

Abstract
The innate immune system is a critical regulator of the adaptive immune responses that lead to allograft rejection. It is increasingly recognized that endogenous molecules released from tissue injury and cell death are potent activators of innate immunity. Mitochondria, ancestrally related to bacteria, possess an array of endogenous innate immune activating molecules. We have recently demonstrated that extracellular mitochondria are abundant in the circulation of deceased organ donors and that their presence correlates with early allograft dysfunction. Here we demonstrate the ability of mitochondria to activate ECs, the initial barrier between a solid-organ allograft and its host. We find that mitochondria exposure leads to the upregulation of EC adhesion molecules and their production of inflammatory cytokines and chemokines. Additionally, mitochondrial exposure causes DCs to upregulate costimulatory molecules. Infusion of isolated mitochondria into heart donors lead to significant increase in allograft rejection in a murine heterotopic heart transplantation model. Finally, co-incubation of human PBMCs with mitochondria treated ECs results in increased numbers of effector (IFN-γ+, TNF-α+) CD8+ T cells. These data indicate that circulating extracellular mitochondria in deceased organ donors may directly activate allograft ECs and promote graft rejection in transplant recipients.
Introduction
The vascular endothelium is a critical regulator of many pathological processes [1–3]. During organ procurement, cold and warm ischemia followed by reperfusion creates an ischemia-reperfusion injury that has the potential to activate vascular endothelial cells (ECs) or cause EC dysfunction in the donor graft [3, 4]. Furthermore, vascular ECs of a donor organ are the first cells to be exposed to the recipient immune system and serve a critical role in systemic immune activation [5]. When activated, ECs upregulate adhesion molecules and secrete cytokines and chemokines that enhance leukocyte adhesion and promote leukocyte migration and effector functions [6]. Activated ECs also upregulate major histocompatibility complex (MHC) molecules, providing a source of antigen presentation from the non-hematopoietic compartment [6]. ECs also participate in the secretion of glycosylases that are key regulators of the dissolution of the vascular glycocalyx permitting T cell adhesion and diapedesis [7]. However, the role early EC activation and subsequent EC dysfunction plays in contributing to transplant organ dysfunction is poorly understood.
In addition to recognizing pathogen-derived molecules, innate immune pattern recognition receptors, such as Toll-like receptors and Nod-like receptors, recognize endogenous molecules released during sterile tissue damage [8, 9]. These endogenous molecules, termed damage-associated molecular patterns (DAMPs) can be potent initiators of the innate immune inflammatory response and include molecules derived from the extracellular matrix as well as cell organelles (e.g. mitochondria), cytoplasm and nucleus [10, 11].
Mitochondria are evolutionarily derived from bacteria that developed an endosymbiotic relationship with eukaryotic cells approximately 2 billion years ago [12–15]. Because of their ancestry, mitochondria have retained molecules of bacterial origin [12, 13]. While the majority of the mitochondrial genome has migrated to the cell nucleus, mitochondria still contain a remnant genome with unmethylated CpG sequences that can serve as TLR9 ligands [16–19]. They also maintain their own protein translational system resulting in the production of thirteen proteins initiated with n-formylated peptides, a potent innate immune activator recognized by the n-formyl peptide receptor family [20]. Multiple inflammasome activators are also derived from mitochondria, such as adenosine triphosphate (ATP), reactive oxygen species (ROS) and cardiolipin [21–25]. Consequently, mitochondria released during cell injury are a source of multiple DAMPs that can cause endogenous inflammatory responses [19, 26–29].
Here we report that purified mitochondria accumulate within ECs, inducing their upregulation of adhesion molecules and secretion of inflammatory cytokines and. Mitochondrial uptake was dependent on scavenger receptors and actin polymerization, subsequently leading to the intracellular co-localization of exogenous mitochondria with endogenous mitochondria. The danger signals generated during mitochondrion-EC interactions augment allospecific memory T cell responses and pre-treatment of allograft donors with mitochondria increases cardiac allograft rejection. Our results indicate that mitochondria directly initiate EC inflammatory responses that provoke alloreactive T cell adhesion and activation, and ultimately increasing allograft rejection.
Materials and Methods
Mice
C57BL/6 (Stock No: 000664) and BALB/c (Stock No: 000651) mice were from The Jackson Laboratory (Bar Harbor, ME). 4C T-cell receptor transgenic (TCR-tg) mice were produced as described previously [30]. The 4C mouse is a CD4+ TCR-tg on the C57BL/6 background with direct allospecificity against the I-Ad MHC class II molecule. All experimental procedures on mice were done in accordance with protocols approved by the Animal Care and Use Committee of Duke University.
Cell lines
Human aortic endothelial cells (HAECs) at passage 2 were purchased from Cell Applications (San Diego, CA) and sub-cultured in EC culture medium (Cell Applications). HAECs were used at passage 6 or 7 for all assays after achieving confluence. LMTK (CCL-1.3), bEnd.3 (CRL-2299), and HeLa cell lines (CCL-2) were purchased from ATCC (Manassas, VA). LMTK and HeLa cell lines stably expressing mitochondrial-targeted DsRed fluorescent protein were developed by expanding single cell clones stably transduced with pLV-mitoDsRed lentivirus, a gift from Pantelis Tsoulfas (Addgene plasmid # 44386). Cell lines were cultured in DMEM media (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (Hyclone™, GE Healthcare Life Sciences, South Logan, UT), and penicillin and streptomycin antibiotics (100 μg/mL each, Sigma, St. Louis, MO).
Antibodies and flow cytometry
Antibodies against human CD3 (HIT3a), CD4 (RPA-T4), CD8 (HIT8a), CD54 (HA58), TNF-α (7N4–1) and INF-γ (B27) were from BD Biosciences (San Jose, CA). Antibodies against human CD106 (STA) and CD62E (P2H3) were from Thermo Fisher Scientific (Canoga Park, CA). Antibodies against mouse CD3 (145–2C11), CD4 (RM4–5), CD8 (53–6.7), CD54 (3E2), CD62E (10E9.6), CD11c (HL3), CD11b (M1/70), CD40 (HM40–3), CD11c (HL3), CD11b (M1/70), CD45.2 (104), CD31 (MEC 13.3), and Ly-6G (1A8), and IFN-γ (XMG1.2) were from BD Biosciences. Anti-mouse CD106 (429) was from Thermo Fisher Scientific. Intracellular IFN-γ and TNF staining was performed as described previously [31]. Flow cytometric data was acquired using an LSRFortessa™ X-20 flow cytometer (BD Biosciences), and events were analyzed using FlowJo software (Version 9.9.6, TreeStar, San Carlos, CA).
Mitochondria purification
Mouse mitochondria were isolated from LMTK cells and human mitochondria were purified from HeLa cells. Purification methods were similar to as described previously [32]. Briefly 1 × 108 LMTK or HeLa cells were homogenized by 10 passages through a 27 G needle in IBc buffer (10 mM Tris-MOPS, 1 mM EGTA, 0.2 M sucrose, pH 7.4) supplemented with and 100 μg/mL DNase I. The homogenate was centrifuged at 1,000xg for 10 min at 4°C in a refrigerated tabletop centrifuge (Beckman Coulter, Brea, CA). The supernatant (cytosol fraction) was then centrifuged at 10,000 xg for 10 min at 4°C in a refrigerated microcentrifuge (Eppendorf, Hamburg, Germany). The pellet was resuspended in 1 mL of 4°C IBc buffer and again pelleted at 10,000 xg for 10 min at 4°C in a refrigerated microcentrifuge. The washed pellet (“mitochondrial fraction”) was re-suspended in ice-cold PBS and protein concentration was measured with Bradford Reagent (Sigma). In experiments involving human mitochondria, anti-TOMM22 MicroBeads (Mitochondria Isolation Kit #130–094-532, Miltenyi Biotec, Auburn, CA) were used to provide further affinity purification of mitochondria (purified mitochondrial fraction).
Verification of mitochondria by western blot
Mitochondria were assessed for purity by western blot analysis using the following antibodies to determine purity and contamination from other cellular compartments: anti-ATP5a (Abcam, ab176569, mitochondria), anti-LAMP2A (Abcam, ab125068, lysosomes), anti-GRP78 (Abcam, ab108613, endosomes), anti-histone H3 (Abcam, ab177184, nucleus), anti-GAPDH (Abcam, ab181602, cytosol). Secondary antibody was peroxidase-conjugated AffiniPure F(ab’)2 fragment goat anti-rabbit IgG (#111–036-114, Jackson ImmunoResearch Laboratories, West Grove, PA). Example of western blot is shown in Figure S1A.
Verification of mitochondria by electron microscopy
Thin sections were obtained from pelleted purified mitochondria fixed with 3% glutaraldehyde in 0.1 M cacodylic acid buffer (pH 7.4, Ladd Research Industries, Williston, VT). The sample was washed 3 times with 0.1 M cacodylic acid buffer and post-stained with 1% osmium tetroxide in cacodylic buffer for 1 hour. Cells were then washed as before and embedded in 1% agarose. The agarose containing the cell sample was then pre-stained with 2% uranyl acetate (Quorum Technologies, Laughton, East Sussex, England) overnight at 4°C. The samples were washed and carried through acetone dehydration steps. Infiltration was done using the Epon embedding kit. Samples were sectioned ultrathin (60–70 nm) on a Reichert Ultracut E ultramicrotome and stained with 2% uranyl acetate in 50% ethanol for 30 minutes and SATO’s Lead stain for 1 minute. Samples were imaged on a Philips CM12 electron microscope. Example of electron micrograph is shown in Figure S1B.
Mouse heart transplantation
MHC-mismatched BALB/c (H2d) to C57BL/6 (H2b) heterotopic cardiac allografts transplants were performed in the setting of costimulation blockade with CTLA4-Ig (250 μg i.p. on POD 0, 2, 4 and 6; Bio-X-Cell, West Lebanon, NH, product #BE0099). Donor BALB/c mice were intravenously injected with third-party murine mitochondria isolated from LMTK cells (300 μg, H-2k) or vehicle solution (VS) one day prior to organ procurement. Mean survival time (MST) of allografts was defined as loss of graft contractions determined by palpation on two consecutive days. In separate experiments, graft histology, CD3 immunohistochemistry and flow cytometric analysis of graft infiltrating lymphocytes (GILs) were performed on POD14.
Mouse endothelial cell (EC) analysis
The mouse endothelial cell line, bEnd.3 was incubated with DsRed-labeled mitochondria purified from LMTK cells stably transduced with pLV-mitoDsRed lentivirus (described above). Uptake of mitochondria was determined by flow cytometry and confocal microscopy. Cytochalasin E (#C2149, Sigma) and Poly-I (#P4154, Sigma) were used in some experiments to determine if mitochondrial uptake was dependent on actin polymerization or scavenger receptors, respectively. Mitochondrial treated bEnd.3 cells were assayed by flow cytometry for the upregulation of adhesion molecules, CD54, CD106 and CD62E.
Mouse dendritic cell (DC) analysis and mixed leukocyte reaction
DCs were generated from BALB/c mice bone marrow cultured in the presence of murine GM-CSF (#1320–03-20, Gold Biotechnology, St. Louis, MO) as previously described [31]. 4C TCR-tg T cells were isolated from the lymph nodes and spleens by affinity purification (#19851, EasySep™ Mouse T cell Isolation Kit, Stemcell Technologies, Cambridge, MA). DCs were co-cultured with 4C TCR-tg T cells for 4 day. Conventional CD11c+, CD11b+ DCs were analyzed by flow cytometry for uptake of DsRed-labeled mitochondria and surface expression of CD40 and CD86 following overnight incubation with mitochondria or vehicle solution. T cells were assayed for IFN-γ production by intracellular cytokine staining following co-incubation with DCs.
T cell adhesion assay
Murine endothelial cells (bEnd.3) were plated overnight in 24 well plates, treated with mitochondria (50 μg/mL) purified from LMTK cells, or murine TNF-α (50 ng/mL) for 5 hr. ECs were then rinsed with DMEM three times and co-incubated with T cells (2×106/well) purified from the 4C TCR-tg mice labeled with CFSE (1 μM, #C34554, Thermo Fisher Scientific) for 1 hr at 37°C. Non-adherent cell were rinsed-off with room temperature DMEM and adherent cells where quantified using a fluorescence plate reader (filter EX485/EM520) based on a standard curve made by measuring the fluorescence from wells containing known quantities of CFSE-labeled cells.
Human endothelial cell analysis
For coincubation experiments, freshly purified mitochondria were added to confluent HAECs in 24-well plates. In selected experiments, purified mitochondria were placed into the upper chambers of Corning transwell 24-well permeable tissue culture plates (0.4 μm pore diameter, Sigma-Aldrich) with confluent HAECs in the bottom wells. At the end of stimulation, HAECs were washed with PBS and released from the tissue culture plates by incubation with PBS containing 20 mM HEPES, 10 mM EDTA, and 0.5% BSA at 4°C for 20 min. HAECs were then surface stained with anti-CD54, anti-CD62E and anti-CD106, and analyzed by flow cytometry. IL-6, IL-8, and MCP-1 in the cell culture supernatant were quantified using cytometric bead assay (BD-Biosciences).
To determine the uptake of mitochondria by HAECs, mitochondria isolated from HeLa cells with mitochondrial-targeted DsRed (described above) were incubated with confluent ECs at 37°C and washed three times with PBS to remove unbound mitochondria. ECs were then analyzed by flow cytometry and by confocal microscopy to access for uptake of DsRed-labeled mitochondria. In some experiments, HAECs were pre-treated with 100 nM MitoTracker Green (Thermo Fisher Scientific) prior to incubation with DsRed-labeled mitochondria.
Human allogeneic mixed-leukocyte reaction
Blood of healthy volunteers who were consented under an Institutional Review Board-approved tissue acquisition protocol was obtained and underwent Percoll (GE17–5445-01, Sigma) gradient centrifugation to isolate PBMCs. Confluent ECs were incubated with 100 μg/mL purified mitochondria or equal volume of vehicle solution for 6 hr and washed three times with PBS to remove unbound mitochondria. PBMCs (0.5 × 106) were then incubated with HAECs for 2 hr, and then 12 hr in the additional presence of 1 μg/mL of GolgiPlug (BD Biosciences). Unstimulated PBMCs were used as controls.
Statistical analysis
Comparisons between the groups were done using a two-tailed Student’s t-test. Comparisons between multiple groups was performed using a Kruskal-Wallis with Dunn’s multiple comparison test. Survival analysis was performed using log rank test. Statistical analyses were performed using Prism v7.0 software (GraphPad Software, La Jolla, CA). For all experiments, a P-value ≤0.05 was considered significant.
Results
Circulating donor mitochondria accelerate costimulation blockade-resistant (CoBR) allograft rejection.
We have previously reported that extracellular mitochondria are abundant in the circulation of deceased human organ donors and that their presence is associated with an inflammatory response in the donor and early allograft dysfunction in transplant recipients [33]. To better understand the role of mitochondria in the circulation of transplant donors, we developed a murine allogeneic cardiac transplant model in which donor mice are treated with mitochondria prior to organ procurement and transplantation (Figure 1A).
Figure 1. Exposure of cardiac allografts to circulating mitochondria increases costimulation blockade resistant rejection.
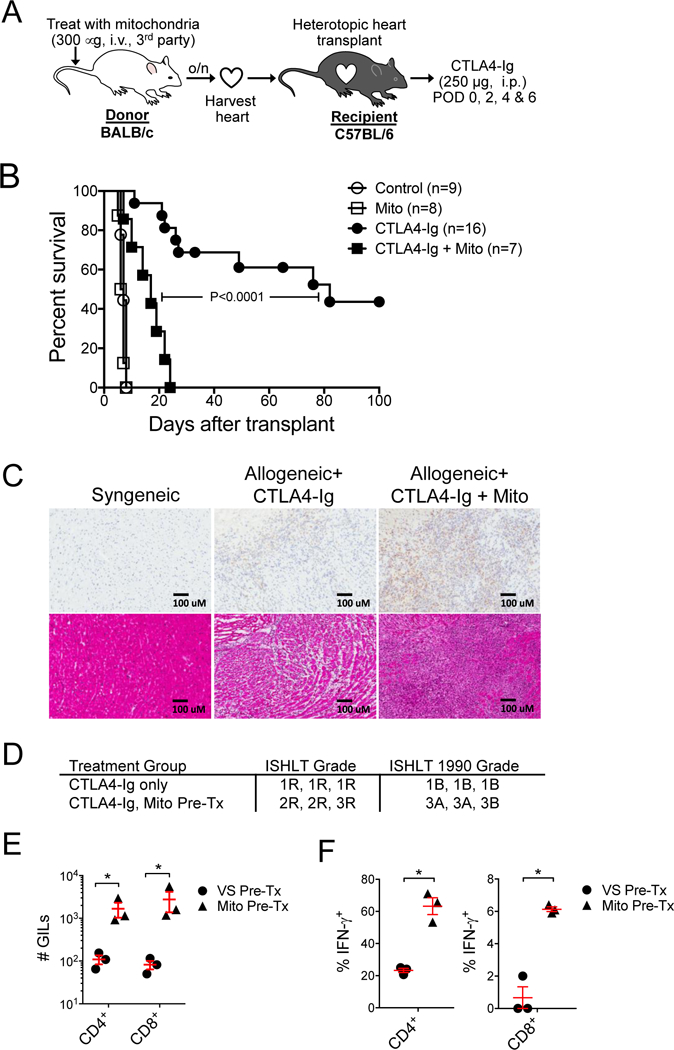
A) Donor hearts were harvested from BALB/c mice (H-2d) following intravenous injection of mitochondria (300 mg) or vehicle solution (control) one day prior. Hearts where then heterotopically transplanted into C57BL/6 mice (H-2b) treated with or without costimulation blockade with murine CTLA4-Ig (250 mg, i.p., POD 0, 2, 4, 6). B) Kaplan-Meier analysis of cardiac allograft survival. Curves were compared by log-rank test. C) CD3 immunohistology (top panels) and H&E staining (lower panels) of 5 mm cryosections from syngeneic or allogeneic cardiac allografts at 2-weeks post-transplantation. Grafts were from donors exposed to mitochondria (Mito) or vehicle (control) one day prior to organ harvest. Allogeneic recipients were treated with CTLA4-Ig. D) Current and 1990 ISHLT histologic grading of cardiac allograft rejection in the allogeneic transplant groups (n=3 per group). E) Quantification of graft-infiltrating CD4+ and CD8+ T cells. Graft infiltrating leukocytes (GILs) were purified from cardiac allografts harvested 2-weeks post-transplantation and assayed by flow cytometry for CD4+ and CD8+ T cells. Allogeneic cardiac allografts were harvested from donor mice exposed to circulating mitochondria (Mito Pre-Tx, n=3) or treated with vehicle solution (VS, n=3) one day prior. F) Percentage of CD4+ and CD8+ GILs producing IFN-g in cardiac allografts in from donors pre-treated with intravenous mitochondria (Mito Pre-Tx, n=3) or VS (n=3). Comparisons were performed using a two-tailed Student’s t-test. *A p-value <0.05 was considered significant.
In this model, BALB/c (H-2d) donor mice were intravenously injected with mitochondria (300 μg) purified from 3rd party cells (LMTK, H-2k), or vehicle solution (VS) one day prior to heart procurement. The hearts were then transplanted into C57BL/6 (H-2b) recipient mice treated with or without costimulation blockade using CTLA4-Ig (250 μg, i.p. on POD 0, 2, 4 and 6). In the absence of costimulation blockade, both VS and mitochondria pre-treated grafts were rejected rapidly with median survival times (MST) of 7 days. However, in the setting of costimulation blockade, cardiac allografts from donors treated with mitochondria rejected significantly earlier than grafts from VS treated heart donors (MST 17 d vs. 82 d, Figure 1B). Amongst the CTLA4-Ig treated cohort, mitochondria-treated donors had significantly greater T cell infiltrates compared to VS treated donors, as detected by CD3 immunohistology at 2-weeks post-transplantation (Figure 1C, top panel). In addition, based on the H&E staining, mitochondria-treated cardiac allografts had higher ISHLT grades of acute rejection (Figures 1C bottom panel & 1D). Analysis of hearts and spleens from healthy mice that underwent systemic injection of purified mitochondria indicates an uptake of mitochondria by potential endothelial cell in the heart and the spleen (Figure S2A). The mitochondria were also taken up by peripheral blood monocytes (CD11b+, Ly6Gneg), splenic monocytes (CD11b+, CD11cneg, Ly6Gneg), splenic dendritic cells (CD11c+, CD11b+, Ly6Gneg), and splenic endothelial cells (CD31+, CD45neg) (Figure S2).
Finally, mitochondria-treated donor grafts had significantly greater numbers of CD4+ and CD8+ graft infiltrating lymphocytes (GILs) at 2-weeks post-transplantation, as quantified by flow cytometry (Figure 1E). The CD4+ and CD8+ GILS in the mitochondria pre-treated donor group also included a significantly higher percentage of IFN-γ producing CD4+ and CD8+ GILs (Figure 1F).
ECs uptake extracellular mitochondria and upregulate vascular adhesion molecules, causing increased allospecific T cell adhesion.
The endothelium is the initial barrier between circulation and a vascularized transplant graft. We hypothesized that exposure of donor allografts to extracellular mitochondria would result in the activation of graft ECs. We investigated the interaction of mitochondria with the ECs. For this purpose, mitochondria were purified from LMTK cells stably expressing mitochondrial-targeted DsRed fluorescent protein and co-incubated with ECs. Interestingly, we observed a dose-dependent uptake of extracellular mitochondria by ECs by flow cytometry (Figure 2A) and determined their localization to be within the EC cytosol by confocal microscopy (Figure 2B). Thus, ECs appear to internalize extracellular mitochondria.
Figure 2. ECs uptake extracellular mitochondria and upregulate vascular adhesion molecules, causing increased T cell adhesion.
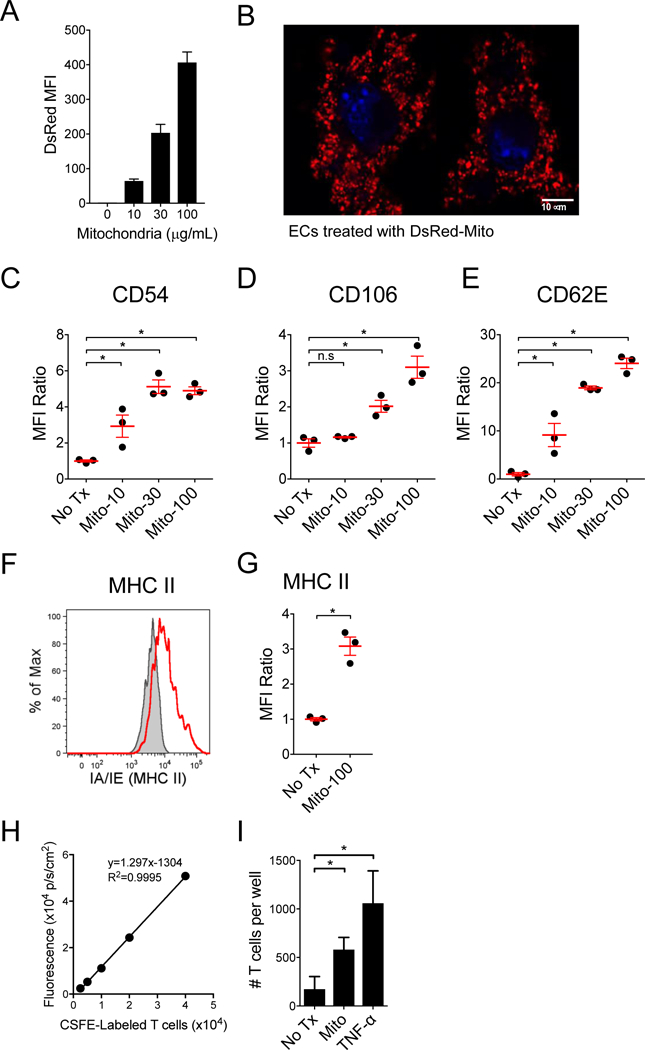
A) Increasing concentrations of purified DsRed expressing murine mitochondrial were incubated with murine ECs and analyzed for mitochondria uptake by FACS. B) Murine ECs incubated with DsRed-mitochondria (30 μg/mL) were imaged with confocal microscopy and demonstrate intracellular localization of the exogenous mitochondria. Nuclei were labeled blue with DAPI. C-E) Upregulation of adhesion molecules, CD54, CD106 and CD62E were assessed by flow cytometry following a 16 hr pulse with mitochondria (n=3 per group). Upregulation of MHC II molecules on ECs following treatment with mitochondria (100 μg/mL). F) Representative flow cytometry plot and D) graph of n=3 per group. Treatment of ECs with mitochondria increases allogeneic T cell adhesion. H) Adherent T cells were quantified using a fluorescent plate reader based on a standard curve of the CFSE-labeled T cells (n=4 per group). I) Mouse ECs (bEnd.3, H-2d) were treated for 5 hr with third-party mitochondria purified from LMTK fibroblasts (H-2k), or with murine TNF-α (50 ng/mL), and then incubated with CFSE-labeled 4C-TCR-tg T cells (direct allo-specificity against I-Ad) for 30 mins. Comparisons were performed using a Kruskal-Wallis with Dunn’s multiple comparison test. *A p-value <0.05 was considered significant, ns=not significant.
EC activation by DAMPs elicits inflammatory responses that result in the upregulation of cell-surface adhesion molecules, including CD54 (ICAM-1), CD106 (VCAM-1) and CD62E (E-selectin), leading to the adhesion of neutrophils, monocytes and lymphocytes [34–37]. To determine whether extracellular mitochondria caused a similar activation of ECs, we incubated murine ECs (bEnd.3 cell line) with mitochondria purified from the LMTK cells, and then tested for the upregulation of adhesion molecules by flow cytometry. We found that treating ECs with increasing concentrations of mitochondria lead to increasing upregulation of CD54, CD106, and CD62E (Figures 2C-E). At the highest mitochondria concentration (100 mg/mL), we also found an upregulation of MHC II molecules (Figure 2F and 2G).
We next examined whether mitochondria-treated ECs had a higher binding affinity for allospecific T cells. CFSE-labeled allospecific T cells purified from the 4C TCR-tg mouse [30] were co-cultured with bEnd.3 cells treated with mitochondria or vehicle solution (VS). T cells from the 4C TCR-tg mouse are CD4+ T cells on the C57BL/6 background with direct allospecificity against MHC class II (I-Ad) present on the bEnd.3 cells, which were originally derived from BALB/c mice. T cell adherence to the EC monolayer was then quantified using a fluorescent plate reader, based on a standard curve derived from known quantities of the CFSE-labeled T cells (Figure 2H). We found that ECs treated with mitochondria demonstrated significantly increased affinity for binding the allospecific T cells (Figure 2I).
Dendritic cells (DCs) uptake extracellular mitochondria, upregulate costimulation molecules and increase IFN-γ production by allospecific T cells.
DCs are major antigen presenting cells within cardiac allografts [38]. We next sought to determine if DCs also uptake extracellular mitochondria and become activated. Similar to experiments with ECs, we observed a dose-dependent uptake of DsRed-labeled murine mitochondria by bone-marrow derived DCs (BMDCs, Figure 3A). Following incubation with purified mitochondria, DCs demonstrated upregulation of costimulation molecules (CD40 and CD86) and antigen presenting molecules (MHC II) (Figure 3B-D). Furthermore, following mitochondrial treatment, DCs were better able to prime allospecific T cells as demonstrated by increased IFN-γ production by 4C TCR-tg T cells in co-culture with mitochondria treated BALB/c BMDCs (Figures 3E & F).
Figure 3. Dendritic cells (DCs) also uptake extracellular mitochondria, causing costimulation molecule upregulation and increased IFN-g production by allospecific T cells.
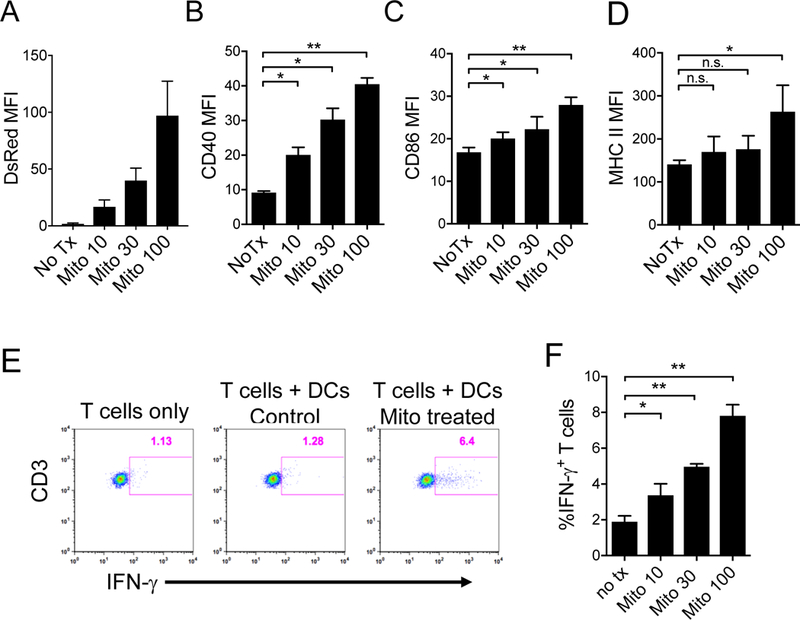
A) Purified DsRed expressing murine mitochondrial were incubated with murine BMDCs and analyzed for mitochondria uptake by flow cytometry (n=3 per group). MFI, mean fluorescence intensity. B-D) Costimulation molecules (CD40 and CD86) and antigen presenting molecules (MHC II) are upregulated on DCs following co-incubation with purified mitochondria. E) Extracellular mitochondria prime DCs to activate allogeneic T cells. BALB/c BMDCs pretreated with mitochondria overnight were co-cultured with 4C TCR-tg T cells for 4 days. IFN-γ producing T cells were then assessed by intracellular cytokine staining and flow cytometry. Representative data from cultures of T cells only, T cells with untreated DCs (control), and T cells with DCs treated with 100 μg/mL mitochondria are shown (n=3 per group). F) Comparison of the percentages of IFN-γ producing allogeneic T cells following 4-day co-culture with DCs pre-treated with 0, 10, 30 and 100 μg/mL purified mitochondria. Comparisons were performed using a Kruskal-Wallis with Dunn’s multiple comparison test. *p-value <0.05, **p-value <0.001.
Extracellular mitochondria activate human ECs.
We next sought to determine if our results with murine ECs translated to human ECs. Primary human aortic endothelial cells (HAECs) were co-cultured with mitochondria purified from HeLa cells, a human cervical cancer cell line rich in mitochondria. Following co-incubation with purified mitochondria, HAEC surface expression of adhesion molecules was quantified by flow cytometry. Mitochondria treatment caused the upregulation of CD54, CD106 and CD62E by HAECs, compared with unstimulated cells in a dose- and time-dependent manner (Figures 4A & B).
Figure 4. Extracellular mitochondria cause human ECs to upregulate vascular adhesion molecules.
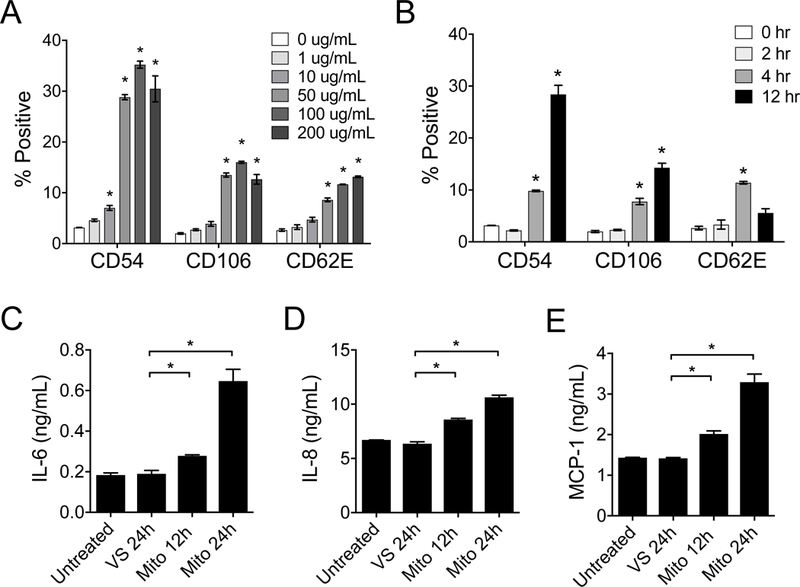
Primary human aortic ECs (HAECs) were treated with mitochondria purified from HeLa cells and analyzed for the upregulation of adhesion molecules CD54, CD106, and CD62E by flow cytometry. A) Dose response of the upregulation of adhesion molecules by HAECs following treatment with purified mitochondria (12 hours). B) Time response of EC upregulation of adhesion molecules following treatment with increasing exposure time of purified mitochondria (100 μg/mL). C-E) HAECs treated with mitochondria (100 μg/mL for 12 hr) demonstrate increased secretion of IL-6, IL-8, and MCP-1. HAECs without treatment or treated VS were used as controls. Comparisons were performed using a Kruskal-Wallis with Dunn’s multiple comparison test. *A p-value <0.05 was considered significant. In A & B, experimental groups were compared to no treatment.
Activated ECs produce inflammatory cytokines and chemokines such as interleukin (IL)-6, IL-8 and monocyte chemoattractant protein-1 (MCP-1) that recruit lymphocytes, neutrophils and monocytes to areas of injury or inflammation [28, 39, 40]. We found that HAECs incubated with purified mitochondria produced significant amounts of IL-6, IL-8 and MCP-1 (Figures 4C-E).
Direct mitochondria contact is necessary for HAEC activation.
It was not known whether direct contact with intact mitochondria was necessary for the observed activation of HAECs. To determine this, we co-cultured HAECs for 12 hr with mitochondria that were separated by a semi-permeable transwell insert of 0.4 μM porosity (with transwell) or in direct contact with mitochondria (without transwell). HAECs were then assayed for the upregulation of adhesion molecules by flow cytometry. We found that with the transwell insert present, HAEC activation was significantly reduced and was similar to vehicle-solution (VS) treatment (Figure 5).
Figure 5. Direct mitochondria contact is necessary for EC activation.
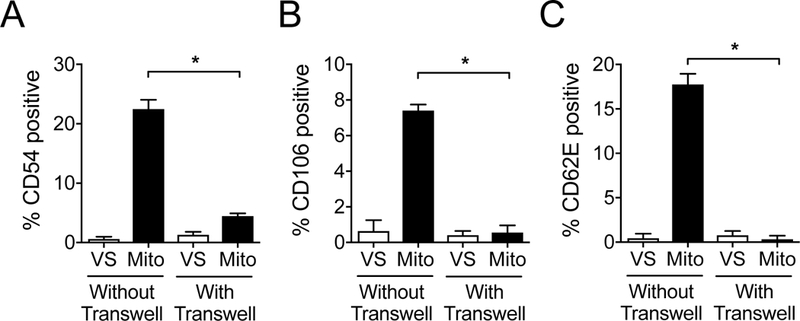
Mitochondria (100 μg/mL) or vehicle solution (VS) were co-cultured for 12 hr with HAEC monolayers that and were either in direct contact with the HAECs (Without Transwell), or separated from the HAEC monolayer by permeable transwell inserts with 0.4 μM pores (With Transwell). Presence of the transwell insert prevented EC upregulation of A) CD54, B) CD106 and C) CD62E in response to mitochondria treatment. Comparisons were performed a two-tailed Student’s t-test. *A p-value <0.05 was considered significant.
HAECs uptake extrinsic mitochondria that can co-localize with endogenous mitochondria.
Considering that direct contact between mitochondria and HAECs was necessary for EC activation, we investigated whether HEACs internalize the mitochondria, similar to the mouse endothelial cell line. HAECs were incubated with mitochondria purified from human HeLa cells stably expressing mitochondrial-targeted DsRed fluorescent protein. The uptake of fluorescent mitochondria was assayed by flow cytometry and HAECs demonstrated a dose-dependent association of mitochondria (Figure 6A). We found that inhibitors of phagocytosis including poly-I (100 nM, class A scavenger receptor inhibitor) and cytochalasin E (10 μg/mL, inhibitor of actin polymerization) prevented mitochondria uptake by ECs (Figure 6B & C).
Figure 6. ECs uptake extrinsic mitochondria that can co-localize with endogenous mitochondria.
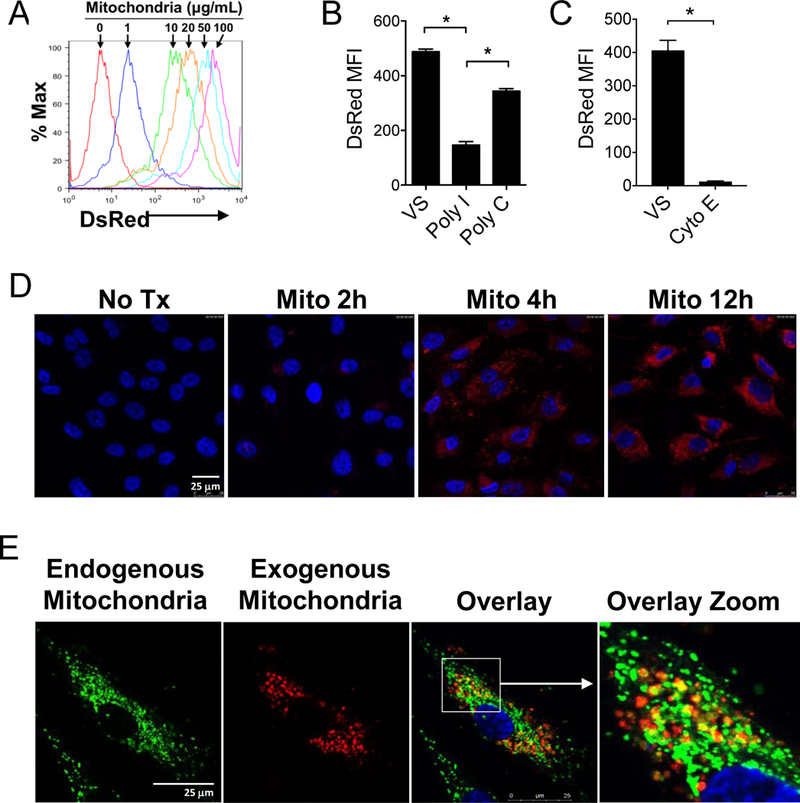
A) Confluent HAECs were incubated for 12 hr with DsRed-labeled human mitochondria at the indicated doses. B) poly-I (100 nM), a class A scavenger receptor inhibitor inhibited the uptake of DsRed-labeled mitochondria by ECs as determined by flow cytometry. Vehicle solution (VS) and Poly-C (100 nM) are negative controls. MFI, mean fluorescence intensity. C) Cytochalasin E (10 mg/mL), an inhibitor of actin polymerization, also inhibited mitochondrial uptake. Comparisons were performed using a two-tailed Student’s t-test. *A p-value <0.05 was considered significant. D) Confocal microscopy of HAECs treated with 30 mg/mL DsRed-labeled mitochondria for increasing periods of time demonstrate intracellular localization of the exogenous mitochondria. Nuclei are labeled with DAPI nuclear stain (Blue). E) HAECs treated with MitoTracker Green to label endogenous mitochondria were then treated with exogenous DsRed-labeled mitochondria (30 μg/mL) for 6 hr. Co-localized endogenous and exogenous mitochondria are yellow in the overlay images.
We next examined the location of the DsRed-labeled mitochondria by confocal microscopy. Similar to findings with the mouse EC line, primary HAECs also internalized extracellular mitochondria (Figure 6D). Interestingly, when endogenous mitochondria in HAECs were first labeled with MitoTracker Green dye, and then the HAECs were incubated with DsRed exogenous mitochondria, co-localization of endogenous and exogenous mitochondria was observed (Figure 6E). This observed co-localization with the endogenous mitochondria is likely in the endocytic compartments of phagosomes and autophagosomes.
Mitochondria-EC interactions do not cause apoptosis.
Mitochondrial dysfunction plays a central role in regulating apoptosis [41–43]. Since cell death can lead to the release of other non-mitochondrial DAMPs, we sought to determine whether exogenous mitochondria induced apoptotic cell-death. ECs stimulated by mitochondria (100 μg/mL for 12 hr) were assessed for apoptosis by staining with live/dead viability dye and flow cytometric analysis for caspase-3 cleavage, a specific marker for apoptotic cell death[44]. We observed no significant EC death or apoptosis following treatment with mitochondria (Figure 7).
Figure 7. Determination of EC viability and apoptosis following mitochondrion–EC interaction.
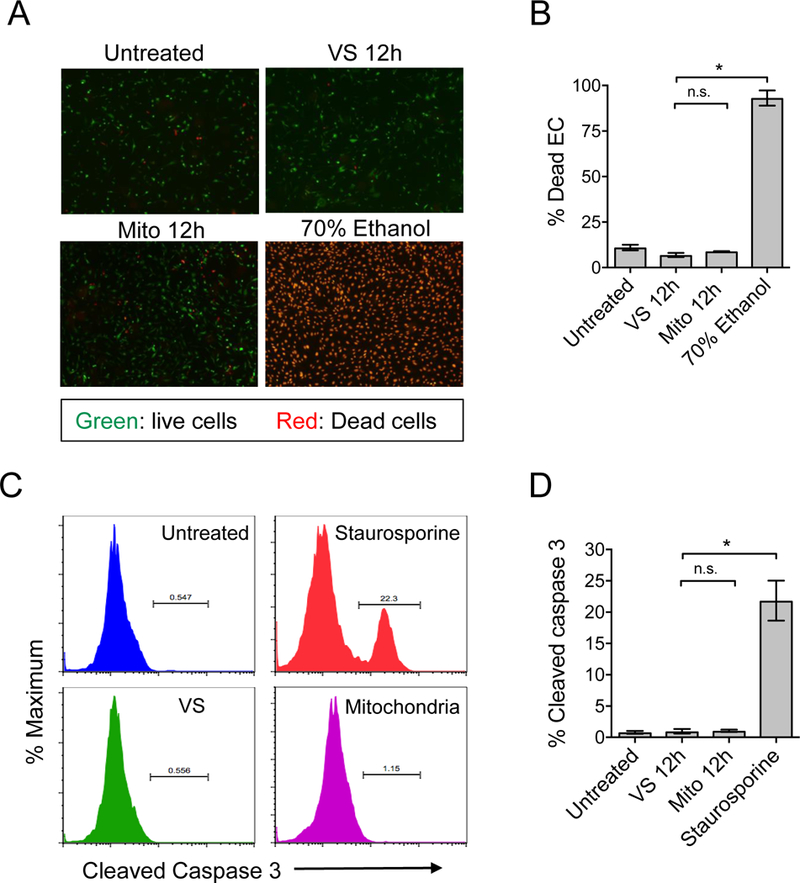
A) The viability of mitochondria-stimulated ECs was evaluated by staining with live/dead cell viability dye. Live cells stain green and dead cells stain red in this assay. ECs treated with staurosporine were used as positive controls. B) No significant increase of cell death was revealed at 12 hours post mitochondrion–EC interaction. C) Detection of EC apoptosis during mitochondrion–EC interaction was determined by detection of cleaved caspase 3. A representative experiment of ECs treated with mitochondria, vehicle solution (VS), or staurosporine (positive control). Following their interaction with exogenous mitochondria, ECs did not undergo significant apoptosis.
Mitochondria-treated ECs increase the human allogeneic T cell response.
ECs express class I and II MHC-peptide complexes, major costimulatory molecules, and adhesion molecules that can directly activate memory T cells [45–48]. We incubated HAEC monolayers with exogenous human mitochondria or vehicle solution (VS) to determine if mitochondria treatment would increase their ability to prime allogeneic T cells. After removal of unbound mitochondria, the ECs were co-incubated with healthy-donor human PBMCs for 16 hr. CD8+ T cells were then analyzed for activation by intracellular FACS analysis for the production of effector-cytokines (TNF-α/IFN-γ, Figure 8A). We found that mitochondria-conditioned ECs stimulated an increased allo-specific CD8+ T cell responses characterized by a significant increase of TNF-α/IFN-γ dual producers when compared with VS treated ECs (Figure 8B). This observation suggests that damage signals from mitochondrial DAMPs can initiate effector T cell immune responses by ECs.
Figure 8. Mitochondria-treated HAECs increase the allogeneic T cell response.
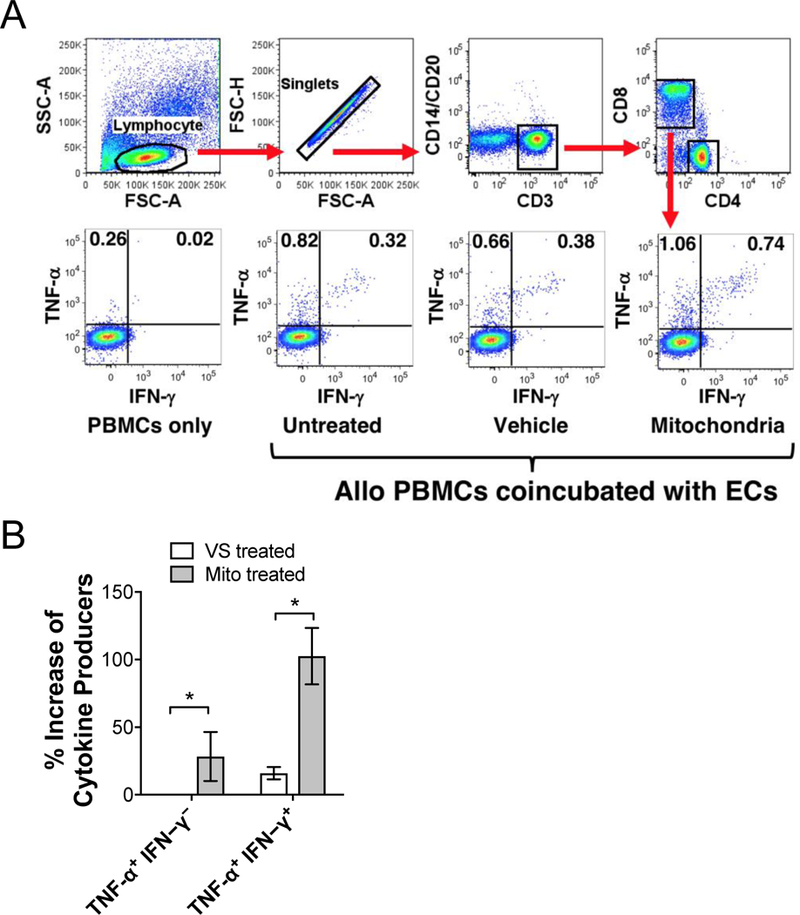
Human PBMCs, isolated from healthy individuals, were stimulated with allogeneic HAECs with or without mitochondrial pre-treatment followed by intracellular cytokine staining. CD8+ T cells were FACS-analyzed based on gating strategy shown in (A) and activated allospecific T cells were identified based on expression of TNF-α and IFN-γ. B) Mitochondria conditioned HAECs resulted in a significant increase in the percentage of TNF-α and IFN-γ producing T cells compared with vehicle solution (VS) treated ECs. Comparisons were performed using a two-tailed Student’s t-test. *A p-value <0.05 was considered significant; n=3 per group.
Discussion
Activation of vascular ECs, an initial barrier between vascularized transplant organ allografts and host immunity, is of paramount importance in initiating cell-mediated acute rejection [5, 37, 49, 50]. Unlike cardiomyocytes or hepatocytes where mitochondria can make up more than 30% of the cell, mitochondria make up less than 10% of the cell volume of ECs, a cell type largely dependent on anaerobic metabolism [51, 52]. An important role of mitochondria in ECs is thought to be cell signaling [53]. During scenarios of endogenous or exogenous stress, ECs can become activated, leading to either EC senescence or long-term EC dysfunction, processes involving EC mitochondria [43, 54]. Previous studies have demonstrated that ECs are capable of receptor-mediated uptake of a number of different molecules including other cells, bacteria and microparticles [55]. In this study, we demonstrate that ECs are also capable of receptor-mediated uptake of extracellular mitochondrial.
Our observations establish the paradigm of mitochondrial DAMPs functioning to augment allospecific T cell immunity. We show that EC interactions with extrinsic mitochondria cause EC activation as demonstrated by the upregulation of adhesion molecules and the generation of inflammatory cytokines, notably without inducing EC apoptosis. Importantly, mitochondrial uptake lead to increased MHC II expression in both ECs and DCs another possible mechanism causing increased rejection. Mitochondria released into the circulation as the result of major trauma and central nervous system injury in organ donors may be an important innate immune stimulus of the adaptive lymphocyte responses in transplant recipients. [56, 57] This eventually translates to allograft rejection driven by increase in GIL infiltration of the allograft caused by EC activation, and increased allo-antigen presentation caused by graft DC activation. Our central hypothesis and results are summarized in Figure 9.
Figure 9. Diagram of hypothesis.
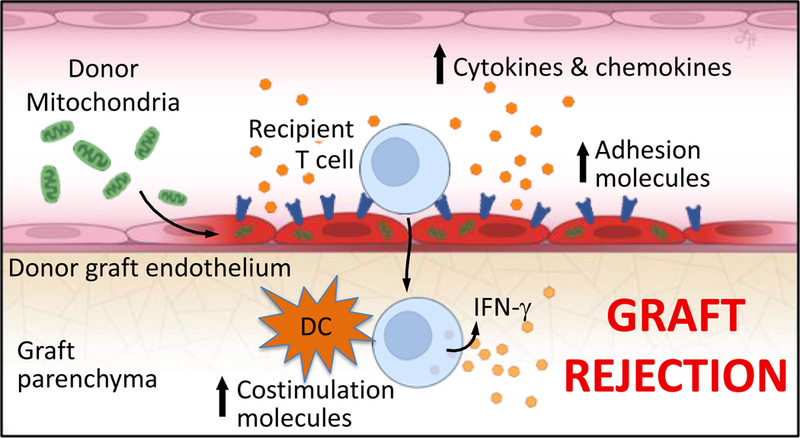
Circulating extracellular mitochondria released in the setting of donor brain death cause vascular EC activation resulting in adhesion molecule upregulation and inflammatory cytokine and chemokine production. Mitochondria-active ECs promote increased T cell adhesion and infiltration into allograft tissue. Mitochondria-primed graft DCs upregulate costimulation molecules and accelerate graft rejection.
For example, our results demonstrate that mitochondrial DAMPs function to augment allospecific T cell immunity. Mitochondria released into the circulation may be a key innate immune trigger of allospecific T cell responses [10, 58–60]. This has been demonstrated with other DAMPs, where DAMPs are generated and released in brain dead donors as well as during reperfusion [60, 61] These DAMPs were shown to activate cytotoxic T cells and increase the generation of donor specific antibodies (DSA) that contribute to allograft rejection and long-term graft dysfunction [10, 57, 60, 62]. Recently, we demonstrated that increased levels of extracellular mitochondria are present in the circulation of deceased donors and that these mitochondria activate cellular innate immune responses, which are associated with early allograft dysfunction in liver transplant recipients [57]. Our study provides additional evidence that circulating extracellular mitochondria in either the organ donor or the organ recipient can increase rates of graft rejection. Of note, though the purified mitochondria used in this study were well-enriched as shown by western blot, they were not entirely pure of non-mitochondrial structures as shown by electron microscopy imaging (Figure S1B). Since the mitochondria were purified by both differential centrifugation and affinity purification, we hypothesize that much of the debris seen on EM are fragments of mitochondria or fusion of mitochondrial fragments with other cellular debris. However, it remains possible that non-mitochondrial components of the purified fraction or mitochondrial constituents could also be responsible for the observed EC activation, including mitochondrial DNA [9].
The immediate uptake of the mitochondria is important, since it highlights that even short exposure of organs to circulating mitochondria in a brain-dead donor can lead to graft EC activation. Our data suggests that in the heart, such mitochondrial uptake is taking place near the coronary vasculature, suggesting endothelial uptake. This route of uptake is most likely phagocytosis. The observed colocalization of exogenous mitochondria with endogenous mitochondria likely occurs in endocytic compartments, perhaps a result of fusion of phagosomes and autophagosomes, or colocalization within lysosomes. The specific mitochondrial DAMPs involved in the EC activation has not yet been elucidated but could involve mitochondria DNA exposing to TLR9 receptors within lysosomes [9].
Therapies directed at blocking the uptake of mitochondria, either through scavenger receptor blockade, affinity depletion of circulating mitochondria, or targeting the receptors of specific mitochondrial DAMPs could be beneficial in the setting of inflammatory responses resulting from sterile injury. In endothelial cells, scavenger receptors play a critical role in foam cell formation through the uptake of low-density lipoproteins (LDL) [63], and bind a wide variety of different ligands and pathogens [64]. Activation of these receptors has been previously demonstrated to cause secondary expression of endothelial adhesion molecules [63]. Similarly, in our study, uptake of purified exogenous mitochondria led to a marked activation of endothelial cells, and upregulation of MHC II, VCAM, ICAM and E-selectin. Curiously, one of the main mechanisms of late graft failure in heart transplantation relates to advanced/diffuse vascular disease termed cardiac allograft vasculopathy (CAV). Our results may help shed light on a possible mechanism causing long-term endothelial dysfunction and foam cell formation.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Drs. Sara Miller and Ricardo Vancini for performing the electron microscopy. This work was funded in part by grants from the National Institutes of Health (AI097423, ADK; AI101263, TVB), (1R38HL143612–01 to MB) and the Roche Organ Transplant Research Foundation (HX).
Abbreviations
- (ATP)
Adenosine triphosphate
- (CAV)
Cardiac allograft vasculopathy
- (CoBR)
Costimulation blockade-resistant
- (DAMPs)
Damage-associated molecular patterns
- (DC)
Dendritic cell
- (DSA)
Donor specific antibodies
- (ECs)
Endothelial cells
- (GILs)
Graft infiltrating lymphocytes
- (HAECs)
Human aortic endothelial cells
- (LDL)
Low-density lipoproteins
- (MHC)
Major histocompatibility complex
- (MST)
Mean survival time
- (MCP-1)
Monocyte chemoattractant protein-1
- (ROS)
Reactive oxygen species
- (VS)
Vehicle solution
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of this article.
References
- 1.Taccone FS, et al. , Endothelium and regulatory inflammatory mechanisms during organ rejection. Angiology, 2014. 65(5): p. 379–87. [DOI] [PubMed] [Google Scholar]
- 2.Mori DN, et al. , Inflammatory triggers of acute rejection of organ allografts. Immunol Rev, 2014. 258(1): p. 132–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cardinal H, Dieude M, and Hebert MJ, Endothelial Dysfunction in Kidney Transplantation. Front Immunol, 2018. 9: p. 1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nordling S, et al. , Enhanced protection of the renal vascular endothelium improves early outcome in kidney transplantation: Preclinical investigations in pig and mouse. Sci Rep, 2018. 8(1): p. 5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briscoe DM, Alexander SI, and Lichtman AH, Interactions between T lymphocytes and endothelial cells in allograft rejection. Curr Opin Immunol, 1998. 10(5): p. 525–31. [DOI] [PubMed] [Google Scholar]
- 6.Jin YP, et al. , HLA Class II-Triggered Signaling Cascades Cause Endothelial Cell Proliferation and Migration: Relevance to Antibody-Mediated Transplant Rejection. J Immunol, 2018. 200(7): p. 2372–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porras G, Ayuso MS, and Gonzalez-Manchon C, Leukocyte-endothelial cell interaction is enhanced in podocalyxin-deficient mice. Int J Biochem Cell Biol, 2018. 99: p. 72–79. [DOI] [PubMed] [Google Scholar]
- 8.Patel MN, et al. , Inflammasome Priming in Sterile Inflammatory Disease. Trends Mol Med, 2017. 23(2): p. 165–180. [DOI] [PubMed] [Google Scholar]
- 9.Lubkin D BM, Barbas A, Brennan TV, Kirk AD, Extracellular Mitochondrial DNA and N-Formyl Peptides in Trauma and Critical Illness: A Systematic Review. Crit Care Med, 2018. In press. [DOI] [PubMed] [Google Scholar]
- 10.Land WG, et al. , Transplantation and Damage-Associated Molecular Patterns (DAMPs). Am J Transplant, 2016. 16(12): p. 3338–3361. [DOI] [PubMed] [Google Scholar]
- 11.Kaczmarek A, Vandenabeele P, and Krysko DV, Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity, 2013. 38(2): p. 209–23. [DOI] [PubMed] [Google Scholar]
- 12.Carvalho DS, et al. , What are the Evolutionary Origins of Mitochondria? A Complex Network Approach. PLoS One, 2015. 10(9): p. e0134988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gray MW, Burger G, and Lang BF, Mitochondrial evolution. Science, 1999. 283(5407): p. 1476–81. [DOI] [PubMed] [Google Scholar]
- 14.Zimmer C, Origins. On the origin of eukaryotes. Science, 2009. 325(5941): p. 666–8. [DOI] [PubMed] [Google Scholar]
- 15.Emelyanov VV, Mitochondrial connection to the origin of the eukaryotic cell. Eur J Biochem, 2003. 270(8): p. 1599–618. [DOI] [PubMed] [Google Scholar]
- 16.Kaniak-Golik A and Skoneczna A, Mitochondria-nucleus network for genome stability. Free Radic Biol Med, 2015. 82: p. 73–104. [DOI] [PubMed] [Google Scholar]
- 17.Hemmi H, et al. , A Toll-like receptor recognizes bacterial DNA. Nature, 2000. 408(6813): p. 740–5. [DOI] [PubMed] [Google Scholar]
- 18.West AP and Shadel GS, Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol, 2017. 17(6): p. 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.West AP, Shadel GS, and Ghosh S, Mitochondria in innate immune responses. Nat Rev Immunol, 2011. 11(6): p. 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Y, Murphy PM, and Wang JM, Formyl-peptide receptors revisited. Trends Immunol, 2002. 23(11): p. 541–8. [DOI] [PubMed] [Google Scholar]
- 21.Prevete N, et al. , Formyl peptide receptors at the interface of inflammation, angiogenesis and tumor growth. Pharmacol Res, 2015. 102: p. 184–91. [DOI] [PubMed] [Google Scholar]
- 22.Zhou R, et al. , A role for mitochondria in NLRP3 inflammasome activation. Nature, 2011. 469(7329): p. 221–5. [DOI] [PubMed] [Google Scholar]
- 23.Zhang JZ, et al. , Mitochondrial DNA induces inflammation and increases TLR9/NF-kappaB expression in lung tissue. Int J Mol Med, 2014. 33(4): p. 817–24. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Amores-Iniesta J, et al. , Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep, 2017. 21(12): p. 3414–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadatomi D, et al. , Mitochondrial function is required for extracellular ATP-induced NLRP3 inflammasome activation. J Biochem, 2017. 161(6): p. 503–512. [DOI] [PubMed] [Google Scholar]
- 26.Mehta MM, Weinberg SE, and Chandel NS, Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol, 2017. 17(10): p. 608–620. [DOI] [PubMed] [Google Scholar]
- 27.Calfee CS and Matthay MA, Clinical immunology: Culprits with evolutionary ties. Nature, 2010. 464(7285): p. 41–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crouser ED, et al. , Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Crit Care Med, 2009. 37(6): p. 2000–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iyer SS, et al. , Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity, 2013. 39(2): p. 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brennan TV, et al. , A new T-cell receptor transgenic model of the CD4+ direct pathway: level of priming determines acute versus chronic rejection. Transplantation, 2008. 85(2): p. 247–55. [DOI] [PubMed] [Google Scholar]
- 31.Brennan TV, et al. , Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. J Clin Invest, 2016. 126(1): p. 207–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartwig S KJ, Lehr S, Isolation and quality control of functional mitochondria, in Methods Molecular Biology. 2015, Springer New York: New York: p. 9–23. [DOI] [PubMed] [Google Scholar]
- 33.Pollara J, et al. , Circulating mitochondria in deceased organ donors are associated with immune activation and early allograft dysfunction. JCI Insight, 2018. 3(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirk AD, Morrell CN, and Baldwin WM 3rd, Platelets influence vascularized organ transplants from start to finish. Am J Transplant, 2009. 9(1): p. 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu H, Dhanireddy KK, and Kirk AD, Human monocytes as intermediaries between allogeneic endothelial cells and allospecific T cells: a role for direct scavenger receptor-mediated endothelial membrane uptake in the initiation of alloimmunity. J Immunol, 2006. 176(2): p. 750–61. [DOI] [PubMed] [Google Scholar]
- 36.Bevilacqua MP, Endothelial-leukocyte adhesion molecules. Annu Rev Immunol, 1993. 11: p. 767–804. [DOI] [PubMed] [Google Scholar]
- 37.Rose ML, Endothelial cells as antigen-presenting cells: role in human transplant rejection. Cell Mol Life Sci, 1998. 54(9): p. 965–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsen CP, Morris PJ, and Austyn JM, Migration of dendritic leukocytes from cardiac allografts into host spleens. A novel pathway for initiation of rejection. J Exp Med, 1990. 171(1): p. 307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaplanski G, et al. , IL-6 and IL-8 production from cultured human endothelial cells stimulated by infection with Rickettsia conorii via a cell-associated IL-1 alpha-dependent pathway. J Clin Invest, 1995. 96(6): p. 2839–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cushing SD, et al. , Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci U S A, 1990. 87(13): p. 5134–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guaragnella N, et al. , The expanding role of yeast in cancer research and diagnosis: insights into the function of the oncosuppressors p53 and BRCA1/2. FEMS Yeast Res, 2014. 14(1): p. 2–16. [DOI] [PubMed] [Google Scholar]
- 42.Tait SW and Green DR, Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol, 2010. 11(9): p. 621–32. [DOI] [PubMed] [Google Scholar]
- 43.Wang C and Youle RJ, The role of mitochondria in apoptosis*. Annu Rev Genet, 2009. 43: p. 95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crowley LC and Waterhouse NJ, Detecting Cleaved Caspase-3 in Apoptotic Cells by Flow Cytometry. Cold Spring Harb Protoc, 2016. 2016(11). [DOI] [PubMed] [Google Scholar]
- 45.Damle NK and Aruffo A, Vascular cell adhesion molecule 1 induces T-cell antigen receptor-dependent activation of CD4+T lymphocytes. Proc Natl Acad Sci U S A, 1991. 88(15): p. 6403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi J, et al. , T lymphocyte-endothelial cell interactions. Annu Rev Immunol, 2004. 22: p. 683–709. [DOI] [PubMed] [Google Scholar]
- 47.Karmann K, et al. , CD40 on human endothelial cells: inducibility by cytokines and functional regulation of adhesion molecule expression. Proc Natl Acad Sci U S A, 1995. 92(10): p. 4342–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kreisel D, et al. , Mouse vascular endothelium activates CD8+ T lymphocytes in a B7-dependent fashion. J Immunol, 2002. 169(11): p. 6154–61. [DOI] [PubMed] [Google Scholar]
- 49.Jane-Wit D, et al. , Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-kappaB signaling in endothelial cells. Circulation, 2013. 128(23): p. 2504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kariya T, et al. , Direct evidence for activated CD8+ T cell transmigration across portal vein endothelial cells in liver graft rejection. J Gastroenterol, 2016. 51(10): p. 985–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dromparis P and Michelakis ED, Mitochondria in vascular health and disease. Annu Rev Physiol, 2013. 75: p. 95–126. [DOI] [PubMed] [Google Scholar]
- 52.Kluge MA, Fetterman JL, and Vita JA, Mitochondria and endothelial function. Circ Res, 2013. 112(8): p. 1171–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quintero M, et al. , Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A, 2006. 103(14): p. 5379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang YC, et al. , Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell, 2018: p. e12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lockett AD, et al. , Scavenger receptor class B, type I-mediated uptake of A1AT by pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol, 2015. 309(4): p. L425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simmons JD, et al. , Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg, 2013. 258(4): p. 591–6; discussion 596–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pollara J ER, Lin L, Bendersky VA, Brennan TV., Circulating mitochondria in deceased organ donors are assoicated with immune activation and early graft dysfunction. JCI-Insight, 2018. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matzinger P, The danger model: a renewed sense of self. Science, 2002. 296(5566): p. 301–5. [DOI] [PubMed] [Google Scholar]
- 59.Land WG, et al. , DAMP-Induced Allograft and Tumor Rejection: The Circle Is Closing. Am J Transplant, 2016. 16(12): p. 3322–3337. [DOI] [PubMed] [Google Scholar]
- 60.Todd JL and Palmer SM, Danger signals in regulating the immune response to solid organ transplantation. J Clin Invest, 2017. 127(7): p. 2464–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weber DJ, et al. , The HMGB1-RAGE axis mediates traumatic brain injury-induced pulmonary dysfunction in lung transplantation. Sci Transl Med, 2014. 6(252): p. 252ra124. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Day JD, Metes DM, and Vodovotz Y, Mathematical Modeling of Early Cellular Innate and Adaptive Immune Responses to Ischemia/Reperfusion Injury and Solid Organ Allotransplantation. Front Immunol, 2015. 6: p. 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Palkama T, et al. , Regulation of endothelial adhesion molecules by ligands binding to the scavenger receptor. Clin Exp Immunol, 1993. 92(2): p. 353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zani IA, et al. , Scavenger receptor structure and function in health and disease. Cells, 2015. 4(2): p. 178–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.