

Abstract
Through a phenotypic high throughput screen (HTS) using a serum response element luciferase (SRE.L) promoter, we identified a novel 5-aryl-1,3,4-oxadiazol-2-ylthiopropionic acid lead inhibitor of Rho/MRTF/SRF-mediated gene transcription with good potency (IC50 = 180 nM). We were able to rapidly improve the cellular potency by 5 orders of magnitude guided by sharply defined and synergistic SAR. The remarkable potency and depth of the SAR, as well as the relatively low molecular weight of the series, suggests, but does not prove, that binding to the unknown molecular target may be occurring through a covalent mechanism. The series nevertheless has no observable cytotoxicity up to 100 μM. Ensuing pharmacokinetic optimization resulted in the development of two potent and orally bioavailable anti-fibrotic agents that were capable of dose-dependently reducing connective tissue growth factor (CTGF) gene expression in vitro as well as significantly reducing the development of bleomycin-induced dermal fibrosis in mice in vivo.
Graphical Abstract
Introduction:
Scleroderma is a chronic connective tissue disease that is characterized by thickening and fibrosis of dermal epithelial tissue and vascular abnormalities. It has been designated as an orphan disease, and, while the exact number affected is unknown, estimates suggest ~100,000–300,000 individuals have some form of scleroderma in the U.S.1 There are two classifications of the disease: localized and systemic.2 Localized scleroderma does not involve internal organs, but systemic sclerosis (SSc) is associated with pathology in multiple organs including skin, gastrointestinal tract, lungs, kidneys, heart, etc.3–4 The incidence of SSc in the U.S. is 9–46 cases per million per year, and roughly 50% of these individuals have a highly aggressive form: diffuse systemic sclerosis (dSSc).1, 5 Patients suffering with dSSc have a higher risk of developing fibrosis within the internal organs, greatly increasing the patient’s mortality risk.2, 5 Current treatments are limited to immunosuppressants and anti-inflammatories that only address the symptoms rather than the underlying causes of the disease.2, 5 Currently in the U.S., the only FDA-approved antifibrotics are pirfenidone and nintedanib for the treatment of idiopathic pulmonary fibrosis (IPF).3, 6
Although the etiology of fibrosis is unknown, the activation of fibroblasts and their transition to myofibroblasts are key characteristics of the fibrotic process (Figure 1).7–8 Myofibroblasts produce excess extracellular matrix (ECM) components (e.g. collagen and fibronectin) and stress fibers, e.g. α-smooth muscle actin (α-SMA).8–9 Many of these components undergo cross-linking and maturation, resulting in a highly stable matrix that accounts for epithelial tissue stiffness.10 Dysregulation of ECM homeostasis is thought to contribute to mortality associated with SSc.1
Figure 1.
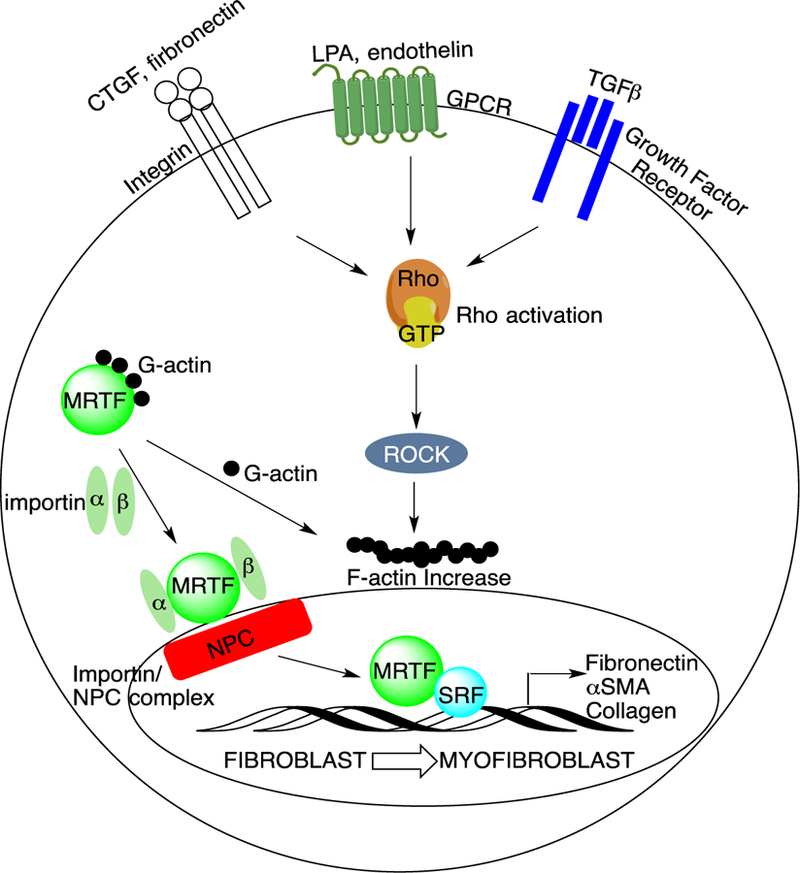
The fibroblast-to-myofibroblast transition is tightly regulated by Rho/MRTF/SRF signaling. Both soluble and mechanical stimuli promote MRTF nuclear translocation in fibroblasts. In the resting state, actin is not polymerized, and MRTF is bound to G-actin and sequestered into the cytoplasm. When stress fibers form in response to these stimuli, G-actin polymerizes into F-actin and MRTF is released. This allows MRTF to translocate into the nucleus via binding to importins followed by MRTF/importin/nuclear pore complex (NPC) formation. The MRTF/SRF coactivator complex increases ECM and pro-fibrotic gene expression, contributing to the fibroblast-to-myofibroblast transition.
There are several known cell surface receptors involved in the fibrotic process, including various integrins, GPCRs, and growth factor receptors that activate the production of ECM components (Figure 1).9, 11–13 These surface receptors, such as those for transforming growth factor β (TGFβ), connective tissue growth factor (CTGF), and lysophosphatidic acid (LPA), activate intracellular signaling through a common mediator (Rho GTPase), which leads to activation of Rho-associated protein kinase (ROCK).11, 14–18 Activation of ROCK induces the polymerization of globular (G)-actin to filamentous (F)-actin, which releases, and thereby promotes, the nuclear localization of myocardin-related transcription factor (MRTF).14 In the nucleus, MRTF binds to the serum response factor (SRF) on serum response elements (SREs). Binding of MRTF/SRF to these promoter regions of DNA activates expression of multiple genes associated with the fibrotic process, including: COL1A1, CTGF, and ACTA2.9, 14, 16
While Rho/MRTF/SRF signaling plays an important role in normal wound healing, over-stimulation or dysregulation of this pathway can lead to pathological fibrosis as well as increased cancer cell migration and metastasis.7, 19–22 Several attempts to target some of the aforementioned surface receptors for this signaling pathway have been made (e.g. STX-100 for integrins, AM152 for LPA receptors, and LY2157299 for TGFβ receptors); unfortunately, therapeutics exploiting these targets have not been successfully developed for fibrotic diseases to date.19 As an alternative approach, a MRTF-A knockout mouse model has demonstrated reduced fibrosis and scarring in heart, lung, dermal, and vascular fibrosis in response to decreased collagen production and tissue stiffness.23 The development of an inhibitor that blocks the Rho/MRTF/SRF transcription pathway downstream of surface receptors, which in principle could block multiple extracellular inputs, is a promising new approach toward the treatment of SSc and other fibrotic diseases.
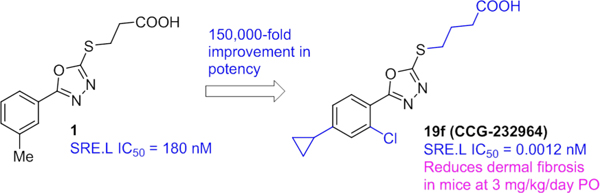
We have previously reported the development and execution of a SRE promoter-driven luciferase (SRE.L) HTS for inhibitors of the Rho/MRTF/SRF pathway.19 Although SRF responds to at least two different co-activator mechanisms, Rho/MRTF and Ras/Ternary Complex Factor (TCF), this SRE.L luciferase assay is selective for MRTF as the TCF sites in the promoter have been eliminated.24 This led to the discovery of CCG-1423, which we subsequently optimized to CCG-203971, CCG-222740, and CCG-257081—modestly potent inhibitors that are active in multiple animal models of fibrosis.19, 22, 25–32 An expanded HTS of 80,000 compounds produced a second, more potent class of Rho/MRTF/SRF inhibitors based on the confirmed hit (3-((5-(m-tolyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid 1 (SRE.L IC50 = 180 nM, Figure 2). The ensuing SAR of this class proved to be remarkably deep (over 5 orders of magnitude range of activity), suggesting that the currently unknown biological target has a very well-defined binding site. Furthermore, we report indirect evidence (from the SAR) that suggests engagement with the unknown target may be covalent; nevertheless, the series appears to have very low cytotoxicity (no impact on cell viability up to 100 µM). Herein, we report the synthesis, biological activity, pharmacokinetic profile, and initial in vivo efficacy of this structurally distinct new series of Rho/MRTF/SRF-mediated gene transcription inhibitors.
Figure 2.
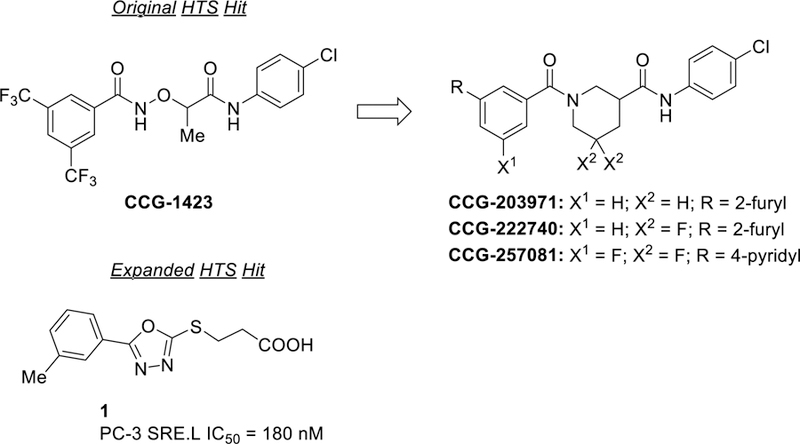
Original hit (CCG-1423) from SRE.L HTS and series development to CCG-257081. New chemotype hit 1 from expanded SRE.L HTS of Rho/MRTF/SRF-pathway.
Chemistry
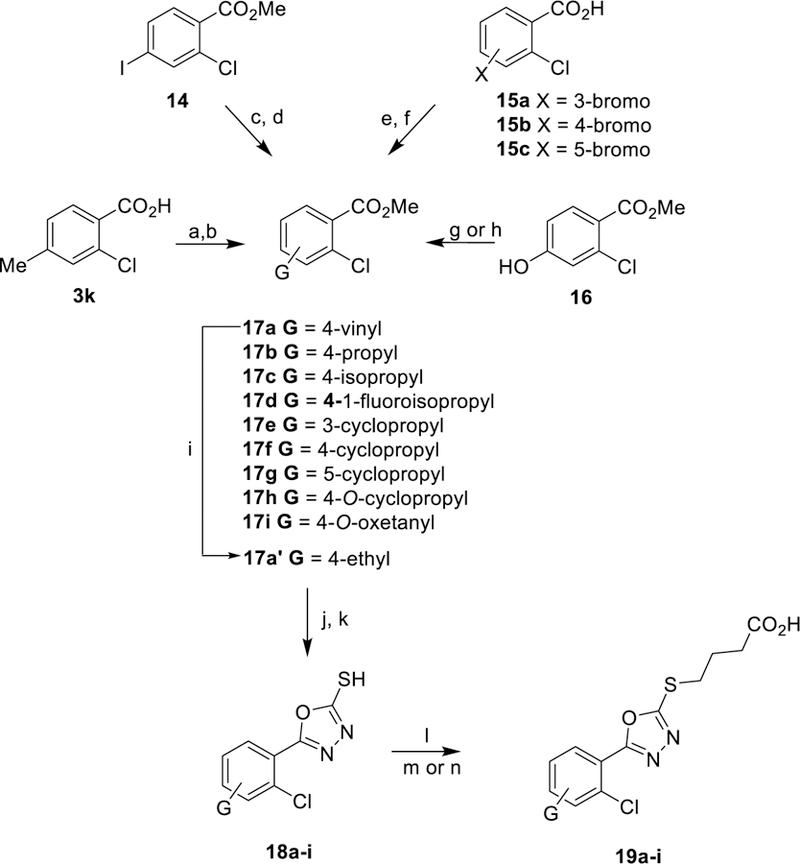
The general synthesis of new 5-aryl-1,3,4-oxadiazol-2-ylthioalkanoic acids and derivatives is summarized in Scheme 1. Various aromatic benzoic acids (2) were converted to their respective methyl esters under standard Fischer esterification conditions, and then converted to the corresponding hydrazides (3) by refluxing in MeOH with excess hydrazine hydrate. Refluxing 3 and carbon disulfide in EtOH under basic conditions, followed by acidic workup, generated 2-mercapto-5-aryl-1,3,4-oxadiazoles (4). S-alkylation with either methyl, ethyl, or t-butyl Ω-bromoalkanoates provided the thioether esters (5–7). Hydrolysis of the esters was accomplished with either trifluoroacetic acid (t-butyl esters) or sodium hydroxide (methyl and ethyl esters) to give final carboxylic acids 8 (Table 1). The tert-butyl esters became the preferred precursor when n = 2 because basic hydrolysis of the methyl ester intermediates resulted in competing retro-Michael elimination to regenerate the 1, 3, 4-oxadiazole-2-thiols 4. Amide 9 was prepared by conversion of the corresponding acid 8 to the acid chloride with oxalyl chloride in DMF/CH2Cl2 at room temperature, followed by treatment with methylamine.
Scheme 1. General Synthesis of 5-Aryl-1,3,4-oxadiazol-2-ylthioalkanoic Acidsa.
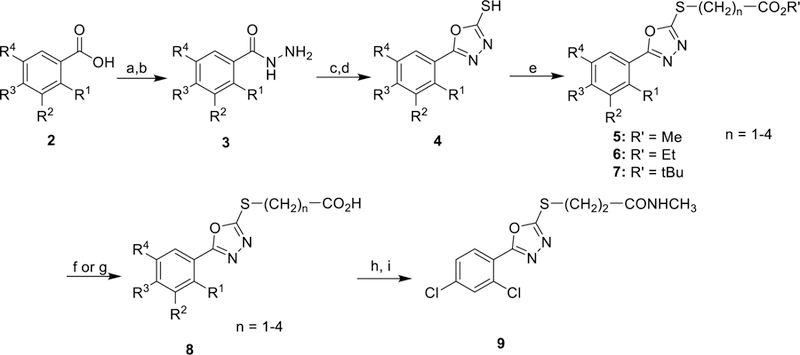
aReagents and conditions: (a) H2SO4/MeOH, 85 °C 6 h. (b) H4N2•H2O, 85 °C 14 h. (c) KOH/H2O/EtOH, CS2, 95 °C 16 h. (d) 1N HCl workup (e) acetone, K2CO3, Br-(CH2)n-CO2R’, 25 °C, 5–24 h. (f) DCM, TFA, 25 °C 3 h. (g) 1M NaOH, THF, 25 °C 16 h. (h) oxalyl chloride, DMF, CH2Cl2, 25 °C, 2 h. (i) MeNH2 in EtOH, 25 °C, 16 h.
Table 1.
SRE.L Activity of 5-Aryl-1,3,4-Oxadiazole-2-thiopropionic acids
 | |||||
|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | R4 | SRE.L IC50 (nM)a, b |
| 8a | H | H | H | H | 3,500 |
| 8b | H | H | Cl | H | 340 |
| 8c | H | H | Me | H | 34 |
| 8d | H | H | OMe | H | 160 |
| 8e | H | Cl | H | H | 1,500 |
| 1 | H | Me | H | H | 180 |
| 8f | H | OMe | H | H | 3,600 |
| 8g | Cl | H | H | H | 25 |
| 8h | Me | H | H | H | 6,300 |
| 8i | OMe | H | H | H | 3,100 |
| 8j | Cl | H | Cl | H | 4 (2c) |
| 8k | Cl | H | Me | H | 1.5 |
| 8l | Cl | H | OMe | H | 2.6 |
| 8m | Cl | Me | Me | H | 0.02c |
| 8n | Cl | H | Me | Me | 0.23c |
| 8o | Cl | H | Cl | Me | 0.32c |
Inhibition of Gα12-stimulated SRE.L activity (mean of n = 3, SEM < 10%) in PC3 cells unless otherwise noted.
Cell viability and proliferation assay with WST-1 < 5% inhibition up to 100 μM inhibitor (mean of n = 3, SEM < 10%).
SRE.L assay performed in HEK293T cells (mean of n = 3, SEM < 10%).
The syntheses of 4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)oxy)butanoic acid (11) and 2-(((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)methyl)thio)acetic acid (13) were accomplished starting with common intermediate 3j. Intermediate 10 was produced through the cyclization/rearrangement of 3j with propylene oxide.33 O-alkylation with t-butyl 4-bromobutyrate followed by acidic hydrolysis with trifluoroacetic acid provided carboxylic acid 11. Alternatively, subjecting 3j to bromoacetic acid and POCl3 afforded intermediate 12.34 Basic S-alkylation with potassium carbonate and 2-mercaptoacetic acid produced carboxylic acid 13.
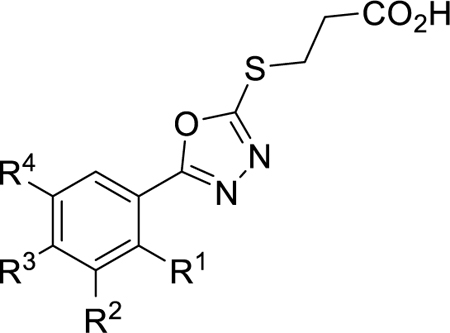
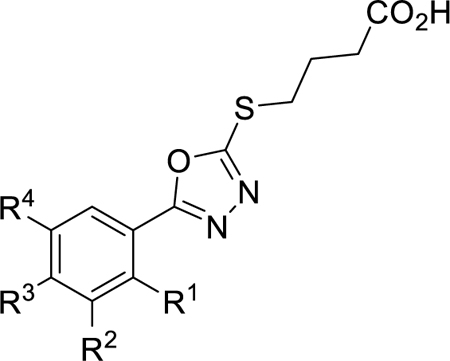
Next, alkyl, fluorinated alkyl, cycloalkyl, alkoxy, and cycloalkoxy derivatives were synthesized as shown in Scheme 3. Routes to obtain vinyl and isopropyl methyl esters 17a and 17c were performed as previously described.35–36 Alkyl methyl ester 17b was synthesized by alkylating 3k with two equivalents of LDA and ethyl iodide, followed by esterification. 4-(1-fluoro)-isopropyl methyl ester 17d was synthesized from 14 by Grignard reaction with acetone followed by fluorination of the tertiary alcohol with diethylaminosulfur trifluoride (DAST). Suzuki coupling of commercially available 15a-c with cyclopropyl boronic acid afforded cycloalkyl methyl esters 17e-g. 4-alkoxy aryl derivatives were formed by O-alkylation of commercially available phenol 16 using cesium carbonate and either DMA or DMSO (17h and 17i, respectively). Reduction of vinyl intermediate 17a with 2-nitrobenzene-1-sulfonyl chloride and hydrazine hydrate resulted in ethyl intermediate 17a’. Using chemistry analogous to Scheme 1, the aryl methyl esters 17a’ and 17b-i were converted to respective hydrazides, 5-aryl-2-mercapto-1,3,4-oxadiazoles (18a-i), thioethers, and final carboxylic acids (19a-i) (Table 3).
Scheme 3. Synthetic Route to Generate Thiobutanoate Analogs 19a-ia.
aReagents and conditions: (a) LDA (2 eq.), THF, 0 °C, 10 min, then ethyl iodide, 0 °C, 1 h. (b) MeOH, H2SO4, 85 °C, 16 h. (c) iPrMgCl, THF, −20 °C for 1.5 h, then acetone, 25 °C for 1.5 h. (d) DAST, DCM, −78 °C to 25 °C 2 h. (e) MeOH, H2SO4, 85 °C, 16 h. (f) Pd(II)OAc2Cl2, K3PO4, P(Cy)3, cyclopropyl boronic acid, toluene, H2O, 100 °C, 3 h. (g) bromocyclopropane, Cs2CO3, DMA, 155 °C, 24 h then MeOH, H2SO4, 85 °C, 16 h. (h) 3-bromooxetane, Cs2CO3, DMSO, 105 °C, 8 h. (i) 2-nitrobenzene-1-sulfonyl chloride, H2N4•H2O, MeCN, 0 °C to 25 °C, 16 h. (j) H4N2•H2O, MeOH, 85 °C, 16 h. (k) KOH/H2O/EtOH, CS2, 95 °C, 16 h., followed by HCl workup (l) 4-bromo-butanoate ester, K2CO3, acetone, 25 °C, 16 h. (m) 1M NaOH, THF, 25 °C, 1 h. (n) TFA, DCM, 25 °C, 1 h.
Table 3.
SRE.L Activity of 5-Aryl-1,3,4-Oxadiazole-2-thiobutanoic Acids.
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | R4 | SRE.L IC50 (nM)a, b | MLM T1/2 (min)b |
| 8q | Cl | H | Cl | H | 0.84 | >60 |
| 8s | Cl | H | Me | H | 0.1c | 28 |
| 8t | Cl | H | MeO | H | 2 | - |
| 19a | Cl | H | Et | H | 0.0007 | - |
| 19b | Cl | H | Pr | H | <0.0001 | - |
| 19c | Cl | H | i-Pr | H | 0.0007 | - |
| 8u | H | H | CF3 | H | 46 | - |
| 8v | CF3 | H | H | H | 10,000 | - |
| 8w | Cl | H | CF3 | H | 0.06 | >60 |
| 19d | Cl | H | CF(CH3)2 | H | 0.5 | - |
| 8x | Cl | H | Br | H | 0.07 | - |
| 8y | Cl | H | Ph | H | 25 | - |
| 19e | Cl | cPr | H | H | 0.019 | - |
| 19f | Cl | H | c-Pr | H | 0.001 (0.0012c) | 47 |
| 19g | Cl | H | H | cPr | 20 | - |
| 19h | Cl | H | O-cyclopropyl | H | 1.3 | >60 |
| 19i | Cl | H | O-oxetanyl | H | 370 | - |
Inhibition of Gα12-stimulated SRE.L activity (mean of n = 3, SEM < 10%) in HEK cells.
Cell viability and proliferation assay with WST-1 < 5% inhibition up to 100 μM inhibitor (mean of n = 3, SEM < 10%).
SRE.L data generated with transfected PC-3 cells (mean of n = 3, SEM < 10%).
Half-life in mouse liver microsomes.
Sulfone 21 was prepared as shown in Scheme 4, which also includes an alternative synthetic route for 8q. 1,3,4-oxadiazole thioether 4j was converted to electrophilic sulfone 20 in two steps. First, thiol 4j was methylated with methyl iodide and triethylamine. This thioether intermediate was oxidized to 20 using either mCPBA or ammonium molybdate tetrahydrate and hydrogen peroxide.37 Nucleophilic displacement of sulfone 20 using 4-mercaptobutanoic acid efficiently produced thiobutanoic acid 8q, which was subsequently oxidized to thiobutanoic acid sulfone analog 21 using mCPBA.
Scheme 4. Synthetic Route to Generate Thiobutanoic Acid Sulfone Analog 21 a.
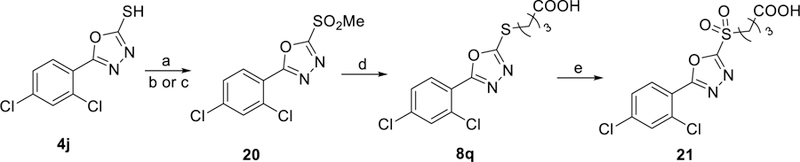
aReagents and conditions: (a) MeI, Et3N, THF, 25 °C, 2 hr (b) (NH4)6Mo7O24, H2O2, EtOH, 25 °C, 2 hr (c) mCPBA, DCM, 25 °C, 2 hr (d) 4-mercaptobutanoic acid, K2CO3, acetone, 25 °C, 1 hr, (e) mCPBA, DCM, 25 °C, 2 hr.
Results and Discussion
We began our SAR investigation by probing the importance of the phenyl-substituted oxadiazole ring system and the carboxylic acid with commercially available compounds (Figure 3). Pyridyl (22 and 23) and furanyl (24) substituted oxadiazole analogs led to complete loss in activity. Attempts to replace the oxadiazole core with other heterocycles (25-27) were similarly unsuccessful. Replacement of the carboxylic acid with an acetal isostere (28) also abolished activity. Based on these strongly negative results, we focused our initial synthetic SAR effort on 5-aryl-1,3,4-oxadiazaole-2-thioalkanoic acids.
Figure 3.
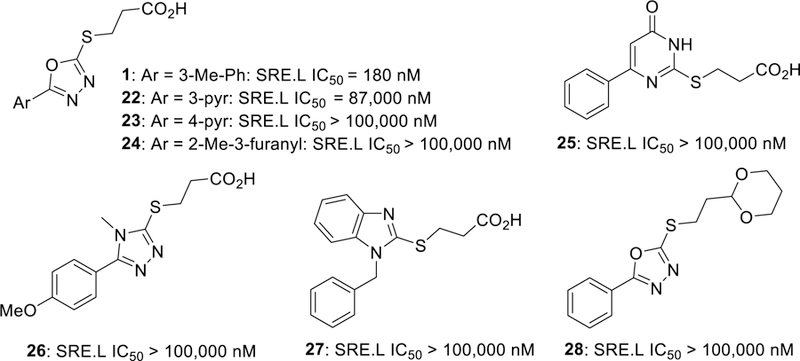
Preliminary SAR investigating the importance of the heterocycle and carboxylic acid. SRE.L activity (mean of n = 3, SEM < 10%).
We started by systematically investigating prototype electron donating (methoxy), withdrawing (chloro) and alkyl (methyl) groups at each of the three aryl positions (8a-i, Table 1). Remarkably, this revealed an over 250-fold span of potency over just these first 9 analogs; SRE.L IC50s ranged from 6300 nM (8h) to 25 nM (8g). Within this small initial group we observed several clear activity cliffs, including 2-Cl 8g (25 nM) vs 3-Cl 8e (1500 nM) and 4-Me 8c (34 nM) vs 3-Me 1 (180 nM) vs 2-Me 8h (6300 nM). We were then interested to see if the individual optimum substitutions would be synergistic. From the data in Table 1, it is clear that, among the four substituents investigated at R1, Cl is optimum. We thus combined this substitution with all three non-H substituents at R3 (8j-l) and observed a significant increase in potency in each case; all three of these analogs had single digit nM SRE.L activities. Despite these significant improvements in SRE.L activity, all analogs tested did not affect cell proliferation/viability up to 100 µM as measured by WST-1, highlighting the series lack of acute cytotoxicity even at high concentrations. For comparison, the clinically approved antifibrotics pirfenidone and nintedanib were tested in the SRE.L assay. Pirfenidone was inactive up to a concentration of 100 μM, above which it was cytotoxic (as determined by CellTiter-Fluor™). Nintedanib had an IC50 of 1.3 μM, but showed some cytotoxicity at 10 uM and an IC50 of under 30 μM.
It should be noted that the phenotypic screening SRE.L assay was originally designed as a HTS for cancer therapeutics, and it was developed using PC-3 cells.19 However, to improve the throughput of the assay, the transfected cell line was switched to HEK-293T cells. To ensure equivalency in SRE.L potency from one cell line to the other, various analogs were tested under both assay conditions (e.g. 8j, 8q, and 19f). We did not observe any notable difference in activity between the two transfected cell lines, so the assay was permanently switched to HEK-293T cells.
We were able to improve activity even further with trisubstituted analogs (8m-o) that combined the best R1, R3-disubstitution analogs (8j & 8k) with the best R2/R4 substituent (Me). This resulted in our first sub-nanomolar analogs with SRE.L potency as high as IC50 = 20 pM (8m). These extremely tight and potent SAR trends suggest that these inhibitors likely interact with a very well-defined binding site on the unknown molecular target.
We then focused on elucidating how the propionic acid sidechain modulated activity (Table 2). Replacing the carboxylic acid functionality with neutral methyl amide (9) was unsuccessful; however, ethyl ester 6 maintained activity, likely due to facile hydrolysis by non-specific esterases in the cells. This is consistent with the lack of activity of neutral acetal 28 (Figure 3), suggesting that the acid functionality is crucial for target engagement. To determine the optimal carbon chain length between the oxadiazole and the carboxylic acid, analogs bearing linkers from one to four methylenes were synthesized (8p-r). We found that the optimal carbon chain length was three (8q), and that activity was completely lost when there was only one carbon separating the oxadiazole and carboxylic acid (8p). This steep SAR is further evidence of the importance of the carboxylic acid group and its position relative to the oxadiazole ring. A three-carbon chain was used for all subsequent analogs.
Table 2.
Carboxylic Acid Sidechain SAR.
 | ||||
|---|---|---|---|---|
| Compd | Z | G | SRE.L IC50 (nM)a, b | MLM T1/2 (min)d |
| 8j | (CH2)2 | CO2H | 4 (2c) | >60 |
| 6 | (CH2)2 | CO2Et | 2.6 | - |
| 9 | (CH2)2 | CONHCH3 | >100,000 | - |
| 8p | CH2 | CO2H | >100,000 | - |
| 8q | (CH2)3 | CO2H | 1.8 (0.84c) | >60 |
| 8r | (CH2)4 | CO2H | 43 | - |
Inhibition of Gα12-stimulated SRE.L (mean of n = 3, SEM < 10%) in PC3 cells.
Cell viability and proliferation assay with WST-1 < 5% inhibition up to 100 μM inhibitor (mean of n = 3, SEM < 10%) up to 100 μM inhibitor.
SRE.L assay performed in HEK293T cells (mean of n = 3, SEM < 10%).
Half-life in mouse liver microsomes.
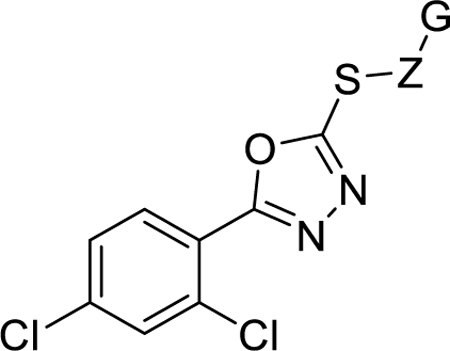
Based on our unsuccessful attempts to replace the oxadiazole core with similar heterocycles (Figure 3), the steep SAR of the series with regard to both aryl substitution and alkanoic acid chain length, and (3) the reported ability of 5-methylsulfonyl-1,3,4-oxadiazole probes to ligate cellular nucleophiles37–39, we hypothesized that the oxadiazole thioether core might be covalently engaging with a binding site nucleophile as illustrated in Figure 4. We therefore carefully selected analogs (11, 13, 29) that are structurally very similar to 8j but predicted to be much less capable of reacting with nucleophiles (Figure 4). In support of our hypothesis, we saw an almost complete loss in activity upon replacement of the S with O or CH2 (11, 29) or migration of the S away from the heterocyclic core (13). Conversely, activity was lost when the system was made more susceptible to nucleophilic attack by oxidizing the sulfide of 8q to the corresponding sulfone (21). Although the inactivity of the sulfone could be explained by simple poor cell permeability (due to lower ClogP) or rapid reaction with abundant cellular nucleophiles, this particular result certainly suggests that oxidation of our sulfide inhibitors in cellulo to irreversibly-binding sulfones is not occurring. Furthermore, it has been reported that a non-covalent 2-benzylthio-1,3,4-oxadiazole inhibitor of glycogen synthase kinase-3β can markedly lose potency with simple changes to the thiomethyl linker analogous to what we made.40 Overall, therefore, our SAR does not prove or disprove a covalent mode of binding of our compounds. Confirmation will require careful proteome labeling studies, which are underway and will be reported in due course.
Figure 4.
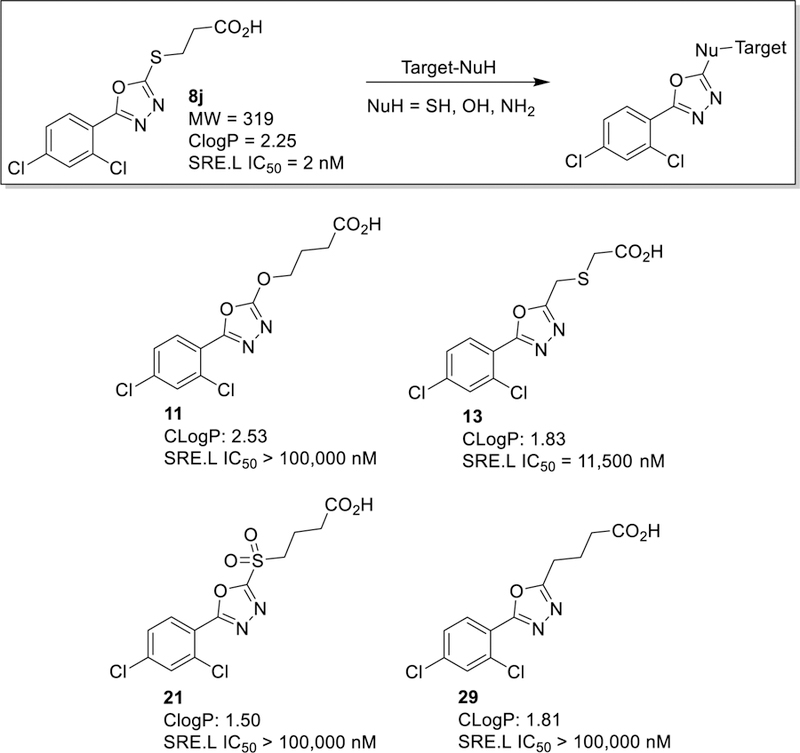
Hypothetical reaction of 8j with a target nucleophile, and close structural analogs designed to be less (11, 13 and 29) or more (21) susceptible to attack by cellular nucleophiles. Indicated below each structure is SRE.L activity (HEK293T cells, mean of n = 3, SEM < 10%).
Finally, we investigated the SAR on the aryl ring of the butanoic acid analog 8q (Table 3). As shown previously in Table 1, incorporation of a small alkyl or alkoxy group at R2/4 or R3 led to marked improvements in potency with the propionic acid series. We therefore explored expanding the size of the alkyl group and, anticipating the potential for metabolic oxidation of the aromatic methyl group, included small cycloalkyl groups, fluoroalkyl groups, and small cycloalkoxy groups that we expected to be less prone to metabolic oxidation. Consistent with what we previously observed, replacement of the 4-Cl group of butanoic acid 8q with Me led to a significant improvement in potency (8s). Larger alkyl butanoic acids (19a-c, 19f) achieved even greater potency, approaching the limit of what our SRE.L assay can accurately determine (< 0.1 pM). While the CF3 analogs 8u,v,w all maintained activity comparable to their corresponding methyl analogs 8c,h,s, the larger fluoro containing derivative, 1-fluoroisopropyl 19d, was less potent than its alkyl comparator 19c, suggesting a possible size limit at R3. Interestingly, replacement of the 4-Cl of 8q with 4-Br (8x) led to a 10-fold improvement in activity, consistent with the greater potency observed when increasing the size of the 4-methyl group to small alkyls. Additional evidence for the size limitation at R3 was provided by the 4-Ph analog 8y. Incorporation of oxygen (e.g. 8t, 19h and 19i) also did not afford the same levels of activity as the corresponding aliphatic analogs, perhaps due to decreased permeability. The narrow SAR we had been observing was once again evident when the cyclopropyl at R3 of 19f was migrated to the R2 (19e) or R4 (19g) positions, leading to significant reductions in SRE.L activity. Finally, with regard to in vitro metabolic stability (as determined by half-life in the presence of mouse liver microsomes (MLM T½)), replacement of the Cl at R3 of 8q with more potent Me (8s) was detrimental as expected, but further replacement of the Me with c-Pr (19f) returned a measure of metabolic stability while improving potency 100-fold.
IV and PO PK analyses were performed on representative analogs 8j, 8q, and 19f (Table 4). Propionic acid 8j demonstrated good oral bioavailability (67%) and a moderate half-life (2 h). Rather surprisingly, the corresponding butanoic acid 8q, despite apparently similar metabolic stability in vitro, had inferior in vivo PK, with a clearance rate almost 3-fold higher than 8j and lower oral bioavailability. Butanoic acid 19f also had a higher in vivo clearance rate, suggesting that the longer alkanoic acid moiety is a primary site of metabolism. It is interesting to note, however, that plasma exposure of 19f was not linear with oral dose, increasing from 11% bioavailability at 3 mg/kg to 29% at 30 mg/kg, suggesting some saturation of first pass clearance.
Table 4.
Physical and Pharmacokinetic Parameters for Selected Rho/MRTF/SRF Inhibitors.
| Compound | SRE. L IC50 (nM) | Solubility (uM) | MLM T½ (min)c | Route, Dose (mg/kg) | T ½ (hr) | AUC (hr*ng/mL) | Cl (mL/min/kg) | Cmax (ng/mL) | F% |
|---|---|---|---|---|---|---|---|---|---|
| 8j | 4 | 682a | >60 | IV, 15 | 2.0 | 47350 | 5.3 | 88550 | - |
| PO, 30 | 2.0 | 63170 | - | 34050 | 67 | ||||
| 8q | 1.8 | 841a | >60 | IV, 10 | 1.4 | 10716 | 15.5 | 35650 | - |
| PO, 20 | 1.2 | 11140 | - | 23150 | 52 | ||||
|
19f (CCG-232964) |
0.001 | >1000b | 47 | IV, 3 | 1.7 | 3255 | 15.4 | 8140 | - |
| PO, 3 | 1.6 | 361 | - | 397 | 11 | ||||
| PO, 10 | 1.8 | 1826 | - | 1433 | 17 | ||||
| PO, 30 | 2.2 | 9496 | - | 10053 | 29 |
Thermodynamic solubility analysis was performed by Analiza Inc. using quantitative nitrogen detection. (www.analiza.com).
Equilibrium solubility in PBS at 25 ¼C.
Half-life in mouse liver microsomes. PK parameters were estimated using non-compartmental analysis with Phoenix/WINONLIN.
To assess direct effects on pro-fibrotic gene expression, primary human dermal fibroblasts were pre-treated with 8j (CCG-58150) or 19f (CCG-232964) for 48 or 72 h (Figure 6). After the pre-incubation, cells were stimulated with LPA—a direct activator or the Rho pathway through Gα12/1341—for 1 h. Interestingly, after 48 h, neither 8j nor 19f had any significant effect on CTGF gene expression; however, after 72 h, both inhibitors dose-dependently reduced CTGF gene expression, with 19f being ~100x more potent than 8j. Under the same assay conditions, nintedanib inhibited CTGF expression at 10−6 M (45% of DMSO control) but not below, and was cytotoxic at 10−5 M. Pirfenidone did not significantly inhibit CTGF expression even at a concentration of 10−5 M (91% control).
Figure 6. 8j and 19f inhibit LPA induced CTGF gene expression.
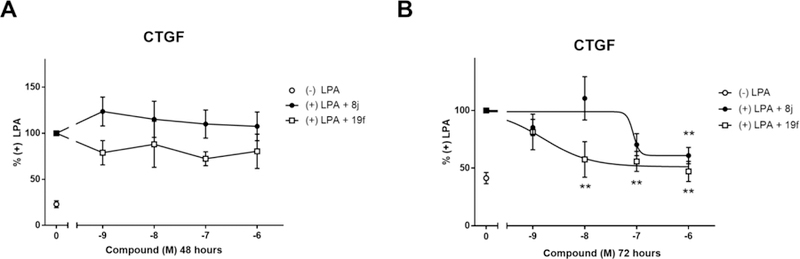
Primary dermal fibroblasts from healthy donors were pre-treated with varying concentrations of 8j (CCG-58150) and 19f (CCG-232964) for (A) 48 h or (B) 72 h and stimulated with 1 µM LPA for 1 h. CTGF mRNA levels were measured by qPCR. Results are expressed as the mean ± SEM. *p<0.05, **p<0.01 vs DMSO control using One-way ANOVA; n>3.
We selected a mouse model of dermal fibrosis that entails inhibition of dermal thickening induced by bleomycin as our preliminary measure of in vivo efficacy of 8j (CCG-58150) and 19f (CCG-232964). This model has been well-described in the literature29, 42–43, and histopathological examination of fibrotic lung and dermal tissue induced by bleomycin treatment in mice reveals a high level of similarity to fibrotic disease progression in humans.44 The protocol entails daily intracutaneous injections of bleomycin with concurrent treatment by drug or vehicle for 14 days. At the end of the dosing period, the mice are sacrificed and sections of skin at the injection site are excised and analyzed for dermal thickness and hydroxyproline—a major component of collagen and an accepted biomarker for collagen content.45
Based on the high Cmax achieved in the mouse PK study for 8j (30 mg/kg PO dosing (107 μM) relative to the SRE.L IC50 (4 nM)), we selected three lower doses for 8j (0.1, 1 and 10 mg/kg/day) by oral gavage during bleomycin treatment. 8j was able to dose-dependently reduce both measures of bleomycin-induced fibrotic effects (dermal thickness and hydroxyproline content), having significant effects at 10 mg/kg vs bleomycin/vehicle control (Figure 7A and 7B). In a subsequent study, two doses were selected for the more potent but less bioavailable compound 19f (3 and 10 mg/kg PO). The lower dose was calculated based on the single dose oral PK data (Table 4) to achieve comparable plasma AUC: SRE.L IC50 ratio as the efficacious 10 mg/kg dose of 8j. At the conclusion of this study, 19f significantly reduced bleomycin-induced dermal fibrosis at both doses, indicating that it is at least 3-fold more potent than 8j in vivo (Figure 7C–E). By way of comparison, nintedanib has been reported by the Distler lab to be effective at preventing bleomycin-induced dermal thickness in mice at a minimum dose of 30 mg/kg/day,46 suggesting that our compounds are significantly more potent in this assay. To our knowledge, pirfenidone has not been tested in this assay for prevention of dermal fibrosis, but has been reported to attenuate bleomycin-induced lung fibrosis in mice at a dose of 300 mg/kg.47
Figure 7. 5-Aryl-1,3,4-oxadiazol-2-ylthioalkanoic Acids Prevent Bleomycin-Induced Dermal Fibrosis.
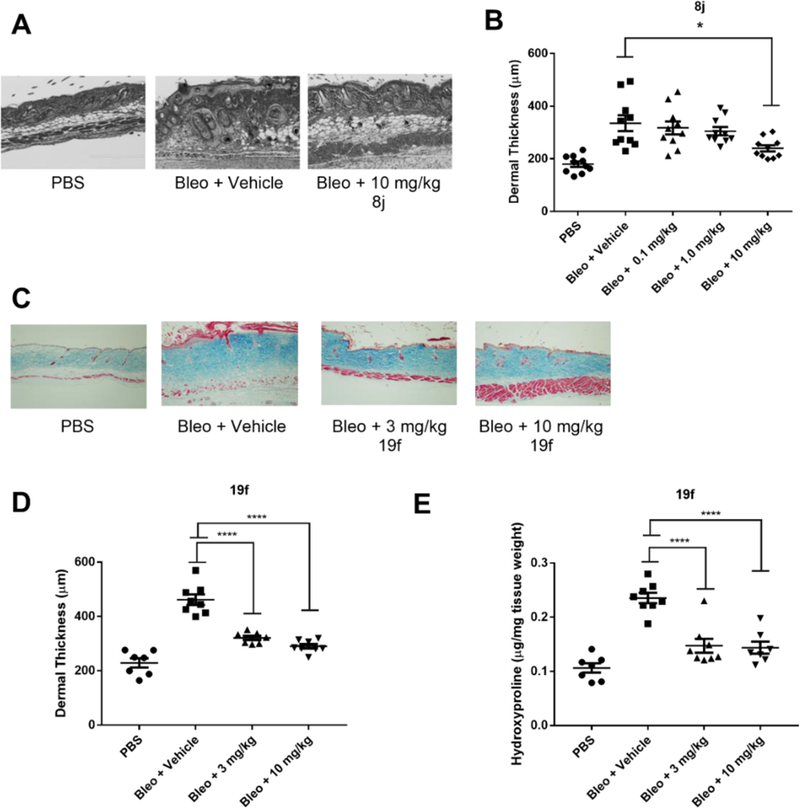
Mice treated with inhibitors displayed significantly reduced skin thickness and collagen content as compared to vehicle control. (A) Masson’s trichrome stained skin sections from PBS (left panel), Bleomycin + Vehicle control (middle panel), and Bleomycin +10 mg/kg 8j (right panel). N=10 per group (B) Quantification of A, by measuring the distance between the subcutaneous fat and the epidermis. *, p<0.05 by using One-way ANOVA (C) Masson’s trichrome stained skin sections for PBS (left panel), Bleomycin+Vehicle (middle left panel), Bleomycin +3 mg/kg 19f (middle right panel) or Bleomcyin +10 mg/kg 19f (right panel). N=7 per group (D) Quantification of C. ****, p<0.0001, using One-way ANOVA. (E) Quantification of collagen content by hydroxyproline measurement. ****, p<0.0001, using One-way ANOVA.
Conclusion:
In conclusion, we have developed a novel and highly potent series of inhibitors of Rho/MRTF/SRF-mediated gene transcription. The hit 1 was discovered during our previously reported phenotypic HTS utilizing a SRE.L expression readout. We systematically explored modifications to the heterocyclic core, the aromatic ring at C-5, and the alkanoic acid sidechain. During this process we observed extremely tight SAR and multiple activity cliffs. We ultimately achieved an astonishing 150,000-fold improvement in SRE.L activity while progressing from hit 1 (SRE.L IC50 = 180 nM) to lead 19f (SRE.L IC50 = 0.001 nM). Based on the structural similarity of our chemical series to previously reported in vivo probes that can label proteins, it is possible that the extreme potency we have achieved is due to the availability of a covalent mode of binding to the unknown molecular target. Our SAR in part supports that hypothesis by showing that simple modification of the potentially reactive thio group largely abolishes activity, but confirmation will require further work to unambiguously demonstrate labeling of cellular proteins. Despite their extraordinarily high potency at inhibiting SRE.L driven gene expression, the compounds have very low acute cytotoxicity up to 100 μM, as measured by WST-1 cell proliferation.
Selected SRE.L inhibitors 8j and 19f were able to reduce expression of the pro-fibrotic gene CTGF in LPA-stimulated human fibroblasts. In improving the series’ activity, we were also able to generate probes with acceptable PK profiles for in vivo studies. The potent and dose-dependent anti-fibrotic effects displayed by both 8j (CCG-58150) and 19f (CCG-232964) introduce orally efficacious 5-aryl-1,3,4-oxadiazol-2-ylthioalkanoic acid Rho/MRTF/SRF-mediate gene transcription inhibitors as potential therapeutics for fibrotic diseases. As part of further development of these inhibitors, identification of the biological target(s) will be key and is under active investigation.
Experimental Section:
SRE.L Luciferase:
For luciferase assays using PC3 cells, 20,000 cells/well were plated in a 96- well plate in 10% FBS+DMEM. The next day, cells were transfected with 56 ng of SRE.L Luciferase reporter and 2 ng/well of either Gα12 Q231L or pcDNA using Lipofectamine 2000 (Life Technologies) and Opti-MEM (Gibco). After 4 h of transfection, media was changed to 0.5% FBS+DMEM and cells were treated with compounds or DMSO control overnight. The next day, viability was measured using WST-1 (Roche) and absorbance was read at 450 nm. After viability was measured, cells were lysed, and luciferase was measured using the Luciferase Assay System (Promega).
For luciferase assays in HEK293T cells, see Hutchings et al.25
qPCR for LPA-stimulated Expression of CTGF:
LPA stimulation
Primary dermal fibroblasts isolated from healthy donors were cultured in DMEM containing 10% FBS and 1% Pen/Strep. Prior to LPA stimulation, cells were seeded in 0.5% FBS containing DMEM and pre-treated with compounds or DMSO control for 48 or 72 h. After the pre-treatment, LPA (1-Oleoyl Lysophosphatidic Acid, Cayman Chemical, Cat #10010093) was added at a final concentration of 10 µM for 1 h in DMEM containing 0.5% FBS and 1% Pen/Strep. After LPA stimulation, cells were lysed in iScript RT-qPCR lysis buffer (Biorad, Hercules CA, Cat #170–8898). Cells used for experiments were passaged no more than 8 times.
qPCR
For qPCR analysis, the iTaq Universal SYBR Green One Step Kit was used (BioRad, Hercules, CA Cat #172–5151) following the manufacturer’s instructions. 1 µL of template lysate was added to the RT-qPCR master mix. Primers used were: CTGF Forward 5’-CAGAGTGGAGCGCCTGTT −3’, CTGF Reverse 5’- TTGTCCGCGAGGTGAGAC-3’, GAPDH forward 5’ GAAGGTGAAGGTCGGAGTCA and GAPDH reverse 5’-TTGAGGTCAATGAAGGGGTC-3’. Fold differences were calculated using the ΔΔCt method with GAPDH as a reference. PCR products were analyzed on a 1% agarose gel.
Metabolic Stability in Mouse Liver Microsomes:
The metabolic stability was assessed using CD-1 mouse liver microsomes. One micromolar of each compound was incubated with 0.5 mg/mL microsomes and 1.7 mM cofactor NADPH in 0.1 M phosphate buffer (pH = 7.4) containing 3.3 mM MgCl2 at 37 °C. The DMSO concentration was less than 0.1% in the final incubation system. At 0, 5, 10, 15, 30, 45, and 60 min of incubation, 40 µL of reaction mixture were taken out, and the reaction is quenched by adding 3-fold excess of cold acetonitrile containing 100 ng/mL of internal standard for quantification. The collected fractions were centrifuged at 15000 rpm for 10 min to collect the supernatant for LC−MS/ MS analysis, from which the amount of compound remaining was determined. The natural log of the amount of compound remaining was plotted against time to determine the disappearance rate and the half-life of tested compounds.
Pharmacokinetic Studies in Mice:
All animal experiments in this study were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine. The abbreviated pharmacokinetics for 8j, 8q, and 19f were determined in female CD-1 mice following intravenous (iv) and/or orally (po). Compound was dissolved in the vehicle containing 15% (v/v) DMSO, 15% (v/v) PEG-400, and 70% (v/v) PBS. Four blood samples (50 μL) were collected over 7 h (at 0.5h, 2h, 4h, and 7h), centrifuged at 3500 rpm for 10 min, and plasma was frozen at −80˚C for later analysis. Plasma concentrations of the compounds were determined by the LC−MS/MS method developed and validated for this study. The LC−MS/MS method consisted of a Shimadzu HPLC system, and chromatographic separation of tested compound which was achieved using a Waters Xbridge-C18 column (5 cm × 2.1 mm, 3.5 μm). An AB Sciex QTrap 4500 mass spectrometer equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. All pharmacokinetic parameters were calculated by non-compartmental methods using WinNonlin, version 3.2 (Pharsight Corporation, Mountain View, CA, USA).
Bleomycin-Induced Antifibrotic Effects in Mice—CCG-58150 (8j):
During intradermal injections and gavage of test substance mice were anesthetized using isoflurane. The back was shaved and the injection site (~1cm2) cleaned with three alternating passes of chlorhexidine and warm, sterile saline or water. The periphery of the injection site was circled using a sharpie marker as a visual aid. Bleomycin was dissolved in sterile PBS at a concentration of 1mg/ml, sterile filtered, and frozen in aliquots appropriately sized for the daily injections prior to the start of the dosing period. 100μl of bleomycin or PBS (Vehicle 1) was administered to anesthetized mice by intradermal injection using a 0.5cc TB syringe with a 27G needle. Following bleomycin or vehicle administration, test substance (8j, CCG-58150) or Vehicle 2 (20% DMSO/30% PEG/50% PBS) was administered by oral gavage at 10μL/gram of body weight. Mice received daily clinical observations, injections of vehicle/bleomycin and oral vehicle/test substance for 14 days. Mice were weighed weekly and were euthanized on Day 15. The bleomycin injection site was visually assessed, and apparent fibrosis was graded as Not Detectable (0), Mild Fibrosis (1), Moderate Fibrosis (2) or Severe Fibrosis (3), based on the appearance of lesion. A terminal blood sample was collected for determination of serum chemistry values. The liver was collected and fixed in neutral buffered formalin, slides prepared and stained with Hematoxolin and eosin (H&E). The skin at the injection site was excised to contain a few mm of normal skin around the perimeter of the fibrotic skin. A small section (~5×2mm) of the fibrotic skin was collected using a scalpel, placed in a microfuge tube, snap frozen in liquid nitrogen, and stored at −80°C for future evaluation by the sponsor. The remaining tissue was pinned flat and fixed in 10% buffered formalin at room temperature. After fixation for at least 24 h, the skin was cut into 1–2mm wide strips and embedded in paraffin with the tissue cross section facing up and all strips oriented in the same direction in the cassette. Slides were prepared and stained with H& E and Masson’s trichrome. All slides and frozen tissue were sent to the Sponsor at study completion.
Bleomycin-Induced Antifibrotic Effects in Mice—CCG-232964 (19f):
C57BL/6J mice were ordered from Jackson labs for delivery at 15–16 weeks of age. Mice were preconditioned with supplemental chow for two weeks prior to start of experiment. Mice were randomized by weight into four groups (vehicle, 19f (3 mg/kg), 19f (10 mg/kg), PBS). Each group consisted of two cages with four mice per cage. Drug stock solutions were aliquoted and stored at 4 °C for the duration of the study. Aliquots were warmed to 37 °C and briefly vortexed prior to filling the gavage syringes. Vehicle and 19f groups received daily bleomycin injections (0.1mL, ID) each afternoon in a defined area of shaved dorsal skin. PBS control group received daily injections (0.1mL, ID) of PBS in the afternoon. Mice were anesthetized with ketamine/xylazine for ID injections. 19f (3 or 10 mg/kg) and the vehicle (20% DMSO/50% PEG-400/30% PBS) were administered (po) in the morning by the University of Michigan’s Unit for Laboratory Animal Medicine In-Vivo Animal Core. Mice were weighed daily to determine gavage dosage volume. Mice were euthanized after fourteen days of treatment by CO2 inhalation/thoracotomy. Skin from the defined area was excised for biochemical and histological analysis. A portion of the skin was fixed in neutral buffered formalin (10%), washed in 70% ethanol, and paraffin embedded for Masson’s trichrome histological staining. Another portion of the skin was snap frozen for hydroxyproline measurement.
Fixed skin was paraffin embedded and sectioned at the University of Michigan Comprehensive Cancer Center Histology Core. Skin sections were stained with Masson’s trichrome (Sigma-Aldrich). Stained sections were analyzed with an Olympus BX51\DP72 microscope. Dermal thickness was determined by measuring the maximal distance between the epidermal-dermal junction and the dermal-subcutaneous fat junction. Three measurements were averaged from each skin section. The measurement was performed using the measurement tool in the cellSens imaging software package (Olympus).
Skin sections were weighed and hydrolyzed in 6M HCl at 120 °C for three h. Hydrolyzed skin supernatant and hydroxyproline standards (Sigma-Aldrich) were transferred to a microplate and dried at 60 °C. Samples and standards were oxidized with Chloramine T oxidation buffer for 5 minutes at room temperature. 4-(Dimethylamino) benzaldehyde was added to the wells and incubated at 60 °C until the standard was well defined. Absorbance was measured at 560 nm using a Synergy HT microplate reader (BioTek Instruments). Hydroxyproline values were normalized to tissue weight.
Chemistry:
All reagents were used without further purification as received from commercial sources unless noted otherwise. 1H NMR and 13C NMR spectra were taken in DMSO-d6, MeOD-d4, or CDCl3 at room temperature on Varian Inova 400 MHz or Varian Inova 500 MHz instruments. Reported chemical shifts for the 1H NMR and 13C NMR spectra were recorded in parts per million (ppm) on the δ scale from an internal standard of residual tetramethylsilane (0 ppm). Mass spectrometry data were obtained on either a Micromass LCT or Agilent Q-TOF. An Agilent 1100 series HPLC with an Agilent Zorbax Eclipse Plus−C18 column (3.5 μM, 4.6 × 100 mm) was used to determine purity of biologically tested compounds. All tested compounds were determined to be >95% pure using a 6-minute gradient of 10–90% acetonitrile in water followed by a 2-minute hold at 90% acetonitrile at a flow rate of 1 mL/min with detection at 254 nm. Purification was accomplished using silica gel 40–63µm 60Å for column chromatography. Purification of some final compounds required Waters semipreparative HPLC with a Vydac protein and peptide C18 reverse phase column, using a linear gradient of 0% solvent B (0.1% TFA in acetonitrile) in solvent A (0.1% TFA in water) to 100% solvent B in solvent A at a rate 1% per minute and monitoring UV absorbance at 230 nm.
Method A: General Procedure for Fisher Esterification of Aryl Carbocyclic Acids
The appropriate aryl acid (1.0 eq) was dissolved in MeOH (2–4 mL/1 mmol), and 98% sulfuric acid (1 eq) was added. The mixture was heated to reflux 4–16 h. The reaction solution was cooled to room temperature and concentrated in vacuo. The resulting crude residue was dissolved in a sat. NaHCO3 (aq) solution and extracted with DCM. The combined organic layers were washed with brine, dried over MgSO4 and concentrated to afford the desired ester. Yields: 50%−100%
Method B: General Procedure for Hydrazine Coupling of Aryl Methyl Carboxylates
In a round-bottomed flask, the starting aryl ester (1.0 eq) was dissolved in MeOH (2–4 mL/mmol), and hydrazine hydrate (10 eq) was added. The solution was heated to reflux (85 °C) for overnight. The reaction was cooled to 25 °C and concentrated in vacuo. The residue was taken up in DCM, partitioned between water, and the product was extracted with DCM (3× 20 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo to obtain white crystals. Yields: 70–100%
Method C: General Procedure for 1, 3, 4-oxadiazole-thiol Cyclization of Aryl Hydrazides
In a round-bottomed flask, the appropriate aryl hydrazide (1eq) was dissolved in a 0.1 M solution of KOH (1 eq) in EtOH (2–4 mL/mol)/H2O (10 eq) and placed under nitrogen. Carbon disulfide (1 eq) was added and the reaction was heated to reflux (95 °C) overnight. Upon completion, most of the EtOH was evaporated in vacuo and the residue was cooled to 0 °C. The product was acidified to pH 1 with 1 N HCl, and the subsequent white precipitate was filtered and dried under vacuum. Yields: 50–90%
Method D: General Procedure for Alkyl Bromide Coupling of 1,3,4-Oxadiazole-2-Thiols
The starting thiol (1 eq), acetone (2–4 mL/1 mmol), and potassium carbonate (1.3 eq) were combined in a round-bottomed flask. The alkyl bromide (1.3 eq mmol) was added to the solution and the reaction was stirred under nitrogen at 25 °C overnight. The crude mixture was concentrated in vacuo, and the residue was partitioned between DCM and water. The product was extracted with DCM (3× 15 mL), washed with brine (30 mL), dried with MgSO4, and concentrated in vacuo. The oil was subjected to silica gel chromatography eluting with 30–80% EtOAc: Hex. The fractions containing product were concentrated; and, after sitting overnight, some of the subsequent oils produced white solid. Yields: 50–95%
Method E: General Procedure for Acidic Ester Hydrolysis of t-Butyl Esters
In a round-bottomed flask the appropriate t-butyl ester (1 eq) was dissolved in DCM (2–4 mL/1 mmol). Trifluoroacetic acid (25 eq) was added and reaction was stirred under nitrogen at 25 °C for 3 h. The crude mixture was concentrated in vacuo and then dissolved in a minimal amount of EtOAc. The product was triturated out with Hex, and the subsequent white precipitate was filtered and dried under vacuum. Yields: 20–90%
Method F: General Procedure for Basic Ester Hydrolysis of Methyl Esters
In a round-bottomed flask, the starting methyl ester (1 eq) was dissolved in THF (1–2 mL/1 mmol). 1 M NaOH (1–2 mL/1 mmol) was added to the solution and the reaction was stirred at 25 °C for 3 h. The THF was evaporated in vacuo, and then the aqueous layer was acidified with 1 N HCl (7 mL). The product was extracted with EtOAc (3× 15 mL), washed with brine (3× 10 mL), dried with MgSO4, and concentrated in vacuo. The residue was dissolved in minimal EtOAc and the product was triturated with Hex, producing a white precipitate that was filtered and dried under vacuum. Yields: 50–90%
Method G: General Procedure for Suzuki Cross Coupling of Aryl Bromides
The appropriate aryl bromide (1 eq) was dissolved in toluene (2–4 mL/1 mmol) and degassed under inert atmosphere. H2O (10 eq), potassium phosphate dibasic (3.5 eq), cyclopropyl boronic acid (1.2 eq), tricyclohexylphosphine (0.1 eq), and palladium (II) acetate (0.15 eq) were added. The mixture was heated at reflux (100 °C) for 3 hr under N2. The mixture was cooled to 25 °C and H2O was added. The product was extracted with EtOAc (3 × 15 mL), washed with brine (2 × 15 mL), dried with MgSO4, and concentrated in vacuo. The yellow residue was subjected to silica gel chromatography eluting with 2.5% EtOAc: 97.5% Hex. Yields: 70–95%
2-chloro-4-methylbenzohydrazide (3k)
Method B starting from methyl 2-chloro-4-methylbenzoate (2k’, 1.0 g) gave 2-chloro-4-methylbenzohydrazide as white powder (0.79 g, 73%); 1H NMR (400 MHz, CDCl3) δ 7.63 (s, 1H) 7.55 (d, J = 7.9 Hz, 1H) 7.22 (d, J = 1.6 Hz, 1H) 7.12 (dd, J = 7.9, 1.6 Hz, 1H) 4.14 (br. s., 2H) 2.36 (s, 3H); HPLC Ret: 3.95 min.
2-chloro-4-methoxybenzohydrazide (3l)
Method A followed by Method B starting from 2-chloro-4-methoxybenzoic acid (2l) gave 2-chloro-4-methoxybenzohydrazide as off-white solid (2-steps, 886 mg, 82%); 1H NMR (500 MHz, DMSO-d6) δ 9.43 (t, J=4.1 Hz, 1H), 7.31 (d, J=8.6 Hz, 1H), 7.04 (d, J=2.5 Hz, 1H), 6.92 (dd, J=8.5, 2.5 Hz, 1H), 4.43 (d, J=4.2 Hz, 2H),3.78 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 166.0, 160.9, 132.0, 130.7, 128.1, 115.4, 113.3, 56.2; MS (ESI+) m/z: 200.9 [M+H]+; HPLC Ret: 4.33 min.
2-chloro-3,4-dimethylbenzohydrazide (3m)
Method A followed by Method B starting from 2-chloro-3,4-dimethylbenzoic acid (2m, 0.25 g) gave 2-chloro-3,4-dimethylbenzohydrazide as white solid (2-steps, 0.27 g, 100%); 1H NMR (500 MHz, DMSO-d6) δ 9.42 (br. s., 1H) 7.16 (d, J = 7.7 Hz, 1H) 7.07 (d, J = 7.7 Hz, 1H) 4.43 (br. s., 2H) 2.32 (s, 3H) 2.29 (s, 3H); HRMS (ESI+) m/z: 199.0633 [M+H]+ (expected 199.0638); HPLC Ret: 4.33 min.
2-chloro-4,5-dimethylbenzohydrazide (3n)
Method A followed by Method B starting from 2-chloro-4,5-dimethylbenzoic acid (2n, 0.25 g) gave 2-chloro-4,5-dimethylbenzohydrazide as white solid (2-steps, 0.24 g, 90%); 1H NMR (500 MHz, DMSO-d6) δ 9.43 (br. s., 1H) 7.26 (s, 1H) 7.16 (s, 1H) 4.44 (br. s., 2H) 2.23 (s, 3H) 2.19 (s, 3H); HRMS (ESI+) m/z: 199.0635 [M+H]+ (expected 199.0638); HPLC Ret: 4.41 min.
2,4-dichloro-5-methylbenzohydrazide (3o)
Method A followed by Method B starting from 2,4-dichloro-5-methylbenzoic acid (2o, 0.09 g) gave 2,4-dichloro-5-methylbenzohydrazide as white solid (2-steps, 0.07 g, 72%); 1H NMR (500 MHz, DMSO-d6) δ 9.56 (br. s., 1H) 7.63 (s, 1H) 7.40 (s, 1H) 4.49 (br. s., 2H) 2.32 (s, 3H); HRMS (ESI+) m/z: 219.0087 [M+H]+ (expected 219.0092); HPLC Ret: 4.74 min.
2-(trifluoromethyl)benzohydrazide (3v)
Method B starting from methyl 2-(trifluoromethyl)benzoate (2v’, 0.5 g) gave 2-(trifluoromethyl)benzohydrazide as white solid (0.38 g, 76%); 1H NMR (500 MHz, CDCl3) δ 7.73 (dd, J = 7.7, 1.3 Hz, 1H) 7.65–7.48 (m, 3H) 6.98 (br. s, 1H) 4.12 (br. s, 2H); HRMS (ESI+) m/z: 205.0585 [M+H]+ (expected 205.0589); HPLC Ret: 3.25 min.
2-chloro-4-(trifluoromethyl)benzohydrazide (3w)
Method B starting from methyl 2-chloro-4-(trifluoromethyl)benzoate (2w’, 0.5 g) gave 2-chloro-4-(trifluoromethyl)benzohydrazide as white solid (0.48 g, 98%); 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.1 Hz, 1H) 7.70 (d, J = 1.7 Hz, 1H) 7.60 (dd, J = 8.0, 1.7 Hz, 1H) 7.50 (br. s., 1H) 3.94 (br. s., 2H); MS (ESI+) m/z: 239.0 [M+H]+; HPLC Ret: 4.48 min.
4-bromo-2-chlorobenzohydrazide (3x)
Method B starting from methyl 4-bromo-2-chlorobenzoate (2x’, 0.5 g) gave 4-bromo-2-chlorobenzohydrazide as white solid (0.46 g, 97%); 1H NMR (400 MHz, DMSO-d6) δ 7.42 (d, J = 8.2 Hz, 1H) 7.31 (s, 1H) 7.19 (dd, J = 8.2, 1.9 Hz, 1H); MS (ESI+) m/z: 249.0 [M+H]+; HPLC Ret: 4.48 min.
3-chloro-[1, 1’-biphenyl]-4-carbohydrazide (3y)
Method B starting from methyl 3-chloro-[1, 1’-biphenyl]-4-carboxylate (2x’, 0.42 g) gave 3-chloro-[1, 1’-biphenyl]-4-carbohydrazide as white solid (0.36 g, 94%); 1H NMR (500 MHz, DMSO-d6) δ 9.62 (d, J = 26.9 Hz, 1H) 7.87–7.61 (m, 3H) 7.57–7.34 (m, 5H) 4.53 (d, J = 24.8 Hz, 2H); MS (ESI+) m/z: 247.0 [M+H]+; HPLC Ret: 5.36 min.
5-(2-chloro-4-methylphenyl)-1,3,4-oxadiaole-2-thiol (4k)
Method C starting from 2-chloro-4-methylbenzohydrazide (3k, 0.3 g) gave 5-(2-chloro-4-methylphenyl)-1,3,4-oxadiaole-2-thiol as white powder (0.29 g, 77%); 1H NMR (400 MHz, DMSO-d6) δ 7.80 (d, J = 8.0 Hz, 1H) 7.55 (s, 1H) 7.37 (d, J = 8.1 Hz, 1H) 2.39 (s, 3H); HPLC Ret: 6.70 min.
5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazole-2-thiol (4l)
Method C starting from 2-chloro-4-methoxybenzohydrazide (886 mg, 3l) gave 5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazole-2-thiol as white solid (912 mg, 85%); 1H NMR (500 MHz, DMSO-d6) δ 7.80 (d, J=8.8 Hz, 1H), 7.24 (d, J=2.5 Hz, 1H), 7.09 (dd, J=8.8, 2.5 Hz, 1H), 3.85 (s, 3H); MS (ESI+) m/z: 242.9 [M+H]+; HPLC Ret: 6.30 min.
5-(2-chloro-3,4-dimethylphenyl)-1,3,4-oxadiazole-2-thiol (4m)
Method C starting from 2-chloro-3,4-dimethylbenzohydrazide (3m, 0.27 g) gave 5-(2-chloro-3,4-dimethylphenyl)-1,3,4-oxadiazole-2-thiol as white solid (0.28 g, 86%); 1H NMR (500 MHz, CDCl3) δ 10.71 (br. s., 1H) 7.62 (d, J = 7.7 Hz, 1H) 7.21 (d, J = 8.0 Hz, 1H) 2.41 (br. s., 6H); HRMS (ESI+) m/z: 241.0192 [M+H]+ (expected 241.0202); HPLC Ret: 6.97 min.
5-(2-chloro-4,5-dimethylphenyl)-1,3,4-oxadiazole-2-thiol (4n)
Method C starting from 2-chloro-4,5-dimethylbenzohydrazide (3n, 0.24 g) gave 5-(2-chloro-4,5-dimethylphenyl)-1,3,4-oxadiazole-2-thiol as white solild (0.22 g, 76%); 1H NMR (500 MHz, CDCl3) δ 11.03 (br. s., 1H) 7.69 (s, 1H) 7.32 (s, 1H) 2.32 (br. s., 6H); MS (ESI+) m/z: 241.0194 [M+H]+ (expected 241.0202); HPLC Ret: 7.00 min.
5-(2,4-dichloro-5-methylphenyl)-1,3,4-oxadiazole-2-thiol (4o)
Method C starting from 2,4-dichloro-5-methylbenzohydrazide (3o, 0.07 g) gave 5-(2,4-dichloro-5-methylphenyl)-1,3,4-oxadiazole-2-thiol as yellow solid (0.07 g, 85%); 1H NMR (500 MHz, CDCl3) δ 10.30 (br. s., 1H) 7.80 (s, 1H) 7.57 (s, 1H) 2.42 (s, 3H); HRMS (ESI+) m/z: 260.9656 [M+H]+ (expected 260.9656); HPLC Ret: 7.23 min.
5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazole-2-thiol (4u)
Method C starting from 4-(trifluoromethyl)benzohydrazide (3u, 0.25 g) gave 5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazole-2-thiol as white solid (0.26 g, 86%); 1H NMR (500 MHz, CDCl3) δ 10.66 (br. s, 1H) 8.07 (d, J = 8.1 Hz, 2H) 7.79 (d, J = 8.1 Hz, 2H); HRMS (ESI-) m/z: 244.9999 [M-H]- (expected 244.9996) HPLC Ret: 6.92 min.
5-(2-(trifluoromethyl)phenyl)-1,3,4-oxadiazole-2-thiol (4v)
Method C starting from 2-(trifluoromethyl)benzohydrazide (3v, 0.38 g) gave 5-(2-(trifluoromethyl)phenyl)-1,3,4-oxadiazole-2-thiol as white powder (0.41 g, 90%); 1H NMR (500 MHz, CDCl3) δ 10.64 (br. s, 1H) 7.98–7.85 (m, 2H) 7.77–7.70 (m, 2H); HPLC Ret: 6.49 min.
5-(2-chloro-4-((trifluoromethyl)phenyl)-1,3,4-oxadiazole-2-thiol (4w)
Method C starting from 2-chloro-4-(trifluoromethyl)benzohydrazide (3w, 0.48 g) gave 5-(2-chloro-4-((trifluoromethyl)phenyl)-1,3,4-oxadiazole-2-thiol as white powder (0.26 g, 59%); 1H NMR (500 MHz, DMSO-d6) δ 8.16–8.09 (m, 2H) 7.95–7.89 (m, 1H); MS (ESI+) m/z: 280.1 [M+H]+; HPLC Ret: 7.14 min.
5-(4-bromo-2-chlorophenyl)-1,3,4-oxadiazole-2-thiol (4x)
Method C starting from 4-bromo-2-chlorobenzohydrazide (3x, 0.46 g) gave 5-(4-bromo-2-chlorophenyl)-1,3,4-oxadiazole-2-thiol as white solid (0.37 g, 81%); 1H NMR (400 MHz, DMSO-d6) δ 7.99 (d, J = 8.2 Hz, 1H) 7.84–7.72 (m, 2H); MS (ESI+) m/z: 292.9 [M+Na]+; HPLC Ret: 7.06 min.
5-(3-chloro-[1, 1’-biphenyl]-4-yl)-1, 3, 4-oxadiazole-2-thiol (4y)
Method C starting from 3-chloro-[1, 1’-biphenyl]-4-carbohydrazide (3y, 0.36 g) gave 5-(3-chloro-[1, 1’-biphenyl]-4-yl)-1, 3, 4-oxadiazole-2-thiol as white solid (0.29 g, 88%); 1H NMR (400 MHz, DMSO-d6) δ 8.02–7.93 (m, 2H) 7.80–7.72 (m, 3H) 7.53–7.40 (m, 3H); MS (ESI+) m/z: 289.0 [M+H]+; HPLC Ret: 7.69 min.
methyl-4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (5q)
Method D starting from 5-(2,4-dichlorophenyl)-1,3,4-oxadiazole-2-thiol (4j, 0.2 g) gave methyl-4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.19 g, 66%); 1H NMR (400 MHz, DMSO-d6) δ 8.00 (d, J = 8.5 Hz, 1H) 7.92 (d, J = 2.1 Hz, 1H) 7.66 (dd, J = 8.5, 2.1 Hz, 1H) 3.60 (s, 3H) 3.35 (t, J = 7.4 Hz, 2H) 2.50 (t, J = 7.4 Hz, 2H) 2.12–1.97 (m, 2H); HRMS (ESI+) m/z: 368.9836 [M+Na]+ (expected 368.9843); HPLC Ret: 7.94 min.
methyl 5-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)pentanoate (5r)
Method D starting from 5-(2,4-dichlorophenyl)-1,3,4-oxadiazole-2-thiol (4j, 0.27 g) gave methyl 5-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)pentanoate as white solid (330 g, 82%); 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J=8.4 Hz, 1H), 7.90 (J=2.1 Hz, 1H), 7.64 (dd, J=8.5, 2.1 Hz, 1H), 3.55 (s, 3H), 3.30 (t, J=7.3 Hz, 2H), 2.35 (t, J=7.3 Hz, 2H), 1.84–1.72 (m, 2H), 1.79–1.63 (m, 2H); MS (ESI+) m/z: 382.9 [M+Na]+; HPLC Ret: 7.95 min.
methyl 4-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (5s)
Method D starting from 5-(2-chloro-4-methyl)-1,3,4-oxadiazole-2-thiol (4k, 0.02 g, 0.10 mmol) gave methyl 4-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (0.02 g, 70%); 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 8.0 Hz, 1H) 7.36 (d, J = 1.2 Hz, 1H) 7.18 (dd, J = 8.0, 1.2 Hz, 1H) 3.69 (s, 3H) 3.36 (t, J = 7.1 Hz, 2H) 2.53 (t, J = 7.2 Hz, 2H) 2.41 (s, 3H) 2.22 (p, J = 7.2 Hz, 2H); HRMS (ESI+) m/z: 349.0403 [M+Na]+ (expected 349.0390); HPLC Ret: 7.62 min.
methyl 4-((5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (5t)
Method D starting from 5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazole-2-thiol (4l, 0.1 g) gave methyl 4-((5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.12 g, 82%); 1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 8.8 Hz, 1H) 7.05 (dd, J = 2.5 Hz, 1H) 6.92 (dd, J = 8.8, 2.5 Hz, 1H) 3.87 (s, 3H) 3.69 (s, 3H) 3.35 (t, J = 7.1 Hz, 2H) 2.53 (t, J = 7.2 Hz, 2H) 2.20 (p, J = 7.2 Hz, 2H); HRMS (ESI+) m/z: 343.0525 [M+H]+ (expected 343.0519); HPLC Ret: 7.31 min.
methyl 4-((5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (5u)
Method D starting from 5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazole-2-thiol (4u, 0.075 g) gave methyl 4-((5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as white solid (0.09 g, 87%); 1H NMR (500 MHz, CDCl3) δ 8.14 (d, J = 8.2 Hz, 2H) 7.77 (d, J = 8.2 Hz, 2H) 3.70 (s, 3H) 3.39 (t, J = 7.2 Hz, 2H) 2.54 (t, J = 7.1 Hz, 2H) 2.22 (p, J = 7.1 Hz, 2H); HRMS (ESI+) m/z: 347.0674 [M+H]+ (expected 347.0677); HPLC Ret: 7.75 min.
methyl 4-((5-(2-chloro-4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (5w)
Method D starting from 5-(2-chloro-4-((trifluoromethyl)phenyl)-1,3,4-oxadiazole-2-thiol (4w, 0.1 g) gave methyl 4-((5-(2-chloro-4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.05 g, 47%); HPLC Ret: 7.98 min.
Methyl 4-((5-(3-chloro-[1, 1’-biphenyl]-4-yl)-1, 3, 4-oxadiazol-2-yl)thio)butanoate (5y)
Method D starting from 5-(3-chloro-[1, 1’-biphenyl]-4-yl)-1, 3, 4-oxadiazole-2-thiol (4y, 0.1 g) gave methyl 4-((5-(3-chloro-[1, 1’-biphenyl]-4-yl)-1, 3, 4-oxadiazol-2-yl)thio)butanoate as white solid (0.08 g, 75%); 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 8.2 Hz, 1H) 7.77 (d, J = 1.8 Hz, 1H) 7.64–7.58 (m, 3H) 7.52–7.39 (m, 3H) 3.70 (s, 3H) 3.38 (t, J = 7.1 Hz, 2H) 2.54 (t, J = 7.2 Hz, 2H) 2.22 (p, J = 7.2 Hz, 2H); MS (ESI+) m/z: 389.0 [M+H]+; HPLC Ret: 8.00 min.
Ethyl 3-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (6)
Method D starting from 5-(2,4-dichlorophenyl)-1,3,4-oxadiazole-2-thiol (4j, 1.2 g) gave Ethyl 3-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate as white solid (1.6 g, 91%); 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J=8.5 Hz, 1H), 7.90 (d, J=2.1 Hz, 1H), 7.64 (dd, J=8.5, 2.1 Hz, 1H), 4.08 (q, J=7.1 Hz, 2H), 3.48 (t, J=6.8 Hz, 2H), 2.90 (t, J=6.8 Hz, 2H), 1.17 (t, J=7.1 Hz, 3H); MS (ESI+) m/z: 346.8 [M +H]+; HPLC Ret: 7.95 min; 98% pure.
tert-butyl 3-((5-(o-tolyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7h)
Method D starting from 5-(2-methylphenyl)-1,3,4-oxadiazole-2-thiol (4h) gave tert-butyl 3-((5-(o-tolyl)-1,3,4-oxadiazol-2-yl)thio)propanoate as colorless oil (313 mg, 94%); 1H NMR (400 MHz, CDCl3) δ 7.87 (dd, J=7.9, 1.4 Hz, 1H), 7.40 (td, J=7.5, 1.4 Hz, 1H), 7.36–7.27 (m, 2H), 3.50 (t, J=6.8 Hz, 2H), 2.87 (t, J=6.8 Hz, 2H), 2.69 (s, 3H), 1.46 (s, 9H).
tert-butyl 3-((5-(2-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7i)
Method D starting from 5-(2-methoxyphenyl)-1,3,4-oxadiazole-2-thiol (4i) gave tert-butyl 3-((5-(2-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate as colorless oil (310 mg, 96%); 1H NMR (400 MHz, CDCl3) δ 7.77 (dd, J=7.9, 1.8 Hz), 7.39 (ddd, J=8.9, 7.5, 1.8 Hz, 1H), 6.95 (dd, J=8.3, 6.2 Hz, 2H), 3.85 (s, 3H), 3.39 (t, J=6.9 Hz, 2H), 2.78 (t, J=6.9 Hz, 2H), 1.38 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 170.3, 164.5, 163.6, 157.6, 133.0, 130.0, 120.6, 112.6, 111.9, 81.2, 55.9, 35.3, 28.0, 27.5; MS (ESI+) m/z: 337.1 [M+H]+.
tert-butyl-3-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7k)
Method D starting from 5-(2-chloro-4-methyl)-1,3,4-oxadiazole-2-thiol (4k, 0.25 g) gave tert-butyl-3-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate as white solid (0.27 g, 70%); 1H NMR (400 MHz, DMSO-d6) δ 7.86 (d, J = 8.1 Hz, 1H) 7.56 (d, J = 1.5 Hz, 1H) 7.37 (d, J = 8.2 Hz, 1H) 3.45 (t, J = 6.8 Hz, 2H) 2.82 (t, J = 6.8 Hz, 2H) 2.40 (s, 3H) 1.41 (s, 9H); MS (ESI+) m/z: 354.0 [M+H]+; HPLC Ret: 8.66 min.
tert-butyl 3-((5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7l)
Method D starting from 5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazole-2-thiol (250 mg, 4l) gave tert-butyl 3-((5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate as colorless oil (276 mg, 90%); 1H NMR (400 MHz, DMSO-d6) δ 7.87 (dd, J=8.8, 2.5 Hz, 1H), 7.26 (d, J=2.5 Hz, 1H), 7.11 (dd, J=8.8, 2.6 Hz, 1H), 3.85 (s, 3H), 3.42 (t, J=6.8 Hz, 2H), 2.79 (t, J=6.8 Hz, 2H), 1.39 (s, 9H).
tert-butyl 3-((5-(2-chloro-3,4-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7m)
Method D starting from 5-(2-chloro-3,4-dimethylphenyl)-1,3,4-oxadiazole-2-thiol (4m, 0.14 g) gave tert-butyl 3-((5-(2-chloro-3,4-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate as colorless oil (0.17 g, 80%); 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 7.9 Hz, 1H) 7.18 (d, J = 7.9 Hz, 1H) 3.49 (t, J = 6.8 Hz, 2H) 3.42 (s, 3H) 2.86 (t, J = 6.9 Hz, 2H) 2.38 (s, 3H) 1.47 (s, 9H); HRMS (ESI+) m/z: 369.1035 [M+H]+ (expected 369.1040); HPLC Ret: 7.63 min.
tert-butyl 3-((5-(2-chloro-4,5-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7n)
Method D starting from 5-(2-chloro-4,5-dimethylphenyl)-1,3,4-oxadiazole-2-thiol (4n, 0.11 g) gave tert-butyl 3-((5-(2-chloro-4,5-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate as colorless oil (0.13 g, 77%); 1H NMR (500 MHz, CDCl3) δ 7.71 (s, 1H) 7.30 (s, 1H) 3.49 (t, J = 6.9 Hz, 2H) 3.32 (s, 3H) 2.86 (t, J = 6.8 Hz, 2H) 2.28 (s, 3H) 1.47 (s, 9H); HRMS (ESI+) m/z: 369.1034 [M+H]+ (expected 369.1040); HPLC Ret: 7.81 min.
tert-butyl 3-((5-(2,4-dichloro-5-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7o)
Method D starting from 5-(2,4-dichloro-5-methylphenyl)-1,3,4-oxadiazole-2-thiol (4o, 0.07 g) gave tert-butyl 3-((5-(2,4-dichloro-5-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate as green oil (0.09 g, 82%); 1H NMR (500 MHz, CDCl3) δ 7.84 (s, 1H) 7.55 (s, 1H) 3.51 (t, J = 6.8 Hz, 2H) 2.86 (t, J = 6.8 Hz, 2H) 2.42 (s, 3H) 1.47 (s, 9H); HRMS (ESI+) m/z: 389.0494 [M+H]+ (expected 389.0493); HPLC Ret: 8.95 min.
tert-butyl 4-((5-(2-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (7v)
Method D starting from 5-(2-(trifluoromethyl)phenyl)-1,3,4-oxadiazole-2-thiol (4v, 0.1 g) gave tert-butyl 4-((5-(2-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.14 g, 88%); 1H NMR (500 MHz, CDCl3) δ 8.06–8.00 (m, 1H) 7.85 (dd, J = 7.1, 1.9 Hz, 1H) 7.74–7.64 (m, 2H) 3.34 (t, J = 7.2 Hz, 2H) 2.43 (t, J = 7.2 Hz, 2H) 2.15 (p, J = 7.2 Hz, 2H) 1.45 (s, 9H); HRMS (ESI+) m/z: 389.1173 [M+H]+ (expected 389.1147); HPLC Ret: 8.46 min.
t-butyl 4-((5-(4-bromo-2-chlorophenyl)-1,3,4-oxadiazole-2-yl)thio)butanoate (7x)
Method D starting from 5-(4-bromo-2-chlorophenyl)-1,3,4-oxadiazole-2-thiol (4x, 0.1 g) gave t-butyl 4-((5-(4-bromo-2-chlorophenyl)-1,3,4-oxadiazole-2-yl)thio)butanoate as colorless oil (0.12 g, 84%); 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 8.4 Hz, 1H) 7.73 (d, J = 1.9 Hz, 1H) 7.54 (dd, J = 8.4, 1.9 Hz, 2H) 3.36 (t, J = 7.2 Hz, 2H) 2.43 (t, J = 7.2 Hz, 2H) 2.16 (p, J = 7.2 Hz, 2H) 1.45 (s, 9H); MS (ESI+) m/z: 434.9 [M+H]+; HPLC Ret: 9.22 min.
3-((5-(o-tolyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid (8h)
Method E starting from tert-butyl 3-((5-(o-tolyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (313 mg, 7h) gave 3-((5-(o-tolyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid as off-white solid (248 mg, 96%); 1H NMR (400 MHz, DMSO-d6) δ 12.52 (s, 1H), 7.83 (dd, J=7.8, 1.5 Hz, 1H), 7.47 (td, J=7.4, 1.5 Hz, 1H), 7.42–7.31 (m, 2H), 3.43 (t, J=6.8 Hz, 2H), 2.82 (t, J=6.8 Hz, 2H), 2.57 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 172.9, 165.7, 163.6, 137.9, 132.1, 131.8, 129.1, 126.9, 122.7, 34.3, 27.9, 21.9; HRMS (ESI+) m/z: 265.0640 [M+H]+ (expected 265.0647); HPLC Ret: 6.08 min; 97% pure.
3-((5-(2-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid (8i)
Method E starting from tert-butyl 3-((5-(2-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (310 mg, 7i) gave 3-((5-(2-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid as white solid (103 mg, 50%); 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 7.79 (dd, J=7.7, 1.8 Hz, 1H), 7.58 (ddd, J=8.9, 7.4, 1.8 Hz, 1H), 7.24 (d, J=8.4 Hz, 1H), 7.10 (t, J=7.5 Hz, 1H), 3.87 (s, 3H), 3.41 (t, J=6.8 Hz, 2H), 2.81 (t, J=6.8 Hz, 2H); HRMS (ESI+) m/z: 281.0592 [M+H]+ (expected 281.0596); HPLC Ret: 5.37 min; 99% pure.
3-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid (8j)
2-(2,4-dichlorophenyl)-5-(methylsulfonyl)-1,3,4-oxadiazole (20, 0.05 g, 0.171 mmol) was dissolved in 2 mL acetone. K2CO3 (0.028 g, 0.205 mmol) and 3-mercaptopropanoic acid (0.022 g, 0.018 mL, 0.205 mmol) were added and the mixture was stirred at 25 °C for 1 h. The acetone was removed in vacuo and the remaining residue was partitioned between water (5 mL) and DCM (5 mL). The product was extracted with DCM (3× 10 mL), washed with brine (10 mL), dried with MgSO4, and concentrated in vacuo. The residue was dissolved in minimal EtOAc and the product was triturated with Hex, producing a white precipitate that was filtered and dried under vacuum. Yield=51%. 1H NMR (500 MHz, CDCl3) δ ppm 7.90 (d, J = 8.5 Hz, 1H) 7.57 (d, J = 2.0 Hz 1H) 7.39 (dd, J = 8.5, 2.1 Hz, 1H) 3.55 (t, J = 6.7 Hz, 2H) 3.05 (t, J = 6.7 Hz, 2H); MS (ESI+) m/z: 318.9 [M+H]+; HPLC Ret: 6.64 min.
3-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid (8k)
Method E starting from t-butyl-3-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7k, 0.27 g) gave 3-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid as white powder (0.13 g, 57%); 1H NMR (400 MHz, DMSO-d6) δ 12.45 (br. s., 1H) 7.85 (d, J = 8.0 Hz, 1H) 7.55 (s, 1H) 7.37 (d, J = 8.1 Hz, 1H) 3.46 (t, J = 6.8 Hz, 2H) 2.84 (t, J = 6.8 Hz, 2H) 2.40 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 172.88, 164.42, 163.75, 144.35, 131.77, 131.26, 128.98, 119.78, 34.26, 27.99, 21.05; HRMS (ESI+) m/z: 321.0070 [M+Na]+ (expected 321.0077); HPLC Ret: 6.31 min; 98% pure.
3-((5-(2-Chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid (8l)
Method E starting from tert-butyl 3-((5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7l) gave 3-((5-(2-Chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid as white solid (130 mg, 75%); 1H NMR (400 MHz, DMSO-d6) δ 12.55 (s, 1H), 7.87 (d, J=8.8,1H),7.26 (d, J=2.5 Hz, 1H), 7.10 (dd, J=8.8, 2.6 Hz, 1H), 3.85 (s, 3H), 3.42 (t, J=6.8 Hz, 2H), 2.81 (t, J=6.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 172.9, 164.0, 163.7, 162.6, 133.3, 132.7, 116.6, 114.9, 114.6, 56.5, 34.3, 28.0; HRMS (ESI+) m/z: 315.0201 [M+H]+ (expected 315.0206); HPLC Ret: 5.91 min; 95% pure.
3-((5-(2-chloro-3,4-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid (8m)
Method E starting from tert-butyl 3-((5-(2-chloro-3,4-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7m, 0.17 g) gave 3-((5-(2-chloro-3,4-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid as white powder (0.11 g, 80%); 1H NMR (500 MHz, CDCl3) δ 10.59 (br. s., 1H) 7.60 (d, J = 7.9 Hz, 1H) 7.17 (d, J = 7.9 Hz, 1H) 3.53 (t, J = 6.8 Hz, 2H) 3.32 (s, 3H) 3.04 (t, J = 6.8 Hz, 2H) 2.28 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 176.37, 165.02, 164.19, 142.27, 136.67, 133.15, 128.14, 120.93, 34.00, 26.99, 21.37, 16.72; HRMS (ESI-) m/z: 311.0258 [M-H]- (expected 311.0257); HPLC Ret: 5.83 min; 95% pure.
3-((5-(2-chloro-4,5-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid (8n)
Method E starting from tert-butyl 3-((5-(2-chloro-4,5-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7n, 0.13 g) gave 3-((5-(2-chloro-4,5-dimethylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid as white powder (0.08 g, 75%); 1H NMR (500 MHz, CDCl3) δ 11.00 (br. s., 1H) 7.71 (s, 1H) 7.29 (s, 1H) 3.54 (t, J = 6.8 Hz, 2H) 3.32 (s, 3H) 3.04 (t, J = 6.8 Hz, 2H) 2.28 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 176.49, 164.54, 164.10, 142.24, 135.99, 131.95, 131.52, 129.77, 119.69, 33.99, 27.00, 19.70, 19.06; HRMS (ESI-) m/z: 311.0259 [M-H]- (expected 311.0257); HPLC Ret: 5.91 min; 99% pure.
3-((5-(2,4-dichloro-5-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid (8o)
Method E starting from tert-butyl 3-((5-(2,4-dichloro-5-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoate (7o, 0.09 g) gave 3-((5-(2,4-dichloro-5-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid as white powder (0.04 g, 65%); 1H NMR (500 MHz, CDCl3) δ 7.83 (s, 1H) 7.54 (s, 1H) 3.55 (t, J = 6.8 Hz, 2H) 3.04 (t, J = 6.8 Hz, 2H) 2.41 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 175.94, 164.62, 163.72, 138.29, 135.72, 132.36, 131.31, 130.62, 129.34, 120.94, 33.84, 27.05, 19.46; HRMS (ESI-) m/z: 330.9710 [M-H]- (expected 330.9711); HPLC Ret: 6.81 min; 95% pure.
4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (8q)
Method F starting from methyl-4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (5q, 0.19 g) gave 4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white powder (0.06 g, 35%); 1H NMR (500 MHz, CDCl3) δ 12.21 (br. s., 1H) 8.00 (d, J = 8.6 Hz, 1H) 7.93 (d, J = 2.1 Hz, 1H) 7.66 (dd, J = 8.6, 2.1 Hz, 1H) 3.34 (t, J = 7.3 Hz, 2H) 2.40 (t, J = 7.3 Hz, 2H) 2.11–1.95 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 174.15, 164.95, 162.95, 137.54, 133.14, 132.75, 131.19, 128.64, 121.70, 32.62, 31.96, 24.96; HRMS (ESI+) m/z: 354.9677 [M+Na]+ (expected 354.9687); HPLC Ret: 6.82 min; 98% pure.
5-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)pentanoic acid (8r)
Modified Method F (product required silica gel column chromatography eluting with 2% MeOH: 98% DCM and semi-prep to obtain > 95% purity) starting from methyl 5-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)pentanoate (5r, 0.33 g) gave 5-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)pentanoic acid as white solid (109 mg, 34%); 1H NMR (400 MHz, DMSO-d6) δ 12.05 (s, 1H), 7.97 (d, J=8.5 Hz, 1H), 7.90 (d, J=2.1 Hz, 1H),7.64, (dd, J=8.5, 2.1 Hz, 1H), 3.30 (t, J=7.3 Hz, 2H), 2.25 (t, J=7.3 Hz, 2H), 1.86–1.69 (m, 2H), 1.80–1.60 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 174.6, 165.1, 162.9, 137.5, 133.1, 132.7, 131.2, 128.6, 121.7, 33.4, 32.2, 28.9, 23.8; MS (ESI-) m/z: 345.0 [M-H]-; HPLC Ret: 7.01 min; 98% pure.
4-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (8s)
Method F starting from methyl 4-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (0.15 g, 0.46 mmol) gave 4-((5-(2-chloro-4-methylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (0.10 g, 69%); 1H NMR (500 MHz, CDCl3) δ 8.67 (br. s., 1H) 7.82 (d, J = 8.0 Hz, 1H) 7.35 (s, 1H) 7.19 (d, J = 7.9 Hz, 1H) 3.38 (t, J = 7.1 Hz, 2H) 2.59 (t, J = 7.1 Hz, 2H) 2.40 (s, 3H) 2.21 (q, J = 7.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 178.02, 164.39, 164.28, 143.42, 132.64, 131.68, 130.67, 127.96, 119.89, 32.31, 31.60, 24.34, 21.24; HRMS (ESI+) m/z: 335.0244 [M+Na]+ (expected 335.0233); HPLC Ret: 6.53 min; 96% pure.
4-((5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (8t)
Method F starting from methyl 4-((5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (5t, 0.1 g) gave 4-((5-(2-chloro-4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white solid (0.09 g, 94%); 1H NMR (500 MHz, CDCl3) δ 10.78 (br. s, 1H) 7.87 (d, J = 8.8 Hz, 1H) 7.04 (d, J = 2.5 Hz, 1H) 6.91 (dd, J = 8.8, 2.5 Hz, 1H) 3.87 (s, 3H) 3.37 (t, J = 7.2 Hz, 2H) 2.59 (t, J = 7.1 Hz, 2H) 2.21 (p, J = 7.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 178.05, 164.18, 164.03, 162.21, 134.12, 131.96, 116.34, 115.12, 113.38, 55.76, 32.32, 31.60, 24.34; HRMS (ESI+) m/z: 329.0386 [M+H]+ (expected 329.0363); HPLC Ret: 6.27 min; 99% pure.
4-((5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (8u)
Method F starting from methyl 4-((5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (5u, 0.09 g) gave 4-((5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white powder (0.06 g, 68%); 1H NMR (500 MHz, CDCl3) δ 10.41 (br. s., 1H) 8.14 (d, J = 8.2 Hz, 2H) 7.77 (d, J = 8.2 Hz, 2H) 3.41 (t, J = 7.2 Hz, 2H) 2.60 (t, J = 7.1 Hz, 2H) 2.22 (p, J = 7.1 Hz, 2H); 19F NMR (500 MHz, CDCl3) δ ppm −63.16 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 177.97, 165.00, 164.64, 133.41, 126.97, 126.13, 124.59, 121.90, 32.22, 31.57, 24.22; HRMS (ESI+) m/z: 333.0352 [M+H]+ (expected 333.0521); HPLC Ret: 6.76 min; 99% pure.
4-((5-(2-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (8v)
Modified Method E (product required column chromatography eluting with 45% EtOAc: 55% Hex: 0.1% AcOH to obtain > 95% purity) starting from tert-butyl 4-((5-(2-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (7v, 0.13 g) gave 4-((5-(2-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as colorless oil (0.10 g, 93%); 1H NMR (500 MHz, CDCl3) δ 10.45 (br. s, 1H) 8.07–8.00 (m, 1H) 7.85 (dd, J = 7.1, 2.0 Hz, 1H) 7.75–7.65 (m, 2H) 3.37 (t, J = 7.2 Hz, 2H) 2.59 (t, J = 7.1 Hz, 2H) 2.21 (p, J = 7.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 178.29, 165.45, 164.05, 132.18, 131.65, 128.78, 127.06, 126.98, 124.19, 122.01, 32.28, 31.55, 24.34; HRMS (ESI+) m/z: 333.0547 [M+H]+ (expected 333.0521); HPLC Ret: 6.31 min; 97% pure.
4-((5-(2-chloro-4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (8w)
Method F starting from methyl 4-((5-(2-chloro-4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (5w, 0.05 g) gave 4-((5-(2-chloro-4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white powder (0.03 g, 81%); 1H NMR (500 MHz, CDCl3) δ 8.12 (d, J = 8.2 Hz, 1H) 7.82 (d, J = 1.6 Hz, 1H) 7.66 (dd, J = 8.3, 1.8 Hz, 1H) 3.41 (t, J = 7.2 Hz, 2H) 2.60 (t, J = 7.1 Hz, 2H) 2.24 (p, J = 7.1 Hz, 2H); 19F NMR (500 MHz, CDCl3) δ ppm −63.24 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 177.45, 165.54, 163.05, 133.60, 131.35, 128.38, 125.99, 123.94, 123.91, 32.16, 31.62, 24.28; HRMS (ESI+) m/z: 367.0022 [M+H]+ (expected 367.0131); HPLC Ret: 6.99 min; 97% pure.
4-((5-(4-bromo-2-chlorophenyl)-1,3, 4-oxadiazole-2-yl)thio)butanoic acid (8x)
Method E starting from t-butyl 4-((5-(4-bromo-2-chlorophenyl)-1,3,4-oxadiazole-2-yl)thio)butanoate (7× 0.12 g) gave 4-((5-(4-bromo-2-chlorophenyl)-1,3, 4-oxadiazole-2-yl)thio)butanoic acid as white solid (0.1 g, 94%); 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 8.4 Hz, 1H) 7.73 (d, J = 1.9 Hz, 1H) 7.54 (dd, J = 8.5, 1.9 Hz, 2H) 3.39 (t, J = 7.2 Hz, 2H) 2.59 (t, J = 7.1 Hz, 2H) 2.22 (p, J = 7.1 Hz, 2H) 13C NMR (125 MHz, CDCl3) δ 176.35, 164.99, 163.50, 134.00, 133.78, 131.66, 130.54, 126.15, 121.75, 32.23, 31.60, 24.31; HRMS (ESI+) m/z: 376.9455 [M+H]+ (expected 376.9362); HPLC Ret: 6.91 min; 95% pure.
4-((5-(3-chloro-[1, 1’-biphenyl]-4-yl)-1, 3, 4-oxadiazol-2-yl)thio)butanoic acid (8y)
Method F starting from methyl 4-((5-(3-chloro-[1, 1’-biphenyl]-4-yl)-1, 3, 4-oxadiazol-2-yl)thio)butanoate (5y, 0.08 g) gave 4-((5-(3-chloro-[1, 1’-biphenyl]-4-yl)-1, 3, 4-oxadiazol-2-yl)thio)butanoic acid (0.05 g, 82%); 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 8.1 Hz, 1H) 7.77 (d, J = 1.8 Hz, 1H) 7.63–7.60 (m, 3H) 7.52–7.39 (m, 3H) 3.40 (t, J = 7.2 Hz, 2H) 2.60 (t, J = 7.1 Hz, 2H) 2.24 (p, J = 7.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 177.29, 164.63, 164.11, 145.45, 138.32, 133.28, 131.16, 129.64, 129.10, 128.75, 127.11, 125.64, 121.23, 32.16, 31.63, 24.37; HRMS (ESI+) m/z: 333.0489 [M+H]+ (expected 375.0570); HPLC Ret: 7.43 min; 98% pure.
3-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)-N-methylpropanamide (9)
To a solution of 3-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)propanoic acid (8j, 200mg, 0.63 mmol) in dichloromethane (5 ml) was added oxalyl chloride (0.060 ml, 0.69 mmol) followed by one drop of DMF. After 2h, the reaction was concentrated and taken into the next step without further purification. 3-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)propanoyl chloride was dissolved in 2mL of 33% methylamine in ethanol. The reaction stirred overnight in a sealed tube. The reaction was concentrated. The crude reaction mixture was purified by column chromatography eluting with 50–80% ethylacetate in hexanes. Isolated 69 mg of a white solid. Yield=66%; 1H NMR (400 MHz, DMSO-d6) δ 7.89 (d, J=8.5 Hz, 1H), 7.57 (d, J=2.0 Hz, 1H), 7.39 (dd, J=8.5, 2.0 Hz, 1H), 5.65 (s, 1H), 3.57 (t, J=6.8 Hz, 2H), 2.82 (m, 5H); 13C NMR (100 MHz, DMSO-d6) δ 170.4, 165.1, 162.9, 137.5, 133.1, 132.7, 131.2, 128.6, 121.7, 35.1, 28.7, 25.9; HRMS (ESI+) m/z: 353.9838 [M+Na]+ (expected 353.9847); HPLC Ret: 6.20 min; 96% pure.
5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-ol (10)
In a 50 mL round bottomed flask, 2,4-dichlorobenzohydrazide (3j, 0.5g, 2.44 mmol), K3PO4 (0.52, 2.44 mmol), and CS2 (0.19g, 0.15 mL, 2.44 mmol) were stirred in 10 mL of H2O for 10 min at 25 °C. The reaction was then stirred at reflux (106 °C) for 2 hr and then cooled to 25 °C. propylene oxide (0.14 g, 2.44 mmol, 0.17 mL) was added and the reaction was stirred at 25 °C for 16 h. The product was extracted with EtOAc (3× 15 mL), washed with brine, dried with MgSO4, and the solvent was evaporated. The residue was subjected to silica gel chromatography eluting with 20% EtOAc: 80% Hex. The fractions containing product were concentrated in vacuo to produce white solid. Yield=23% (2 steps); 1H NMR (500 MHz, CDCl3) δ 9.18 (br. s., 1H) 7.83–7.73 (m, 1H) 7.57–7.55 (m, 1H) 7.41–7.38 (m, 1H); MS (ESI-) m/z: 228.9 [M-H]-; HPLC Ret: 6.39 min.
tert-butyl 4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)oxy)butanoate (10’)
5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-ol (10, 0.06 g, 0.251 mmol) was dissolved in DMF (2 mL). K2CO3 (0.045 g, 0.33 mmol) and tert-butyl 4-bromobutanoate (0.07 g, 0.33 mmol, 0.06 mL) were added and the reaction was stirred at 90 °C for 1 h. The reaction was cooled to room temperature and the solvent was removed in vacuo. The residue was partitioned between H2O (10 mL) and DCM (10 mL), and the product was extracted with DCM (3× 10 mL), washed with brine, dried with MgSO4, and the solvents removed in vacuo. The oil was subjected to silica gel chromatography eluting with 10% EtOAc: 90% Hex. The fractions containing product were concentrated in vacuo to produce white solid. Yield=62%; 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.5 Hz, 1H) 7.54 (d, J = 2.1 Hz, 1H) 7.37 (dd, J = 8.5, 2.1 Hz, 1H) 3.88 (t, J = 6.8 Hz, 2H) 2.36 (t, J = 7.3 Hz, 2H) 2.10 (p, J = 7.1 Hz, 2H) 1.45 (s, 9H); MS (ESI+) m/z: 396.9 [M+Na]+; HPLC Ret: 8.72 min.
4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)oxy)butanoic acid (11)
Modified Method E (crystallization not needed) starting from tert-butyl 4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)oxy)butanoate (10’, 0.06 g, 0.16 mmol) gave 4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)oxy)butanoic acid (0.04 g, 89%); 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.5 Hz, 1H) 7.59 (br. s., 1H) 7.54 (d, J = 2.1 Hz, 1H) 7.37 (dd, J = 8.5, 2.1 Hz, 1H) 3.93 (t, J = 6.7 Hz, 2H) 2.53 (t, J = 7.2 Hz, 2H) 2.17 (p, J = 7.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 177.90, 153.08, 150.84, 137.89, 133.40, 131.38, 130.53, 127.56, 121.02, 45.15, 30.61, 23.22; HRMS (ESI+) m/z: 338.9912 [M+Na]+ (expected 338.9915); HPLC Ret: 6.52 min.
2-(bromomethyl)-5-(2,4-dichlorophenyl)-1,3,4-oxadiazole (12)
In a 100 mL round bottomed flask, 2,4-dichlorobenzohydrazide (3j, 0.5 g, 2.44 mmol) and bromoacetic acid (0.34 g, 2.44 mmol) were combined under inert atmosphere. The reagents were dissolved in POCl3 (5.0 mL) and stirred at reflux (106 °C) for 16 h. The reaction was cooled to room temperature and poured over ice water (5 mL). The solution was neutralized to pH ~ 9 with NaHCO3 (20% to sat), causing a yellow precipitate to form. The product was extracted with EtOAc (3× 25 mL), washed with brine, dried with MgSO4, and the solvents were evaporated in vacuo producing brown oil. The impurities were triturated with EtOAc:Hex (tan solid) and the mother liquor was evaporated to provide the desired product as white/tan solid. Yield=49%.; 1H NMR (500 MHz, CDCl3) δ 7.97 (d, J = 8.3, 1H) 7.60 (s, 1H) 7.42 (dd, J = 8.3, 2.2 Hz, 1H) 4.62 (s, 2H); MS (ESI+) m/z: 359.9 [M+ MeOH+Na]+; HPLC Ret: 7.42 min.
2-(((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)methyl)thio)acetic acid (13)
2-mercaptoacetic acid (0.13 g, 1.36 mmol, 0.10 mL) was dissolved in acetone (5 mL), and K2CO3 (0.19 g, 1.36 mmol) and 2-(bromomethyl)-5-(2,4-dichlorophenyl)-1,3,4-oxadiazole (12, 0.35 g, 1.14 mmol) were added. The reaction was stirred at 25 °C for 24 hr and then the solvent was removed in vacuo. The residue was diluted with H2O (2 mL) and acidified to pH ~ 1 with 1 N HCl (3 mL). The product was extracted with DCM (3× 15 mL), washed with brine, dried with MgSO4, and the solvent was evaporated in vacuo. The residue was subjected to silica gel chromatography eluting with 5% MeOH: 95% DCM: 0.1% AcOH. The fractions containing product were concentrated and the residue was crystalized with DCM. The product was filtered, producing white crystals. Yield=20%.; 1H NMR (400 MHz, DMSO-d6) δ 12.73 (br. s., 1 H) 7.96 (d, J = 8.4 Hz, 1H) 7.89 (d, J = 2.1 Hz, 1H) 7.65 (dd, J = 8.5, 2.1 Hz, 1H) 4.62 (s, 2H) 3.46 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 171.17, 165.24, 162.28, 137.57, 133.27, 132.91, 131.20, 128.81, 121.93, 33.93, 24.95; HRMS (ESI+) m/z: 340.9524 [M+Na]+ (expected 340.9530); HPLC Ret: 6.13 min.
methyl 2-chloro-4-(2-hydroxypropan-2-yl)benzoate (14’)
In a 100 mL round-bottomed flask, methyl 2-chloro-4-iodobenzoate (14, 1.0 g, 3.4 mmol) was dissolved in 10 mL THF and cooled to −20 °C. 2M iPrMgCl in THF (1.7 mL, 3.4 mmol) was added dropwise and the reaction was stirred at −20 °C for 1.5 h. Acetone (0.29 g, 0.37 mL, 5.1 mmol) was then added and the reaction was stirred at 25 °C for 1.5 h. The reaction was quenched with MeOH and then diluted with H2O. The product was extracted with EtOAc (3 × 15 mL), and the organic layers were combined, washed with brine, dried with MgSO4, and concentrated in vacuo. The subsequent oil was subjected to silica gel chromatography eluting with 30% EtOAc: 70% Hex. The fractions containing product were concentrated in vacuo to produce colorless oil. Yield=53%; 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.2 Hz, 1H) 7.59 (d, J = 1.7 Hz, 1H) 7.41 (dd, J = 8.2, 1.8 Hz, 1H) 3.93 (s, 3H) 1.58 (s, 6H); HPLC Ret: 6.09 min.
methyl 3-bromo-2-chlorobenzoate (15a’)
Method A starting from 3-bromo-2-chlorobenzoic acid (0.5 g) gave methyl 3-bromo-2-chlorobenzoate as colorless oil (15a, 0.52 g, 98%); 1H NMR (500 MHz, CDCl3) δ 7.77 (dd, J = 8.0, 0.8 Hz, 1H) 7.68 (dd, J = 7.7, 0.8, 1H) 7.19 (td, J = 7.9, 0.8 Hz, 1H) 3.94 (s, 3H); HPLC Ret: 7.44 min.
methyl 4-bromo-2-chlorobenzoate (15b’)
Method A starting from 4-bromo-2-chlorobenzoic acid (15b, 1.0 g) gave methyl 4-bromo-2-chlorobenzoate as yellow oil (0.93 g, 87%); 1H NMR (400 MHz, CDCl3) δ 7.72 (dd, J = 8.4, 1.1 Hz, 1H) 7.64 (s, 1H) 7.46 (dd, J = 8.4, 1.6 Hz, 1H) 3.93 (s, 3H); HPLC Ret: 7.71 min.
methyl 2-chloro-4-ethylbenzoate (17a’)
In a flamed dried 25 mL round bottomed flask, freshly prepared methyl 2-chloro-4-vinylbenzoate (17a, 0.12 g, 0.59 mmol) as previously described36 and 2-nitrobenzene-1-sulfonyl chloride (0.26 g, 1.19 mmol) were dissolved in 3 mL dry MeCN. The solution was cooled to 0 °C and hydrazine hydrate (0.08 g, 0.07 mL, 2.4 mmol) was slowly added. The reaction was warmed to 25 °C and stirred vigorously for 16 hr under N2. Upon completion, the reaction was quenched with water (15 mL) and the product was extracted with EtOAc (4 × 20 mL). The layers were combined, dried with MgSO4, and concentrated in vacuo. The yellow residue was subjected to silica gel chromatography eluting with 2.5% EtOAc: 97.5% Hex. The fractions containing product were concentrated in vacuo to produce yellow oil. Yield=66%. 1H NMR (500 MHz, CDCl3) δ ppm 7.78 (dd, J = 8.0, 1.0 Hz, 1H) 7.29 (d, J = 1.6 Hz, 1H) 7.13 (dd, J = 8.2, 1.3 Hz, 1H) 3.92 (s, 3H) 2.66 (q, J = 7.6 Hz, 2H) 1.25 (t, J = 7.6 Hz, 3 Hz); HRMS (ESI+) m/z: 199.0691 [M+H]+ (expected 199.0526); HPLC Ret: 7.73 min.
2-chloro-4-methylbenzohydrazide (17a’’)
Method B starting from methyl 2-chloro-4-ethylbenzoate (17a’, 0.08 g) gave 2-chloro-4-ethylbenzohydrazide as white solid (0.075 g, 98%); 1H NMR (500 MHz, CDCl3) δ 7.61 (dd, J = 7.9, 1.1 Hz, 1H) 7.45 (br. s, 1H) 7.26 (d, J = 1.1 Hz, 1H) 7.17 (dd, J = 8.4, 1.4 Hz, 1H) 4.13 (br. s, 2H) 2.66 (q, J = 7.6 Hz, 2H) 1.24 (t, J = 7.6 Hz, 3H); HRMS (ESI+) m/z: 199.0633 [M+H]+ (expected 199.0638); HPLC Ret: 4.61 min.
methyl 2-chloro-4-propylbenzoate (17b)
In a flame dried 25 mL round-bottomed flask, 2-chloro-4-methylbenzoic acid (3k, 0.1 g, 0.59 mmol) was dissolved in THF (1.0 mL). The solution was cooled to 0 °C and freshly prepared LDA (0.13 g, 1.2 mmol, 2.5 mL) was added. The reaction was stirred at 0 °C for 10 mins, and then ethyl iodide (0.14 g, 0.88 mmol, 0.07 mL) was added. After stirring at 0 °C for 1 h, the reaction was quenched with 1N HCl (10 mL). The product was extracted with EtOAc (3 × 20 mL), washed with brine, dried with MgSO4, and concentrated in vacuo to produce white solid. HPLC Ret: 6.87 min. The subsequent oil was subjected to Method A to produce methyl 2-chloro-4-propylbenzoate as colorless oil (0.03 g, 25%, 2-steps) after silica gel chromatography eluting with 2% EtOAc: 98% Hex; 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J = 8.0 Hz, 1H) 7.27 (d, J = 1.6 Hz, 1H) ) 7.11 (dd, J = 8.1, 1.6 Hz, 1H) 3.92 (s, 3H) 2.59 (t, J = 7.4 Hz, 2H) 1.65 (h, J = 7.4 Hz, 2H) 0.94 (t, J = 7.4 Hz, 3H); HPLC Ret: 8.22 min.
2-chloro-4-propylbenzohydrazide (17b’)
Method B starting from methyl 2-chloro-4-propylbenzoate (17b, 0.03 g) gave 2-chloro-4-propylbenzohydrazide as white solid (0.03 g, quant.); HPLC Ret: 5.00 min.
methyl 2-chloro-4-(2-fluoropropan-2-yl)benzoate (17d)
In a flame dried 25-mL round bottomed flask, methyl 2-chloro-4-(2-hydroxypropan-2-yl)benzoate (14’, 0.11 g, 0.48 mmol) was dissolved in DCM (2 mL) and cooled to −78 °C. DAST (0.08 g, 0.06 mL, 0.48 mmol) was added and the solution was stirred at 25 °C for 2 h. The reaction was diluted with H2O and the product was extracted with DCM (3 × 20 mL). The organic layers were combined, washed with brine (2 × 15 mL), dried with MgSO4, and concentrated in vacuo. The oil was subjected to silica gel chromatography eluting with 2.5% EtOAc: 97.5% Hex to 5% EtOAc: 95% Hex. The fractions containing product were concentrated to produce colorless oil. Yield=51%; 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 8.2 Hz, 1H) 7.47 (d, J = 1.7 Hz, 1H) 7.30 (dd, J = 8.2, 1.7 Hz, 1H) 3.93 (s, 3H) 1.70 (d, J = 1.5 Hz, 3H) 1.65 (d, J = 1.5 Hz, 3H); 19F NMR (100 MHz, CDCl3) δ −139.4 (hept., 1H); HRMS (ESI+) m/z: 231.0583 [M+H]+ (expected 231.0588); HPLC Ret: 8.02 min.
2-chloro-4-(2-fluoropropan-2-yl)benzohydrazide (17d’)
Method B starting from methyl 2-chloro-4-(2-fluoropropan-2-yl)benzoate (17d, 0.25 g) gave 2-chloro-4-(2-fluoropropan-2-yl)benzohydrazide as white solid (0.24 g, 96%); 1H NMR (500 MHz, CDCl3) δ 7.67 (d, 8.1 Hz, 1H) 7.47–7.42 (m, 2H) 7.32 (dd, J = 8.1, 1.7 Hz, 1H) 4.15 (d, J = 4.3 Hz, 2H) 1.69 (s, 3H) 1.65 (s, 3H); HRMS (ESI+) m/z: 231.0697 [M+H]+ (expected 231.0700); HPLC Ret: 4.76 min.
methyl 2-chloro-3-cyclopropylbenzoate (17e)
Method G starting from methyl 3-bromo-2-chlorobenzoate (15a’, 0.52 g) gave methyl 2-chloro-3-cyclopropylbenzoate as yellow oil (0.43 g, 97%); 1H NMR (500 MHz, CDCl3) δ 7.51 (dd, J = 7.7, 1.7 Hz, 1H) 7.21 (t, J = 7.7 Hz, 1H) 7.10 (dd, J = 7.8, 1.6 Hz, 1H) 3.94 (s, 3H) 2.29–2.21 (m, 1H) 1.09–0.96 (m, 2H) 0.71–0.66 (m, 2H); HRMS (ESI+) m/z: 211.0521 [M+H]+ (expected 211.0526); HPLC Ret: 7.31 min.
2-chloro-3-cyclopropylbenzohydrazide (17e’)
Method B starting from methyl 2-chloro-3-cyclopropylbenzoate (17e, 0.43 g) gave 2-chloro-3-cyclopropylbenzohydrazide as white solid (0.41 g, 95%); 1H NMR (500 MHz, CDCl3) δ 7.33 (dd, J = 7.3, 1.4 Hz, 1H) 7.24 (t, J = 7.5 Hz, 1H) 7.06 (dd, J = 7.8, 1.6 Hz, 1H) 4.14 (d, J = 4.2 Hz, 2H) 2.20 (tt, J = 8.6, 5.3 Hz, 1H) 1.09–0.96 (m, 2H) 0.74–0.63 (m, 2H); HRMS (ESI+) m/z: 211.0641 [M+H]+ (expected 211.0638); HPLC Ret: 4.55 min.
methyl 2-chloro-4-cyclopropylbenzoate (17f)
Method G starting from methyl 4-bromo-2-chlorobenzoate (15b’, 0.27 g) gave methyl 2-chloro-4-cyclopropylbenzoate as yellow oil (0.18 g, 51%); 1H NMR (500 MHz, CDCl3) δ 7.81–7.70 (m, 1H) 7.15–7.12 (m, 1H) 7.03–6.94 (m, 1H) 3.90 (s, 3H) 1.95–1.84 (m, 1H) 1.11–1.00 (m, 2H) 0.81–0.70 (m, 2H); MS (ESI+) m/z: 211.0 [M+H]+; HPLC Ret: 7.29 min.
2-chloro-4-cyclopropylbenzohydrazide (17f’)
Method B starting from methyl 2-chloro-4-cyclopropylbenzoate (17f, 0.12 g) gave 2-chloro-4-cyclopropylbenzohydrazide as white solid (0.10 g, 85%); 1H NMR (500 MHz, CDCl3) δ 7.63–7.52 (m, 1H) 7.09 (s, 1H) 7.02–7.00 (m, 1H) 4.12 (br. s., 2H) 1.93–1.84 (m, 1H) 1.05 (dtd, J = 6.4, 4.8, 1.3 Hz, 2H) 0.74 (dtd, J = 6.4, 4.8, 1.3 Hz, 2H); MS (ESI+) m/z: 211.0 [M+H]+; HPLC Ret: 4.64 min.
methyl 2-chloro-5-cyclopropylbenzoate (17g)
Method G starting from methyl 5-bromo-2-chlorobenzoate (15c’, 0.5 g) gave methyl 2-chloro-5-cyclopropylbenzoate as yellow oil (0.35 g, 83%); 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 1.6 Hz, 1H) 7.33 (d, J = 8.2 Hz, 1H) 7.11 (dd, J = 7.9, 1.5 Hz, 1H) 3.93 (s, 3H) 1.93–1.86 (m, 1H) 1.05–0.98 (m, 2H) 0.73–0.68 (m, 2H); HRMS (ESI+) m/z: 211.0521 [M+H]+ (expected 211.0526); HPLC Ret: 7.31 min.
2-chloro-5-cyclopropylbenzohydrazide (17g’)
Method B starting from methyl 2-chloro-5-cyclopropylbenzoate (17g, 0.35 g) gave 2-chloro-5-cyclopropylbenzohydrazide as white solid (0.33 g, 96%); 1H NMR (500 MHz, CDCl3) δ 7.41 (br. s., 1H) 7.36 (d, J = 2.3 Hz, 1H) 7.28 (dd, J = 8.3, 0.8 Hz, 1H) 7.08 (dd, J = 8.3, 2.3 Hz, 1H) 1.89 (tt, J = 8.6, 5.0 Hz, 1H) 1.08–0.94 (m, 2H) 0.76–0.64 (m, 2H); HRMS (ESI+) m/z: 211.0646 [M+H]+ (expected 211.0638); HPLC Ret: 4.59 min.
methyl 2-chloro-4-cyclopropoxybenzoate (17h)
methyl 2-chloro-4-hydroxybenzoate (16, 0.25 g, 1.34 mmol) was dissolved in 2.0 mL DMA. Bromocyclopropane (0.54 g, 4.42 mmol) along with Cs2CO3 (1.4 g, 4.42 mmol) were added and the reaction was stirred at 155 °C for 24 h. The reaction was diluted with 10 mL H2O and washed with EtOAc (3× 10 mL). The aqueous layer was acidified to pH ~1 with 1N HCl and the product was extracted with EtOAc (3× 15 mL). The organic layers were combined, washed with brine (3× 10 mL), dried with MgSO4, and evaporated in vacuo. The subsequent oil was subjected to silica gel chromatography eluting with 45% EtOAc: 55% Hex: 0.1% AcOH. The fractions containing product were concentrated in vacuo to produce white solid. Yield=65%; 1H NMR (500 MHz, DMSO-d6) δ 13.00 (br. s, 1H) 7.84 (d, 8.7 Hz, 1H) 7.20 (d, J = 2.5 Hz, 1H) 6.99 (dd, J = 8.8, 2.5 Hz, 1H) 3.98 (tt, J = 6.1, 2.9 Hz, 1H) 0.87–0.79 (m, 2H) 0.70 (tdd, J = 5.9, 3.0, 1.5, 2H); MS (ESI+) m/z: 213.0 [M+H]+; HPLC Ret: 6.33 min. Method A starting from 2-chloro-4-cyclopropoxybenzoic acid (0.3 g) gave methyl 2-chloro-4-cyclopropoxybenzoate as colorless oil (17h, 0.21 g, 66%); 1H NMR (500 MHz, CDCl3) δ 7.86 (d, 8.5 Hz, 1H) 7.14 (d, J = 2.5 Hz, 1H) 6.94 (dd, J = 8.8, 2.4 Hz, 1H) 3.90 (s, 3H) 3.77 (tt, J = 6.4, 3.0 Hz, 1H) 0.88–0.75 (m, 4H); MS (ESI+) m/z: 226.1 [M+H]+; HPLC Ret: 7.67 min.
2-chloro-4-cyclopropoxybenzohydrazide (17h’)
Method B starting from methyl 2-chloro-4-cyclopropoxybenzoate (17h, 0.21 g) gave 2-chloro-4-cyclopropoxybenzohydrazide as white solid (0.21 g, 98%); 1H NMR (500 MHz, CDCl3) δ 7.69 (d, 8.7 Hz, 1H) 7.54 (br. s, 1H) 7.09 (d, J = 2.4 Hz, 1H) 6.99 (dd, J = 8.7, 2.4 Hz, 1H) 4.13 (br. s, 2H) 3.80–3.72 (m, 1H) 0.88–0.73 (m, 4H); HRMS (ESI+) m/z: 227.0581 [M+H]+ (expected 227.0587); HPLC Ret: 4.57 min.
methyl 2-chloro-4-(oxetan-3-yloxy)benzoate (17i)
In a 50 mL round-bottomed flask, methyl 2-chloro-4-hydroxybenzoate (16, 0.3 g, 1.6 mmol) was dissolved in 6 mL anhydrous DMSO. Cs2CO3 (0.68 g, 2.09 mmol) and 3-bromooxetane (0.29 g, 0.17 mL, 2.09 mmol) were added and the reaction was heated at 105 °C for 8 h. The reaction was cooled, diluted with H2O, and extracted with EtOAc (3 × 20 mL). The organic layers were combined, washed with brine, and concentrated in vacuo. The yellow residue was subjected to silica gel chromatography eluting with 30% EtOAc: 70% Hex. The fractions containing product were concentrated in vacuo to produce yellow solid. Yield=70%.; 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 8.7 Hz, 1H) 6.76 (d, J = 2.5 Hz, 1H) 6.64 (dd, J = 8.8, 2.5 Hz, 1H) 5.28–5.20 (m, 2H) 5.02–4.95 (m, 2H) 4.78–4.72 (m, 2H) 3.90 (s, 3H); HPLC Ret: 6.39 min.
2-chloro-4-(oxetan-3-yloxy)benzohydrazide (17i’)
Method B starting from methyl 2-chloro-4-(oxetan-3-yloxy)benzoate (17i, 0.27 g) gave 2-chloro-4-(oxetan-3-yloxy)benzohydrazide as white solid (0.20 g, 72%); 1H NMR (500 MHz, CDCl3) δ 7.69 (d, 8.7 Hz, 1H) 7.51 (br. s, 1H) 6.74 (d, J = 2.5 Hz, 1H) 6.67 (dd, J = 8.7, 2.5 Hz, 1H) 5.22 (pd, 6.0, 1.0 Hz, 1H) 4.98 (t, J = 7.1 Hz, 2H) 4.74 (dd, J = 7.3, 5.1 Hz, 2H) 4.12 (d, J = 4.1 Hz, 2H); HRMS (ESI+) m/z: 243.0533 [M+H]+ (expected 243.0536); HPLC Ret: 3.10 min.
5-(2-chloro-4-ethylphenyl)-1,3,4-oxadiazole-2-thiol (18a)
Method C starting from 2-chloro-4-ethylbenzohydrazide (17a’’, 0.075 g) gave 5-(2-chloro-4-ethylphenyl)-1,3,4-oxadiaole-2-thiol as white powder (0.08 g, 87%); 1H NMR (500 MHz, CDCl3) δ 10.44 (br. s, 1H) 7.83 (dd, J = 8.1, 1.5 Hz, 1H) 7.40 (d, J = 1.5 Hz, 1H) 7.25 (dd, J = 8.1, 1.3 Hz, 1H) 2.71 (q, J = 7.6 Hz, 2H) 1.28 (t, J = 7.6 Hz, 3H); HPLC Ret: 7.16 min.
tert-butyl 4-((5-(2-chloro-4-ethylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18a’)
Method D starting from 5-(2-chloro-4-ethylphenyl)-1,3,4-oxadiazole-2-thiol (18a, 0.05 g) gave tert-butyl 4-((5-(2-chloro-4-ethylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.07 g, 85%); 1H NMR (500 MHz, CDCl3) δ 7.85 (dd, J = 8.0, 1.5 Hz, 1H) 7.37 (d, J = 1.5 Hz, 1H) 7.22 (dd, J = 8.0, 1.6 Hz, 1H) 3.34 (t, J = 7.2 Hz, 2H) 2.70 (q, J = 7.6 Hz, 2H) 2.43 (t, J = 7.2 Hz, 2H) 2.15 (p, J = 7.2 Hz, 2H) 1.45 (s, 9H) 1.27 (t, J = 7.6 Hz, 3H); HRMS (ESI+) m/z: 405.1009 [M+Na]+ (expected 405.1016); HPLC Ret: 9.28 min.
5-(2-chloro-4-propylphenyl)-1,3,4-oxadiazole-2-thiol (18b)
Modified Method C (post acidification, product was extracted with EtOAc (3 × 15 mL), washed with brine, dried with MgSO4, concentrated in vacuo) starting from 2-chloro-4-propylbenzohydrazide (17b’, 0.03 g) gave 5-(2-chloro-4-propylphenyl)-1,3,4-oxadiazole-2-thiol as white solid (0.03 g, 71%); 1H NMR (500 MHz, CDCl3) δ 10.36 (br. s., 1H) 7.82 (d, J = 8.1 Hz, 1H) 7.37 (d, J = 1.5 Hz, 1H) 7.26 (dd, J = 8.0, 1.5 Hz, 1H) 2.64 (t, J = 7.6 Hz, 2H) 1.66 (h, J = 7.4 Hz, 2H) 0.96 (t, J = 7.5 Hz, 3H); HRMS (ESI+) m/z: 255.0349 [M+H]+ (expected 255.0359); HPLC Ret: 7.63 min.
tert-butyl 4-((5-(2-chloro-4-propylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18b’)
Method D starting from 5-(2-chloro-4-propylphenyl)-1,3,4-oxadiazole-2-thiol (18b, 0.03 g) gave tert-butyl 4-((5-(2-chloro-4-propylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.03 g, 62%); 1H NMR (500 MHz, CDCl3) δ 7.85 (dd, J = 8.0, 1.3 Hz, 1H) 7.27 (s, 1H) 7.20 (dd, J = 8.0, 1.5 Hz, 1H) 3.34 (t, J = 7.2 Hz, 2H) 2.63 (t, J = 7.6 Hz, 2H) 2.43 (t, J = 7.3 Hz, 2H) 2.16 (p, J = 7.3 Hz, 2H) 1.68 (h, J = 7.5 Hz, 2H) 1.46 (s, 9H) 0.96 (t, J = 7.5 Hz, 3H); HRMS (ESI+) m/z: 397.1359 [M+H]+ (expected 397.1353); HPLC Ret: 9.81 min.
5-(2-chloro-4-isopropylphenyl)-1,3,4-oxadiazole-2-thiol (18c)
Method B starting from mixture of methyl 2-chloro-4-isopropylbenzoate (17c) and methyl-butyl 4-isopropylbenzoate (0.05 g) prepared as previously described35 gave the mixture of 2-chloro-4-isopropylbenzohydrazide and 4-isopropylbenzohydrazide as white solid (0.05 g, quant.) followed by modified Method C (post acidification, product was extracted with EtOAc (3 × 15 mL), washed with brine, dried with MgSO4, concentrated in vacuo) gave 5-(2-chloro-4-isopropylphenyl)-1,3,4-oxadiazole-2-thiol and 5-(4-isopropylphenyl)-1,3,4-oxadiazole-2-thiol as yellow solid (0.04 g, 88%); 1H NMR (500 MHz, CDCl3) δ 10.43 (br. s, 2H) 7.88–7.82 (m, 3H) 7.43–7.26 (m, 4H) 3.03–2.91 (m, 2H) 1.29–1.25 (m, 12H); HRMS (ESI+) m/z: 255.0351 and 221.0749 [M+H]+ (expected 255.0359 and 221.0749); HPLC Ret: 7.53 & 7.31 min.
tert-butyl 4-((5-(2-chloro-4-isopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18c’)
Method D starting from a mixture of 5-(2-chloro-4-isopropylphenyl)-1,3,4-oxadiazole-2-thiol (18c) and 5-(4-isopropylphenyl)-1,3,4-oxadiazole-2-thiol (0.04 g) gave the mixture of tert-butyl 4-((5-(2-chloro-4-isopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate and tert-butyl 4-((5-(4-isopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.03 g, 60%); 1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 8.3Hz, 2H) 7.86 (d, J = 8.0 Hz, 0.5H) 7.41–7.32 (m, 2.5H) 7.25 (dd, J = 8.0, 1.6 Hz, 0.5H) 3.34 (t, J = 7.2 Hz, 3H) 3.02–2.90 (m, 1.5H) 2.43 (t, J = 7.2 Hz, 3H) 2.15 (p, J = 7.2 Hz, 3H) 1.45 (s, 13.5H) 1.30–1.26 (m, 9H); HRMS (ESI+) m/z: 397.1355 & 363.1757 [M+H]+ (expected 397.1353 and 363.1742); HPLC Ret: 9.70 & 9.40 min.
5-(2-chloro-4-(2-fluoropropan-2-yl)phenyl)-1,3,4-oxadiazole-2-thiol (18d)
Method C starting from 2-chloro-4-(2-fluoropropan-2-yl)benzohydrazide (17d’, 0.24 g) gave 5-(2-chloro-4-(2-fluoropropan-2-yl)phenyl)-1,3,4-oxadiazole-2-thiol as white solid (0.25 g, 88%); 1H NMR (500 MHz, CDCl3) δ 10.62 (br. s, 1H) 7.91 (d, 8.3 Hz, 1H) 7.59 (d, J = 1.4 Hz, 1H) 7.41 (dd, J = 8.3, 1.4 Hz, 1H) 1.73 (d, J = 1.1 Hz, 3H) 1.68 (d, J = 1.1 Hz, 3H); HRMS (ESI-) m/z: 271.0110 [M-H]- (expected 271.0108); HPLC Ret: 7.15 min.
methyl 4-((5-(2-chloro-4-(2-fluoropropan-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18d’)
Method D starting from 5-(2-chloro-4-(2-fluoropropan-2-yl)phenyl)-1,3,4-oxadiazole-2-thiol (18d, 0.08 g) gave methyl 4-((5-(2-chloro-4-(2-fluoropropan-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.09 g, 86%); 1H NMR (500 MHz, CDCl3) δ 7.94 (d, J = 8.2 Hz, 1H) 7.56 (d, J = 1.7 Hz, 1H) 7.38 (dd, J = 8.3, 1.8 Hz, 1H) 3.70 (s, 3H) 3.37 (t, J = 7.1 Hz, 2H) 2.54 (t, J = 7.2 Hz, 2H) 2.22 (p, J = 7.2 Hz, 2H) 1.72 (s, 3H) 1.68 (s, 3H); HRMS (ESI+) m/z: 373.0785 [M+H]+ (expected 373.0789); HPLC Ret: 7.96 min.
5-(2-chloro-3-cyclopropylphenyl)-1,3,4-oxadiazole-2-thiol (18e)
Method C starting from 2-chloro-3-cyclopropylbenzohydrazide (17e’, 0.2 g) gave 5-(2-chloro-3-cyclopropylphenyl)-1,3,4-oxadiazole-2-thiol as yellow powder (0.13 g, 55%); 1H NMR (500 MHz, CDCl3) δ 10.56 (br. s., 1H) 7.69 (dd, J = 7.8, 1.6 Hz, 1H) 7.32 (t, J = 7.8 Hz, 1H) 7.19 (dd, J = 7.8, 1.6 Hz, 1H) 2.27 (ddd, J = 13.9, 8.6, 5.4 Hz, 1H) 1.15–1.03 (m, 2H) 0.77–0.65 (m, 2H); HRMS (ESI-) m/z: 251.0050 [M-H]- (expected 251.0046); HPLC Ret: 7.16 min.
tert-butyl 4-((5-(2-chloro-3-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18e’)
Method D starting from 5-(2-chloro-3-cyclopropylphenyl)-1,3,4-oxadiazole-2-thiol (18e, 0.05 g) gave tert-butyl 4-((5-(2-chloro-3-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.06 g, 82%); 1H NMR (500 MHz, CDCl3) δ 7.69 (dd, J = 7.7, 1.6 Hz, 1H) 7.29 (t, J = 7.8 Hz, 1H) 7.15 (dd, J = 7.9, 1.6 Hz, 1H) 3.35 (t, J = 7.2 Hz, 2H) 2.43 (t, J = 7.2 Hz, 2H) 2.26 (tt, J = 8.6, 5.4 Hz, 1H) 2.16 (p, J = 7.2 Hz, 2H) 1.46 (s, 9H) 1.13–1.02 (m, 2H) 0.77–0.66 (m, 2H); HRMS (ESI+) m/z: 395.1208 [M+H]+ (expected 395.1196); HPLC Ret: 9.19 min.
5-(2-chloro-4-cyclopropylphenyl)-1,3,4-oxadiazole-2-thiol (18f)
Method C starting from 2-chloro-4-cyclopropylbenzohydrazide (17f’, 0.1 g) gave 5-(2-chloro-4-cyclopropylphenyl)-1,3,4-oxadiazole-2-thiol as yellow powder (0.08 g, 70%); 1H NMR (500 MHz, CDCl3) δ 10.53 (br. s., 1H) 7.81–7.73 (m, 1H) 7.23 (s, 1H) 7.08 (dd, J = 8.2, 1.8 Hz, 1H) 1.96–1.91 (m, 1H) 1.12 (dt, J = 6.8, 4.8, Hz, 2H) 0.81 (dt, J = 6.8, 4.8, Hz, 2H); MS (ESI+) m/z: 253.0 [M+H]+; HPLC Ret: 7.14 min.
methyl 4-((5-(2-chloro-4-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18f’)
Method D starting from 5-(2-chloro-4-cyclopropylphenyl)-1,3,4-oxadiazole-2-thiol (18f, 0.08 g) gave methyl 4-((5-(2-chloro-4-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as white oil (0.07 g, 64%); 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.2 Hz, 1H) 7.19 (s, 1H) 7.05 (dd, J = 8.2, 1.7 Hz, 1H) 3.69 (s, 3H) 2.37 (t, J = 7.2 Hz, 2H) 2.58 (t, J = 7.1 Hz, 2H) 2.20 (p, J = 7.2 Hz, 2H) 1.96–1.91 (tt, J = 8.4, 5.0 Hz, 1H) 1.08 (dt, J = 6.9, 4.9 Hz, 2H) 0.78 (dt, J = 6.9, 4.9 Hz, 2H); HRMS (ESI+) m/z: 375.0540 [M+Na]+ (expected 375.0546); HPLC Ret: 8.01 min.
5-(2-chloro-5-cyclopropylphenyl)-1,3,4-oxadiazole-2-thiol (18g)
Method C starting from 2-chloro-5-cyclopropylbenzohydrazide (17g’, 0.15 g) gave 5-(2-chloro-5-cyclopropylphenyl)-1,3,4-oxadiazole-2-thiol as white solid (0.15 g, 81%); 1H NMR (500 MHz, CDCl3) δ 10.58 (br. s., 1H) 7.59 (d, J = 2.3 Hz, 1H) 7.42 (d, J = 8.3 Hz, 1H) 7.18 (dd, J = 8.4, 2.3 Hz, 1H) 1.94 (ddd, J = 13.4, 8.4, 5.1 Hz, 1H) 1.11–0.99 (m, 2H) 0.79–0.68 (m, 2H); HRMS (ESI-) m/z: 251.0053 [M-H]- (expected 251.0046); HPLC Ret: 7.19 min.
tert-butyl 4-((5-(2-chloro-5-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18g’)
Method D starting from 5-(2-chloro-5-cyclopropylphenyl)-1,3,4-oxadiazole-2-thiol (18g, 0.05 g) gave tert-butyl 4-((5-(2-chloro-5-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.07 g, 90%); 1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 2.3 Hz, 1H) 7.39 (d, J = 8.4 Hz, 1H) 7.14 (dd, J = 8.3, 2.3 Hz, 1H) 3.35 (t, J = 7.2 Hz, 2H) 2.43 (t, J = 7.2 Hz, 2H) 2.16 (p, J = 7.2 Hz, 2H) 1.93 (ddd, J = 13.5, 8.6, 5.1 Hz, 1H) 1.45 (s, 9H) 1.07–0.98 (m, 2H) 0.75–0.64 (m, 2H); HRMS (ESI+) m/z: 395.1208 [M+H]+ (expected 395.1196); HPLC Ret: 9.21 min.
5-(2-chloro-4-cyclopropoxyphenyl)-1,3,4-oxadiazole-2-thiol (18h)
Method C starting from 2-chloro-4-cyclopropoxybenzohydrazide (17h’, 0.2 g) gave 5-(2-chloro-4-cyclopropoxyphenyl)-1,3,4-oxadiazole-2-thiol as yellow solid (0.18 g, 78%); 1H NMR (500 MHz, CDCl3) δ 10.36 (br. s, 1H) 7.84 (d, 8.8 Hz, 1H) 7.24 (d, J = 2.5 Hz, 1H) 7.06 (dd, J = 8.8, 2.5 Hz, 1H) 3.85–3.77 (m, 1H) 0.92–0.79 (m, 4H); HRMS (ESI+) m/z: 269.0149 [M+H]+ (expected 269.0152); HPLC Ret: 7.13 min.
methyl 4-((5-(2-chloro-4-cyclopropoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18h’)
Method D starting from 5-(2-chloro-4-cyclopropoxyphenyl)-1,3,4-oxadiazole-2-thiol (18h, 0.1 g) gave methyl 4-((5-(2-chloro-4-cyclopropoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.11 g, 78%); 1H NMR (500 MHz, CDCl3) δ 7.86 (d, 8.8 Hz, 1H) 7.24 (d, J = 2.5 Hz, 1H) 7.04 (dd, J = 8.8, 2.5 Hz, 1H) 3.83–3.76 (m, 1H) 3.69 (s, 3H) 3.35 (t, J = 7.2 Hz, 2H) 2.53 (t, J = 7.2 Hz, 2H) 2.20 (p, J = 7.2 Hz, 2H) 0.90–0.78 (m, 4H); HRMS (ESI+) m/z: 369.0674 [M+H]+ (expected 369.0676); HPLC Ret: 7.94 min.
5-(2-chloro-4-(oxetan-3-yloxy)phenyl)-1,3,4-oxadiazole-2-thiol (18i)
Method C starting from 2-chloro-4-(oxetan-3-yloxy)benzohydrazide (17i’, 0.2 g) gave 5-(2-chloro-4-(oxetan-3-yloxy)phenyl)-1,3,4-oxadiazole-2-thiol as white solid (0.19 g, 81%); 1H NMR (500 MHz, CDCl3) δ 10.61 (br. s, 1H) 7.85 (d, 8.8 Hz, 1H) 6.87 (d, J = 2.5 Hz, 1H) 6.74 (dd, J = 8.9, 2.6 Hz, 1H) 5.27 (pd, 6.0, 1.0 Hz, 1H) 5.02 (t, J = 7.1 Hz, 2H) 4.78 (dd, J = 7.3, 5.1 Hz, 2H); HRMS (ESI-) m/z: 282.9945 [M-H]- (expected 282.9944); HPLC Ret: 6.03 min.
methyl 4-((5-(2-chloro-4-(oxetan-3-yloxy)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18i’)
Method D starting from 5-(2-chloro-4-(oxetan-3-yloxy)phenyl)-1,3,4-oxadiazole-2-thiol (18i, 0.08 g) gave methyl 4-((5-(2-chloro-4-(oxetan-3-yloxy)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate as colorless oil (0.07 g, 70%); 1H NMR (500 MHz, CDCl3) δ 7.88 (d, 8.8 Hz, 1H) 6.85 (d, J = 2.6 Hz, 1H) 6.73 (dd, J = 8.8, 2.6 Hz, 1H) 5.26 (pd, 6.0, 1.0 Hz, 1H) 5.01 (t, J = 7.1 Hz, 2H) 4.77 (dd, J = 7.3, 5.1 Hz, 2H) 3.69 (s, 3H) 3.35 (t, J = 7.2 Hz, 2H) 2.53 (t, J = 7.2 Hz, 2H) 2.20 (p, J = 7.2 Hz, 2H); HRMS (ESI+) m/z: 285.0621 [M+H]+ (expected 385.0625); HPLC Ret: 6.03 min.
4-((5-(2-chloro-4-ethylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (19a)
Method E starting from tert-butyl 4-((5-(2-chloro-4-ethylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18a’, 0.07 g) gave 4-((5-(2-chloro-4-ethylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white solid (0.05 g, 83%); 1H NMR (500 MHz, CDCl3) δ 9.52 (br. s, 1H) 7.85 (d, J = 8.0, 1H) 7.37 (d, J = 1.6 Hz, 1H) 7.22 (dd, J = 8.1, 1.6 Hz, 1H) 3.38 (t, J = 7.1 Hz, 2H) 2.70 (q, J = 7.6 Hz, 2H) 2.59 (t, J = 7.1 Hz, 2H) 2.23 (p, J = 7.2 Hz, 2H) 1.27 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 178.04, 164.39, 164.29, 149.58, 132.76, 130.81, 130.54, 126.78, 120.06, 32.32, 31.60, 28.51, 24.34, 14.87; HRMS (ESI+) m/z: 349.0385 [M+Na]+ (expected 349.0390); HPLC Ret: 6.96 min; 98% pure.
4-((5-(2-chloro-4-propylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (19b)
Modified Method E (product required silica gel column chromatography eluting with 2% MeOH: 98% DCM and semi-prep to obtain > 95% purity) starting from tert-butyl 4-((5-(2-chloro-4-propylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18b’, 0.03 g) gave 4-((5-(2-chloro-4-propylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as colorless oil (0.01 g, 47%); 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 8.0, 1H) 7.35 (s, 1H) 7.20 (dd, J = 8.1, 1.6 Hz, 1H) 3.38 (t, J = 7.2 Hz, 2H) 2.64–2.57 (m, 4H) 2.22 (p, J = 7.2 Hz, 2H) 1.68 (h, J = 7.4 Hz, 2H) 0.96 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 177.82, 164.39, 164.31, 148.13, 132.68, 131.10, 131.06, 130.72, 127.35, 120.08, 37.52, 32.27, 31.59, 24.35, 23.95, 13.65; HRMS (ESI+) m/z: 341.0738 [M+H]+ (expected 341.0727); HPLC Ret: 7.43 min; 97% pure.
4-((5-(2-chloro-4-isopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (19c)
Modified Method E (product required semi-prep to obtain > 95% purity) starting from a mixture of tert-butyl 4-((5-(2-chloro-4-isopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (19c’) and tert-butyl 4-((5-(4-isopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (0.03 g) gave 4-((5-(2-chloro-4-isopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white oil (0.01 g, 36%); 1H NMR (500 MHz, MeOH-d4) δ 7.86 (d, J = 8.1 Hz, 1H) 7.49 (d, J = 1.7 Hz, 1H) 7.38 (dd, J = 8.1, 1.7 Hz, 1H) 3.38 (t, J = 7.2 Hz, 2H) 2.99 (hept, J = 7.0 Hz, 1H) 2.51 (t, J = 7.2 Hz, 2H) 2.14 (p, J = 7.2 Hz, 2H) 1.29 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, MeOH-d4) δ 174.11, 165.07, 164.11, 154.83, 132.45, 130.74, 128.88, 125.45, 119.83, 33.80, 31.86, 31.34, 24.65, 22.39; HRMS (ESI+) m/z: 341.0902 [M+H]+ (expected 341.0727); HPLC Ret: 7.32 min; 99% pure.
4-((5-(2-chloro-4-(2-fluoropropan-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (19d)
Modified Method F (product required column chromatography eluting with 50% EtOAc: 50% Hex: 0.1% AcOH to obtain > 95% purity) starting from methyl 4-((5-(2-chloro-4-(2-fluoropropan-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18d’, 0.09 g) gave 4-((5-(2-chloro-4-(2-fluoropropan-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as colorless oil (0.06 g, 76%); 1H NMR (500 MHz, CDCl3) δ 7.94 (d, J = 8.2 Hz, 1H) 7.56 (d, J = 1.7 Hz, 1H) 7.38 (dd, J = 8.3, 1.7 Hz, 1H) 3.39 (t, J = 7.1 Hz, 2H) 2.60 (t, J = 7.1 Hz, 2H) 2.23 (p, J = 7.1 Hz, 2H) 1.72 (s, 3H) 1.68 (s, 3H); 19F NMR (100 MHz, CDCl3) δ −139.2 (hept., 1H); 13C NMR (125 MHz, CDCl3) δ 178.08, 164.73, 163.90, 150.88, 133.03, 130.94, 126.80, 122.72, 121.58, 32.30, 31.60, 29.05, 28.85, 24.31; HRMS (ESI+) m/z: 359.0627 [M+H]+ (expected 359.0632); HPLC Ret: 6.95 min; 98% pure.
4-((5-(2-chloro-3-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (19e)
Modified Method E (product required column chromatography eluting with 5% MeOH: 95% DCM to obtain > 95% purity) starting from tert-butyl-4-((5-(2-chloro-3-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18e’, 0.06 g) gave 4-((5-(2-chloro-3-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white oil (0.02 g, 39%); 1H NMR (500 MHz, CDCl3) δ 7.68 (dd, J = 7.7, 1.6 Hz, 1H) 7.29 (t, J = 7.8 Hz, 1H) 7.15 (dd, J = 7.8, 1.6 Hz, 1H) 3.39 (t, J = 7.2 Hz, 2H) 2.59 (t, J = 7.1 Hz, 2H) 2.31–2.18 (m, 3H) 1.13–0.98 (m, 2H) 0.77–0.66 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 175.45, 164.64, 143.05, 134.30, 129.45, 128.56, 126.72, 123.34, 109.99, 32.30, 31.60, 24.37, 14.02, 8.05; HRMS (ESI+) m/z: 339.0571 [M+H]+ (expected 339.0570); HPLC Ret: 6.94 min; 95% pure.
4-((5-(2-chloro-4-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (19f)
Modified Method F (product required column chromatography eluting with 5% MeOH: 95% DCM to obtain > 95% purity) starting from methyl-4-((5-(2-chloro-4-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18f’, 0.07 g) gave 4-((5-(2-chloro-4-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as colorless oil (0.07 g, 92%); 1H NMR (500 MHz, CDCl3) δ 8.11 (br. s., 1H) 7.81 (d, J = 8.2 Hz, 1H) 7.19 (s, 1H) 7.05 (dd, J = 8.2, 1.7 Hz, 1H) 2.58 (t, J = 7.1 Hz, 2H) 2.37 (t, J = 7.2 Hz, 2H) 2.20 (p, J = 7.2 Hz, 2H) 1.96–1.91 (tt, J = 8.4, 5.0 Hz, 1H) 1.08 (dt, J = 6.9, 4.9 Hz, 2H) 0.78 (dt, J = 6.9, 4.9 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 177.84, 164.36, 164.26, 150.10, 132.77, 130.66, 128.12, 124.27, 119.43, 32.36, 31.61, 24.37, 15.40, 10.41; HRMS (ESI+) m/z: 361.0383 [M+Na]+ (expected 361.0390); HPLC Ret: 6.93 min; 97% pure.
4-((5-(2-chloro-5-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (19g)
Modified Method E (product required column chromatography eluting with 5% MeOH: 95% DCM to obtain > 95% purity) starting from tert-butyl-4-((5-(2-chloro-5-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18g’, 0.07 g) gave 4-((5-(2-chloro-5-cyclopropylphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white oil (0.03 g, 55%); 1H NMR (500 MHz, CDCl3) δ 8.90 (br. s, 1H) 7.63 (d, J = 2.2 Hz, 1H) 7.39 (d, J = 8.3 Hz, 1H) 7.14 (dd, J = 8.4, 2.3 Hz, 1H) 3.38 (t, J = 7.1 Hz, 2H) 2.59 (t, J = 7.1 Hz, 2H) 2.22 (p, J = 7.1 Hz, 2H) 1.93 (tt, J = 8.4, 5.0 Hz, 1H) 1.08–0.98 (m, 2H) 0.73 (dt, 6.8, 4.9 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 177.92, 164.64, 164.35, 143.59, 130.93, 129.86, 129.51, 128.16, 122.37, 32.32, 31.62, 24.35, 14.89, 9.56; HRMS (ESI+) m/z: 339.0573 [M+H]+ (expected 339.0570); HPLC Ret: 6.98 min; 97% pure.
4-((5-(2-chloro-4-cyclopropoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (19h)
Method F starting from methyl 4-((5-(2-chloro-4-cyclopropoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18h’, 0.1 g) gave 4-((5-(2-chloro-4-cyclopropoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white powder (0.07 g, 75%); 1H NMR (500 MHz, CDCl3) δ 11.07 (br. s, 1H) 7.86 (d, J = 8.8 Hz, 1H) 7.22 (d, J = 2.4 Hz, 1H) 7.04 (dd, J = 8.8, 2.5 Hz, 1H) 3.80 (tt, J = 6.2, 3.0 Hz, 1H) 3.37 (t, J = 7.1 Hz, 2H) 2.59 (t, J = 7.1 Hz, 2H) 2.21 (p, J = 7.1 Hz, 2H) 0.92–0.77 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 177.72, 164.22, 164.01, 133.99, 131.86, 131.78, 117.37, 115.47, 114.41, 51.62, 32.22, 31.60, 24.35, 6.28; HRMS (ESI+) m/z: 355.0555 [M+H]+ (expected 355.0519); HPLC Ret: 6.90 min; 97% pure.
4-((5-(2-chloro-4-(oxetan-3-yloxy)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (19i)
Method F starting from methyl 4-((5-(2-chloro-4-(oxetan-3-yloxy)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoate (18i’, 0.07 g) gave 4-((5-(2-chloro-4-(oxetan-3-yloxy)phenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid as white powder (0.05 g, 79%); 1H NMR (500 MHz, CDCl3) δ 7.88 (d, 8.8 Hz, 1H) 6.85 (d, J = 2.5 Hz, 1H) 6.73 (dd, J = 8.8, 2.5 Hz, 1H) 5.27 (p, 6.0 Hz, 1H) 5.02 (t, J = 7.1 Hz, 2H) 4.78 (dd, J = 7.3, 5.1 Hz, 2H) 3.37 (t, J = 7.1 Hz, 2H) 2.58 (t, J = 7.1 Hz, 2H) 2.21 (p, J = 7.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 177.64, 164.26, 163.89, 159.11, 134.38, 132.27, 117.00, 116.16, 113.67, 77.44, 70.69, 32.25, 31.60, 24.33; HRMS (ESI+) m/z: 371.0307 [M+H]+ (expected 371.0468); HPLC Ret: 5.85 min; 98% pure.
2-(2,4-dichlorophenyl)-5-(methylthio)-1,3,4-oxadiazole (20’)
5-(2,4-dichlorophenyl)-1,3,4-oxadiazole-2-thiol (4j, 0.25 g, 1.01 mmol) was dissolved in 5 mL THF. Triethylamine (0.12 g, 0.17 mL, 1.21 mmol) and iodomethane (0.16 g, 0.07 mL, 1.11 mmol) were added and the mixture was stirred at 25 °C for 2 h. The reaction was quenched with 5 mL H2O. The product was extracted with EtOAc (3x, 15 mL), washed with brine (20 mL), dried with MgSO4, and concentrated in vacuo. The residue was dissolved in minimal EtOAc and the product was triturated with Hex, producing a white precipitate that was filtered and dried under vacuum. Yield=81%. 1H NMR (400 MHz, CDCl3) δ ppm 7.91 (d, J = 8.5 Hz, 1H) 7.57 (d, J = 2.1 Hz, 1H) 7.39 (dd, J = 8.5, 2.1 Hz, 1H) 2.79 (s, 3H); MS (ESI+) m/z: 260.9 [M+H]+; HPLC Ret: 7.57 min.
2-(2,4-dichlorophenyl)-5-(methylsulfonyl)-1,3,4-oxadiazole (20)
Route 1: 2-(2,4-dichlorophenyl)-5-(methylthio)-1,3,4-oxadiazole (20’, 0.22 g, 0.823 mmol) was dissolved in 5 mL EtOH and cooled to 0 °C. Ammonium molybdate tetrahydrate (0.20 g, 0.165 mmol) and hydrogen peroxide (0.28 g, 0.25 mL, 2.47 mmol) were added and stirred at 25 °C for 2 h. The reaction was quenched with 10% sodium thiosulfate (10 mL). The product was extracted with EtOAc (3x, 15 mL), washed with brine (20 mL), dried with MgSO4, and concentrated in vacuo. The yellow residue was subjected to silica gel chromatography eluting with 20% EtOAc: 80% Hex. The fractions containing product were concentrated in vacuo to produce white solid. Yield=54%. 1H NMR (400 MHz, CDCl3) δ ppm 8.02 (d, J = 8.5 Hz 1H) 7.64 (d, J = 2.0 Hz 1H) 7.47 (dd, J = 8.5, 2.0 Hz, 1H) 3.55 (s, 3H); MS (ESI+) m/z: 292.9 [M+H]+; HPLC Ret: 6.96 min.
Route 2: 2-(2,4-dichlorophenyl)-5-(methylthio)-1,3,4-oxadiazole (20’, 0.3 g, 1.15 mmol) was dissolved in 5 mL DCM. mCPBA (0.57 g, 2.30 mmol) was added and the reaction was stirred at 25 °C for 2 h. The solvent was evaporated, and the yellow residue was subjected to silica gel chromatography eluting with 20% EtOAc: 80% Hex. The fractions containing product were concentrated in vacuo to produce white solid. Yield=64%.
4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)sulfonyl)butanoic acid (21).
4-((5-(2,4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)butanoic acid (8q, 0.05 g, 0.15 mmol) was dissolved in 1 mL DCM. 70% mCPBA (0.08 g, 0.33 mmol) was added and the reaction was stirred at 25 °C for 4 h. The solvent was evaporated, and the yellow residue was subjected to silica gel chromatography eluting with 5% MeOH: 95% DCM. The fractions containing product were concentrated in vacuo to produce white solid. The solid was crystalized with EtOAc/Hex to produce white powder. Yield=58%. 1H NMR (500 MHz, DMSO-d6) δ ppm 12.31 (br. s, 1H) 8.10 (d, J = 8.5 Hz, 1H) 8.00 (J = 2.0 Hz, 1H) 7.74 (dd, J = 8.5, 2.1 Hz, 1H) 3.83 (t, J = 7.3 Hz, 2H) 2.43 (t, J = 7.3 Hz, 2H) 2.01 (p, J = 7.5 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ ppm 173.73, 164.02, 161.87, 138.74, 133.93, 133.60, 131.45, 128.83, 120.84, 54.32, 31.71, 17.93; HRMS (ESI-) m/z: 362.9609 [M-H]- (expected 362.9609); HPLC Ret: 6.56 min; 99% pure.
Scheme 2. Synthesis of Thioalkanoate-modified Analogs 11 and 13a.
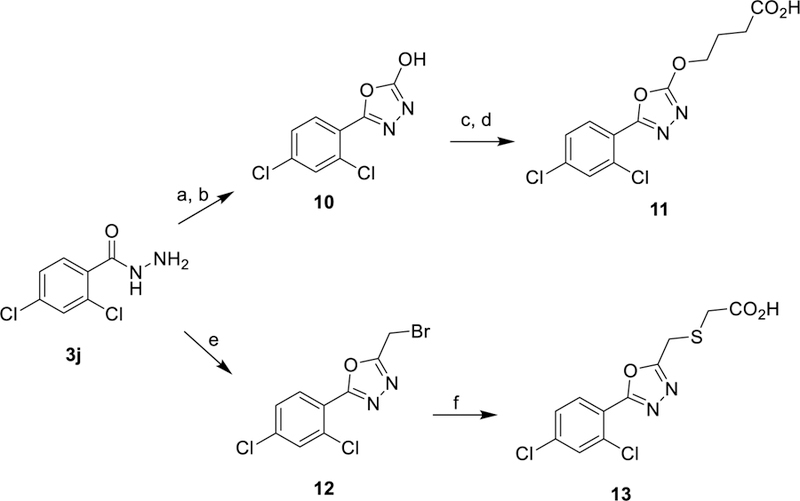
aReagents and conditions: (a) K3PO4, CS2, H2O, 106 °C, 2 h. (b) propylene oxide, 25 °C, 16 h. (c) DMF, K2CO3, Br-(CH2)3-CO2tBu, 90 °C, 1 h. (d) DCM, TFA, 25 °C, 1 h. (e) POCl3, bromoacetic acid, 106 °C, 16 h. (f) 2-mercaptoacetic acid, acetone, K2CO3, 25 °C, 2 h.
Acknowledgements:
This research was supported by NIH NIAMS award R01AR066049 (SDL) and NIH NIGMS award R01GM115459-01A1 (RRN). Support was also provided in part by a University of Michigan Mi-TRAC award (SDL and RRN) sponsored by the Michigan Economic Development Corporation.
Abbreviations Used:
- SSc
scleroderma
- MRTF
myocardin-related transcription factor
- SRF
serum response factor
- SRE
serum response element
- CTGF
connective tissue growth factor
- LPA
lysophosphatidic acid
- dSSc
diffuse scleroderma
- IPF
idiopathic pulmonary fibrosis
- ECM
extracellular matrix
- α-SMA
α-smooth muscle actin
- TGFβ
transforming growth factor β
- ROCK
Rho-associated protein kinase
- G
globular
- F
filamentous
- NPC
nuclear pore complex
- DAST
diethylaminosulfur trifluoride
- mCPBA
m-chloroperoxybenzoic acid
- DMEM
Dulbecco’s Modified Eagle Medium
Footnotes
Supporting Information
Molecular formula strings (CSV)
References:
- 1.Varga J, Systemic Sclerosis (Scleroderma) and Related Disorders. In Harrison’s Principles of Internal Medicine, 20e, Jameson JL; Fauci AS; Kasper DL; Hauser SL; Longo DL; Loscalzo J, Eds. McGraw-Hill Education: New York, NY, 2017. [Google Scholar]
- 2.Distler O; Cozzio A, Systemic sclerosis and localized scleroderma-current concepts and novel targets for therapy. Semin Immunopathol 2016, 38 (1), 87–95. [DOI] [PubMed] [Google Scholar]
- 3.Allanore Y; Simms R; Distler O; Trojanowska M; Pope J; Denton CP; Varga J, Systemic sclerosis. Nat Rev Disease Primers 2015, 1, 1–21. [DOI] [PubMed] [Google Scholar]
- 4.Careta MF; Romiti R, Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol 2015, 90 (1), 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pattanaik D; Brown M; Postlethwaitel BC; Postlethwaite AE, Pathogenesis of systemic sclerosis. Front Immunol 2015, 6, 1–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fala L, Ofev (Nintedanib): First tyrosine kinase inhibitor approved for the treatment of patients with Idiopathic Pulmonary Fibrosis. Am Health Drug Benefits 2015, 8 (Spec Feature), 101–104. [PMC free article] [PubMed] [Google Scholar]
- 7.Nanthakumar CB; Hatley RJ; Lemma S; Gauldie J; Marshall RP; Macdonald SJ, Dissecting fibrosis: therapeutic insights from the small-molecule toolbox. Nat Rev Drug Discov 2015, 14 (10), 693–720. [DOI] [PubMed] [Google Scholar]
- 8.Thannickal VJ; Henke CA; Horowitz JC; Noble PW; Roman J; Sime PJ; Zhou Y; Wells RG; White ES; Tschumperlin DJ, Matrix biology of idiopathic pulmonary fibrosis: a workshop report of the national heart, lung, and blood institute. Am J Pathol 2014, 184 (6), 1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinz B; Phan SH; Thannickal VJ; Prunotto M; Desmouliere A; Varga J; De Wever O; Mareel M; Gabbiani G, Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 2012, 180 (4), 1340–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang X; Yang N; Fiore VF; Barker TH; Sun Y; Morris SW; Ding Q; Thannickal VJ; Zhou Y, Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol 2012, 47 (3), 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsou PS; Haak AJ; Khanna D; Neubig RR, Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: focus on Rho GTPase-regulated gene transcription. Am J Physiol Cell Physiol 2014, 307 (1), C2–C13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wipff PJ; Rifkin DB; Meister JJ; Hinz B, Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol 2007, 179 (6), 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wynn TA, Cellular and molecular mechanisms of fibrosis. J Pathol 2008, 214 (2), 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandbo N; Dulin N, Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl Res 2011, 158 (4), 181–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandbo N; Lau A; Kach J; Ngam C; Yau D; Dulin NO, Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-beta. Am J Physiol Lung Cell Mol Physiol 2011, 301 (5), L656–L666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandbo N; Ngam C; Torr E; Kregel S; Kach J; Dulin N, Control of myofibroblast differentiation by microtubule dynamics through a regulated localization of mDia2. J Biol Chem 2013, 288 (22), 15466–15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomasek JJ; Vaughan MB; Kropp BP; Gabbiani G; Martin MD; Haaksma CJ; Hinz B, Contraction of myofibroblasts in granulation tissue is dependent on Rho/Rho kinase/myosin light chain phosphatase activity. Wound Repair Regen 2006, 14 (3), 313–320. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Y; Huang X; Hecker L; Kurundkar D; Kurundkar A; Liu H; Jin TH; Desai L; Bernard K; Thannickal VJ, Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest 2013, 123 (3), 1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evelyn CR; Wade SM; Wang Q; Wu M; Iniguez-Lluhi JA; Merajver SD; Neubig RR, CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther 2007, 6 (8), 2249–2260. [DOI] [PubMed] [Google Scholar]
- 20.Marcucci F; Stassi G; De Maria R, Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nature Reviews Drug Discovery 2016, 15 (5), 311–325. [DOI] [PubMed] [Google Scholar]
- 21.Rosenbloom J; Mendoza FA; Jimenez SA, Strategies for anti-fibrotic therapies. Biochim Biophys Acta 2013, 1832 (7), 1088–1103. [DOI] [PubMed] [Google Scholar]
- 22.Sisson TH; Ajayi IO; Subbotina N; Dodi AE; Rodansky ES; Chibucos LN; Kim KK; Keshamouni VG; White ES; Zhou Y; Higgins PD; Larsen SD; Neubig RR; Horowitz JC, Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol 2015, 185 (4), 969–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu SW; Stratton R; Nikitorowicz-Buniak J; Ahmed-Abdi B; Ponticos M; Denton C; Abraham D; Takahashi A; Suki B; Layne MD; Lafyatis R; Smith BD, A Role of Myocardin Related Transcription Factor-A (MRTF-A) in Scleroderma Related Fibrosis. Plos One 2015, 10 (5), e0126015/1–e0126015/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hill CS; Wynne J; Treisman R, Serum-regulated transcription by serum response factor (SRF): a novel role for the DNA binding domain. EMBO J 1994, 13 (22), 5421–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutchings KM; Lisabeth EM; Rajeswaran W; Wilson MW; Sorenson RJ; Campbell PL; Ruth JH; Amin A; Tsou PS; Leipprandt JR; Olson SR; Wen B; Zhao T; Sun DX; Khanna D; Fox DA; Neubig RR; Larsen SD, Pharmacokinetic optimitzation of CCG-203971: Novel inhibitors of the Rho/MRTF/SRF transcriptional pathway as potential antifibrotic therapeutics for systemic scleroderma. Bioorg Med Chem Lett 2017, 27 (8), 1744–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell JL; Haak AJ; Wade SM; Kirchhoff PD; Neubig RR; Larsen SD, Optimization of novel nipecotic bis(amide) inhibitors of the Rho/MKL1/SRF transcriptional pathway as potential anti-metastasis agents. Bioorg Med Chem Lett 2013, 23 (13), 3826–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evelyn CR; Bell JL; Ryu JG; Wade SM; Kocab A; Harzdorf NL; Showalter HDH; Neubig RR; Larsen SD, Design, synthesis and prostate cancer cell-based studies of analogs of the Rho/MKL1 transcriptional pathway inhibitor, CCG-1423. Bioorg Med Chem Lett 2010, 20 (2), 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haak AJ; Appleton KM; Lisabeth EM; Misek SA; Ji YJ; Wade SM; Bell JL; Rockwell CE; Airik M; Krook MA; Larsen SD; Verhaegen M; Lawlor ER; Neubig RR, Pharmacological inhibition of Myocardin-related Transcription Factor pathway blocks bung metastases of RhoC-overexpressing melanoma. Mol Cancer Ther 2017, 16 (1), 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haak AJ; Tsou PS; Amin MA; Ruth JH; Campbell P; Fox DA; Khanna D; Larsen SD; Neubig RR, Targeting the myofibroblast genetic switch: inhibitors of Myocardin-Related Transcription Factor/Serum Response Factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. J Pharmacol Exp Ther 2014, 349 (3), 480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin WZ; Goldfine AB; Boes T; Henry RR; Ciaraldi TP; Kim EY; Emecan M; Fitzpatrick C; Sen A; Shah A; Mun E; Vokes M; Schroeder J; Tatro E; Jimenez-Chillaron J; Patti ME, Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. Journal of Clinical Investigation 2011, 121 (3), 918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakai N; Chun J; Duffield JS; Wada T; Luster AD; Tager AM, LPA(1)-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. Faseb J 2013, 27 (5), 1830–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu-Wai-Man C; Spencer-Dene B; Lee RMH; Hutchings K; Lisabeth EM; Treisman R; Bailly M; Larsen SD; Neubig RR; Khaw PT, Local delivery of novel MRTF/SRF inhibitors prevents scar tissue formation in a preclinical model of fibrosis. Sci Rep 2017, 7:518,1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan X; Zhou S; Wang YQ; Ge ZM; Cheng TM; Li RT, Propylene oxide assisted one-pot, tandem synthesis of substituted-1,3,4-oxadiazole-2(3H)-ones in water. Tetrahedron 2012, 68 (38), 7978–7983. [Google Scholar]
- 34.Sun J; Cao N; Zhang XM; Yang YS; Zhang YB; Wang XM; Zhu HL, Oxadiazole derivatives containing 1,4-benzodioxan as potential immunosuppressive agents against RAW264.7 cells. Bioorgan Med Chem 2011, 19 (16), 4895–4902. [DOI] [PubMed] [Google Scholar]
- 35.Albrecht BK; Audia JE; Gagnon A; Harmange J; Naveschuk CG “Modulators of methyl modifying enzymes, compositions and uses thereof”, WO/2013/075084, 2013.
- 36.Shen D-M; Sinz CJ; Crespo A; Wilson JE; McCracken T; Xu S; Li H “Triazolyl pyrimidinone compounds as PDE2 inhibitors”, WO/2016/145614, 2016.
- 37.Toda N; Asano S; Barbas CF, Rapid, stable, chemoselective labeling of thiols with Julia-Kocienski-like reagents: A serum-stable alternative to maleimide-based protein conjugation. Angew Chem Int Edit 2013, 52 (48), 12592–12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adumeau P; Davydova M; Zeglis BM, Thiol-reactive bifunctional chelators for the creation of site-selectively modified radioimmunoconjugates with improved stability. Bioconjug Chem 2018, 29 (4), 1364–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrukh A; Paez JI; Salierno M; Fan W; Berninger B; Del Campo A, Bifunctional poly(acrylamide) hydrogels through orthogonal coupling chemistries. Biomacromolecules 2017, 18 (3), 906–913. [DOI] [PubMed] [Google Scholar]
- 40.Saitoh M; Kunitomo J; Kimura E; Hayase Y; Kobayashi H; Uchiyama N; Kawamoto T; Tanaka T; Mol CD; Dougan DR; Textor GS; Snell GP; Itoh F, Design, synthesis and structure-activity relationships of 1,3,4-oxadiazole derivatives as novel inhibitors of glycogen synthase kinase-3beta. Bioorg Med Chem 2009, 17 (5), 2017–2029. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki N; Hajicek N; Kozasa T, Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals 2009, 17 (1), 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang MR; Lv JY; Zou LL; Yang W; Xiong YL; Chen XJ; Guan M; He R; Zou HJ, A modified murine model of systemic sclerosis: bleomycin given by pump infusion induced skin and pulmonary inflammation and fibrosis. Lab Invest 2015, 95 (3), 342–350. [DOI] [PubMed] [Google Scholar]
- 43.Batteux F; Kavian N; Servettaz A, New insights on chemically induced animal models of systemic sclerosis. Curr Opin Rheumatol 2011, 23 (6), 511–518. [DOI] [PubMed] [Google Scholar]
- 44.Avouac J, Mouse model of experimental dermal fibrosis: The bleomycin-induced dermal fibrosis. Methods Mol Biol 2014, 1142, 91–98. [DOI] [PubMed] [Google Scholar]
- 45.Srivastava AK; Khare P; Nagar HK; Raghuwanshi N; Srivastava R, Hydroxyproline: A potential biochemical marker and its role in the pathogenesis of different diseases. Curr Protein Pept Sc 2016, 17 (6), 596–602. [DOI] [PubMed] [Google Scholar]
- 46.Huang J; Beyer C; Palumbo-Zerr K; Zhang Y; Ramming A; Distler A; Gelse K; Distler O; Schett G; Wollin L; Distler JH, Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis 2016, 75 (5), 883–890. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y; Lu F; Kang L; Wang Z; Wang Y, Pirfenidone attenuates bleomycin-induced pulmonary fibrosis in mice by regulating Nrf2/Bach1 equilibrium. BMC Pulm Med 2017, 17:63, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]