5 and that not all HDL particles contribute equally to efflux, with minor HDL subfractions responsible for most of the effect.4,6 Another concept suggests that the capacity of HDL to support cholesterol efflux reflects HDL functional properties rather than its concentration.7 Finally, we have suggested that the efficiency of reverse cholesterol transport (RCT) may primarily depend on the flow of cholesterol through this pathway, which is not fully reflected by the concentration of HDL or any of its subfractions.8 These concepts have yet to be proven.
We have recently shown that infusion of rHDL in patients with type 2 diabetes enhanced the capacity of plasma to support cholesterol efflux ex vivo.9 The increase in cholesterol efflux, however, did not correlate with changes in HDL-C or apoA-I levels in the plasma and its causes and the mechanisms remained unclear. The present study was undertaken to elucidate potential mechanisms through which changes in HDL level and composition translate into changes in cholesterol efflux.
Methods
Detailed methods are presented in the online supplement.
Clinical trial
The clinical trial infusing rHDL into patients with type 2 diabetes has been described previously.9 In brief, this was a randomized, placebo (saline) controlled cross-over clinical trial in 13 male patients with diagnosed type 2 diabetes receiving 80 mg/kg of rHDL (CSL-111, CSL) as a single 4 h intravenous infusion.
Assays
The methodology of cholesterol efflux assay has been described by us previously.10 Distribution of apoA-I among HDL subfractions was analysed by non-denaturing PAGE followed by immunoblotting using antibody against human apoA-I as described previously.11
Statistics
In vitro experiments were carried out in quadruplicate. Mean ± SD are shown. A repeated measure ANOVA (Bonferroni corrected) was used to determine statistical significance of the differences between the various time points; differences between the groups in the in vitro experiments were assessed by Student t-test; the threshold for statistical significance for both tests was 0.05 (two-sided). Correlations were calculated using Pearson Product Moment Correlations. SigmaStat was used for statistical calculations.
Results
Infusion of rHDL caused cholesterol efflux from tissues
As was reported previously,9 infusion of rHDL over 4 h resulted in a gradual increase in plasma levels of apoA-I (Figure 1A) and HDL-C (Figure 1B); there was no change in either parameter after infusion of saline. The rise in apoA-I concentration was consistent with a cumulative process throughout the duration of rHDL infusion. Reconstituted HDL does not contain cholesterol, therefore increases in HDL-C that matched apoA-I indicate that rHDL rapidly acquired cholesterol. The was no statistically significant change in the concentration of low density lipoprotein cholesterol (LDL-C) and only a slight increase in the concentration of very low-density cholesterol (VLDL-C) after rHDL infusion (Figure 1C and D) indicating that LDL and VLDL were an unlikely source to have donated cholesterol to HDL-C. Furthermore, total plasma cholesterol concentration rose quantitatively similarly to HDL-C (Figure 1E) and there was no change in non-HDL cholesterol (see Supplementary Data) suggesting that most, if not all, additional HDL-C was derived from tissues. That is infused rHDL actively promoted cholesterol efflux.
Figure 1.
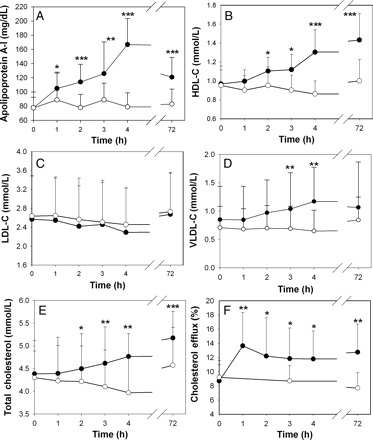
Changes in lipid and lipoprotein levels and capacity of plasma to support cholesterol efflux during and after reconstituted HDL infusion. (A) Changes in plasma concentration of apolipoprotein A-I. (B) Changes in plasma concentrations of HDL-C. (C) Changes in plasma concentrations of LDL-C. (D) Changes in plasma concentrations of VLDL-C. (E) Changes in plasma total cholesterol concentrations. (F) Changes in the capacity of patient plasma to support cholesterol efflux. Closed symbols, reconstituted HDL infusion arm; open symbols, placebo infusion arm. Mean ± SD are shown; n = 13; *P < 0.050; **P < 0.005; ***P < 0.001 (vs. pre-infusion values). Data presented at panels A, B and F were reported previously.9
To confirm that cholesterol efflux capacity of plasma was increased by rHDL infusion, we evaluated the capacity of plasma to support cholesterol efflux from THP-1 human macrophages. Efflux was found to be elevated (Figure 1F), and this occurred as early as 1 h after commencement of the infusion and then remained at this level in samples taken over the next 3 h. This suggests that a near constant level of cholesterol efflux occurred during infusion of rHDL despite the progressive elevation in the levels of apoA-I and HDL-C. That occurred despite the concentration of plasma in the ex vivo assay (2%) being well within the proportional part of the dose-dependence curve (not shown) ruling out saturation. Thus, the efflux ex vivo did not correlate with the plasma concentrations of apoA-I or HDL-C. One conclusion that could be drawn is that the increased concentration of HDL-C after infusion of rHDL was the result of the higher capacity of plasma to induce cholesterol efflux from tissues, rather than its cause.
Requirement for non-HDL plasma components
It has been suggested that while HDL is the primary acceptor of cholesterol from cells, most free cholesterol is then temporarily transferred into LDL to boost ‘storage’ capacity for a cholesterol pool prior to a relatively slow esterification step.12 To test this possibility we precipitated apoB-containing lipoproteins from patient plasma and tested plasma devoid of LDL and VLDL in a cholesterol efflux assay. Consistent with our previous findings3 removal of apoB-containing lipoproteins reduced the capacity of plasma to support cholesterol efflux; surprisingly however elevation of plasma capacity to support cholesterol efflux after infusion of rHDL disappeared after precipitation of LDL/VLDL (Figure 2A). To ascertain that the precipitation procedure did not result in losses of apoA-I containing lipoproteins, we measured apoA-I and preβ1-HDL content before and after precipitation of LDL/VLDL. The apoA-I content was reduced by precipitation by ∼4% (likely due to precipitation of apoA-I in VLDL), which was not statistically significant; there was no statistically significant difference in preβ1-HDL content (seeSupplementary Data). When we compared the size distribution of HDL subfractions, this was also unaffected by the precipitation procedure (Figure 2B), with one exception. The abundance of a small band of apoA-I containing particles with the size 7.2 nm (for comparison, the size of lipid-free apoA-I is 6.5 nm) was consistently reduced by precipitation (Figure 2B, arrow). In a separate experiment we probed this band with anti-preβ1-HDL antibody and found no staining demonstrating that this band was neither lipid-free apoA-I nor preβ1-HDL; the exact identity of this particle is not clear.
Figure 2.
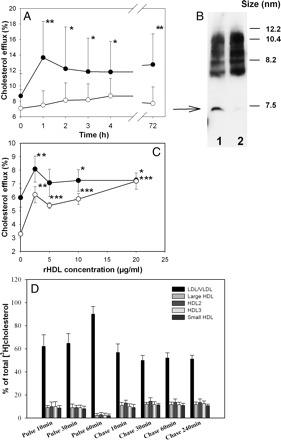
Contribution of apoB-containing lipoproteins to enhanced capacity of plasma to support cholesterol efflux. (A) Changes in the capacity of whole patient plasma and apoB-depleted plasma to support cholesterol efflux. Mean ± SD are shown; n = 13. *P < 0.050, **P < 0.005. (B) Distribution of apoA-I among HDL subfractions before (1) and after (2) precipitation of apoB-containing lipoproteins. (C) Dose dependence of cholesterol efflux from THP-1 cells to whole human plasma (closed symbols) or apoB-depleted plasma (open symbols) supplemented with indicated concentrations of rHDL. Mean ± SD of quadruplicate determinations are shown. *P < 0.050, **P < 0.015, ***P < 0.001 (vs. before infusion). (D) The distribution of labelled cholesterol between lipoprotein fractions after pulse-chase experiment. The values shown are percentages of labelled cholesterol in each fraction relative to the total amount of released [3H]cholesterol (this normalization was necessary to permit comparisons among samples with a wide range of total labelled cholesterol). Means ± SD are shown; n = 13.
Further, we attempted to reproduce these findings in an in vitro ‘infusion’ system to test if the mere presence of LDL/VLDL is sufficient for the effects of rHDL on cholesterol efflux. Reconstituted HDL was added to whole human plasma or plasma after precipitation of apoB-containing lipoproteins and tested for cholesterol efflux from THP-1 cells. The difference between the capacities of whole and LDL/VLDL-depleted plasma was diminished with increasing concentrations of added rHDL and at 20 µg/ml disappeared completely (Figure 2C). Importantly, the capacity of rHDL added in vitro to enhance the capacity of plasma to induce cholesterol efflux was not affected by removal of LDL/VLDL. Thus, presence of LDL/VLDL in vivo is essential for increasing the capacity of plasma to support cholesterol efflux after infusion of rHDL.
Finally, we performed ex vivo pulse-chase experiments to follow the movement of labelled cholesterol between lipoprotein fractions in the patients’ plasma. In these experiments plasma samples were incubated for 60 min with [3H]cholesterol-labelled THP-1 cells within the conditions described for the efflux experiments (pulse) and then transferred to wells with unlabelled cells (chase). Aliquots of the medium were taken during pulse and chase incubations, lipoproteins separated using non-denaturing electrophoresis and bands corresponding to the individual lipoprotein fractions were excised from the gel and radioactivity counted. The distribution of labelled cholesterol among lipoprotein fractions is shown in Figure 2D. When plasma samples at 2.5 h post-rHDL infusion were tested, most of the labelled cholesterol released to the medium during pulse phase was recovered in the LDL/VLDL fraction and the proportion of cholesterol in this fraction was found to be rising over time at the expense of HDL subfractions. During chase incubation the amount of labelled cholesterol in LDL/VLDL rapidly declined, while that in HDL increased, the distribution of labelled cholesterol stabilizing after 30 min of chase incubation. Similar relationships were observed with plasmas from the patients after placebo infusion, except that the amount of labelled cholesterol in all subfractions was proportionally less (not shown). Thus, cholesterol released from cells accumulated initially mainly in LDL/VLDL.
Contribution of preβ1-HDL or lipid-free apoA-I
It has been suggested that a minor HDL subfraction, preβ1-HDL, or lipid-free apoA-I may be responsible for most of the cholesterol efflux capacity of HDL.4,13 While rHDL used in this study is not analogous to preβ1-HDL, upon entering the circulation it may bind to lipoprotein particles causing their remodelling and release of preβ1-HDL or lipid-free apoA-I. These particles may be responsible for the initial increase in the capacity of plasma to induce cholesterol efflux, but after acquiring cholesterol they may convert into other less potent particles types.
The time course of changes in preβ1-HDL level in plasma after infusing rHDL is shown in Figure 3A: it was sharply elevated after 1 h of the rHDL infusion and remained at this level throughout the study, no such elevation being observed during the saline infusion (Figure 3A). There was a statistically significant correlation between the preβ1-HDL level and the capacity of plasma to support cholesterol efflux (r = 0.86, P < 0.030).
Figure 3.
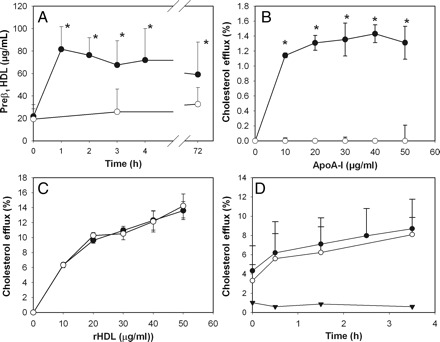
Contribution of lipid-free apoA-I and preβ1-HDL to the enhanced capacity of plasma to support cholesterol efflux. (A) Changes in plasmas concentrations of preβ1-HDL. Mean ± SD are shown; n = 13; *P < 0.001 (vs. pre-infusion values). (B) Cholesterol efflux from HeLa-ABCA1 cells (closed symbols) and HeLa-mock cells (open symbols) to lipid-free apoA-I; mean ± SD of quadruplicate determinations are shown; *P < 0.001 (vs. mock). (C) Cholesterol efflux from HeLa-ABCA1 cells (closed symbols) and HeLa-mock cells (open symbols) to rHDL; mean ± SD of quadruplicate determinations are shown. (D) Changes in the capacity of patient plasma to support cholesterol efflux from HeLa-ABCA1 cells (closed symbols) and HeLa-mock cells (open symbols); triangles denote an ‘ABCA1-dependent’ component of the efflux (i.e. the difference in the efflux from HeLa-ABCA1 and HeLa-mock cells). Mean ± SD are shown; n = 13.
To clarify the issue further we employed a functional assay to investigate if preβ1-HDL or apoA-I are responsible for the elevated capacity of plasma to support cholesterol efflux. The assay is based on the fact that cholesterol efflux to preβ1-HDL and lipid-free apoA-I is predominantly determined by ABCA1.6 We used HeLa cells stably transfected with ABCA1 or mock-transfected cells. Lipid-free apoA-I dose dependently promoted the efflux from HeLa-ABCA1 cells, but not from HeLa-mock cells (Figure 3B). On the other hand, when rHDL was used as an acceptor, there was no difference in efflux from HeLa-ABCA1 and HeLa-Mock cells (Figure 3C). The cholesterol efflux to plasma samples taken at various times after commencement of the rHDL infusion is presented in Figure 3D. Cholesterol efflux gradually increased over the period of rHDL infusion, but there was no difference in the efflux from HeLa-ABCA1 and HeLa-Mock cells. We conclude that although rHDL infusion leads to the formation of preβ1-HDL, this is unlikely to significantly contribute to changes in cholesterol efflux.
Contribution of mature HDL particles
Another possibility was that after entering the circulation rHDL causes remodelling of HDL particles with formation of a mature HDL subfraction that has increased efficacy in cholesterol efflux. To assess remodelling of HDL particles after rHDL infusion, plasma was separated using non-denaturing PAGE and apoA-I containing particles were identified by western blotting as described previously.11 Four subfractions were identified in plasma before infusion of rHDL: HDL2 (8.8–12.0 nm), HDL3 (7.0–8.8 nm) small HDL (<7.0 nm) and large HDL (>12 nm) (Figure 4A). The time course of HDL remodelling during and after rHDL infusion is shown in Figure 4B. Infusions of rHDL led to the increase in concentration of larger HDL (HDL2 and large HDL) with concomitant transient decline in HDL3 concentration; concentration of small HDL was not affected by rHDL infusion. It is unclear if this remodelling is a result of fusion of rHDL to HDL3 converting them into larger HDL, or fusion of rHDL to large HDL increasing their concentration without significantly affecting their size. Quantitatively, the increase in the concentration of large HDL cannot be fully accounted for by the decline in HDL3 and it is likely that both remodelling and fusion took place. There was however no statistically significant correlation between changes in concentration of individual subfractions of mature HDL and capacity of plasma to support cholesterol efflux (r < 0.15; P > 0.194).
Figure 4.
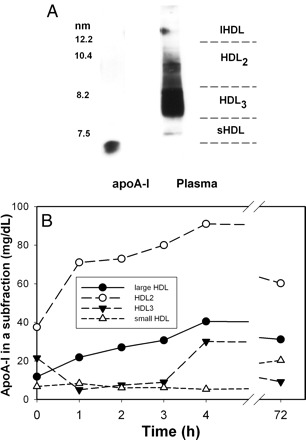
Dynamics of remodelling of HDL subfractions after infusion of reconstituted HDL. (A) Distribution of apoA-I in HDL subfraction in a sample of pooled plasma. (B) Time course of changes in the abundance of HDL subfractions after rHDL infusion. The concentration of individual subfractions were calculated from relative abundance of the subfraction and apoA-I concentration in the sample. Means are shown; n = 13.
To further investigate the role of mature HDL in the capacity of plasma to support cholesterol efflux we again employed a functional assay: the efflux from HeLa cells stably transfected with ABCG1 and BHK cells stably transfected with SR-B1, two transporters responsible for cholesterol efflux to mature HDL. rHDL dose dependently promoted efflux from both HeLa-ABCG1 and HeLa-Mock cells (Figure 5A), however cholesterol efflux from HeLa-ABCG1 cells was higher, indicating a presence of an ABCG1-dependent component of the efflux (circles, Figure 5A). Consistent with our previous findings,10 the contribution of ABCG1-dependent efflux from macrophages was relatively small. We then tested efflux to plasma samples collected before and after rHDL infusion. After infusion of rHDL the capacity of plasma to promote cholesterol efflux from both HeLa-ABCG1 and HeLa-Mock cells increased, but the increment in cholesterol efflux after rHDL infusion was similar for HeLa-ABCG1 and HeLa-Mock cells after 30 and 210 min of infusion (Figure 5B). Reconstituted HDL also dose dependently promoted the efflux from both BHK-SR-B1 and BHK-Mock cells (Figure 5C), however cholesterol efflux from BHK-SR-B1 cells was much higher, indicating a significant contribution of SR-B1 to the efflux to rHDL. When the efflux to plasma samples collected before and after rHDL infusion was tested, the capacity of plasma to promote cholesterol efflux from both BHK-SR-B1 and BHK-Mock cells increased substantially, but, again, the increment in cholesterol efflux after rHDL infusion was similar for BHK-SR-B1 and BHK-Mock cells after 30 and 210 min of infusion (Figure 5D). Thus, efflux to mature HDL through ABCG1-and SR-B1-dependent pathways was unlikely to contribute to the elevated capacity of plasma to support cholesterol efflux. Taking these data together, we conclude that remodelling of mature HDL particles did not contribute significantly to the changes in cholesterol efflux after rHDL infusion.
Figure 5.
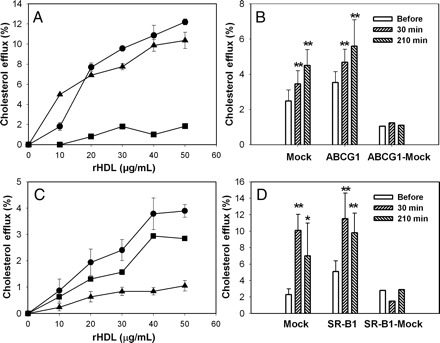
Contribution of mature HDL to enhanced capacity of plasma to support cholesterol efflux. (A) Cholesterol efflux from HeLa-ABCG1 cells (circles) and HeLa-mock cells (triangles) to reconstituted HDL; mean ± SD of quadruplicate determinations are shown; squares denote an ‘ABCG1-dependent’ component of the efflux (i.e. the difference in the efflux from HeLa-ABCG1 and HeLa-mock cells). (B) Changes in the capacity of patient plasma to support cholesterol efflux from HeLa-ABCG1 cells and HeLa-mock cells. Mean ± SD are shown; n = 13. (C) Cholesterol efflux from BHK-SR-B1 cells (circles) and BHK-mock cells (triangles) to rHDL; mean ± SD of quadruplicate determinations are shown; squares denote an ‘SR-B1-dependent’ component of the efflux (i.e. the difference in the efflux from BHK-SR-B1 and BHK-mock cells). (B) Changes in the capacity of patient plasma to support cholesterol efflux from BHK-SR-B1 cells and BHK-mock cells. Mean ± SD are shown; n = 13. *P < 0.010; **P < 0.001 (vs. before infusion).
Contribution of rHDL itself
Another possibility was that the increase in cholesterol efflux was caused by rHDL itself. Reconstituted HDL alone was capable of supporting robust cholesterol efflux from THP-1 cells in vitro (Figure 6A). Using values of apoA-I concentration before and after 1 h infusion, we estimated that the maximum level of rHDL after 1 h of infusion would be 250 µg/ml. This is almost certainly an overestimation, as a proportion of rHDL would have undergone remodelling or catabolism during this time. As plasma was added to the cells at the final concentration of 2%, the amount of rHDL in the efflux medium would be 5.0 µg/ml which would be predicted to add a maximum of 2% to the efflux, or 40% of what was observed after rHDL infusion in vivo. It is possible, however, that the efflux capacity of rHDL alone is different from that when added in plasma, as it may require plasma components to participate in cholesterol efflux. To examine this possibility, we added various amounts of rHDL to fresh human plasma, followed by adding the mixture to THP-1 cells at a final concentration of 2% and measuring efflux. The amount of rHDL that caused a 5% increase in cholesterol efflux when introduced in vivo, caused only a 1% increase in efflux when added in vitro (Figure 6B).
Figure 6.
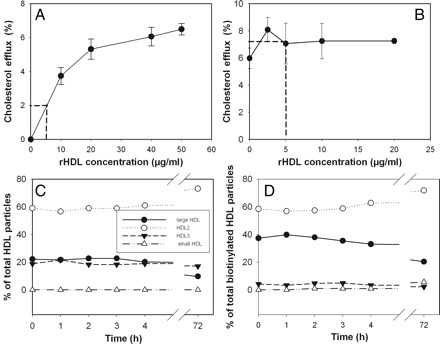
Contribution of rHDL to enhanced capacity of plasma to support cholesterol efflux. (A) Dose dependence of cholesterol efflux from THP-1 cells to rHDL. (B) Dose dependence of cholesterol efflux from THP-1 cells to human plasma supplemented with indicated concentrations of rHDL; mean ± SD of quadruplicate determinations is shown in panels A and B. (C) Time course of remodelling of HDL subfractions after addition of rHDL to plasma in vitro. (D) Time course of redistribution of rHDL between HDL subfractions after addition of rHDL to plasma in vitro.
To ascertain the reasons for such a remarkable difference in stimulation of cholesterol efflux ex vivo and in vitro, we analysed the time course of remodelling of apoA-I containing particles after introduction of rHDL in vitro. Unexpectedly, in sharp contrast to the in vivo situation (Figure 4), there was no remodelling of HDL when rHDL was added in vitro to plasma at a concentration of 2 mg/ml (the maximum concentration that would be reached if all rHDL infused remained intact in the circulation) (Figure 6C). It must be recognized that these experiments were designed to compare with the remodelling of HDL occurring in vivo and that remodeling events which may have occurred and re-equilibrated within 1 h would be undetected.
We then used this model to test the capacity of rHDL to interact with various HDL particles. Reconstituted HDL was labelled by biotinylation and recovery of biotin in various HDL subfractions was assessed with anti-apoA-I antibody or streptavidin. Binding of rHDL to HDL2 and small HDL was proportional to their concentration in the plasma; however, rHDL showed a disproportionally higher capacity to bind to large HDL and showed almost no binding to HDL3 (Figure 6D). The proportion of rHDL associated with individual subfractions did not change over time.
Thus, rHDL itself can account for only a proportion of the increased cholesterol efflux after rHDL infusion. Interestingly, remodelling of HDL which was apparent in vivo, was not reproducible in vitro.
Discussion
The major finding of this study is that infusion of rHDL in humans stimulates cholesterol efflux most likely by enhancing the flow of cholesterol through the RCT pathway. Based on our findings we suggest the following model of how rHDL stimulates cholesterol efflux (see Supplementary Data). Upon introduction into circulation, rHDL, which are cholesterol-free phospholipid-containing particles, causes rapid efflux of cholesterol from tissues. It cannot be ruled out that at this stage some cholesterol temporarily moves to rHDL from other lipoproteins. This flux of cholesterol causes remodelling of rHDL particles and possibly of VLDL particles and release of lipid-free apoA-I and/or preβ1-HDL. Experiments with model cells expressing or not ABCA1 suggest that preβ1-HDL is unlikely to contribute significantly to cholesterol efflux. However, the exact source of additional cholesterol after rHDL infusion is not known and mechanisms of cholesterol efflux from cells donating this cholesterol may be different to the model cells precluding completely ruling out a role for preβ1-HDL. rHDL are good acceptors of cholesterol, however, their capacity to store unesterified cholesterol is limited and they do not carry lecithin cholesterol acyltransferase (LCAT); consequently, after saturating rHDL particles, cholesterol flows to the reservoir with the biggest capacity to store unesterified cholesterol, LDL/VLDL particles, allowing rHDL to receive more cholesterol from tissues. Removing LDL/VLDL reservoir renders rHDL incapable of accepting more cholesterol from cells. Simultaneously, rHDL fuses with HDL particles forming larger HDL particles with much higher capacity for free cholesterol and containing LCAT. These new particles do not add to the capacity for cholesterol efflux per se, but receive cholesterol back from the LDL/VLDL pool and esterify it, forming HDL2 and entering the RCT pathway. The transfer of cholesterol to LDL/VLDL is transient explaining why we did not observe a rise in LDL-C level. Importantly, flux of cholesterol between lipoprotein particles only occurs in the intact circulation, no remodelling was observed in the in vitro system despite the presence of all particles and plasma factors. This model is consistent with an earlier finding by von Eckardstein12 and from our laboratory14 showing that a continuous flux of cholesterol between HDL and LDL/VLDL is necessary to maintain the capacity of HDL to efflux cholesterol from cells. This is also consistent with our previous suggestion that a major determinant of the efficiency of RCT is a flow of cholesterol through various lipoprotein particles, rather than the concentration of a specific particle.8 Similar findings were recently reported by Cuchel et al. from experiments on rHDL infusion in mice.15
This raises the question as to what exactly occurs in the circulation to maintain the ability of HDL to promote cholesterol efflux which apparently does not happen in vitro. One can speculate that rapid uptake of free cholesterol associated with LDL and/or cholesteryl esters associated with HDL by the liver are essential to maintain the capacity of plasma to accommodate cholesterol released from tissues, a hypothesis similar to that suggested by Dobiasova and Frohlich.16 Remodelling of HDL may also require factors present on cells, such as hepatocytes, or unstable factors that disappear from plasma rapidly unless constantly replenished in vivo.
Another unanswered question is what are the cellular mechanisms responsible for the flux of the extra cholesterol into the RCT system. We demonstrated that neither ABCA1, ABCG1 nor SR-B1-dependent efflux contribute significantly to the increment of cholesterol flux. Lack of involvement of ABCA1 and ABCG1 is consistent with findings of Cuchel et al.,15 however these investigators found that SR-B1 contributes to the efflux; the reasons for this discrepancy are not clear apart from possible species-specific differences. Our results however are consistent with a suggestion that the initial cholesterol efflux step through whatever mechanism it may occur is not the rate-limiting step for RCT. Instead, the capacity of the RCT system to store and transport additional cholesterol may be rate-limiting. Re-wording this hypothesis in terms of a ‘shuttle–sink’ model,17 our findings are consistent with a ‘sink’ (which includes a flow that maintains sink capacity) being more important than ‘shuttle’ for the efficiency of RCT. If confirmed, this model would explain why changes in HDL concentrations in the range well above saturating concentration for cholesterol efflux may affect efficiency of RCT as the size of the reservoir may saturate at a different level. The frequently observed lack of correlation (or even negative correlation) between HDL level and capacity of plasma to support cholesterol efflux may be explained by hypothesizing that isolated elevation of the level of the shuttle without a concomitant elevation of the capacity of sink or of a flow from the sink, may not be sufficient to enhance cholesterol efflux. This hypothesis is also relevant to the development of new strategies for HDL therapy. Approaches that only increase HDL concentration without consideration to the flow of cholesterol through RCT (a situation likely to occur with the combination of CETP inhibitors and statins) may not be effective. It appears that approaches utilizing influx of extra HDL, such as rHDL infusion or use of apoA-I mimetic peptides, may lead to a successful enhancement in the capacity of RCT to mobilize tissue cholesterol.
Supplementary material
Funding
This study was supported by grants #526614 (to D.S.) and #418920 (to B.K.) from the National Health and Medical Research Council of Australia. D.S. and B.K. are Fellows of the National Health and Medical Research Council of Australia. B.G.D. is a Post-Doctoral Training Fellow of the National Health and Medical Research Council of Australia. rHDL for the original clinical trial was supplied by CSL Behring.
Conflict of interest: none declared.
Supplementary Material
References
- 1.Shaw JA, Bobik A, Murphy A, Kanellakis P, Blombery P, Mukhamedova N, Woollard K, Lyon S, Sviridov D, Dart AM. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103:1084–1091. doi: 10.1161/CIRCRESAHA.108.182063. [DOI] [PubMed] [Google Scholar]
- 2.Nicholls SJ, Tuzcu EM, Sipahi I, Schoenhagen P, Crowe T, Kapadia S, Nissen SE. Relationship between atheroma regression and change in lumen size after infusion of apolipoprotein A-I Milano. J Am Coll Cardiol. 2006;47:992–997. doi: 10.1016/j.jacc.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 3.Sviridov D, Hoang A, Ooi E, Watts G, Barrett PHR, Nestel P. Indices of reverse cholesterol transport in subjects with metabolic syndrome after treatment with rosuvastatin. Atherosclerosis. 2008;197:732–739. doi: 10.1016/j.atherosclerosis.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 4.de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nanjee MN, Cooke CJ, Olszewski WL, Miller NE. Concentrations of electrophoretic and size subclasses of apolipoprotein A-I-containing particles in human peripheral lymph. Arterioscler Thromb Vasc Biol. 2000;20:2148–2155. doi: 10.1161/01.atv.20.9.2148. [DOI] [PubMed] [Google Scholar]
- 6.Asztalos BF, de la Llera-Moya M, Dallal GE, Horvath KV, Schaefer EJ, Rothblat GH. Differential effects of HDL subpopulations on cellular ABCA1- and SR-BI-mediated cholesterol efflux. J Lipid Res. 2005;46:2246–2253. doi: 10.1194/jlr.M500187-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Sviridov D, Mukhamedova N, Remaley AT, Chin-Dusting J, Nestel P. Antiatherogenic functionality of high density lipoprotein: how much vs. how good. J Atheroscler Thromb. 2008;15:52–62. doi: 10.5551/jat.e571. [DOI] [PubMed] [Google Scholar]
- 8.Sviridov D, Nestel P. Dynamics of reverse cholesterol transport; protection against atherosclerosis. Atherosclerosis. 2002;161:245–254. doi: 10.1016/s0021-9150(01)00677-3. [DOI] [PubMed] [Google Scholar]
- 9.Patel S, Drew BG, Nakhla S, Duffy SJ, Murphy AJ, Barter PJ, Rye K-A, Chin-Dusting J-, Hoang A, Sviridov D, Celermajer DS, Kingwell BA. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with Type 2 diabetes. J Am Coll Cardiol. 2009;53:962–971. doi: 10.1016/j.jacc.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Mukhamedova N, Escher G, D'Souza W, Tchoua U, Grant A, Krozowski Z, Bukrinsky M, Sviridov D. Enhancing apolipoprotein A-I-dependent cholesterol efflux elevates cholesterol export from macrophages in vivo. J Lipid Res. 2008;49:2312–2322. doi: 10.1194/jlr.M800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sviridov D, Chin-Dusting J, Nestel P, Kingwell B, Hoang A, Olchawa B, Starr J, Dart A. Elevated HDL cholesterol is functionally ineffective in cardiac transplant recipients: evidence for impaired reverse cholesterol transport. Transplantation. 2006;81:361–366. doi: 10.1097/01.tp.0000197556.83675.a6. [DOI] [PubMed] [Google Scholar]
- 12.Huang Y, von Eckardstein A, Assmann G. Cell-derived unesterified cholesterol cycles between different HDLs and LDL for its effective esterification in plasma. Arterioscler Thromb. 1993;13:445–458. doi: 10.1161/01.atv.13.3.445. [DOI] [PubMed] [Google Scholar]
- 13.Castro GR, Fielding CJ. Early incorporation of cell-derived cholesterol into pre-beta-migrating high-density lipoprotein. Biochemistry. 1988;27:25–29. doi: 10.1021/bi00401a005. [DOI] [PubMed] [Google Scholar]
- 14.Sviridov D, Fidge N. Pathway of cholesterol efflux from human hepatoma cells. Biochim Biophys Acta. 1995;1256:210–230. doi: 10.1016/0005-2760(95)00028-b. [DOI] [PubMed] [Google Scholar]
- 15.Cuchel M, Lund-Katz S, de la Llera-Moya M, Millar JS, Chang D, Fuki I, Rothblat GH, Phillips MC, Rader DJ. Pathways by which reconstituted high-density lipoprotein mobilizes free cholesterol from whole body and from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:526–532. doi: 10.1161/ATVBAHA.109.196105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobiasova M, Frohlich JJ. Advances in understanding of the role of lecithin cholesterol acyltransferase (LCAT) in cholesterol transport. Clin Chim Acta. 1999;286:257–271. doi: 10.1016/s0009-8981(99)00106-0. [DOI] [PubMed] [Google Scholar]
- 17.Atger VM, de la Llera Moya M, Stoudt GW, Rodrigueza WV, Phillips MC, Rothblat GH. Cyclodextrins as catalysts for the removal of cholesterol from macrophage foam cells. J Clin Invest. 1997;99:773–780. doi: 10.1172/JCI119223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.