

Abstract
Traumatic brain injury (TBI) is a devastating condition which often initiates a sequel of neurological disorders that can last throughout lifespan. From metabolic perspective, TBI also compromises systemic physiology including the function of body organs with subsequent malfunctions in metabolism. The emerging panorama is that the effects of TBI on the periphery strike back on the brain and exacerbate the overall TBI pathogenesis. An increasing number of clinical reports are alarming to show that metabolic dysfunction is associated with incidence of long-term neurological and psychiatric disorders. The autonomic nervous system, associated hypothalamic-pituitary axis, and the immune system are at the center of the interface between brain and body and are central to the regulation of overall homeostasis and disease. We review the strong association between mechanisms that regulate cell metabolism and inflammation which has important clinical implications for the communication between body and brain. We also discuss the integrative actions of lifestyle interventions such as diet and exercise on promoting brain and body health and cognition after TBI.
Keywords: Metabolic Syndrome, TBI, Inflammation, Autonomic Nervous System, brain plasticity
1. Introduction
Traumatic brain injury (TBI) is a devastating condition which often initiates a sequel of neurological and psychiatric disorders that can last for considerable time. TBI affects millions of Americans each year, resulting in approximately 2.5 million emergency department visits in 2013 (Taylor et al., 2017). Based on a series of large-scale population studies, a TBI incidence of 823.7 per 100,000 people is reported in USA. The situation in the European Union is also alarming with an overall incidence rate of 81.0-643.5 for admitted TBI per 100,000 people (Majdan et al., 2016). Most of the efforts to understand the TBI pathology have been centered on the brain, and we know that processes such as ischemia, hypoxemia, oxidative stress, and inflammation are intrinsic aspects of the TBI pathology (Ayton et al., 2014). However, we are just starting to understand that TBI also compromises systemic physiology and the function of body organs including liver, pancreas, and spleen with subsequent failures in metabolic and immune functions (Tables 1 and 2) (Plesnila, 2016). The emerging panorama is that the effects of TBI on the periphery initiate a pathological loop that can strike back on the brain and exacerbate the overall TBI pathogenesis. The consequences of dysfunctional cell metabolism on the outcome of brain trauma are particularly alarming for the Western society given the explosion of metabolic disorders. The magnitude of the problem is dreadful when considering that the incidence of TBI and associated cognitive disorders is on the rise, as is the prevalence of metabolic disease (Roozenbeek et al., 2013). A recent meta-analysis concluded that metabolic disorders such as diabetes and obesity complicate the healing prospect of TBI patients (Gharib et al., 2015). Therefore, it is crucial to understand how TBI impacts systemic physiology to have a thorough dimension of the TBI pathogenesis, which is necessary for the design of effective treatments to cope with the long-term burden of living with TBI.
Table 1.
Summary of clinical studies and Main Outcomes Addressing the effect of TBI on Peripheral Metabolism
| Study (year) |
Subjects | Purpose | Measurements | Main results |
|---|---|---|---|---|
| (Silva et al., 2018) (Silva et al., 2018) |
Mechanicall y-ventilated patients with severe TBI | Relationship between neuromuscular electrophysiological disorders (NED) and muscle atrophy in TBI patients. | The muscle structure (thickness and echogenicity) was assessed by B-mode ultrasound. | Mechanically-ventilated patients with TBI developed NED and muscle atrophy |
| (Shibahashi et al., 2017) (Shibahashi et al., 2017) |
Olderpatients (age ≥ 60 years) with TBI | Evaluate skeletal muscle mass as predictive marker for TBI outcome. | Skeletal muscle mass and clinical outcomes (Glasgow scale) | Reduced skeletal muscle mass was associated with poorer outcome after TBI |
| (Rizoli et al., 2017) (Rizoli et al., 2017) |
Patients with isolated moderate-to-severe TBI | Association between catecholamine levels post-trauma and functional outcome. | Epinephrine (Epi) and norepinephrine (NE) and clinical outcomes (Glasgow scale) | Elevated circulating catecholamines, are independently associated with functional outcome and mortality after TBI |
| (Czorlich et al., 2017) (Czorlich et al., 2017) |
Patients with severe TBI | Evaluate the impact of body mass index (BMI) on mortality and early neurologic outcome | Patients were categorized into underweight, normal, pre-obese and obese based on BMI. Early neurologic outcome was classified using the Glasgow Outcome Scale. | The BMI ≥35 is an independent predictor of mortality and is associated with an inferior early functional neurologic outcome. |
| (Mossberg et al., 2017) (Mossberg et al., 2017) |
Patients with isolated moderate to severe TBI | The effects of recombinant human growth hormone (rhGH) replacement on physical and cognitive functioning in TBI patients. | Peak cardiorespiratory capacity, body composition, muscle force testing and neuropsychological tests. | The rhGH replacement has a positive impact on cardiorespiratory fitness and a positive impact on perceptual fatigue in survivors of TBI with altered GH secretion. |
| (Rau et al., 2017) (Rau et al., 2017) |
Patients with isolated moderate to severe TBI | Analyze whether hyperglycemia is associated with higher morbidity and mortality in TBI patients. | TBI patients were allocated into four groups: Stress-induced hyperglycemia (SIH), diabetic hyperglycemia (DH), diabetes normoglycemia, and non-diabetic normoglycemia (NDN) | Patients with SIH and DH had significantly higher mortality than patients with NDN. The mortality was significantly higher in patients with SIH and but not with DH. |
| (Lu et al., 2017) (Lu et al., 2017) |
Patients with isolated moderate to severe TBI | Determine if TBI patients have a higher risk of myocardial dysfunction than the general population and to identify the risk factors of myocardial dysfunction in TBI patients. | Patients who visited ambulatory care centers or were hospitalized with a diagnosis of TBI. | Diabetes, hypertension, peptic ulcer disease, chronic liver disease and chronic renal disease were risk factors of myocardial dysfunction in TBI patients |
| (Hendrick et al., 2016) (Hendrick et al., 2016) |
Nonhead injured trauma patients | Impact of beta-blockers on Nonhead injured trauma patients | Mortality, length stay in intensive care unit (ICU) | Beta-blockers had not effect on mortality and ICU in TBI patients |
| (Park et al., 2016) (Park et al., 2016) |
Patients with diffuse axonal injury. | Investigate the regional cerebral metabolism related to growth hormone deficiency (GHD) after traumatic brain injury (TBI) | Patients underwent brain F-18 FDG PET study and an insulin tolerance test (ITT). | Compared with subjects with TBI but normal GH, patients with GHD after TBI showed decreased cerebral glucose metabolism. |
| (Giuliano et al., 2017) (Giuliano et al., 2017) |
Patients with complicated mild TBI | Evaluate whether mild TBI patients with GH deficiency had developed alterations in the glycolipid profile and clinical indices of injury severity and neurological outcome. | GH deficiency was investigated by the combined test (GH releasing hormone + arginine). The glycolipid and clinical outcomes (Glasgow scale) were also evaluated. | TBI Patients had high occurrence of isolated GH deficiency, which was associated with visceral adiposity and metabolic alterations. |
| (Di Battista et al., 2016) (Di Battista et al., 2016) |
Patients with moderate-to-severe TBI | Early dynamic profile of circulating inflammatory cytokines/chemokines and interrelationships between these mediators with catecholamines and clinical indices of injury severity and neurological outcome. | Plasma cytokine, chemokin, catecholamines. Neurological outcome was assessed using the extended Glasgow outcome scale (GOSE) | Positive association between catecholamines , cytokines and chemokine with poor outcome at 6 months after TBI. |
| (Ko et al., 2016) (Ko et al., 2016) |
Patients with moderate-to-severe TBI | The effect of β-Adrenergic receptor blockers (BBs) on TBI-induced cascade of immune and inflammatory. | Patients who received early propranolol after TBI (Feldman et al.) were compared with those who did not (non-EPAT). Data including demographics, hospital length of stay (LOS) and mortality were collected. | Early administration of propranolol after TBI was associated with improved survival. |
| (Majdan et al., 2015) (Majdan et al., 2015) |
Patients with severe TBI | Analyze whether BMI, height and weight of patients were related to severity, patterns and outcomes of TBI caused by low level falls. | Patients were categorized into underweight, normal, pre-obese and obese based on BMI and demographic characteristics, injury severity, patterns and outcomes were compared. | The patients in all BMI groups were of similar injury severity and neurological status. Obese and pre-obese patients required longer stay at ICU. |
Table 2.
Summary of preclinical studies and Main Outcomes Addressing the effect of TBI on Peripheral Metabolism
| Study (year) |
Experimental model/sample |
Purpose | Main results |
|---|---|---|---|
| (Shahidi et al., 2018) (Shahidi et al., 2018) |
Controlled cortical impact model (CCI) | investigate skeletal muscle–related changes (atrophy and degeneration/regeneration) resulting from CCI | CCI induces degeneration in Soleus and atrophy in tibialis anterior muscle. |
| (Ma et al., 2017b) (Ma et al., 2017b) |
Controlled cerebral blast injury model | Correlation between cytokines and hepatic cytochrome P450 (CYP450) enzyme superfamily after TBI | The cytokines in serum have a negative correlation with the expressions of CYP450 enzymes. |
| (de Castro et al., 2017) | Fluid Percussion Injury model (FPI) | Investigate whether a peripheral oxidative/inflammatory response contributes to neuronal dysfunction after TBI, as well as the prophylactic role of exercise training. | Exercise training alters the profile of oxidative-inflammatory status in liver and protects against acute hyperglycaemia and a cerebral inflammatory response after TBI |
| (Lang et al., 2015) (Lang et al., 2015) |
weight drop model of TBI | Evaluate the impact of body mass index (BMI) on mortality and early neurologic outcome in patients suffering from severe TBI. | Beta-adrenergic blockade reduced TBI-induced sympathetic hyperactivity, and prevented histopathological intestinal injury, gut permeability after TBI |
| (Sun et al., 2015) (Sun et al., 2015) |
Feeney's weight-drop method | The effects of probiotic Lactob acillus acidophilus on the intestinal smooth muscle contraction in TBI mouse model. | PKC/MLCK/MLC signaling pathway plays an important role in Lactobacillus acidophilus-mediated improvement of contractile properties of intestinal smooth muscle after TBI. . |
| (Villapol et al., 2015) (Villapol et al., 2015b) |
Controlled cerebral blast injury model | Investigate whether systemic response to trauma is associated with the hepatic acute-phase response. | TBI induces an increase in expression of the acute-phase protein, SAA1, and also AT1R mRNA, together with several other inflammatory changes in liver. |
| (Anderson et al., 2015) (Anderson et al., 2015) |
Cortical contusion impact (CCI) injury model. | Determine the effects of TBI alone on the gene expression of hepatic inflammatory proteins, drug-metabolizing enzymes, and transporters in CCI model | In contrast to clinical TBI, there was not a significant effect of experimental TBI on CYP or UGT27B7 metabolic enzymes. |
| (Zhang et al., 2014) (Zhang and Jiang, 2015a) |
Fluid Percussion Injury model(FPI) | The investigate the expression of Resistin in subcutaneous adipose tissue of rats with traumatic brain injury | FPI increased the gene expression of Resistin in subcutaneous fat 12 h, 24 h, 72 h, 1 week, 2 weeks, and 4 weeks after TBI. |
| (Jin et al., 2014) (Jin et al., 2014) |
Fluid Percussion Injury model(FPI) | The investigate the expression of Resistin in muscle of rats after TBI | Compared with control, the muscular resistin expression in FPI increased the gene expression of Resistin in muscle |
| (Zhu et al., 2014) (Zhu et al., 2014) |
Feeney's weight-drop method | Alterations in rat enterocyte mitochondrial respiratory function and enzyme activities in gastrointestinal after traumatic brain injury (TBI). | Rat enterocyte mitochondrial respiratory function and enzyme activities are inhibited following TBI. |
| (Keshavarzi et al., 2014) (Keshavarzi et al., 2014) |
Controlled cerebral blast injury model | Assess the alteration of gastric function and barrier function of gastrointestinal (GI) tract following TBI. | TBI induced Inflammation, congestion, ulcer and intragastric pressure reduction |
| (Hu et al., 2013)(Hu et al., 2013) | Cortical contusion impact (CCI) injury model | Effect of TBI on intestinal expression pattern of CD40 | The positive relationship between the expression of CD40 and that of TNF-α, VCAM-1, and ICAM-1 in jejunum after TBI. |
| (Olsen et al., 2013) (Olsen et al., 2013) |
Controlled cortical impact injury (TBI) | Determine whether TBI affects intestinal smooth muscle function. | TBI decreased intestinal contractile activity, delayed transit that is attributed to inflammatory injury in the intestinal smooth. |
| (Chu et al., 2013) (Chu et al., 2013) |
Controlled cortical impact injury (TBI) | The effect of TBI on molecular mechanisms in spleen and local brain inflammation. | Immediate splenectomy down-regulates the MAPKYNF-JB signaling pathway in rat brain after severe TBI. |
| (Larson et al., 2012) (Larson et al., 2012) |
Fluid Percussion Injury model (FPI) | The effect of β-adrenergic blockade on blood pressure and left ventricle contractility after TBI | Treatment with propranolol protected against TBI-induced blood pressure, cardiac contractility and ROS generation increase. |
| (Zlotnik et al., 2012) (Zlotnik et al., 2012) |
Controlled cortical impact injury (TBI) | The effect of nonselective β-adrenergic block antagonists on blood glutamate levels and on the neurological outcomes of rats after TBI | Systemic glutamate reduction after TBI is the result of a stress response and of the activation of the sympathetic nervous system through the β2 adrenergic receptors. |
| (Bansal et al., 2009) (Bansal et al., 2009b) |
Weight drop TBI model | The effect of vagal nerve stimulation intestinal permeability after TBI. | The central vagal stimulation regulates intestinal permeability after TBI. |
| (Moinard et al., 2008) (Moinard et al., 2008) |
Fluid Percussion Injury model (FPI) | The effect of TBI on liver energy homeostasis. | TBI is responsible for an impairment of liver energy homeostasis. |
The autonomic nervous system (ANS), associated hypothalamic-pituitary axis, and the immune system are at the center of the interface between brain and body and are central to the regulation of overall body homeostasis; therefore, they are primary intermediate for the action of TBI on systemic physiology (Figure 1). Indeed, clinical evidence indicates that injury-related alterations of the hypothalamic-pituitary system can have devastating consequences for body physiology. Here we discuss how the interaction between the ANS and the neuroendocrine system influences peripheral metabolism and the function of organ systems after TBI, and how the peripheral pathophysiology can exacerbate brain pathology. We also examine the strong association between mechanisms that regulate energy metabolism and inflammation given their implications for the communication between body and brain (Anthony and Pitossi, 2013). In addition, we discuss how the capacity of lifestyle to modulate brain-gut interation can be pivotal for the prevention of secondary complications, and healing of patients affected by TBI.
Figure 1.

The diagram illustrates the main organ components for the interaction between the brain and periphery after TBI. TBI induces central inflammation and triggers a multiorgan inflammatory response, involving the action of the autonomic nervous system and hypothalamic-pituitary axis. Autonomic dysregulation and subsequent secretion of catecholamines into the periphery are major players on the generalized host stress response after TBI. For instance: Sympathetic hyperactivity after TBI results in an elevated heart rate and blood pressure. Catecholamine signal through α- and β-adrenergic receptors which are expressed in immune organs like lymph nodes, spleen and bone marrow have the potential to influence the production of inflammatory mediators that alter the redox status in the liver and increase the intestinal permeability after TBI. The generalized sepsis response caused by translocation of bacteria induces a systemic inflammatory sequel with subsequent secondary inflammation in the brain. The liver reacts to sympathetic activation after TBI by displaying a systemic acute-phase response, involving leukocyte mobilization, and increase in cytokines that penetrate the leaky BBB, and become a major player of the secondary brain damage. Indeed, the muscle wasting during the initial phase of injury has been associated with ACTH and catecholamine release since muscle release of amino acids are used for inflammatory protein synthesis in the liver. The Catecholamine and cortisol effects on immune organs like lymph nodes, spleen and bone marrow also contribute to secondary damage after TBI.
2. Central Role of Autonomic Nervous System (ANS) and Hypothalamic-Pituitary Axis (HPA) in Maintaining Homeostasis
The HPA is often compromised in TBI patients and imbalances in the body’s hormone homeostasis are one of the most important metabolic consequences of TBI (Bosarge et al., 2015; Tan et al., 2017). A large proportion of severe TBI patients (20-55%) present significant endocrine dysfunction, which reduces quality of life, rehabilitative outcome, and life expectancy (Roquilly et al., 2013). Post-traumatic hypopituitarism is a major sequel of TBI, in which hypophyseal vessels are particularly affected by direct injury or occlusion resulting in disruption of blood supply to the pituitary gland (Fernandez-Rodriguez et al., 2011). Endocrine dysfunction after TBI can result in neurobehavioral deficits that can last for years. For example, chronic hypopituitarism associated with mild TBI has been associated with the pathogenesis of post-traumatic stress disorder (Undurti et al., 2018).
An increasing line of clinical and experimental evidence indicates that the disruptive effects of TBI on autonomic function are major contributors to the multiorgan failure that exacerbates the TBI pathophysiology. The ANS innervates smooth muscle and glands, and regulates all bodily functions such as circulation, digestion, body fluids, urination, and sexual arousal. The hypothalamus integrates diverse physiological processes involving the autonomic nervous system and the HPA, such that dysfunction of the hypothalamus is a major factor for the loss of body homeostasis (Shi et al., 2016).
2.1. TBI Promotes Dysfunction of the Hypothalamic-Pituitary-Adrenal Axis
Damage to the pituitary-adrenal axis can alter blood levels of catecholamines, cortisol, glucagon and growth hormone (GH), which in turn disrupt glycogenolysis and glucose production (Bulger et al., 2012) (Bosarge et al., 2015). The catecholamines epinephrine and norepinephrine, produced in the adrenal medulla, are involved in a fast stress response. Massive secretion of catecholamines into the circulation is a major player on the stress response to TBI (Desborough, 2000; Woolf, 1987), and a direct effect of sympathetic hyperactivation. As discussed below, a growing body of evidence supports a causative effect of sympathetic hyperactivity on the development of systemic organ dysfunction after TBI (Hinson et al., 2017; Lang et al., 2015). Epinephrine and Norepinephrine signal through α- and β-adrenergic receptors which are also expressed in leukocytes, which indicates that leukocytes have the potential to influence the production of inflammatory agents under stress conditions (see Figure 1) (Bierhaus et al., 2003; van der Poll and van Deventer, 1999). β-blocker therapy improves outcome in TBI patients (Alali et al., 2017), and attenuates the inflammatory response in animal studies (Villapol et al., 2015a). It is noteworthy that elevated stress has consequences for the regulation of other hormonal systems such as the hypothalamic-pituitary-gonadal (HPG). HPG dysfunction involves an adaptive process often involving an excess of stress hormones such as cortisol, and occurring at the early phase post-TBI (Ntali and Tsagarakis, 2019). Furthermore, prospective and longitudinal studies have shown high frequency (15% to 68%) of sex steroid hormone deficiency among TBI survivors. Izzo et al., (2016) demonstrated that the hypogonadism-induced sex steroid deficiency has implications beyond psychosexual function and fertility for survivors of TBI (Izzo et al., 2016). Chronic hypogonadism also induces muscle weakness and osteoporosis exacerbating the immobility after neuronal injury (Agha and Thompson, 2004). Neuroendocrine dysfunction can last for many years after the original onset of brain injury (Alavi et al., 2016).
The adrenal cortex produces the glucocorticoid cortisol which is involved in regulation of metabolism and immune function, and the mineralocorticoid aldosterone which is important for blood pressure control. Adrenal insufficiency can lead to life-threatening complications chiefly related to a drop in cortisol levels (Cohan et al., 2005; Molaie and Maguire, 2018). It is considered adrenal insufficiency when cortisol levels are lower than 15-18μg/dL in acutely ill patients or lower than 10 μg/dL in chronic TBI patients (Kleindienst et al., 2009). Patients with adrenal insufficiency lose their ability to maintain normal blood pressure and cardiovascular function (Quinkler et al., 2018).
The acute effects of TBI on the hypothalamic-pituitary-adrenal axis also can disrupt metabolic balance by the release of glucocorticoids, and sustained elevation in cortisol is associated with poor clinical outcomes (Santarsieri et al., 2014), particularly in TBI patients (Wagner et al., 2011). Hypercortisolemia increases the release of glucose from the liver, together with inhibiting insulin release from pancreatic beta cells (Lambillotte et al., 1997) and weakening the insulin-dependent transport of glucose into cells (Cree et al., 2010). It is noteworthy that the therapeutic benefit of targeting the action of glucocorticoids in TBI is controversial as while they can reduce inflammation (Campolo et al., 2013), they can also impair synaptic plasticity and promote neuronal degeneration (Rothman and Mattson, 2013).
2.2. The Impact of TBI on the Hypothalamic-Pituitary-growth hormone (GH) Axis
The hypothalamic-growth hormone axis has been one of the most studied in the context of TBI (Giuliano et al., 2017). GH produced by the anterior pituitary has a crucial action on the control of immune function, nitrogen and mineral retention, and lipid and protein synthesis, in addition to regulation of IGF-1 release by the liver (Simsek et al., 2015). Chronic growth hormone deficiency (GHD) is common among survivors of TBI which can last for several months or years following the original incident (Giuliano et al., 2017). GHD is associated with reductions in lean body mass (Mossberg et al., 2017), exercise capacity (Gonzalez et al., 2018) cardiac function (Cittadini et al., 2009), and bone mineral density (Bogdan et al., 2016). In addition, chronic GHD is characterized by metabolic abnormalities including visceral obesity and prevalence of non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH) (see Figure 2) (Takahashi, 2017). GHD is associated with poor quality of life, being particularly detrimental for the well-being of TBI patients based on sleep disturbances, social isolation, and reduced physical capacity (Giuliano et al., 2017; Ranke and Wit, 2018). It is noteworthy that the GH and gonadotrophin systems are highly susceptible to TBI, as gonadotrope and somatotrope cells are proximal to the vascular territory of the long hypophyseal portal vessels. Indeed, HPG dysfunction can affect up to 80% of TBI patients (Alavi et al., 2016).
Figure 2.

Proposed mechanism by which TBI alters hypothalamic-pituitary growth hormone (GH) Axis. TBI-induced chronic growth hormone deficiency (GHD) is characterized by metabolic abnormalities associated with decreased IGF-1 synthesis. The presence of low plasma GH decreases the hepatic IGF-1 synthesis via dysregulation of the GH receptor, JAK2 and STAT5 pathways. Early hepatic inflammation after TBI also activates the hepatic toll like receptors (TLR-4) that leads to nuclear translocation of the transcription factor NF-κB, TNF-αreceptor upregulation and TNF-α increase. This systemic inflammation could induce insulin resistance by stimulating cytokines receptors and JNK pathway that leads to serine/threonine phosphorylation of IRS1, TNF-α and mitochondrial ROS production. It is recognized that the growth hormone GH/IGF system is involved in metabolism manifestations. In turn, TBI-induced GH/IGF-1 deficiency may be associated with visceral obesity that leads to systemic inflammation and ROS production. Low plasma IGF-1 induces β-cell exhaustion and consequently peripheral insulin resistance by dysregulation of the IGF-1 receptor, PDE and PKA pathway.
IGF-1 is largely produced in the liver (Tanriverdi and Kelestimur, 2015) and TBI patients often exhibit reduced circulating IGF-1 levels (Aimaretti et al., 2004b). A meta-analysis study showed a positive correlation between blood IGF-1 levels and cognitive function, in which adequate levels of IGF-1 seem necessary to maintain cognitive function (Fernandez and Torres-Aleman, 2012). Although the brain also synthesizes IGF-1, IGF-1 from the body can cross the blood-brain barrier and affect brain tissue. Contrary to peripheral IGF-1, brain IGF-1 levels tend to increase after TBI such that IGF-1 mRNA levels increase in the brain within days after brain injury in adult rats (Feeney et al., 2017) and developmental rats (Schober et al., 2012). IGF-1 has various isoforms that respond to TBI with a different time course and appear to have distinctive functions (Aperghis et al., 2004). For example, IGF-1A mRNA (encoding for the Eb peptide) peaks by 3 days post-TBI (Schober et al., 2012), whereas IGF-1B mRNA (encoding the Eb peptide) peaks by 2 days post-TBI.
TBI patients with hypopituitarism present alterations in glucose and lipid metabolism (Karamouzis et al., 2016) that are within the range of action of GH (Bartke, 2011b). GH/IGF-1 deficiency is associated with multiple physical and metabolic manifestations including diminished lean body mass, disrupted lipoprotein and carbohydrate metabolism (Mavalli et al., 2010). In addition, it has been shown that IGF-1 levels can vary as a function of hepatic insulin resistance and pancreatic beta cell dysfunction (Friedrich et al., 2012). Peripheral resistance to GH action is manifested by elevated plasma GH and low plasma IGF-1 (Agha et al., 2004). Considering that excessive amount of GH opposes to the effects of insulin in the liver and peripheral tissue, the TBI-induced down regulation of peripheral IGF-1 decreases the anabolic effects of GH, resulting in nitrogen wasting and hyperglycemia (see Figure 2) (Takahashi, 2017). The possibility that GH replacement can improve patient’s outcome during the acute phase after TBI is still controversial (Garrahy et al., 2017). For example, GH may exacerbate pulmonary dysfunction, intracranial hypertension, and hyperglycemia which are common in TBI patients (Garrahy et al., 2017).
2.3. The Impact of TBI on the Hypothalamic-Neurohypophysial Axis: Dysfunctional Antidiuretic System Disrupts Water Homeostasis
Brain trauma often leads to dysfunction of the hypothalamic neurons secreting antidiuretic hormone (ADH) into the posterior pituitary gland. Proper action of the ADH system is vital for maintaining water homeostasis (Capatina et al., 2015). Inadequate ADH secretion after TBI leads to varying degrees of water dysregulation, and can evolve into several clinical manifestations (Behan et al., 2008). Continuous secretion of ADH causes reabsorption and retention of water by the kidneys, leading to concentrated urine and hyponatremia. Hyponatremia (low levels of sodium in blood) after TBI is the most common electrolyte problem resulting in edema and seizures (Capatina et al., 2015). In turn, post-traumatic diabetes insipidus (PTDI) is usually caused by damage to the posterior part of the pituitary gland from where ADH is secreted (Silva et al., 2015), such that patients with PTDI lose their ability to concentrate urine (polyuria). The lack of fluid consumption can result in severe dehydration and hypovolemic hypernatremia. Dehydration characterized by decreased skin turgor, dry mucous membranes, hypotension, tachypnea, tachycardia occurs most frequently in the first few days following the trauma episode posing elevated risk of mortality (Capatina et al., 2015). Hypernatraemic dehydration has been shown to increase morbidity and to reduce recovery after brain injury (Garrahy et al., 2017). PTDI occurs in up to 16% of all brain-injured patients usually 5 to 10 days after trauma (Hadjizacharia et al., 2008) and generally lasts for about a month; however, it can prolong up to 3 years post-TBI (Aimaretti et al., 2004a).
3. TBI Disrupts Function of Organ Systems (Figure 3)
Figure 3.

Proposed mechanism by which TBI alters the brain-gut axis, and in turn, how peripheral pathological signals impact the brain. Right after TBI, dysfunctional hypothalamic-pituitary-adrenal axis disrupts metabolic balance and increases stress hormones such adrenocorticotrophic and cortisol. It is postulated that the ACTH-induced Epinephrine (E) and norepinephrine (NE) by adrenal gland together with efferent vagal output promote splanchnic hypoperfusion, thereby increasing Cytokines and chemokines. The secreted TNF-α by spleen macrophages after TBI binds TNF receptors (TNFRs) on intestinal epithelial cells and activates several pathways, including the NF-κB pathway that upregulates genes encoding pro-inflammatory cytokines and myosin light chain kinase (MLCK). These signals enhance tight junction permeability in the intestine involving damage-associated molecular patterns (DAMPs), Pathogen-associated molecular patterns (PAMPs), and cytokines. The Cytokines produced increases intestinal permeability and then penetrate the leaky BBB to contribute to secondary brain damage characterized by fatigue, and learning and memory dysfunctions.
3.1. TBI and Brain-Liver Axis.
The function of the liver in detoxification, synthesis of lipids and proteins is essential for the maintenance of homeostasis, such that liver dysfunction can have devastating consequences for overall performance of the whole organism. Experimental concussive brain injury has been shown to induce early inflammation by disrupting mitochondrial function and redox status in the liver (de Castro et al., 2017; Villapol et al., 2015c). In turn, signals derived from the liver inflammatory reaction can exacerbate brain pathology (de Castro et al., 2017; Villapol et al., 2015c). Concussive brain injury in rodents causes major reductions in liver weight and protein production, with concomitant effects on body physiology (Moinard et al., 2008; Moinard et al., 2005). It seems plausibly that these deficits in protein homeostasis and degradation in liver are responsible for the muscle athrophy and locomotor deficits in TBI patients (Shahidi et al., 2018; Wright et al., 2017). The muscle wasting during the initial phase of injury has also been associated with muscle release of amino acids that are used for inflammatory protein synthesis in the liver (Moinard et al., 2005). In addition, brain-injured patients undergo rapid weight loss, with negative nitrogen balance and enhanced whole-body protein breakdown that seem secondary to inflammation and sympathetic activation (See Figure 1) (Mansoor et al., 1996).
The liver also reacts to TBI by displaying a systemic acute-phase response, involving leukocyte mobilization, and increase in cytokines which may be viewed as an attempt to regain homeostasis after trauma; however, this response may also be perceived as a way to enhance the multiorgan dysfunction syndrome. In particular, the post-injury hepatic inflammation results in higher levels of serum amyloid A1 (SAA1) and proinflammatory cytokines, infiltration of neutrophils and macrophages with detrimental effects on brain cells (Washington et al., 2016). Indeed, the production of cytokines and chemokines by the liver seems to act as an amplifier of the local brain injury inflammatory response (see Figure 1) (Anthony et al., 2012). The liver is highly susceptible to changes in energy metabolism and overproduction of reactive oxygen species based on its high demands for energy (Ray, 2012). Accordingly, the combination of high oxidative metabolism and ATP depletion in the liver has been shown to collapse the mitochondria machinery and to precipitate hepatic cells to a necrotic death (Sullivan et al., 2018). It has been recently reported that TBI collapses the ion gradient homeostasis followed by dysglycemia in the liver (de Castro et al., 2017). In addition, the involvement of liver in the events by which TBI disrupts glucose regulation emphasizes the risk posed by liver dysfunction for development of metabolic disorders such as type-2 diabetes (Tsatsoulis et al., 2013).
3.2. TBI and Brain-Spleen Axis: Inflammation and Sepsis
The spleen is deeply involved in synthesis and storage of immune cells and plays an important role in the systemic response to TBI, which is characterized by a reduction in splenic mass and an increase of immune cells in circulation (Das et al., 2011). The sepsis response to TBI is also reflected in translocation of bacteria to multiple organs with a subsequent increase in systemic inflammatory response and organ failure (Bansal et al., 2009b). As part of the inflammatory/sepsis response, elevated levels of tumor necrosis factor alpha (TNF-α), interleukin 1b (IL-1b), and IL-6 can affect tight junction permeability in the intestine (Al-Sadi et al., 2014). In turn, the secreted TNF-α binds TNF receptors (TNFRs) on intestinal epithelial cells and upregulates molecular pathways related to pro-inflammatory cytokines such as NF-kB. Increased sympathetic activity post-TBI results in splanchnic hypoperfusion with subsequent alterations in the intestinal tight junction proteins ZO-1, occludin, and increased intestinal permeability (see Figure 1) (Zhang and Jiang, 2015b). Aberrant intestinal permeability can be evidenced as elevated ratio of orally ingested lactulose (a marker of paracellular permeability) to mannitol (a marker of transcellular permeability) in urine (McHugh et al., 2007) of TBI patients. The disruption of the intestinal barrier blocks flow of ions, solutes, proteins, and bacterial products across the intestinal wall (Nighot et al., 2017), and may influence an array of metabolic functions (De-Souza and Greene, 2005).
Regarding the effect of TBI on periphery alterations, the spleen is important organ in the flux of monocytes and T cells into the peripheral circulation following injury (Rasouli et al., 2011). Microvesicles (MVs)/exosomes-based therapies offer promise in promoting neuroprotection by mitigating central neuroinflammation as well as modulating peripheral inflammation characterized by spleen-induced macrophages and monocytes release into circulation (Patel et al., 2018). This in turn lessens the infiltration of pro-inflammatory molecules into the brain and prevents the contribution of peripheral immune cells to the secondary injury.
3.3. TBI and Cardiovascular/heart interaction between brain and body
Loss of cardiovascular function is a common sequel in severe TBI patients occurring in 80- 90% of patients admitted to intensive-care units, and is associated with high incidency in hospital mortality (Zygun et al., 2006); (Eric Nyam et al., 2019). The communication between the brain and heart is a highly integrative process involving neuroendocrine and immune factors under autonomic regulation. TBI patients exhibit paroxysmal sympathetic hyperactivity, characterized by periodic episodes of increased heart rate, blood pressure, and systemic inflammation (Schwulst et al., 2013). Pre-clinical evidence indicates the involvement of progressive cardiomyocyte oxidative stress and expression of pro-inflammatory chemokines in TBI-induced apoptosis (Chen et al., 2018). In turn, the spleen transfers resident leukocytes and secretes proinflammatory cytokines into the systemic circulation (Ajmo et al., 2008). Consequently, the immune cells and inflammatory factors may promote collagen deposition, proliferation of cardiac fibroblasts and cardiomyocyte death after TBI. In agreement with this idea, mice subjected to splenectomy after controlled cortical impact (CCI) showed significantly improved cardiac function, and decreased cardiac fibrosis, oxidative stress, cardiomyocyte apoptosis, and decreased infiltration of immune cells and inflammatory factors to the heart. (Zhao et al., 2019). Considering that systemic inflammatory response syndrome in TBI patients is associated with systolic cardiac dysfunction (Deepika et al., 2018) (Hasen et al., 2019), it is plausible that a paroxysmal sympathetic hyperactivity followed by immune response after brain injury may play a vital role in mediating brain-heart interaction. However, existing information with regards to the effects of TBI on myocardial function (Cuisinier et al., 2016);(Venkata and Kasal, 2018) is controversial given possible confounding factors such as multiple trauma, associated co-morbidities and hemorrhagic shock that may induce a cardiac failure independently of the brain injury.
3.4. The Multiple Effects of TBI on the Gastro-Intestinal System
The bidirectional communication between the central and the enteric nervous systems is crucial for modulation of gastrointestinal functions such as motility, secretion, blood flow, intestinal permeability, mucosal immune activity, and visceral sensation including pain (Rhee et al., 2009). One commonly overlooked aspect of TBI is the disruption of the brain-gut axis (Ma et al., 2017a; Sundman et al., 2017). In the first few weeks after TBI, most patients have reduced intestinal contractile activity and absorption, which is manifested by vomiting and abdominal distension (Sundman et al., 2017).
3.4.1. Alterations in Intestinal Permeability
Evidence from human and rodent studies indicate that acute TBI can disrupt the intestinal wall that functions to filter the flow of select ions, proteins, bacteria and their subproducts (Jin et al., 2010). When the gastrointestinal (GI) tract is compromised, substances can aberrantly penetrate the intestine leading to a chronic inflammatory sequel (Anthony et al., 2012). As discussed above, TNF-α is a central player in the regulation of intestinal permeability following TBI. TNF-α expression in mammals is induced by Toll-like receptor (TLR) signaling pathways in macrophages that are part of the defense mechanisms involved in the innate immune response (Verstrepen et al., 2008). Secreted TNF-α binds to TNF receptors (TNFRs) on intestinal epithelial cells and activates pro-inflammatory cytokines resulting in enhanced tight junction permeability in the intestine (Stoecklein et al., 2012). TBI elicits a positive feedback loop involving TNF-α and damage-associated molecular patterns (DAMPs, nucleic acids released from apoptotic and necrotic cells), which disrupt the intestinal barrier function (Anthony et al., 2012). It has been shown that treatment of mice with the hormone ghrelin blocks the TBI-related increase in TNF-α expression along with disruptions in intestinal permeability (Bansal et al., 2010). TBI also induces production of catecholamines which can lead to gastrointestinal dysfunction such that the sedative Propofol, that inhibits catecholamine release, counteracts the increase of TNF-α and intestinal permeability in rats (see Figure 3) (Jin et al., 2010).
TBI patients show an increase in the ratio of orally ingested lactulose (marker of paracellular permeability) to mannitol (marker of transcellular permeability) in urine (Hernandez et al., 2007). The use of the lactulose-mannitol test as well as a dye permeability test have revealed the effects of controlled cortical impact (CCI) in increasing paracellular permeability in rodents (Bansal et al., 2009b). TBI patients also show decreased levels of occludin and increased levels of myosin light chain kinase in the intestine (Bansal et al., 2009b; Zhang and Jiang, 2015b). Interestingly the pathogenesis of type 2 diabetes involves increased intestinal permeability (De-Souza and Greene, 2005), which encompasses with the higher clinical complications and mortality rates observed in brain trauma patients with diabetes or obesity (Ditillo et al., 2014). The altered gastrointestinal motility (Lu et al., 1997), splanchnic ischemia (Hernandez et al., 2007), gut hyperpermeability (Hang et al., 2003), followed by hyperglycemia, reinforce a role for glucose in a positive feedback loop after TBI.
3.4.2. Alterations in Intestinal Contractility
Preclinical and clinical studies indicate that inflammatory processes play a role on the alterations of contractility of the gut observed in the TBI pathogenesis (Sundman et al., 2017). TBI causes a delayed decrease in intestinal contractile activity which disturbs food intake and proper nutrition (Rauch et al., 2012). Experimental brain injury in rodents results in an exaggerated immune response to lipopolysaccharide (LPS) challenge, as manifested by elevated levels of inflammatory cytokines IL-1β, TNF-α, and IL-6, and altered microglia phenotype (Ritzel et al., 2018). In addition, the LPS challenge can cause depressive behavior in mice previously exposed to brain injuries (Fenn et al., 2014; Shitaka et al., 2011). The fact that these depressive symptoms emerge after thirty days post-injury (Fenn et al., 2014) suggests an involvement of chronic inflammation (Mayer et al., 2006). In agreement with this view, pre-clinical studies have shown that dysfunctional contractility of the smooth muscle within the GI tract correlates with both increased systemic inflammation and brain atrophy (Sun et al., 2015). In particular, it has been reported that a dysfunction in colon mucosal barrier develops over time after TBI, as evidenced by reduced expression of claudin-1 and increased activation of sub-epithelial enteric glial cells (EGCs) which regulate mucosal barrier homeostasis (Ma et al., 2017a). Interestingly, these alterations have been associated with inflammatory and degenerative processes in the injured cerebral cortex (see Figure 3) (Ma et al., 2017a).
3.4.3. Microbiota: Minding the Gap between Brain and Gut
As discussed above, gastrointestinal disorders are a notable complication in the TBI pathogenesis (Bansal et al., 2009a; Hang et al., 2003; Katzenberger et al., 2015). An increasing line of evidence indicates that gut microbiota plays an important role in brain-gut interaction and behavioral outcome by producing metabolites, hormones and immune factors (Schmidt, 2015). On the other hand, products of carbohydrate fermentation by gut bacteria such as the short chain fatty acid (SCFA) butyrate, have been shown to regulate brain plasticity and function, including hippocampal neurogenesis, synaptic plasticity, and neuronal repair. The gut microbes also synthesize a vast array of neuroactive molecules including neurotransmitters such as GABA, which have effects on the CNS (Patterson et al., 2014). The intestinal microbiota also affects the intestinal epithelium, local mucosal immune system, enteric nervous system and spinal and vagal nerves. Microbiome sub-products modulate CNS function using hormones such as cortisol and catecholamines, immune regulators such as cytokines, and neurotransmitters such as acetylcholine and serotonin (see Figure 3) (Schmidt, 2015). Gut microbiota also controls the amount of energy that is taken from foods playing an important role in weight regulation, particularly, increasing energy harvest from food in obesity (Ley, 2010). Since TBI patients develop signs of hyperglycemia (Shi et al., 2016) and gut microbiota are associated with metabolic alterations such as fat distribution and adipose tissue inflammation (Boulange et al., 2016), it is plausible that changes in the microbiota composition can contribute to glucose dysregulation after TBI. However, further experimental and longitudinal studies are needed to better understand causal or temporal associations between these events. Further understanding of the role of the gut microbiome and gut-brain axis during TBI may result in the novel application of probiotics, dietary therapeutics and pharmacological compounds in the prevention or reversal of secondary complications of TBI.
3.4.4. Role of microvesicles in the interplay between brain and body
Another way for cells and systems to communicate across body and brain is via microvesicles (MVs)/exosomes. Microvesicles (MVs)/exosomes are a heterogeneous group of extracellular vesicles (EVs) less than 1 μm in diameter released into the extracellular environment from virtually all cell types (Greening and Simpson, 2018). A number of interesting studies, using animal’s models of TBI, and/or clinical samples suggest that (MVs)/exosomes serve as carriers of many bioactive molecules including cytosolic proteins, nucleic acids (mRNA, miRNA), permitting, among others, the acquisition of new functional properties by recipient cells in the systemic TBI-induced pathophysiology (Shlosberg et al., 2010); (Nekludov et al., 2014); (Andrews et al., 2016); (Mondello et al., 2018). (MVs)/exosomes modulate inflammation, neuronal function and plasticity, BBB permeability and cellular responses to brain injury (Walker et al., 2012); (Zhang et al., 2015); (Kim et al., 2016). Furthermore, the higher stability followed by less invasiveness, easy delivery, low or no immunogenicity and tumorigenicity reinforces the potential of microvesicles-derived exosomes (i.e. cell-free exosome-based therapy) for the treatment of TBI pathophysiology (Mondello et al., 2018).
4. The Coalition between Metabolic dysfunction and Inflammation Fuels the TBI Pathogenesis
4.1. Disruption of Glucose Metabolism
Clinical evidence indicates that TBI patients often develop signs of hyperglycemia even on the absence of preexisting diabetes (Shi et al., 2016), and excessive glucose production has been correlated with the severity of the injury and clinical outcome (Asehnoune et al., 2017). TBI patients with hypopituitarism frequently present metabolic alterations in glucose levels, insulin resistance and hypertriglyceridemia (Tanriverdi et al., 2015). Animal studies have shown that hyperglycemia leads to a reduction of immune and bioenergetic functions resulting in inflammation and elevated susceptibility to infections (de Castro et al., 2017). In addition, systemic hyperglycemia contributes to anaerobic metabolism in the brain following acute injury, resulting in lactic acidosis and tissue damage (Kim et al., 2012). Animals (de Castro et al., 2017) and clinical (Asehnoune et al., 2017) studies have shown an association between hyperglycemia and increased morbidity and mortality after TBI. Hyperglycemia and insulin resistance are very common in critically ill patients with TBI, even affecting patients with no history of diabetes (Shi et al., 2016). Along this view, normalization of blood glucose levels in patients with severe TBI has been shown to decrease mortality and morbidity (including infection rate), to reduce stays in Intensive Care Unit, and to improve neurological outcome (Van den Berghe et al., 2006). In turn, hypoglycemia (<40 mg/dl or 2.2 mmol/l blood glucose level) induced by i.v. application of insulin increases stress hormones such as ACTH, cortisol, and GH (Hoffman et al., 1994). The insulin tolerance test (ITT) is often useful to diagnose dysregulation of the hypothalamic–pituitary–adrenal axis. As discussed above, GH plays a major role in the regulation of glucose metabolism (see Figure 2) (Bartke, 2011a).
4.2. Insulin Action across Body and Brain
Insulin produced by the pancreas gland is a major modulator of the effects of TBI on systemic and central physiology. Besides its hormonal action as main regulator of glucose metabolism and other aspects of body physiology (Bedinger and Adams, 2015; Gralle, 2017), insulin is surfacing as an important modulator of brain function and plasticity (Lima et al., 2002). The effect of insulin on neural cells is mediated by a family of receptors located in brain regions particularly associated with synaptic plasticity and cognitive processing such as the hippocampus (Agrawal et al., 2016c). Reduced insulin receptor signaling in the hippocampus impairs long-term potentiation and recognition memory (Nistico et al., 2012). Insulin receptor signaling promotes neurogenesis, increases neuronal survival and reduces neuroinflammation (Adzovic et al., 2015; Bateman and McNeill, 2006). Experimental evidence showing that insulin influences mitochondrial function (Szendroedi et al., 2012) provides a general explanation for how insulin can influence a range of cellular processes highly dependent on metabolic energy. Indeed, reduced sensitivity to the action of insulin is considered a predictor of poor clinical outcome in TBI patients (Mowery et al., 2009b). Manipulation of the insulin signaling is getting recognition as a potential therapeutic target to reduce the burden of brain injury. For example, the anti-inflammatory effects of insulin (van der Heide et al., 2006) have led to the implementation of insulin sensitizing treatment to attenuate cell damage and to promote recovery following CNS insults (Eakin et al., 2013).
Disease states characterized by insulin resistance, such as obesity and type 2 diabetes, are risk factors for TBI complications (Agrawal et al., 2016a; Ley et al., 2011). In addition, insulin resistance is associated with increased mortality after TBI (Majdan et al., 2015). TBI patients suffering type 2 diabetes (T2D) or insulin-dependent T2D have a higher mortality rate compared to severe TBI patients without T2D (Majdan et al., 2015). These results indicate that insulin deficiency may contribute to the increased mortality observed in TBI patients, and that T2D may be an independent predictor of poor outcome and mortality after TBI (Ditillo et al., 2014). In addition, as discussed below, experimental metabolic syndrome (MetS) in rodents elicited by overconsumption of dietary fructose reduces signaling through the insulin receptor in the hippocampus and potentiates the effects of TBI on behavioral dysfunction (Agrawal et al., 2016c). These results additionally emphasize the metabolic impact of diet on TBI outcome (see below).
4.3. Diabetes and Obesity Exacerbate TBI Pathology
Increasing evidence indicates that metabolic disorders such as obesity and T2D that disrupt the regulatory action of insulin on glucose metabolism in the body can also influence the brain. For example, the occurrence of MetS, characterized by elevated levels of serum triglyceride and low glucose tolerance, reduces hippocampal insulin receptor signaling, which is commensurable to poor learning and memory performance (Agrawal and Gomez-Pinilla, 2012). This also implies that metabolic disorders such as obesity and diabetes have the potential to exacerbate the TBI pathology as the inability of brain to metabolize glucose is an intrinsic aspect of the TBI pathology (Ditillo et al., 2014).
It has been shown that obese Zucker rats have an exaggerated hyperglycemic response and insulin dysregulation after trauma as compared to lean rats (Xiang et al., 2014). These results in animals are consistent with clinical evidence that obese critically ill patients have an increased need for insulin (Pieracci et al., 2008). In addition, obese patients that suffer brain trauma have more clinical complications and higher mortality rates than lean patients with similar injuries (Stein et al., 2012). Clinical reports suggest that metabolic dysfunction is a predictor of poor outcome in TBI patients and can increase incidence of long-term neurological and psychiatric disorders (Ley et al., 2011; Mowery et al., 2009a; Stein et al., 2012). The occurrence of obesity has been associated with several neurological abnormalities including cell degeneration, reduced neurogenesis, and increased cognitive/mood disorders (Yehuda et al., 2011). Experimental evidence indicates that obese mice exposed to TBI have higher levels of anxiety as determined in the open field test (Sherman et al., 2016). A separate line of studies denotes a negative correlation between obesity and neurocognitive function in collegiate and professional athletes (Mathews and Wagner, 2008). For example, a study of 38 overweight and 38 normal weight retired National Football League players found that the overweight group had greater cognitive impairment and lower blood flow in the prefrontal cortex as well as hypometabolism in the temporal pole (Willeumier et al., 2012).
4.4. Dysfunctional Metabolism Fosters a Pro-Inflammatory Milieu Across Body and Brain
There is a close association between metabolic dysfunction and inflammation in the TBI pathogenesis. For example, dysglycemia is followed by an increase in circulating cytokines, opening of the blood brain barrier, neutrophils infiltration and neuronal inflammation (de Castro et al., 2017). In addition, TNF-α seems to disturb functionality of glucose metabolism by affecting levels of adipocyte-specific genes and contributing to insulin resistance and hyperglycemia (see Figure 2) (Ruan et al., 2002). The inflammatory reaction after TBI also increases the level of corticotrophin-releasing hormone (CRH) which stimulates the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary, with subsequent elevation of blood glucose. Nitric oxide (NO), which is part of the inflammatory response, participates in signal transduction pathways that lead to the release of corticosterone from the adrenal gland, and subsequent hyperglycemia. While hyperglycemia is generally related to low Glasgow Coma Scale (GCS) score (Kobata et al., 2017), the association between hyperglycemia and poor outcome in TBI is transient (Zhou et al., 2017). Management of hyperglycemia with insulin protocol seems critical for improving TBI outcome, but further studies are needed to better determine the level of serum glucose that is harmful for patients with TBI. As discussed below, consumption of caloric diets, which produce inflammation and disrupt glucose metabolism, have been shown to reduce brain plasticity and cognitive ability (see Figure 4). Acute hyperglycemia is associated with a pro-inflammatory and pro-oxidant cell environment (Dandona et al., 2004b; Kadhim et al., 2008; Zhang et al., 2005). As discussed above, hypothalamus-pituitary axis dysfunction following experimental TBI results in long-term depletion in serum GH and persistent inflammation (Kasturi and Stein, 2009). Early hepatic inflammation in rodent TBI is characterized by an increase of TLR 4, JNK, TNF α, JKK (Villapol et al., 2015c), leading to reduced hepatic insulin sensitivity and loss of GH sensitivity (see Figure 2) (Berryman et al., 2013).
Figure 4.
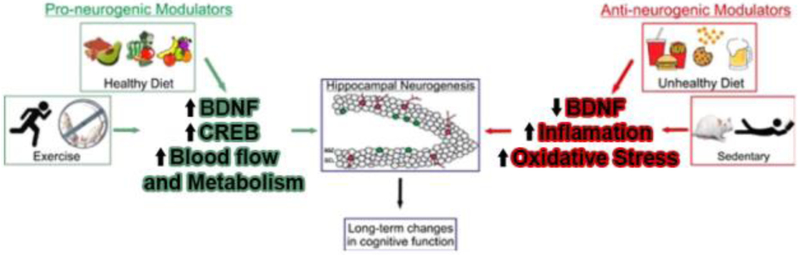
Lifestyle factors influence neuronal plasticity such as neurogenesis, and cognition. Elevated levels of inflammatory metabolic agents elicited by Unhealthy Diet and sedentary lifestyle can negatively influence hippocampal neurogenesis. Since Reactive oxygen species (ROS) play an important role in the progression of inflammatory disorders, the ROS induced by chronic {Yamin, 2008 #396}{Yamin, 2008 #396} exposure to inflammatory mediators disrupts the endothelial barrier and allows the transfer of immune cells and pro-inflammatory cytokines into the brain parenchyma that, in turn, disrupts production of BDNF and the delicate balance needed for synaptic plasticity. This has detrimental consequences for neural precursor cells (neurogenesis), as well as for the normal neuronal functioning. On the other hand, Healthy diet and Physical exercise has pro-neurogenic properties through a variety of mechanisms, particularly by increasing blood flow, cell metabolism, and synaptic plasticity. These pro-neurogenic modulators promote hippocampal plasticity and long-term changes in cognitive function.
Widespread inflammation is becoming recognized as a common component of the neuropathophysiology of obesity (Dandona et al., 2004a; Kadhim et al., 2008). In humans, levels of fibrinogen (a marker of inflammation) in the amygdala have been shown to correlate with overweight and obesity (Cazettes et al., 2011). Adipose tissue collected from obese animals and humans show larger amount of pro-inflammatory factors (Shi et al., 2016), and this suggests that obese individuals are more prone to the inflammatory consequences of brain trauma. Indeed, obese individuals have higher levels of cytokines relative to lean subjects after trauma, which emphasizes the risk posed by metabolic dysfunction on TBI outcome (Andruszkow et al., 2013; Li and Sirko, 2018). In addition, animal studies have shown that the obesity-associated release of pro-inflammatory cytokines is accompanied by activation of the immune system and a local inflammatory response (Kadhim et al., 2008).
5. Diet and Exercise: Green Technology for Managing TBI Pathophysiology
5.1. The Impact of the Epidemic of Metabolic Syndrome (MetS) on the Brain
As discussed above, a particular aspect of the TBI pathophysiology is the inability of the brain to metabolize energy (Lakshmanan et al., 2010; Vespa et al., 2005). TBI patients experience sudden abnormalities in the control of brain glucose metabolism (Kato et al., 2007), which can increase the risk of secondary brain damage (Griesdale et al., 2009; Liu-DeRyke et al., 2009). Clinical reports indicate that metabolic dysfunction is a predictor of poor outcome in TBI patients (Klose and Feldt-Rasmussen, 2018). Overload of an already disrupted brain metabolic regulation may exacerbate pathophysiology (Di Pietro et al., 2010), and may increase incidence of long-term neurological and psychiatric disorders. As discussed below, the fact that the incidence of TBI (Roozenbeek et al., 2013) and metabolic disease (Padwal, 2014) are on the rise, makes the magnitude of the problem even worse.
MetS, characterized by abdominal obesity, hypertriglyceridemia, increased blood pressure, and elevated glucose level (Grundy, 2006) is a major and escalating public-health burden worldwide (Alberti et al., 2009). Insulin resistance is the hallmark of MetS, characterized by a decrease in sensitivity to the action of insulin and associated with oxidative stress and inflammation, is largely enhanced by sugary diets and physical inactivity (Booth et al., 2002). Insulin penetrates the brain blood barrier and has an impact on various neurological events such as feeding behavior, and learning and memory. In addition, animal studies indicate that the insulin resistance observed in the periphery also occurs in brain tissue (see Figure 4) (Agrawal and Gomez-Pinilla, 2012).
5.2. Fructose Consumption Exacerbates the Pathophysiology of TBI
Fructose consumption is considered an important contributor to the epidemic of MetS (Malik and Hu, 2012). Studies in rodents have shown that a high fructose diet results in hepatic oxidative damage and altered lipid (Kelley et al., 2004) and glucose (Agrawal et al., 2016b; Agrawal and Gomez-Pinilla, 2012) metabolism. In addition, high fructose consumption is portraying as a suitable animal model to assess the influence of metabolic disorders on the brain (Agrawal and Gomez-Pinilla, 2012). Fructose consumption increases the levels of the glucose transporter 5 (GLUT5) in glial cells in the hippocampus and cortex in rats (Gomez-Pinilla and Tyagi, 2013). The fact that GLUT5 is considered a specific transporter of fructose into cells, the results of the latter indicate that fructose facilitates its own transport into the brain. The fructose-induced Mets disrupts signaling through insulin receptors which are localized to brain areas involved in cognition processing such hippocampus (Cisternas et al., 2015). Research showing that a high fructose diet disrupts insulin signaling in the brain (Agrawal and Gomez-Pinilla, 2012), suggests that insulin is part of the pathway by which fructose impacts neuronal function. Indeed, insulin activates regulators of mitochondrial biogenesis, such as the peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1α). PGC-1α is a member of a family of transcription co-activators (Ventura-Clapier et al., 2008), which in conjunction with the mitocho2ndrial transcription factor A (TFAM) (Campbell et al., 2012; Ekstrand et al., 2004), and sirtuin 1 (SIRT1) regulate cellular energy metabolism. PGC-1α can interact with brain-derived neurotrophic factor (BDNF) (Cheng et al., 2012), and reactive oxygen species in the regulation of brain plasticity (Chen et al., 2011).
It has been reported (Agrawal et al., 2016b; Agrawal et al., 2016c) that overconsumption of dietary fructose for duration sufficient to disrupt peripheral metabolism exacerbates cognitive dysfunction following TBI. These effects are concomitant to reductions in levels of proteins related to synaptic plasticity and cellular energy metabolism (Agrawal et al., 2016c). For example, fructose consumption aggravates the effects of TBI on molecular systems engaged in cell energy homeostasis (SIRT1, PGC-1α) and synaptic plasticity (BDNF, TrkB, CREB, synaptophysin) in the hippocampus (Agrawal et al., 2016c). Fructose also aggravates the effects of TBI on spatial memory in association with a decrease in hippocampal insulin receptor signaling. Additionally, fructose consumption and TBI have been shown to promote plasma membrane lipid peroxidation, based on elevated protein and phenotypic expression of 4HNE. The results of this study indicate that high fructose consumption exacerbates the pathology of brain trauma by further disrupting energy metabolism and brain plasticity (see Figure 5) (Agrawal et al., 2016c).
Figure 5.

Proposed mechanism by which metabolic disorders like the one elicited by high fructose consumption may aggravate the pathophysiology of TBI. In CNS, fructose increases levels of the fructose transporter GLUT5 suggesting that fructose consumption enhances its own transport into the brain. We propose that intracellular fructose accumulation induces the formation of acetyl-CoA and acyl-CoA. High levels of acyl-CoA can be converted to diacylglycerol (DAG) that activates the protein kinase C epsilon (PKCε), which, in turn, activates the protein c-jun-N terminal kinase-1 (JNK1). This protein leads to insulin resistance through the phosphorylation of IRS-1 on Serine307 residue (IRS-1Ser307). It is well know that the TrkB BDNF receptor and InR receptors are involved in regulation of cell metabolism and synaptic plasticity in neurons. Along this line of thought, the actions of fructose and TBI converge and inhibit pathways associated with management of cell energy metabolism like cAMP response element-binding protein (CREB) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α). Considering the interactive actions of PGC-1α and mitochondrial transcription factor A (TFAM) on mitochondrial biogenesis, it is plausible to propose that fructose and TBI decrease oxidative phosphorylation and bioenergetics. It is import to note fructosylation releases large amounts of superoxide anions, leading to disproportionate formation of reactive oxygen species in mitochondria. Moreover, the loss in energy homeostasis results in ROS, and the harmful by-product of lipid peroxidation 4-hydroxynonenal (4HNE), thereby compromising plasma membrane function. Therefore, a high fructose diet and TBI disrupt the interplay between energy metabolism and synaptic plasticity with profound consequences for brain function.
5.3. Dietary Balance Essential to Preserve Metabolic Homeostasis
Consumption of healthy dietary components is a productive strategy to counteract metabolic dysfunction and to protect mental health. In particular, docosahexaenoic acid (DHA; C22:6n-3) is one of the major n-3 polyunsaturated fatty acids which is an integral component of plasma membranes in the brain. DHA is important for brain development and plasticity, and provides support to learning and memory in animal models of Alzheimer’s disease (Hashimoto et al., 2002; Lim et al., 2005) and brain injury (Yin et al., 2018). The action of DHA has been associated to counteracting several peripheral metabolic disturbances such as diabetes (Coste et al., 2003). It has been found that fructose consumption, particularly under conditions of DHA deficiency, increases hippocampal insulin resistance, as evidenced by a decrease in the insulin receptor signaling (Agrawal and Gomez-Pinilla, 2012). Phosphorylation of insulin receptor and its signaling molecules Akt are diminished under conditions of n-3 deficiency, and these effects are aggravated by fructose consumption. These results indicate the importance of dietary DHA for maintaining proper insulin signaling in brain, and the necessity of adequate levels of n-3 in diet to cope with challenges imposed by fructose (Agrawal and Gomez-Pinilla, 2012). DHA supplementation in rodents exposed to TBI has been shown to normalize levels of BDNF and related synaptic and cell-metabolic modulators, in conjunction with improving learning ability. DHA may help to counteract the effects of TBI by providing resistance to oxidative stress and preserving plasma membrane homeostasis. In support of the neuroprotective action of diet, the supplementation of the Indian curry spice curcumin into the diet for 3 weeks before or after (Sharma et al., 2009) experimental concussive injury can lessen the consequences of the injury on synaptic plasticity markers and cognitive tasks. In addition to having profound antioxidant and anti-inflammatory effects, curcumin prevents a reduction of DHA content in the brain following brain trauma (Wu et al., 2014).
5.4. Exercise Fosters Metabolic Homeostasis in the TBI Pathology
Together with overconsumption of high caloric diets, the lack of physical activity is considered an important contributor to the MetS epidemic. The lack of physical activity contributes to 6% of the burden of coronary heart disease, 7% of T2DM, 10% of breast cancer and 10% of colon cancer around the world (Geiss et al., 2017). According to the Centre for Disease Control and Prevention (CDC) and the American College of Sports Medicine, sedentary subjects are defined as those who do not engage in at least 150 minutes of physical activities per week (Pratt et al., 2016). Sedentary lifestyle has been shown to increase the risk of neurological disorders such as stroke, dementia and depression (Booth et al., 2012). The physical inactivity-induced blood pressure, HDL cholesterol, plasma fibrinogen and platelet aggregation, which are biomarkers of T2DM and obesity, are also important risk factors for stroke (Hu et al., 2001). On the other hand, regular exercise exerts a broad range of beneficial effects, including improvements in cardio-vascular function and provides resistance to several neurological diseases (Radak et al., 2008). A large amount of evidence in humans and animals indicates that physical activity enhances cognitive abilities (Gomez-Pinilla and Hillman, 2013). Systematic reviews and meta-analyses studies have provided compelling evidence that physical activity promotes low-to-moderate risk reductions of dementia (Hamer and Chida, 2009) (Stigger et al., 2018). The fact that obesity is associated with low levels of dopamine may provide cues to understand why obese individuals have a tendency to practice physical activity less frequently (Ruegsegger and Booth, 2017). Physical inactivity also tends to correlate with hippocampal atrophy in advanced age and may lead to cognitive and memory dysfunction (Vivar and van Praag, 2017). The fact that hippocampal atrophy is one of the biomarkers for Alzheimer disease (AD) is congruent with the view that physical inactivity is considered a crucial risk factor for AD (see Figure 4) (Gomez-Pinilla and Hillman, 2013).
Physical exercise facilitates endogenous repair mechanisms in the brain and enhances functional recovery after experimental TBI (Archer et al., 2012; Griesbach, 2011). Neuronal injury results in overproduction of reactive oxygen species that compromise cell function (McKee and Lukens, 2016). Aerobic physical exercise has neuroprotective properties through a large variety of mechanisms particularly by counteracting elevated oxidative stress (da Silva Fiorin et al., 2016; Lima et al., 2009) and by increasing production of BDNF (Gomez-Pinilla and Hillman, 2013). Physical activity enhances neuronal functions and delays or prevents cell damage and neurobehavioral disability after TBI (da Silva Fiorin et al., 2016; Silva et al., 2013). The effects of physical exercise on increasing antioxidant defenses (Marques-Aleixo et al., 2012), improving mitochondrial biogenesis (Navarro and Boveris, 2009), and upregulating the metabotrophin BDNF (Gomez-Pinilla and Hillman, 2013) are a vivid demonstration of the capacity of exercise to promote metabolic homeostasis. Interestingly, experimental studies in rodents have shown that exercise works in concert with a DHA-rich diet to influence molecular systems underlying cognitive function (Chytrova et al., 2010). A possible mechanism for this complementary action of exercise is exerted by acting on molecular systems associated with control of cell metabolism, plasma membrane integrity, and synaptic plasticity, which are necessary for supporting synaptic plasticity and cognition (see Figure 4) (Wu et al., 2014)
5.5. Cell Metabolism and Neuronal Plasticity Provides Grounds for the Impact of Lifestyle on TBI Outcome
Bioenergetics abnormalities are getting recognition as a common feature of neurological disorders (Mattson et al., 2008), and a failure in mitochondrial function is as a major sequel of TBI (Singh et al., 2006). An increasing body of evidence indicates that diet-induced metabolic disease poses a threat for brain function and can increase the risk for neurological and psychiatric disorders (Farooqui et al., 2012). This comes to no surprise if we consider that energy conservation and bioenergetics are driving forces for biological adaptation and brain evolution (Gomez-Pinilla and Yang, 2018). A growing body of evidence indicates that mechanisms that regulate cell metabolism closely interact with those that regulate brain plasticity (Gomez-Pinilla and Yang, 2018). As discussed above, this association is illustrated by results showing that fructose and TBI affect the actions of key elements in the BDNF signaling cascade (Agrawal et al., 2016c). Disruption in BDNF function has been implicated in the pathophysiology of several psychiatric disorders such as depression (Levada and Troyan, 2018) and schizophrenia (Angelucci et al., 2005). Both signaling through the BDNF receptor trkB and the insulin receptor (Lee et al., 2005) have been reported to involve PI3K/Akt/mTOR pathways, which are essential for synaptic plasticity and cognition. CREB is an important step in BDNF signaling and a point of convergence of many signaling pathways regulating synaptic activity and learning and memory (Alonso et al., 2002). Through TrkB receptor, BDNF leads to the activation of CREB, which is a potent activator of PGC-1α (Herzig et al., 2001). PGC-1α is a transcription regulator deeply involved in energy metabolism and mitochondrial function (Romero et al., 2014). The interaction between CREB and PGC-1α is reflected by results showing that phosphorylation of CREB changes in proportion to levels of PGC-1α in response to TBI. Taking all together, the mechanisms underlying the actions of diet and exercise on the brain use common molecular elements of cell bioenergetics and plasticity (Gomez-Pinilla and Yang, 2018). Interestingly, the same molecular elements and systems involved with the positive actions of exercise and diet, are also part of the molecular machinery underlying cognitive functional recovery following TBI (see Figure 4) (Meng et al., 2017).
5.6. Epigenetic Alterations Prolong the Effects of TBI and Lifestyle on the Brain
An increasing line of evidence indicates that the action of lifestyle factors such as diet and exercise can be saved as epigenetic modifications with long-term consequences for neural resilience (Tyagi et al., 2015). In particular, early exposure to dietary omega-3 fatty acids appeared to be saved as changes in DNA methylation of the Bdnf gene. Methylation is one of the most stable forms of epigenetic variability. Results suggest that such epigenetic alterations may serve to create a reservoir of neuroplasticity that can protect the brain when needed, i.e., against the deleterious effects of switching to a western diet. It appears that exposure to a diet rich in omega-3 fatty acids during brain formation can help to build an “epigenetic memory” that confers resilience to metabolic perturbations occurring in adulthood. Systems biology approaches have been used in rodents to gain a thorough view of the impact of TBI on fundamental aspects of gene regulation, which have the potential to alter the course of the TBI pathogenesis (Meng et al., 2017). Studies have shown that TBI perturbs epigenomic programming, transcriptional activities (expression level and alternative splicing) events in the hippocampus which are involved in neuronal signaling, metabolism, and inflammation. Many TBI signature genes and network regulators identified in the rodent model have been causally associated with brain disorders with link to TBI such as Alzheimer’s disease, as shown by human genome-wide association studies. Fructose consumption has also been shown to impact several of the genes affected by TBI (Meng et al., 2017).
6. Adverse Consequences of Autonomic dysfunction in the TBI pathology (Table 2)
As discussed above, TBI compromises the function of body organs including liver, pancreas, and spleen with subsequent failures in metabolic and immune functions (Plesnila, 2016). The emerging panorama is that TBI initiates a pathological loop on systemic physiology that can exacerbate the TBI pathogenesis. Most TBI patients experience enhanced activity of the Sympathetic Nervous System (Meyfroidt et al., 2017) with subsequent episodes of increased heart rate and blood pressure, sweating, hyperthermia, and motor posturing. Sympathetic activation immediately post-TBI is essential for survival as early hypotension (systolic blood pressure <90 mm Hg) is associated with high mortality rate (Krishnamoorthy et al., 2017). Similarly, late hypotension is associated with 11-fold higher risk of death after severe TBI (Geeraerts et al., 2008). In addition, there is strong association between high catecholamine levels and severity of the brain injury, duration of mechanical ventilation, myocardial damage, endocrine abnormalities, length of hospital stay, and functional outcome (Rizoli et al., 2017; Woolf, 1987).
As a compensatory mechanism to restore vital homeostasis in the face of TBI, there is an early activation of the hypothalamic-pituitary axis which elevates blood levels of cortisol, glucagon and growth hormone, resulting on glycogenolysis, hypermetabolism, and excessive glucose production (Bosarge et al., 2015; Bulger et al., 2012). Sympathetic activation also results in a massive secretion of catecholamines into the periphery as part of a generalized stress response to trauma (Di Battista et al., 2016). By functioning on islet beta cells' alpha 2 receptor, the sympathetic activation can also stimulate glucagon production and decrease insulin secretion (López-Gambero et al., 2018). While sympathetic activation is an essential compensatory response to brain injury, excessive or prolonged hyperadrenergic state may have a negative impact on outcome. Excessive release of catecholamines can inhibit glucose transport via inhibition of insulin binding, leading to transient insulin resistance and glucose homeostasis impairment (Reilly and Saltiel, 2017). The secretion of glucocorticoids and catecholamines induced by excessive sympathetic activity is also linked to exacerbation of secondary brain injury and contributes to unfavorable patient outcome and mortality (Di Battista et al., 2016). Excessive catecholamines increases cytokine production and reduces GH and IGF-I production after TBI, and they are part of the metabolic changes observed in the acute phase of TBI characterized by hypermetabolism, hypercatabolism, refractory nitrogen wasting, and immunosuppression (Hatton et al., 2006). Reduced levels of GH and IGF-1 in TBI patients is associated with poor recovery (Schneider et al., 2017), and increase in neuropsychiatric manifestations (Agha et al., 2004; Kelly et al., 2006).
7. Concluding Remarks
Clinical and experimental evidence reveals the pervasive effects of TBI on systemic physiology that can strike back on the brain with subsequent effects on brain plasticity and behavior. TBI disrupts the function of the hypothalamic-pituitary axis and autonomic nervous system with profound alterations in body physiology and brain function. TBI elicits a multiorgan inflammatory response including metabolic alterations, bacteria dysbiosis and translocation, immune dysfunction, anomalous protein synthesis and hormonal status -- which in turn, exacerbate the TBI pathogenesis in the brain. The failure of the brain to perform metabolic homeostasis after TBI increases the consequences of secondary brain damage. The magnitude of the burden posed by disrupted metabolism is exacerbated by lifestyle-induced metabolic disorders such as diabetes and obesity. An increasing body of evidence indicates that diet-induced metabolic dysfunction and the lack of exercise pose a threat for brain function and can exacerbate TBI pathogenesis. Clinical reports indicate that metabolic dysfunction can increase incidence of long-term neurological and psychiatric disorders, and that metabolic dysfunction can be used as predictor of poor outcome in TBI patients. Although alterations in the functions of the autonomic nervous system and hypothalamic-pituitary axis can have devastating consequences for proper maintenance of body homeostasis, their functions are often unreported in TBI patients, and poorly studied in animal models of TBI. Therefore, it is critical to better understand the function of the autonomic nervous system on the pathophysiology of TBI in order to help prevention and treatment of TBI.
Highlights.
Brain pathology impacts the body via autonomic nervous system, endocrine and immune pathways
Peripheral metabolic dysfunction after TBI exacerbates brain pathology
Metabolic dysfunction increases incidence of long-term neurological disorders after TBI
Metabolic dysfunction is a predictor of poor outcome in TBI patients
Acknowledgements
This work was supported by The National Institute of Health award R01 NS50465 to FGP, and Conselho Nacional de Desenvolvimento Cientifico e Tecnologico award CNPq: #307382/2017-6 to LFR. Authors would like to thanks Rafael Parcianello Cipolat for reviewing the color artwork (Figures).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
8. References
- Adzovic L, Lynn AE, D'Angelo HM, Crockett AM, Kaercher RM, Royer SE, Hopp SC, Wenk GL, 2015. Insulin improves memory and reduces chronic neuroinflammation in the hippocampus of young but not aged brains. J Neuroinflammation 12, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agha A, Rogers B, Mylotte D, Taleb F, Tormey W, Phillips J, Thompson CJ, 2004. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin Endocrinol (Oxf) 60, 584–591. [DOI] [PubMed] [Google Scholar]
- Agrawal D, Raghavendran K, Schaubel DE, Mishra MC, Rajajee V, 2016a. A Propensity Score Analysis of the Impact of Invasive Intracranial Pressure Monitoring on Outcomes after Severe Traumatic Brain Injury. J Neurotrauma 33, 853–858. [DOI] [PubMed] [Google Scholar]
- Agrawal J, Kumar R, Malhi P, Dayal D, 2016b. Prevalence of psychosocial morbidity in children with type 1 diabetes mellitus: a survey from Northern India. J Pediatr Endocrinol Metab 29, 893–899. [DOI] [PubMed] [Google Scholar]
- Agrawal R, Gomez-Pinilla F, 2012. 'Metabolic syndrome' in the brain: deficiency in omega-3 fatty acid exacerbates dysfunctions in insulin receptor signalling and cognition. J Physiol 590, 2485–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal R, Noble E, Vergnes L, Ying Z, Reue K, Gomez-Pinilla F, 2016c. Dietary fructose aggravates the pathobiology of traumatic brain injury by influencing energy homeostasis and plasticity. J Cereb Blood Flow Metab 36, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimaretti G, Ambrosio MR, Benvenga S, Borretta G, De Marinis L, De Menis E, Di Somma C, Faustini-Fustini M, Grottoli S, Gasco V, Gasperi M, Logoluso F, Scaroni C, Giordano G, Ghigo E, Italian Society of, E., 2004a. Hypopituitarism and growth hormone deficiency (GHD) after traumatic brain injury (TBI). Growth Horm IGF Res 14 Suppl A, S114–117. [DOI] [PubMed] [Google Scholar]
- Aimaretti G, Ambrosio MR, Di Somma C, Fusco A, Cannavo S, Gasperi M, Scaroni C, De Marinis L, Benvenga S, degli Uberti EC, Lombardi G, Mantero F, Martino E, Giordano G, Ghigo E, 2004b. Traumatic brain injury and subarachnoid haemorrhage are conditions at high risk for hypopituitarism: screening study at 3 months after the brain injury. Clin Endocrinol (Oxf) 61, 320–326. [DOI] [PubMed] [Google Scholar]
- Ajmo CT Jr., Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR, 2008. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res 86, 2227–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sadi R, Ye D, Boivin M, Guo S, Hashimi M, Ereifej L, Ma TY, 2014. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS One 9, e85345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alali AS, Gomez D, McCredie V, Mainprize TG, Nathens AB, 2017. Understanding Hospital Volume-Outcome Relationship in Severe Traumatic Brain Injury. Neurosurgery 80, 534–542. [DOI] [PubMed] [Google Scholar]
- Alavi SA, Tan CL, Menon DK, Simpson HL, Hutchinson PJ, 2016. Incidence of pituitary dysfunction following traumatic brain injury: A prospective study from a regional neurosurgical centre. Br J Neurosurg 30, 302–306. [DOI] [PubMed] [Google Scholar]
- Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC Jr., International Diabetes Federation Task Force on, E., Prevention, Hational Heart, L., Blood, I., American Heart, A., World Heart, F., International Atherosclerosis, S., International Association for the Study of, O., 2009. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120, 1640–1645. [DOI] [PubMed] [Google Scholar]
- Alonso M, Vianna MR, Izquierdo I, Medina JH, 2002. Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. Cell Mol Neurobiol 22, 663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GD, Peterson TC, Vonder Haar C, Farin FM, Bammler TK, MacDonald JW, Kantor ED, Hoane MR, 2015. Effect of Traumatic Brain Injury, Erythropoietin, and Anakinra on Hepatic Metabolizing Enzymes and Transporters in an Experimental Rat Model. AAPS J 17, 1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews AM, Lutton EM, Merkel SF, Razmpour R, Ramirez SH, 2016. Mechanical Injury Induces Brain Endothelial-Derived Microvesicle Release: Implications for Cerebral Vascular Injury during Traumatic Brain Injury. Front Cell Neurosci 10, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruszkow H, Veh J, Mommsen P, Zeckey C, Hildebrand F, Frink M, 2013. Impact of the body mass on complications and outcome in multiple trauma patients: what does the weight weigh? Mediators Inflamm 2013, 345702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F, Brene S, Mathe AA, 2005. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry 10, 345–352. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Couch Y, Losey P, Evans MC, 2012. The systemic response to brain injury and disease. Brain Behav Immun 26, 534–540. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Pitossi FJ, 2013. Special issue commentary: the changing face of inflammation in the brain. Mol Cell Neurosci 53, 1–5. [DOI] [PubMed] [Google Scholar]
- Aperghis M, Johnson IP, Cannon J, Yang SY, Goldspink G, 2004. Different levels of neuroprotection by two insulin-like growth factor-I splice variants. Brain Res 1009, 213–218. [DOI] [PubMed] [Google Scholar]
- Archer T, Svensson K, Alricsson M, 2012. Physical exercise ameliorates deficits induced by traumatic brain injury. Acta Neurol Scand 125, 293–302. [DOI] [PubMed] [Google Scholar]
- Asehnoune K, Balogh Z, Citerio G, Cap A, Billiar T, Stocchetti N, Cohen MJ, Pelosi P, Curry N, Gaarder C, Gruen R, Holcomb J, Hunt BJ, Juffermans NP, Maegele M, Midwinter M, Moore FA, O'Dwyer M, Pittet JF, Schochl H, Schreiber M, Spinella PC, Stanworth S, Winfield R, Brohi K, 2017. The research agenda for trauma critical care. Intensive Care Med 43, 1340–1351. [DOI] [PubMed] [Google Scholar]
- Ayton S, Zhang M, Roberts BR, Lam LQ, Lind M, McLean C, Bush AI, Frugier T, Crack PJ, Duce JA, 2014. Ceruloplasmin and beta-amyloid precursor protein confer neuroprotection in traumatic brain injury and lower neuronal iron. Free Radic Biol Med 69, 331–337. [DOI] [PubMed] [Google Scholar]
- Bansal D, Ave P, Kerneis S, Frileux P, Boche O, Baglin AC, Dubost G, Leguern AS, Prevost MC, Bracha R, Mirelman D, Guillen N, Labruyere E, 2009a. An ex-vivo human intestinal model to study Entamoeba histolytica pathogenesis. PLoS Negl Trop Dis 3, e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V, Costantini T, Kroll L, Peterson C, Loomis W, Eliceiri B, Baird A, Wolf P, Coimbra R, 2009b. Traumatic brain injury and intestinal dysfunction: uncovering the neuro-enteric axis. J Neurotrauma 26, 1353–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V, Ryu SY, Blow C, Costantini T, Loomis W, Eliceiri B, Baird A, Wolf P, Coimbra R, 2010. The hormone ghrelin prevents traumatic brain injury induced intestinal dysfunction. J Neurotrauma 27, 2255–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke A, 2011a. Growth hormone, insulin and aging: the benefits of endocrine defects. Exp Gerontol 46, 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke A, 2011b. Pleiotropic effects of growth hormone signaling in aging. Trends Endocrinol Metab 22, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman JM, McNeill H, 2006. Insulin/IGF signalling in neurogenesis. Cell Mol Life Sci 63, 1701–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedinger DH, Adams SH, 2015. Metabolic, anabolic, and mitogenic insulin responses: A tissue-specific perspective for insulin receptor activators. Mol Cell Endocrinol 415, 143–156. [DOI] [PubMed] [Google Scholar]
- Behan LA, Phillips J, Thompson CJ, Agha A, 2008. Neuroendocrine disorders after traumatic brain injury. J Neurol Neurosurg Psychiatry 79, 753–759. [DOI] [PubMed] [Google Scholar]
- Berryman DE, Glad CA, List EO, Johannsson G, 2013. The GH/IGF-1 axis in obesity: pathophysiology and therapeutic considerations. Nat Rev Endocrinol 9, 346–356. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, Ferstl R, von Eynatten M, Wendt T, Rudofsky G, 2003. A mechanism converting psychosocial stress into mononuclear cell activation. Proceedings of the National Academy of Sciences 100, 1920–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan Y, Tornetta P 3rd, Einhorn TA, Guy P, Leveille L, Robinson J, Bosse MJ, Haines N, Horwitz D, Jones C, Schemitsch E, Sagi C, Thomas B, Stahl D, Ricci W, Brady M, Sanders D, Kain M, Higgins TF, Collinge C, Kottmeier S, Friess D, 2016. Healing Time and Complications in Operatively Treated Atypical Femur Fractures Associated With Bisphosphonate Use: A Multicenter Retrospective Cohort. J Orthop Trauma 30, 177–181. [DOI] [PubMed] [Google Scholar]
- Booth FW, Chakravarthy MV, Gordon SE, Spangenburg EE, 2002. Waging war on physical inactivity: using modern molecular ammunition against an ancient enemy. J Appl Physiol (1985) 93, 3–30. [DOI] [PubMed] [Google Scholar]
- Booth FW, Roberts CK, Laye MJ, 2012. Lack of exercise is a major cause of chronic diseases. Compr Physiol 2, 1143–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosarge PL, Shoultz TH, Griffin RL, Kerby JD, 2015. Stress-induced hyperglycemia is associated with higher mortality in severe traumatic brain injury. J Trauma Acute Care Surg 79, 289–294. [DOI] [PubMed] [Google Scholar]
- Boulange CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME, 2016. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger EM, Guffey D, Guyette FX, MacDonald RD, Brasel K, Kerby JD, Minei JP, Warden C, Rizoli S, Morrison LJ, Nichol G, Resuscitation Outcomes Consortium I, 2012. Impact of prehospital mode of transport after severe injury: a multicenter evaluation from the Resuscitation Outcomes Consortium. J Trauma Acute Care Surg 72, 567–573; discussion 573-565; quiz 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CT, Kolesar JE, Kaufman BA, 2012. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim Biophys Acta 1819, 921–929. [DOI] [PubMed] [Google Scholar]
- Campolo M, Ahmad A, Crupi R, Impellizzeri D, Morabito R, Esposito E, Cuzzocrea S, 2013. Combination therapy with melatonin and dexamethasone in a mouse model of traumatic brain injury. J Endocrinol 217, 291–301. [DOI] [PubMed] [Google Scholar]
- Capatina C, Paluzzi A, Mitchell R, Karavitaki N, 2015. Diabetes Insipidus after Traumatic Brain Injury. J Clin Med 4, 1448–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazettes F, Cohen JI, Yau PL, Talbot H, Convit A, 2011. Obesity-mediated inflammation may damage the brain circuit that regulates food intake. Brain Res 1373, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-D, Yang D-I, Lin T-K, Shaw F-Z, Liou C-W, Chuang Y-C, 2011. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia. International journal of molecular sciences 12, 7199–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Pan Z, Fang Z, Lin W, Wu S, Yang F, Li Y, Fu H, Gao H, Li S, 2018. Omega-3 polyunsaturated fatty acid attenuates traumatic brain injury-induced neuronal apoptosis by inducing autophagy through the upregulation of SIRT1-mediated deacetylation of Beclin-1. J Neuroinflammation 15, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, Mattson MP, 2012. Involvement of PGC-1alpha in the formation and maintenance of neuronal dendritic spines. Nat Commun 3, 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu W, Li M, Li F, Hu R, Chen Z, Lin J, Feng H, 2013. Immediate splenectomy down-regulates the MAPK-NF-kappaB signaling pathway in rat brain after severe traumatic brain injury. J Trauma Acute Care Surg 74, 1446–1453. [DOI] [PubMed] [Google Scholar]
- Chytrova G, Ying Z, Gomez-Pinilla F, 2010. Exercise contributes to the effects of DHA dietary supplementation by acting on membrane-related synaptic systems. Brain Res 1341, 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisternas P, Salazar P, Serrano FG, Montecinos-Oliva C, Arredondo SB, Varela-Nallar L, Barja S, Vio CP, Gomez-Pinilla F, Inestrosa NC, 2015. Fructose consumption reduces hippocampal synaptic plasticity underlying cognitive performance. Biochim Biophys Acta 1852, 2379–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cittadini A, Saldamarco L, Marra AM, Arcopinto M, Carlomagno G, Imbriaco M, Del Forno D, Vigorito C, Merola B, Oliviero U, Fazio S, Sacca L, 2009. Growth hormone deficiency in patients with chronic heart failure and beneficial effects of its correction. J Clin Endocrinol Metab 94, 3329–3336. [DOI] [PubMed] [Google Scholar]
- Cohan P, Wang C, McArthur DL, Cook SW, Dusick JR, Armin B, Swerdloff R, Vespa P, Muizelaar JP, Cryer HG, Christenson PD, Kelly DF, 2005. Acute secondary adrenal insufficiency after traumatic brain injury: a prospective study. Crit Care Med 33, 2358–2366. [DOI] [PubMed] [Google Scholar]
- Coste TC, Gerbi A, Vague P, Pieroni G, Raccah D, 2003. Neuroprotective effect of docosahexaenoic acid-enriched phospholipids in experimental diabetic neuropathy. Diabetes 52, 2578–2585. [DOI] [PubMed] [Google Scholar]
- Cree MG, Paddon-Jones D, Newcomer BR, Ronsen O, Aarsland A, Wolfe RR, Ferrando A, 2010. Twenty-eight-day bed rest with hypercortisolemia induces peripheral insulin resistance and increases intramuscular triglycerides. Metabolism 59, 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuisinier A, Maufrais C, Payen JF, Nottin S, Walther G, Bouzat P, 2016. Myocardial function at the early phase of traumatic brain injury: a prospective controlled study. Scand J Trauma Resusc Emerg Med 24, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czorlich P, Dreimann M, Emami P, Westphal M, Lefering R, Hoffmann M, 2017. Body Mass Index >35 as Independent Predictor of Mortality in Severe Traumatic Brain Injury. World Neurosurg 107, 515–521. [DOI] [PubMed] [Google Scholar]
- da Silva Fiorin F, de Oliveira Ferreira AP, Ribeiro LR, Silva LF, de Castro MR, da Silva LR, da Silveira ME Jr., Zemolin AP, Dobrachinski F, Marchesan de Oliveira S, Franco JL, Soares FA, Furian AF, Oliveira MS, Fighera MR, Freire Royes LF, 2016. The Impact of Previous Physical Training on Redox Signaling after Traumatic Brain Injury in Rats: A Behavioral and Neurochemical Approach. J Neurotrauma 33, 1317–1330. [DOI] [PubMed] [Google Scholar]
- Dandona P, Aljada A, Bandyopadhyay A, 2004a. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 25, 4–7. [DOI] [PubMed] [Google Scholar]
- Dandona P, Chaudhuri A, Aljada A, 2004b. Endothelial dysfunction and hypertension in diabetes mellitus. Med Clin North Am 88, 911–931, x-xi. [DOI] [PubMed] [Google Scholar]
- Das M, Leonardo CC, Rangooni S, Pennypacker KR, Mohapatra S, Mohapatra SS, 2011. Lateral fluid percussion injury of the brain induces CCL20 inflammatory chemokine expression in rats. J Neuroinflammation 8, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De-Souza DA, Greene LJ, 2005. Intestinal permeability and systemic infections in critically ill patients: effect of glutamine. Crit Care Med 33, 1125–1135. [DOI] [PubMed] [Google Scholar]
- de Castro MRT, Ferreira A.P.d.O., Busanello GL, da Silva LRH, da Silveira MEP Junior, Fiorin F.d.S., Arrifano G, Crespo-López ME, Barcelos RP, Cuevas MJ, 2017. Previous physical exercise alters the hepatic profile of oxidative-inflammatory status and limits the secondary brain damage induced by severe traumatic brain injury in rats. The Journal of physiology 595, 6023–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deepika A, Devi BI, Shukla D, Sathyaprabha TN, Christopher R, Ramesh SS, 2018. Neuroimmunology of Traumatic Brain Injury: A Longitudinal Study of Interdependency of Inflammatory Markers and Heart Rate Variability in Severe Traumatic Brain Injury. J Neurotrauma 35, 1124–1131. [DOI] [PubMed] [Google Scholar]
- Desborough JP, 2000. The stress response to trauma and surgery. Br J Anaesth 85, 109–117. [DOI] [PubMed] [Google Scholar]
- Di Battista AP, Rizoli SB, Lejnieks B, Min A, Shiu MY, Peng HT, Baker AJ, Hutchison MG, Churchill N, Inaba K, 2016. Sympathoadrenal activation is associated with acute traumatic coagulopathy and endotheliopathy in isolated brain injury. Shock (Augusta, Ga.) 46, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pietro V, Amin D, Pernagallo S, Lazzarino G, Tavazzi B, Vagnozzi R, Pringle A, Belli A, 2010. Transcriptomics of traumatic brain injury: gene expression and molecular pathways of different grades of insult in a rat organotypic hippocampal culture model. J Neurotrauma 27, 349–359. [DOI] [PubMed] [Google Scholar]
- Ditillo M, Pandit V, Rhee P, Aziz H, Hadeed S, Bhattacharya B, Friese RS, Davis K, Joseph B, 2014. Morbid obesity predisposes trauma patients to worse outcomes: a National Trauma Data Bank analysis. J Trauma Acute Care Surg 76, 176–179. [DOI] [PubMed] [Google Scholar]
- Eakin K, Li Y, Chiang YH, Hoffer BJ, Rosenheim H, Greig NH, Miller JP, 2013. Exendin-4 ameliorates traumatic brain injury-induced cognitive impairment in rats. PLoS One 8, e82016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG, 2004. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 13, 935–944. [DOI] [PubMed] [Google Scholar]
- Eric Nyam TT, Ho CH, Chio CC, Lim SW, Wang JJ, Chang CH, Kuo JR, Wang CC, 2019. Traumatic Brain Injury Increases the Risk of Major Adverse Cardiovascular and Cerebrovascular Events: A 13-Year, Population-Based Study. World Neurosurg 122, e740–e753. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Farooqui T, Panza F, Frisardi V, 2012. Metabolic syndrome as a risk factor for neurological disorders. Cell Mol Life Sci 69, 741–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney C, Sharp DJ, Hellyer PJ, Jolly AE, Cole JH, Scott G, Baxter D, Jilka S, Ross E, Ham TE, Jenkins PO, Li LM, Gorgoraptis N, Midwinter M, Goldstone AP, 2017. Serum insulin-like growth factor-I levels are associated with improved white matter recovery after traumatic brain injury. Ann Neurol 82, 30–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman SF, Lapidus N, Dorival C, Diallo A, Amri I, Fontaine H, Pol S, Carrat F, group, A.A.H.s., 2018. Signal detection on a patient cohort: A disproportionality analysis of the ANRS CO22 HEPATHER cohort to identify associations between direct acting antivirals and adverse events in patients with hepatitis C virus chronic infection. Pharmacoepidemiol Drug Saf 27, 797–805. [DOI] [PubMed] [Google Scholar]
- Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP, 2014. Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry 76, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Rodriguez E, Bernabeu I, Castro AI, Kelestimur F, Casanueva FF, 2011. Hypopituitarism following traumatic brain injury: determining factors for diagnosis. Front Endocrinol (Lausanne) 2, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AM, Torres-Aleman I, 2012. The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci 13, 225–239. [DOI] [PubMed] [Google Scholar]
- Friedrich N, Thuesen B, Jorgensen T, Juul A, Spielhagen C, Wallaschofksi H, Linneberg A, 2012. The association between IGF-I and insulin resistance: a general population study in Danish adults. Diabetes Care 35, 768–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrahy A, Sherlock M, Thompson CJ, 2017. MANAGEMENT OF ENDOCRINE DISEASE: Neuroendocrine surveillance and management of neurosurgical patients. Eur J Endocrinol 176, R217–R233. [DOI] [PubMed] [Google Scholar]
- Geeraerts T, Friggeri A, Mazoit JX, Benhamou D, Duranteau J, Vigue B, 2008. Posttraumatic brain vulnerability to hypoxia-hypotension: the importance of the delay between brain trauma and secondary insult. Intensive Care Med 34, 551–560. [DOI] [PubMed] [Google Scholar]
- Geiss LS, Kirtland K, Lin J, Shrestha S, Thompson T, Albright A, Gregg EW, 2017. Changes in diagnosed diabetes, obesity, and physical inactivity prevalence in US counties, 2004-2012. PLoS One 12, e0173428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharib M, Kaul S, LoCurto J, Perez M, Hajri T, 2015. The obesity factor in critical illness: Between consensus and controversy. J Trauma Acute Care Surg 78, 866–873. [DOI] [PubMed] [Google Scholar]
- Giuliano S, Talarico S, Bruno L, Nicoletti FB, Ceccotti C, Belfiore A, 2017. Growth hormone deficiency and hypopituitarism in adults after complicated mild traumatic brain injury. Endocrine 58, 115–123. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Tyagi E, 2013. Diet and cognition: interplay between cell metabolism and neuronal plasticity. Current opinion in clinical nutrition and metabolic care 16, 726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Yang X, 2018. SYSTEM BIOLOGY APPROACH INTERSECTING DIET AND CELL METABOLISM WITH PATHOGENESIS OF BRAIN DISORDERS. Progress in neurobiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Hillman C, 2013. The influence of exercise on cognitive abilities. Comprehensive Physiology 3, 403–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez S, Sathyapalan T, Javed Z, Atkin SL, 2018. Effects of Growth Hormone Replacement on Peripheral Muscle and Exercise Capacity in Severe Growth Hormone Deficiency. Front Endocrinol (Lausanne) 9, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralle M, 2017. The neuronal insulin receptor in its environment. J Neurochem 140, 359–367. [DOI] [PubMed] [Google Scholar]
- Greening DW, Simpson RJ, 2018. Understanding extracellular vesicle diversity - current status. Expert Rev Proteomics 15, 887–910. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, 2011. Exercise after traumatic brain injury: is it a double-edged sword? PM R 3, S64–72. [DOI] [PubMed] [Google Scholar]
- Griesdale DE, Tremblay MH, McEwen J, Chittock DR, 2009. Glucose control and mortality in patients with severe traumatic brain injury. Neurocrit Care 11, 311–316. [DOI] [PubMed] [Google Scholar]
- Grundy SM, 2006. Atherogenic dyslipidemia associated with metabolic syndrome and insulin resistance. Clin Cornerstone 8 Suppl 1, S21–27. [DOI] [PubMed] [Google Scholar]
- Hadjizacharia P, Beale EO, Inaba K, Chan LS, Demetriades D, 2008. Acute diabetes insipidus in severe head injury: a prospective study. Journal of the American College of Surgeons 207, 477–484. [DOI] [PubMed] [Google Scholar]
- Hamer M, Chida Y, 2009. Physical activity and risk of neurodegenerative disease: a systematic review of prospective evidence. Psychol Med 39, 3–11. [DOI] [PubMed] [Google Scholar]
- Hang CH, Shi JX, Li JS, Wu W, Yin HX, 2003. Alterations of intestinal mucosa structure and barrier function following traumatic brain injury in rats. World J Gastroenterol 9, 2776–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasen M, Almojuela A, Zeiler FA, 2019. Autonomic Dysfunction and Associations with Functional and Neurophysiological Outcome in Moderate/Severe Traumatic Brain Injury: A Scoping Review. J Neurotrauma. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Hossain S, Shimada T, Sugioka K, Yamasaki H, Fujii Y, Ishibashi Y, Oka J, Shido O, 2002. Docosahexaenoic acid provides protection from impairment of learning ability in Alzheimer's disease model rats. J Neurochem 81, 1084–1091. [DOI] [PubMed] [Google Scholar]
- Hatton J, Kryscio R, Ryan M, Ott L, Young B, 2006. Systemic metabolic effects of combined insulin-like growth factor–I and growth hormone therapy in patients who have sustained acute traumatic brain injury. Journal of neurosurgery 105, 843–852. [DOI] [PubMed] [Google Scholar]
- Hendrick LE, Schroeppel TJ, Sharpe JP, Alsbrook D, Magnotti LJ, Weinberg JA, Johnson BP, Lewis RH, Clement LP, Croce MA, Fabian TC, 2016. Impact of Beta-Blockers on Nonhead Injured Trauma Patients. Am Surg 82, 575–579. [PubMed] [Google Scholar]
- Hernandez G, Hasbun P, Velasco N, Wainstein C, Bugedo G, Bruhn A, Klaassen J, Castillo L, 2007. Splanchnic ischemia and gut permeability after acute brain injury secondary to intracranial hemorrhage. Neurocrit Care 7, 40–44. [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M, 2001. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183. [DOI] [PubMed] [Google Scholar]
- Hinson HE, Schreiber MA, Laurie AL, Baguley IJ, Bourdette D, Ling GS, 2017. Early fever as a predictor of paroxysmal sympathetic hyperactivity in traumatic brain injury. Journal of Head Trauma Rehabilitation 32, E50–E54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DM, O'Sullivan AJ, Baxter RC, Ho KK, 1994. Diagnosis of growth-hormone deficiency in adults. Lancet 343, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Hu G, Pekkarinen H, Hanninen O, Tian H, Guo Z, 2001. Relation between commuting, leisure time physical activity and serum lipids in a Chinese urban population. Ann Hum Biol 28, 412–421. [DOI] [PubMed] [Google Scholar]
- Hu YC, Wang F, Zhang DD, Sun Q, Li W, Dai YX, Zhou ML, Hang CH, 2013. Expression of intestinal CD40 after experimental traumatic brain injury in rats. J Surg Res 184, 1022–1027. [DOI] [PubMed] [Google Scholar]
- Izzo G, Tirelli A, Angrisani E, Cannaviello G, Cannaviello L, Puzziello A, Vatrella A, Vitale M, 2016. Pituitary dysfunction and its association with quality of life in traumatic brain injury. Int J. Surg Suppl 1:S103–8. [DOI] [PubMed] [Google Scholar]
- Jin P, Zhu L, Zhang J, Xie S, Pan D, Wen H, Meng W, Lin L, Chen D, 2014. A correlation study of the expression of resistin and glycometabolism in muscle tissue after traumatic brain injury in rats. Chin J Traumatol 17, 125–129. [PubMed] [Google Scholar]
- Jin W, Ni H, Dai Y, Wang H, Lu T, Wu J, Jiang J, Liang W, 2010. Effects of tert-butylhydroquinone on intestinal inflammatory response and apoptosis following traumatic brain injury in mice. Mediators Inflamm 2010, 502564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadhim HJ, Duchateau J, Sebire G, 2008. Cytokines and brain injury: invited review. J Intensive Care Med 23, 236–249. [DOI] [PubMed] [Google Scholar]
- Karamouzis I, Pagano L, Prodam F, Mele C, Zavattaro M, Busti A, Marzullo P, Aimaretti G, 2016. Clinical and diagnostic approach to patients with hypopituitarism due to traumatic brain injury (TBI), subarachnoid hemorrhage (SAH), and ischemic stroke (IS). Endocrine 52, 441–450. [DOI] [PubMed] [Google Scholar]
- Kasturi BS, Stein DG, 2009. Traumatic brain injury causes long-term reduction in serum growth hormone and persistent astrocytosis in the cortico-hypothalamo-pituitary axis of adult male rats. J Neurotrauma 26, 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Nakayama N, Yasokawa Y, Okumura A, Shinoda J, Iwama T, 2007. Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J Neurotrauma 24, 919–926. [DOI] [PubMed] [Google Scholar]
- Katzenberger RJ, Ganetzky B, Wassarman DA, 2015. The gut reaction to traumatic brain injury. Fly (Austin) 9, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley GL, Allan G, Azhar S, 2004. High dietary fructose induces a hepatic stress response resulting in cholesterol and lipid dysregulation. Endocrinology 145, 548–555. [DOI] [PubMed] [Google Scholar]
- Kelly DF, McArthur DL, Levin H, Swimmer S, Dusick JR, Cohan P, Wang C, Swerdloff R, 2006. Neurobehavioral and quality of life changes associated with growth hormone insufficiency after complicated mild, moderate, or severe traumatic brain injury. J Neurotrauma 23, 928–942. [DOI] [PubMed] [Google Scholar]
- Keshavarzi Z, Khaksari M, Shahrokhi N, 2014. The effects of cyclooxygenase inhibitors on the gastric emptying and small intestine transit in the male rats following traumatic brain injury. Iran J Basic Med Sci 17, 406–410. [PMC free article] [PubMed] [Google Scholar]
- Kim DK, Nishida H, An SY, Shetty AK, Bartosh TJ, Prockop DJ, 2016. Chromatographically isolated CD63+CD81+ extracellular vesicles from mesenchymal stromal cells rescue cognitive impairments after TBI. Proc Natl Acad Sci U S A 113, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GS, Jung JE, Narasimhan P, Sakata H, Chan PH, 2012. Induction of thioredoxin-interacting protein is mediated by oxidative stress, calcium, and glucose after brain injury in mice. Neurobiology of disease 46, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleindienst A, Brabant G, Bock C, Maser-Gluth C, Buchfelder M, 2009. Neuroendocrine function following traumatic brain injury and subsequent intensive care treatment: a prospective longitudinal evaluation. J Neurotrauma 26, 1435–1446. [DOI] [PubMed] [Google Scholar]
- Klose M, Feldt-Rasmussen U, 2018. Chronic endocrine consequences of traumatic brain injury - what is the evidence? Nat Rev Endocrinol 14, 57–62. [DOI] [PubMed] [Google Scholar]
- Ko A, Harada MY, Barmparas G, Thomsen GM, Alban RF, Bloom MB, Chung R, Melo N, Margulies DR, Ley EJ, 2016. Early propranolol after traumatic brain injury is associated with lower mortality. J Trauma Acute Care Surg 80, 637–642. [DOI] [PubMed] [Google Scholar]
- Kobata H, Sugie A, Suehiro E, Dohi K, Kaneko T, Fujita M, Oda Y, Kuroda Y, Yamashita S, Maekawa T, 2017. Association between Blood Glucose Levels the Day after Targeted Temperature Initiation and Outcome in Traumatic Brain Injury: A Post-Hoc Analysis of the B-HYPO Study. J Neurotrauma 34, 987–995. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy V, Rowhani-Rahbar A, Chaikittisilpa N, Gibbons EF, Rivara FP, Temkin NR, Quistberg A, Vavilala MS, 2017. Association of Early Hemodynamic Profile and the Development of Systolic Dysfunction Following Traumatic Brain Injury. Neurocrit Care 26, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan R, Loo JA, Drake T, Leblanc J, Ytterberg AJ, McArthur DL, Etchepare M, Vespa PM, 2010. Metabolic crisis after traumatic brain injury is associated with a novel microdialysis proteome. Neurocrit Care 12, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambillotte C, Gilon P, Henquin JC, 1997. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest 99, 414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang Y, Fu F, Sun D, Xi C, Chen F, 2015. Labetalol prevents intestinal dysfunction induced by traumatic brain injury. PLoS one 10, e0133215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson BE, Stockwell DW, Boas S, Andrews T, Wellman GC, Lockette W, Freeman K, 2012. Cardiac reactive oxygen species after traumatic brain injury. J Surg Res 173, e73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Huang CC, Wu MY, Hsu KS, 2005. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem 280, 18543–18550. [DOI] [PubMed] [Google Scholar]
- Levada OA, Troyan AS, 2018. Poststroke Depression Biomarkers: A Narrative Review. Front Neurol 9, 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley EJ, Srour MK, Clond MA, Barnajian M, Tillou A, Mirocha J, Salim A, 2011. Diabetic patients with traumatic brain injury: insulin deficiency is associated with increased mortality. J Trauma 70, 1141–1144. [DOI] [PubMed] [Google Scholar]
- Ley RE, 2010. Obesity and the human microbiome. Curr Opin Gastroenterol 26, 5–11. [DOI] [PubMed] [Google Scholar]
- Li M, Sirko S, 2018. Traumatic Brain Injury: At the Crossroads of Neuropathology and Common Metabolic Endocrinopathies. J Clin Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Calon F, Morihara T, Yang F, Teter B, Ubeda O, Salem N Jr., Frautschy SA, Cole GM, 2005. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci 25, 3032–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima FD, Oliveira MS, Furian AF, Souza MA, Rambo LM, Ribeiro LR, Silva LF, Retamoso LT, Hoffmann MS, Magni DV, Pereira L, Fighera MR, Mello CF, Royes LF, 2009. Adaptation to oxidative challenge induced by chronic physical exercise prevents Na+,K+- ATPase activity inhibition after traumatic brain injury. Brain Res 1279, 147–155. [DOI] [PubMed] [Google Scholar]
- Lima MH, Ueno M, Thirone AC, Rocha EM, Carvalho CR, Saad MJ, 2002. Regulation of IRS-1/SHP2 interaction and AKT phosphorylation in animal models of insulin resistance. Endocrine 18, 1–12. [DOI] [PubMed] [Google Scholar]
- Liu-DeRyke X, Collingridge DS, Orme J, Roller D, Zurasky J, Rhoney DH, 2009. Clinical impact of early hyperglycemia during acute phase of traumatic brain injury. Neurocrit Care 11, 151–157. [DOI] [PubMed] [Google Scholar]
- López-Gambero A, Martínez F, Salazar K, Cifuentes M, Nualart F, 2018. Brain Glucose-Sensing Mechanism and Energy Homeostasis. Molecular Neurobiology, 1–28. [DOI] [PubMed] [Google Scholar]
- Lu K, Liang CL, Li PC, Liliang PC, Huang CY, Lee YC, Wang KW, Yang SN, Sun YT, Wang HK, 2017. Risk factors for myocardial dysfunction after traumatic brain injury: A one-year follow-up study. Injury 48, 1794–1800. [DOI] [PubMed] [Google Scholar]
- Lu YC, Liu S, Gong QZ, Hamm RJ, Lyeth BG, 1997. Inhibition of nitric oxide synthase potentiates hypertension and increases mortality in traumatically brain-injured rats. Mol Chem Neuropathol 30, 125–137. [DOI] [PubMed] [Google Scholar]
- Ma EL, Smith AD, Desai N, Cheung L, Hanscom M, Stoica BA, Loane DJ, Shea-Donohue T, Faden AI, 2017a. Bidirectional brain-gut interactions and chronic pathological changes after traumatic brain injury in mice. Brain Behav Immun 66, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Wang J, Cheng J, Xiao W, Fan K, Gu J, Yu B, Yin G, Wu J, Ren J, Hou J, Jiang Y, Tan Y, Jin W, 2017b. Impacts of Blast-Induced Traumatic Brain Injury on Expressions of Hepatic Cytochrome P450 1A2, 2B1, 2D1, and 3A2 in Rats. Cell Mol Neurobiol 37, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdan M, Brazinova A, Wilbacher I, Rusnak M, Mauritz W, 2015. The impact of body mass index on severity, patterns and outcomes after traumatic brain injuries caused by low level falls. Eur J Trauma Emerg Surg 41, 651–656. [DOI] [PubMed] [Google Scholar]
- Majdan M, Plancikova D, Brazinova A, Rusnak M, Nieboer D, Feigin V, Maas A, 2016. Epidemiology of traumatic brain injuries in Europe: a cross-sectional analysis. Lancet Public Health 1, e76–e83. [DOI] [PubMed] [Google Scholar]
- Malik VS, Hu FB, 2012. Sweeteners and Risk of Obesity and Type 2 Diabetes: The Role of Sugar-Sweetened Beverages. Curr Diab Rep. [DOI] [PubMed] [Google Scholar]
- Mansoor O, Beaufrere B, Boirie Y, Ralliere C, Taillandier D, Aurousseau E, Schoeffler P, Arnal M, Attaix D, 1996. Increased mRNA levels for components of the lysosomal, Ca2+-activated, and ATP-ubiquitin-dependent proteolytic pathways in skeletal muscle from head trauma patients. Proc Natl Acad Sci U S A 93, 2714–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Aleixo I, Oliveira PJ, Moreira PI, Magalhaes J, Ascensao A, 2012. Physical exercise as a possible strategy for brain protection: evidence from mitochondrial-mediated mechanisms. Prog Neurobiol 99, 149–162. [DOI] [PubMed] [Google Scholar]
- Mathews EM, Wagner DR, 2008. Prevalence of overweight and obesity in collegiate American football players, by position. J Am Coll Health 57, 33–38. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A, 2008. Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavalli MD, DiGirolamo DJ, Fan Y, Riddle RC, Campbell KS, van Groen T, Frank SJ, Sperling MA, Esser KA, Bamman MM, Clemens TL, 2010. Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J Clin Invest 120, 4007–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer LS, Bay RC, Politis A, Steinberg M, Steele C, Baker AS, Rabins PV, Lyketsos CG, 2006. Comparison of three rating scales as outcome measures for treatment trials of depression in Alzheimer disease: findings from DIADS. Int J Geriatr Psychiatry 21, 930–936. [DOI] [PubMed] [Google Scholar]
- McHugh GS, Engel DC, Butcher I, Steyerberg EW, Lu J, Mushkudiani N, Hernandez AV, Marmarou A, Maas AI, Murray GD, 2007. Prognostic value of secondary insults in traumatic brain injury: results from the IMPACT study. J Neurotrauma 24, 287–293. [DOI] [PubMed] [Google Scholar]
- McKee CA, Lukens JR, 2016. Emerging Roles for the Immune System in Traumatic Brain Injury. Front Immunol 7, 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Zhuang Y, Ying Z, Agrawal R, Yang X, Gomez-Pinilla F, 2017. Traumatic brain injury induces genome-wide transcriptomic, Methylomic, and network perturbations in brain and blood predicting neurological disorders. EBioMedicine 16, 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyfroidt G, Baguley IJ, Menon DK, 2017. Paroxysmal sympathetic hyperactivity: the storm after acute brain injury. Lancet Neurol 16, 721–729. [DOI] [PubMed] [Google Scholar]
- Moinard C, Gupta S, Besson V, Morio B, Marchand-Leroux C, Chaumeil JC, Cynober L, Charrueau C, 2008. Evidence for impairment of hepatic energy homeostasis in head-injured rat. J Neurotrauma 25, 124–129. [DOI] [PubMed] [Google Scholar]
- Moinard C, Neveux N, Royo N, Genthon C, Marchand-Verrecchia C, Plotkine M, Cynober L, 2005. Characterization of the alteration of nutritional state in brain injury induced by fluid percussion in rats. Intensive Care Med 31, 281–288. [DOI] [PubMed] [Google Scholar]
- Molaie AM, Maguire J, 2018. Neuroendocrine Abnormalities Following Traumatic Brain Injury: An Important Contributor to Neuropsychiatric Sequelae. Front Endocrinol (Lausanne) 9, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondello S, Thelin EP, Shaw G, Salzet M, Visalli C, Cizkova D, Kobeissy F, Buki A, 2018. Extracellular vesicles: pathogenetic, diagnostic and therapeutic value in traumatic brain injury. Expert Rev Proteomics 15, 451–461. [DOI] [PubMed] [Google Scholar]
- Mossberg KA, Durham WJ, Zgaljardic DJ, Gilkison CR, Danesi CP, Sheffield-Moore M, Masel BE, Urban RJ, 2017. Functional Changes after Recombinant Human Growth Hormone Replacement in Patients with Chronic Traumatic Brain Injury and Abnormal Growth Hormone Secretion. J Neurotrauma 34, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowery NT, Carnevale RJ, Gunter OL, Norris PR, Dossett LA, Dortch MJ, Morris JA Jr., May AK, 2009a. Insulin resistance heralds positive cultures after severe injury. Surg Infect (Larchmt) 10, 503–509. [DOI] [PubMed] [Google Scholar]
- Mowery NT, Gunter OL, Guillamondegui O, Dossett LA, Dortch MJ, Morris JA Jr., May AK, 2009b. Stress insulin resistance is a marker for mortality in traumatic brain injury. J Trauma 66, 145–151; discussion 151-143. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A, 2009. Brain mitochondrial dysfunction and oxidative damage in Parkinson's disease. J Bioenerg Biomembr 41, 517–521. [DOI] [PubMed] [Google Scholar]
- Nekludov M, Mobarrez F, Gryth D, Bellander BM, Wallen H, 2014. Formation of microparticles in the injured brain of patients with severe isolated traumatic brain injury. J Neurotrauma 31, 1927–1933. [DOI] [PubMed] [Google Scholar]
- Nighot M, Al-Sadi R, Guo S, Rawat M, Nighot P, Watterson MD, Ma TY, 2017. Lipopolysaccharide-Induced Increase in Intestinal Epithelial Tight Permeability Is Mediated by Toll-Like Receptor 4/Myeloid Differentiation Primary Response 88 (MyD88) Activation of Myosin Light Chain Kinase Expression. Am J Pathol 187, 2698–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistico R, Cavallucci V, Piccinin S, Macri S, Pignatelli M, Mehdawy B, Blandini F, Laviola G, Lauro D, Mercuri NB, D'Amelio M, 2012. Insulin receptor beta-subunit haploinsufficiency impairs hippocampal late-phase LTP and recognition memory. Neuromolecular Med 14, 262–269. [DOI] [PubMed] [Google Scholar]
- Ntali G, Tsagarakis S, 2019. Traumatic brain injury induced neuroendocrine changes: acute hormonal changes of anterior pituitary function. Pituitary. [DOI] [PubMed] [Google Scholar]
- Olsen AB, Hetz RA, Xue H, Aroom KR, Bhattarai D, Johnson E, Bedi S, Cox CS Jr., Uray K, 2013. Effects of traumatic brain injury on intestinal contractility. Neurogastroenterol Motil 25, 593–e463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padwal RS, 2014. Obesity, diabetes, and the metabolic syndrome: the global scourge. Canadian Journal of Cardiology 30, 467–472. [DOI] [PubMed] [Google Scholar]
- Park KD, Lim OK, Yoo CJ, Kim YW, Lee S, Park Y, Lee JK, 2016. Voxel-based statistical analysis of brain metabolism in patients with growth hormone deficiency after traumatic brain injury. Brain Inj 30, 407–413. [DOI] [PubMed] [Google Scholar]
- Patel NA, Moss LD, Lee JY, Tajiri N, Acosta S, Hudson C, Parag S, Cooper DR, Borlongan CV, Bickford PC, 2018. Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. J Neuroinflammation 15, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson E, Cryan JF, Fitzgerald GF, Ross RP, Dinan TG, Stanton C, 2014. Gut microbiota, the pharmabiotics they produce and host health. Proc Nutr Soc 73, 477–489. [DOI] [PubMed] [Google Scholar]
- Pieracci F, Hydo L, Eachempati S, Pomp A, Shou J, Barie PS, 2008. Higher body mass index predicts need for insulin but not hyperglycemia, nosocomial infection, or death in critically ill surgical patients. Surg Infect (Larchmt) 9, 121–130. [DOI] [PubMed] [Google Scholar]
- Plesnila N, 2016. The immune system in traumatic brain injury. Curr Opin Pharmacol 26, 110–117. [DOI] [PubMed] [Google Scholar]
- Pratt SI, Jerome GJ, Schneider KL, Craft LL, Buman MP, Stoutenberg M, Daumit GL, Bartels SJ, Goodrich DE, 2016. Increasing US health plan coverage for exercise programming in community mental health settings for people with serious mental illness: a position statement from the Society of Behavior Medicine and the American College of Sports Medicine. Transl Behav Med 6, 478–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinkler M, Ekman B, Zhang P, Isidori AM, Murray RD, Investigators E-A, 2018. Mortality data from the European Adrenal Insufficiency Registry-Patient characterization and associations. Clin Endocrinol (Oxf) 89, 30–35. [DOI] [PubMed] [Google Scholar]
- Radak Z, Chung HY, Koltai E, Taylor AW, Goto S, 2008. Exercise, oxidative stress and hormesis. Ageing Res Rev 7, 34–42. [DOI] [PubMed] [Google Scholar]
- Ranke MB, Wit JM, 2018. Growth hormone - past, present and future. Nat Rev Endocrinol 14, 285–300. [DOI] [PubMed] [Google Scholar]
- Rasouli J, Lekhraj R, Ozbalik M, Lalezari P, Casper D, 2011. Brain-Spleen Inflammatory Coupling: A Literature Review. Einstein J Biol Med 27, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau CS, Wu SC, Chen YC, Chien PC, Hsieh HY, Kuo PJ, Hsieh CH, 2017. Stress-Induced Hyperglycemia, but Not Diabetic Hyperglycemia, Is Associated with Higher Mortality in Patients with Isolated Moderate and Severe Traumatic Brain Injury: Analysis of a Propensity Score-Matched Population. Int J Environ Res Public Health 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch S, Krueger K, Turan A, You J, Roewer N, Sessler DI, 2012. Use of wireless motility capsule to determine gastric emptying and small intestinal transit times in critically ill trauma patients. J Crit Care 27, 534 e537–512. [DOI] [PubMed] [Google Scholar]
- Ray K, 2012. Liver: Activation of NF-kappaB signaling in hepatocytes induces liver fibrosis. Nat Rev Gastroenterol Hepatol 9, 244. [DOI] [PubMed] [Google Scholar]
- Reilly SM, Saltiel AR, 2017. Adapting to obesity with adipose tissue inflammation. Nature Reviews Endocrinology 13, 633. [DOI] [PubMed] [Google Scholar]
- Rhee SH, Pothoulakis C, Mayer EA, 2009. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat Rev Gastroenterol Hepatol 6, 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RM, Doran SJ, Barrett JP, Henry RJ, Ma EL, Faden AI, Loane DJ, 2018. Chronic Alterations in Systemic Immune Function after Traumatic Brain Injury. J Neurotrauma 35, 1419–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizoli SB, Jaja BN, Di Battista AP, Rhind SG, Neto AC, da Costa L, Inaba K, da Luz LT, Nascimento B, Perez A, Baker AJ, de Oliveira Manoel AL, 2017. Catecholamines as outcome markers in isolated traumatic brain injury: the COMA-TBI study. Crit Care 21, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero MC, Valero A, Navarro MC, Hierro I, Baron SD, Martin-Sanchez J, 2014. Experimental demonstration of pathogenic potential of Anisakis physeteris and Anisakis paggiae in Wistar rats. Parasitol Res 113, 4377–4386. [DOI] [PubMed] [Google Scholar]
- Roozenbeek B, Maas AI, Menon DK, 2013. Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol 9, 231–236. [DOI] [PubMed] [Google Scholar]
- Roquilly A, Vourc'h M, Cinotti R, Asehnoune K, 2013. A new way of thinking: hydrocortisone in traumatic brain-injured patients. Crit Care 17, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Mattson MP, 2013. Activity-dependent, stress-responsive BDNF signaling and the quest for optimal brain health and resilience throughout the lifespan. Neuroscience 239, 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan H, Miles PD, Ladd CM, Ross K, Golub TR, Olefsky JM, Lodish HF, 2002. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: implications for insulin resistance. Diabetes 51, 3176–3188. [DOI] [PubMed] [Google Scholar]
- Ruegsegger GN, Booth FW, 2017. Running from Disease: Molecular Mechanisms Associating Dopamine and Leptin Signaling in the Brain with Physical Inactivity, Obesity, and Type 2 Diabetes. Front Endocrinol (Lausanne) 8, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarsieri M, Niyonkuru C, McCullough EH, Dobos JA, Dixon CE, Berga SL, Wagner AK, 2014. Cerebrospinal fluid cortisol and progesterone profiles and outcomes prognostication after severe traumatic brain injury. J Neurotrauma 31, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, 2015. Mental health: thinking from the gut. Nature 518, S12–15. [DOI] [PubMed] [Google Scholar]
- Schneider KJ, Leddy JJ, Guskiewicz KM, Seifert T, McCrea M, Silverberg ND, Feddermann-Demont N, Iverson GL, Hayden A, Makdissi M, 2017. Rest and treatment/rehabilitation following sport-related concussion: a systematic review. Br J Sports Med 51, 930–934. [DOI] [PubMed] [Google Scholar]
- Schober ME, Ke X, Xing B, Block BP, Requena DF, McKnight R, Lane RH, 2012. Traumatic brain injury increased IGF-1B mRNA and altered IGF-1 exon 5 and promoter region epigenetic characteristics in the rat pup hippocampus. J Neurotrauma 29, 2075–2085. [DOI] [PubMed] [Google Scholar]
- Schwulst SJ, Trahanas DM, Saber R, Perlman H, 2013. Traumatic brain injury-induced alterations in peripheral immunity. J Trauma Acute Care Surg 75, 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidi B, Shah SB, Esparza M, Head BP, Ward SR, 2018. Skeletal Muscle Atrophy and Degeneration in a Mouse Model of Traumatic Brain Injury. J Neurotrauma 35, 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Zhuang Y, Ying Z, Wu A, Gomez-Pinilla F, 2009. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience 161, 1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman M, Liu MM, Birnbaum S, Wolf SE, Minei JP, Gatson JW, 2016. Adult obese mice suffer from chronic secondary brain injury after mild TBI. J Neuroinflammation 13, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Dong B, Mao Y, Guan W, Cao J, Zhu R, Wang S, 2016. Review: Traumatic brain injury and hyperglycemia, a potentially modifiable risk factor. Oncotarget 7, 71052–71061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahashi K, Sugiyama K, Kashiura M, Hamabe Y, 2017. Decreasing skeletal muscle as a risk factor for mortality in elderly patients with sepsis: a retrospective cohort study. J Intensive Care 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitaka Y, Tran HT, Bennett RE, Sanchez L, Levy MA, Dikranian K, Brody DL, 2011. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol 70, 551–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlosberg D, Benifla M, Kaufer D, Friedman A, 2010. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 6, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LF, Hoffmann MS, Gerbatin Rda R, Fiorin Fda S, Dobrachinski F, Mota BC, Wouters AT, Pavarini SP, Soares FA, Fighera MR, Royes LF, 2013. Treadmill exercise protects against pentylenetetrazol-induced seizures and oxidative stress after traumatic brain injury. Journal of neurotrauma 30, 1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva PE, Maldaner V, Vieira L, de Carvalho KL, Gomes H, Melo P, Babault N, Cipriano G Jr., Durigan JLQ, 2018. Neuromuscular electrophysiological disorders and muscle atrophy in mechanically-ventilated traumatic brain injury patients: New insights from a prospective observational study. J Crit Care 44, 87–94. [DOI] [PubMed] [Google Scholar]
- Silva PP, Bhatnagar S, Herman SD, Zafonte R, Klibanski A, Miller KK, Tritos NA, 2015. Predictors of Hypopituitarism in Patients with Traumatic Brain Injury. J Neurotrauma 32, 1789–1795. [DOI] [PubMed] [Google Scholar]
- Simsek Y, Karaca Z, Tanriverdi F, Unluhizarci K, Selcuklu A, Kelestimur F, 2015. A comparison of low-dose ACTH, glucagon stimulation and insulin tolerance test in patients with pituitary disorders. Clin Endocrinol (Oxf) 82, 45–52. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED, 2006. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab 26, 1407–1418. [DOI] [PubMed] [Google Scholar]
- Stein DM, Lindel AL, Murdock KR, Kufera JA, Menaker J, Scalea TM, 2012. Use of serum biomarkers to predict secondary insults following severe traumatic brain injury. Shock 37, 563–568. [DOI] [PubMed] [Google Scholar]
- Stigger F, Marcolino MAZ, Portela KM, Plentz RDM, 2018. Effects of Exercise on Inflammatory, Oxidative and Neurotrophic Biomarkers on Cognitively Impaired Individuals Diagnosed with Dementia or Mild Cognitive Impairment: A Systematic Review and Meta-Analysis. J Gerontol A Biol Sci Med Sci. [DOI] [PubMed] [Google Scholar]
- Stoecklein VM, Osuka A, Lederer JA, 2012. Trauma equals danger--damage control by the immune system. J Leukoc Biol 92, 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EM, Pennington ER, Green WD, Beck MA, Brown DA, Shaikh SR, 2018. Mechanisms by Which Dietary Fatty Acids Regulate Mitochondrial Structure-Function in Health and Disease. Advances in Nutrition 9, 247–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Hu C, Fang H, Zhu L, Gao N, Zhu J, 2015. The effects of Lactobacillus acidophilus on the intestinal smooth muscle contraction through PKC/MLCK/MLC signaling pathway in TBI mouse model. PloS one 10, e0128214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundman MH, Chen NK, Subbian V, Chou YH, 2017. The bidirectional gut-brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease. Brain Behav Immun 66, 31–44. [DOI] [PubMed] [Google Scholar]
- Szendroedi J, Frossard M, Klein N, Bieglmayer C, Wagner O, Pacini G, Decker J, Nowotny P, Muller M, Roden M, 2012. Lipid-induced insulin resistance is not mediated by impaired transcapillary transport of insulin and glucose in humans. Diabetes 61, 3176–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, 2017. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H, Yang W, Wu C, Liu B, Lu H, Wang H, Yan H, 2017. Assessment of the role of intracranial hypertension and stress on hippocampal cell apoptosis and hypothalamic-pituitary dysfunction after TBI. Sci Rep 7, 3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanriverdi F, Kelestimur F, 2015. Neuroendocrine Disturbances after Brain Damage: An Important and Often Undiagnosed Disorder. J Clin Med 4, 847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanriverdi F, Schneider HJ, Aimaretti G, Masel BE, Casanueva FF, Kelestimur F, 2015. Pituitary dysfunction after traumatic brain injury: a clinical and pathophysiological approach. Endocr Rev 36, 305–342. [DOI] [PubMed] [Google Scholar]
- Taylor BC, Hagel Campbell E, Nugent S, Bidelspach DE, Kehle-Forbes SM, Scholten J, Stroupe KT, Sayer NA, 2017. Three Year Trends in Veterans Health Administration Utilization and Costs after Traumatic Brain Injury Screening among Veterans with Mild Traumatic Brain Injury. J Neurotrauma 34, 2567–2574. [DOI] [PubMed] [Google Scholar]
- Tsatsoulis A, Mantzaris MD, Bellou S, Andrikoula M, 2013. Insulin resistance: an adaptive mechanism becomes maladaptive in the current environment - an evolutionary perspective. Metabolism 62, 622–633. [DOI] [PubMed] [Google Scholar]
- Tyagi E, Zhuang Y, Agrawal R, Ying Z, Gomez-Pinilla F, 2015. Interactive actions of Bdnf methylation and cell metabolism for building neural resilience under the influence of diet. Neurobiology of disease 73, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undurti A, Colasurdo EA, Sikkema CL, Schultz JS, Peskind ER, Pagulayan KF, Wilkinson CW, 2018. Chronic hypopituitarism associated with increased postconcussive symptoms is prevalent after blast-induced mild traumatic brain injury. Frontiers in neurology 9, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berghe G, Wilmer A, Milants I, Wouters PJ, Bouckaert B, Bruyninckx F, Bouillon R, Schetz M, 2006. Intensive insulin therapy in mixed medical/surgical intensive care units: benefit versus harm. Diabetes 55, 3151–3159. [DOI] [PubMed] [Google Scholar]
- van der Heide LP, Ramakers GM, Smidt MP, 2006. Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol 79, 205–221. [DOI] [PubMed] [Google Scholar]
- van der Poll T, van Deventer SJ, 1999. Cytokines and anticytokines in the pathogenesis of sepsis. Infectious disease clinics of North America 13, 413–426. [DOI] [PubMed] [Google Scholar]
- Venkata C, Kasal J, 2018. Cardiac Dysfunction in Adult Patients with Traumatic Brain Injury: A Prospective Cohort Study. Clin Med Res 16, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier R, Garnier A, Veksler V, 2008. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 79, 208–217. [DOI] [PubMed] [Google Scholar]
- Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R, 2008. TLR-4, IL-1R and TNF-R signaling to NF-kappaB: variations on a common theme. Cell Mol Life Sci 65, 2964–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vespa P, Bergsneider M, Hattori N, Wu H-M, Huang S-C, Martin NA, Glenn TC, McArthur DL, Hovda DA, 2005. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. Journal of Cerebral Blood Flow & Metabolism 25, 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Balarezo MG, Affram K, Saavedra JM, Symes AJ, 2015a. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 138, 3299–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Kryndushkin D, Balarezo MG, Campbell AM, Saavedra JM, Shewmaker FP, Symes AJ, 2015b. Hepatic expression of serum amyloid A1 is induced by traumatic brain injury and modulated by telmisartan. The American journal of pathology 185, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Kryndushkin D, Balarezo MG, Campbell AM, Saavedra JM, Shewmaker FP, Symes AJ, 2015c. Hepatic expression of serum amyloid A1 is induced by traumatic brain injury and modulated by telmisartan. Am J Pathol 185, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivar C, van Praag H, 2017. Running changes the brain: the long and the short of it. Physiology 32, 410–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AK, Amin KB, Niyonkuru C, Postal BA, McCullough EH, Ozawa H, Dixon CE, Bayir H, Clark RS, Kochanek PM, Fabio A, 2011. CSF Bcl-2 and cytochrome C temporal profiles in outcome prediction for adults with severe TBI. J Cereb Blood Flow Metab 31, 1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker PA, Shah SK, Jimenez F, Aroom KR, Harting MT, Cox CS Jr., 2012. Bone marrow-derived stromal cell therapy for traumatic brain injury is neuroprotective via stimulation of non-neurologic organ systems. Surgery 152, 790–793. [DOI] [PubMed] [Google Scholar]
- Washington PM, Villapol S, Burns MP, 2016. Polypathology and dementia after brain trauma: Does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol 275 Pt 3, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willeumier K, Taylor DV, Amen DG, 2012. Elevated body mass in National Football League players linked to cognitive impairment and decreased prefrontal cortex and temporal pole activity. Transl Psychiatry 2, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, 1987. Excitatory amino acids increase glycogen phosphorylase activity in the rat spinal cord. Neurosci Lett 73, 209–214. [DOI] [PubMed] [Google Scholar]
- Wright DK, Liu S, van der Poel C, McDonald SJ, Brady RD, Taylor L, Yang L, Gardner AJ, Ordidge R, O'Brien TJ, Johnston LA, Shultz SR, 2017. Traumatic Brain Injury Results in Cellular, Structural and Functional Changes Resembling Motor Neuron Disease. Cereb Cortex 27, 4503–4515. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F, 2014. Dietary strategy to repair plasma membrane after brain trauma: implications for plasticity and cognition. Neurorehabil Neural Repair 28, 75–84. [DOI] [PubMed] [Google Scholar]
- Xiang L, Lu S, Mittwede PN, Clemmer JS, Hester RL, 2014. Inhibition of NADPH oxidase prevents acute lung injury in obese rats following severe trauma. Am J Physiol Heart Circ Physiol 306, H684–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda H, Szuchman-Sapir A, Khatib S, Musa R, Tamir S, 2011. Human atherosclerotic plaque lipid extract promotes expression of proinflammatory factors in human monocytes and macrophage-like cells. Atherosclerosis 218, 339–343. [DOI] [PubMed] [Google Scholar]
- Yin Y, Li E, Sun G, Yan HQ, Foley LM, Andrzejczuk LA, Attarwala IY, Hitchens TK, Kiselyov K, Dixon CE, Sun D, 2018. Effects of DHA on Hippocampal Autophagy and Lysosome Function After Traumatic Brain Injury. Mol Neurobiol 55, 2454–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Dong F, Ren J, Driscoll MJ, Culver B, 2005. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol 191, 318–325. [DOI] [PubMed] [Google Scholar]
- Zhang X, Jiang X, 2015a. Effects of enteral nutrition on the barrier function of the intestinal mucosa and dopamine receptor expression in rats with traumatic brain injury. Journal of Parenteral and Enteral Nutrition 39, 114–123. [DOI] [PubMed] [Google Scholar]
- Zhang X, Jiang X, 2015b. Effects of enteral nutrition on the barrier function of the intestinal mucosa and dopamine receptor expression in rats with traumatic brain injury. JPEN J Parenter Enteral Nutr 39, 114–123. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chopp M, Meng Y, Katakowski M, Xin H, Mahmood A, Xiong Y, 2015. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J Neurosurg 122, 856–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Yan T, Li L, Chopp M, Venkat P, Qian Y, Li R, Wu R, Li W, Lu M, Zhang T, Chen J, 2019. Immune Response Mediates Cardiac Dysfunction after Traumatic Brain Injury. J Neurotrauma 36, 619–629. [DOI] [PubMed] [Google Scholar]
- Zhou J, Burns MP, Huynh L, Villapol S, Taub DD, Saavedra JM, Blackman MR, 2017. Temporal Changes in Cortical and Hippocampal Expression of Genes Important for Brain Glucose Metabolism Following Controlled Cortical Impact Injury in Mice. Front Endocrinol (Lausanne) 8, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu KJ, Huang H, Chu H, Yu H, Zhang SM, 2014. Alterations in enterocyte mitochondrial respiratory function and enzyme activities in gastrointestinal dysfunction following brain injury. World J Gastroenterol 20, 9585–9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnik A, Klin Y, Gruenbaum BF, Gruenbaum SE, Ohayon S, Leibowitz A, Kotz R, Dubilet M, Boyko M, Shapira Y, Teichberg VI, 2012. beta2 adrenergic-mediated reduction of blood glutamate levels and improved neurological outcome after traumatic brain injury in rats. J Neurosurg Anesthesiol 24, 30–38. [DOI] [PubMed] [Google Scholar]
- Zygun DA, Zuege DJ, Boiteau PJ, Laupland KB, Henderson EA, Kortbeek JB, Doig CJ, 2006. Ventilator-associated pneumonia in severe traumatic brain injury. Neurocrit Care 5, 108–114. [DOI] [PubMed] [Google Scholar]