

Summary
New therapies, including the anti‐cytotoxic T lymphocyte antigen (CTLA)‐4 antibody, ipilimumab, is approved for metastatic melanoma. Prognostic biomarkers need to be identified, because the treatment has serious side effects. Serum samples were obtained before and during treatment from 56 patients with metastatic or unresectable malignant melanoma, receiving treatment with ipilimumab in a national Phase IV study (NCT0268196). Expression of a panel of 17 inflammatory‐related markers reflecting different pathways including extracellular matrix remodeling and fibrosis, vascular inflammation and monocyte/macrophage activation were measured at baseline and the second and/or third course of treatment with ipilimumab. Six candidate proteins [endostatin, osteoprotegerin (OPG), C‐reactive protein (CRP), pulmonary and activation‐regulated chemokine (PARC), growth differentiation factor 15 (GDF15) and galectin‐3 binding‐protein (Gal3BP)] were persistently higher in non‐survivors. In particular, high Gal3BP and endostatin levels were also independently associated with poor 2‐year survival after adjusting for lactate dehydrogenase, M‐stage and number of organs affected. A 1 standard deviation increase in endostatin gave 1·74 times [95% confidence interval (CI) = 1·10–2·78, P = 0·019] and for Gal3BP 1·52 times (95% CI = 1·01–2·29, P = 0·047) higher risk of death in the adjusted model. Endostatin and Gal3BP may represent prognostic biomarkers for patients on ipilimumab treatment in metastatic melanoma and should be further evaluated. Owing to the non‐placebo design, we could only relate our findings to prognosis during ipilimumab treatment.
Keywords: anti‐CTLA‐4, endostatin, Gal3BP, immunotherapy, melanoma
Introduction
Metastatic melanoma (MM) often affects younger people and is the most devastating form of cancer regarding life‐years lost. Dacarbazine (DTIC) has traditionally been the standard therapeutic drug for patients with MM, but the response rates are low (approximately 10%), and the effect not sustained 1. In Europe, until 2011 no new drugs had been approved for the treatment of MM for more than 30 years. Since then new therapeutic approaches have been authorized, including immunotherapy with ipilimumab (anti‐CTLA‐4 antibody), as well as nivolumab and pembrolizumab [programmed cell death 1 (PD)‐1 inhibitors] and the combination of ipilimumab and nivolumab, resulting in improved overall survival 2, 3, 4, 5, 6, 7, 8. At present, the first line of systemic immunotherapy for MM includes ipilimumab/nivolumab combination or nivolumab or pembrolizumab alone. Second‐line treatment depends on the choice in the first line, but includes ipilimumab, and PD‐1 inhibitors only or in combination with other new immune checkpoint inhibitors in clinical trials. At present, however, immunotherapies in MM lack prognostic biomarkers. As these therapeutic options also have serious side effects, markers could help to select patients that will be in particular benefit of such therapy.
Overcoming tumor immune tolerance can be induced by monoclonal antibodies (mAbs) blocking negative signaling receptors, such as cytotoxic T lymphocyte antigen 4 (CTLA‐4), which is a central regulation mechanism for T cell activation. Ipilimumab is a human CTLA‐4 mAb that blocks the interaction with its ligands CD80/CD86, and can lead to long‐lasting anti‐tumor effects and disease control. In MM, approximately 20% of patients experience long‐term survival after treatment with ipilimumab 4, 9, but 20–27% of patients treated develop serious side effects (grades 3–4) 10. Access to biomarkers defining patients likely to respond and survive has been long awaited 11.
The development and progression of MM are characterized by chronic inflammation 12, 13 which has been recognized as a driving force for both epidermal cell transformation as well as malignant progression. Thus, a wide range of inflammatory and related mediators, including cytokines, chemokines, regulators of the extracellular matrix (ECM), reactive oxygen species and angiogenic factors, are present in the tumor microenvironment. Such factors may be derived from infiltrating inflammatory cells and/or from the tumor cells or interactions between these cells, and may influence the rate of tumor progression and the outcome of cancer treatment 14. These inflammatory mediators may be quantified in the circulation and utilized as biomarkers 11, 15, 16, 17. Thus, whereas MM in itself is associated with enhanced inflammation, immunotherapy such as ipilimumab will further activate certain inflammatory pathways. To characterize the inflammatory phenotype in MM as well as how this phenotype changes during successful ipilimumab therapy may be of major importance to selected patients who will benefit from such treatment.
Based on these issues, we examined markers reflecting different inflammatory pathways that could give novel information on prognosis during immunotherapy (i.e. ipilimumab) in MM. We selected markers on the basis that they reflected activation of pathways that are relevant for melanoma progression 18 as well as the effects of ipilimumab 19. Against this background we examined general downstream markers of inflammation [i.e. C‐reactive protein (CRP), soluble tumor necrosis factor receptor type 1 (sTNFR1)], markers of endothelial cell activation and vascular inflammation [i.e. pentraxin 3 (PTX3), osteoprotegerin (OPG), von Willebrand factor (vWF), the chemokine CXCL16 and the notch ligand delta‐like 1 (DLL1)], angiogenetic factors [i.e. Axl, endothelial cell protein C receptor (ePCR) and endostatin], markers related to fibrosis and extracellular matrix (ECM) remodeling [i.e. growth differentiation factor 15 (GDF‐15), galectin 3 binding protein (Gal3BP), cathepsin S (CatS/CD147)] and markers reflecting monocyte/macrophage activation [the chemokine CCL18 and the soluble (s) markers sCD163 and activated leukocyte cell adhesion molecule (ALCAM)/sCD166]) at three time‐points in melanoma patients treated with ipilimumab and evaluated their association with 2 years’ survival. As this were non‐randomized real‐world data, the identified markers could only be assigned prognostic value on ipilimumab treatment rather than being of predictive value for response to ipilimumab therapy.
Materials and methods
Patient population
From January 2014 to March 2015, 69 patients with unresectable or metastatic malignant melanoma were included and started treatment with ipilimumab as part of a national Phase IV study (NCT0268196/EudraCT2013‐002408‐15) at the Department of Oncology, Oslo University Hospital, The Norwegian Radium Hospital. The patients received treatment with up to four doses of ipilimumab 3 mg/kg intravenously every third week. Six patients had ≤ two doses of ipilimumab due to very rapid disease progression, eight patients had three courses due to high‐grade side effects, and the remainder had all four doses as planned. A total number of 56 patients from whom fresh‐frozen serum samples were obtained at baseline (before the start of treatment), week 4 (before the second treatment) and/or week 7 (before the third treatment) were included in this substudy. The 13 patients who were not reported were due to screening failure (four patients) and lack of blood tests taken (nine patients). Thus, none of the patients who were not reported showed rapid deterioration during follow‐up. The patients started therapy on week 1 and were followed‐up every third week during treatment. They had the first computerized tomography (CT) scan for evaluation at week 12, and the next scan at week 16 to evaluate pseudoprogression. Further regular visits and CT scans were at week 24 and every third month after that for 3 years or until progression. The study was approved by the regional ethics committee and conducted by the Declaration of Helsinki. All patients gave written informed consent to participate in the study.
Blood sampling and biochemical analyses
Peripheral venous blood was drawn into pyrogen‐free tubes without any additives. After coagulation at room temperature, tubes were centrifuged at 1500 g for 10 min, and serum was stored at –80°C in multiple aliquots. Serum levels of CRP (Cat. no. DY1707), PTX3 (Cat. no. DY1826), sTNFR1 (Cat. no. DY225), OPG (Cat. no. DY805), DLL1 (Cat. no. DY1818), CXCL16 (Cat. no. DY1164), Axl (Cat. no. DY154), ePCR (Cat. no. DY2245), endostatin (Cat. no. DY1098), GDF‐15 (Cat. no. DY957), CatS (Cat. no. DY1183), CD147 (Cat. no. DY972), CCL18 (Cat. no. DY394), Gal3BP (Cat. no. DY2226), sCD163 (Cat. no. DY1607) and sCD166 (Cat. no. DY656) were measured in duplicate by enzyme‐linked immunosorbent assay with antibodies obtained from R&D Systems (Minneapolis, MN, USA) in a 384 format using a combination of a CyBi SELMA (CyBio, Jena, Germany), EL406 washer/dispenser (Biotek, Winooski, VT, USA) and Synergy H2 microplate reader (Biotek). vWF was measured by the same method with antibodies obtained from DakoCytomation (Glostrup, Denmark). The intra‐ and interindividual coefficients of variation were <10%.
Statistical analysis
As the inflammatory markers were skewed, data were analyzed using the Mann–Whitney U‐test and χ2 correlation. Receiver operating characteristic (ROC) curves of inflammatory markers at baseline were created, and the area under the curve (AUC) was used to evaluate the prognostic effect of each parameter. Where appropriate, Kaplan–Meier curved on dichotomized levels, multivariable logistic regression analysis and Cox proportional hazards regression [adjusted for lactate dehydrogenase (LDH) levels, M stage and number of organs affected] were performed in order to assess the relationship between inflammatory markers [log‐transformed with hazard ratios expressed per standard deviation (s.d.) change], clinical parameters and the outcome of survival after 2 years. Survival at 2 years was defined from trial inclusion. Two‐sided P‐values of <0·05 were considered significant. spss version 23 software (IBM, Armonk, NY, USA) was used for all statistical analyses.
Results
Patient characteristics
Table 1 presents the baseline characteristics of the MM patient population according to survival status. Twenty‐three (41%) of the patients were alive and thirty‐three (59%) were dead 2 years after inclusion in the Phase IV study. All non‐survivors died from disease progression of melanoma. As expected, non‐survivors had a more advanced disease stage and higher LDH serum levels than survivors. Twelve of the survivors and thirteen of the non‐survivors had a BRAF V600 mutated tumor. Most patients had one or no systemic treatment before they were included in the study.
Table 1.
Clinical characteristics of the study population according to survival
| Total | Survival 2 years | |
|---|---|---|
| Yes (alive) | No (dead) | |
| 23 (41) | 33 (59) | |
| Gender | ||
| Men | 11 (20) | 22 (39) |
| Women | 12 (21) | 11 (20) |
| Age, years, Median (range) | 60 (27–83) | 67 (38–84) |
| M stage M1a/b | 9 (38) | 4 (14) |
| M1c | 14 (62) | 29 (86)* |
| Organs involved ≤ 2 | 16 (29) | 13 (23) |
| >2 | 7 (12) | 20 (36) |
| BRAF v600 Mutation, No | 8 (14) | 17 (30) |
| Yes | 12 (21) | 13 (23) |
| Unknown | 3 (5) | 3 (5) |
| LDH, normal | 19 (34) | 14 (25) |
| >ULN† | 4 (7) | 19 (34)** |
| Number of treatments before inclusion, 0 | 11 (20) | 18 (32) |
| 1 | 9 (16) | 13 (23) |
| 2 | 2 (4) | 1 (1·7) |
| 3 | 1 (1·7) | 1 (1·7) |
| ANC# | 4·7 (1·7) | 4·8 (1·4) |
| AEC# | 0·1 (0·1) | 0·1 (0·1) |
| ALC# | 1·6 (0·5) | 1·4 (0·6) |
| Increase AEC | 13 (57) | 18 (55) |
| Increase ALC | 17 (74) | 16 (49) |
| irAE | 8 (35) | 15 (46) |
Data are shown as number of patients (%) unless otherwise indicated.
ULN = upper limit of normal,
mean (standard deviation).
P < 0·05;
P < 0·01 versus survivors.
ANC = absolute neutrophil count (109/l); AEC = absolute eosinophil count (109/l); ALC = absolute lymphocyte count (109/l); irAE = immune‐related adverse event; LDH = lactate dehydrogenase (U/l); M stage = metastatic stage (AJCC cancer staging manual).
In contrast to what has been reported by others 20, 21, 22, 23, we found no correlation between peripheral blood biomarkers such as absolute neutrophil count (ANC), absolute eosinophil count (AEC), absolute lymphocyte count (ALC) or increase in AEC or ALC and outcome of ipilimumab treatment.
Patients included in this study had a broad spectrum and grade of different immune‐related side effects (irAE). However, the number of patients in our cohort was somewhat low (n = 56), and we therefore did not group the irAE according to grade or type of irAE in statistical analyses. When we included all irAE (grades 1–4) regularly registered during the treatment visits in the IPI4 study, we found no association between any immune‐related side effect and 2 years of survival.
Biomarkers during treatment
Median and interquartile levels of the examined markers before treatment (i.e. baseline), at second treatment (i.e. 4 weeks) and at third treatment (i.e. 7 weeks) with ipilimumab are presented in Table 2. Several of the markers demonstrated higher levels in serum from non‐survivors compared to survivors at several time‐points, i.e. CRP, sTNFR1, PTX3, OPG, CXCL16, Axl, endostatin, GDF15, Gal3BP, CatS, CCL18/PARC and sCD163. Of these, CRP, endostatin, GDF15, OPG, PARC and Gal3BP were consistently elevated in non‐survivors at all time‐points and were therefore selected for further analyses.
Table 2.
Serum levels (quartiles) of biomarkers before and during treatment with ipilimumab stratified survival status
| Survival | Baseline | Week 4 | Week 7 | |
|---|---|---|---|---|
| Inflammation in general | ||||
| CRP (μg/ml) | Alive | 0·8 (0·2, 2·3) | 1·0 (0·5, 3·5) | 1·5 (0·6, 3·5) |
| Dead | 2·8 (1·4, 5·4)** | 4·8 (2·4, 6·4)** | 3·8 (2·5, 7·0)** | |
| sTNF‐R1 (ng/ml) | Alive | 2·2 (1·8, 2·6) | 2·3 (2·0, 3·1) | 2·5 (2·0, 2·8) |
| Dead | 2·2 (1·9, 3·3) | 3·2 (2·6, 4·7)** | 3·1 (2·5, 5·3)** | |
| Vascular inflammation | ||||
| PTX3 (ng/ml) | Alive | 1·1 (0·6, 1·7) | 1·2 (0·6, 1·9) | 1·3 (0·8, 1·9) |
| Dead | 1·9 (1·0, 2·5) | 2·3 (1·6, 3·6)** | 2·2 (1·4, 4·1)** | |
| OPG (ng/ml) | Alive | 3·5 (3·0, 4·5) | 3·7 (2·9, 4·8) | 3·8 (3·4, 5·0) |
| Dead | 5·1 (3·8, 5·6)** | 5·5 (4·0, 6·5)* | 6·2 (4·2, 6·8)** | |
| vWF (AU) | Alive | 58 (48, 71) | 61 (51, 87) | 64 (37, 92) |
| Dead | 68 (51, 87) | 73 (59, 113) | 65 (33, 126) | |
| CXCL16 (ng/ml) | Alive | 2·3 (2·1, 2·7) | 2·4 (2·1, 2·8) | 2·1 (2·0, 2·6) |
| Dead | 2·7 (2·3, 3·3) | 2·6 (2·2, 3·2) | 2·7 (2·3, 3·1)** | |
| DLL1 (ng/ml) | Alive | 13·7 (11·3, 16·4) | 14·8 (12·9, 20·0) | 14·4 (12·1, 15·8) |
| Dead | 13·6 (12·1, 15·9) | 15·1(13·5, 17·8) | 17·0 (12·9, 18·9) | |
| Angiogenesis | ||||
| Axl (ng/ml) | Alive | 5·2 (4·1, 5·6) | 5·2 (4·3, 5·7) | 5·3 (4·5, 6·1) |
| Dead | 5·3 (4·6, 6·4) | 5·9 (5·4, 6·5)** | 6·0 (5·4, 8·6)** | |
| ePCR (ng/ml) | Alive | 33 (29, 43) | 32 (27, 39) | 33 (27, 38) |
| Dead | 36 (28, 41) | 35 (28, 41) | 37 (32, 43) | |
| Endostatin (ng/ml) | Alive | 134 (113, 154) | 132 (114·5, 147·5) | 133 (115, 162) |
| Dead | 158 (137, 189)* | 170·5 (155, 202)* | 181 (148, 221)** | |
| ECM/fibrosis | ||||
| GDF15 (ng/ml) | Alive | 0·4 (0·2, 0·7) | 0·4 (0·3, 0·8) | 0·4 (0·3, 0·8) |
| Dead | 0·8 (0·4, 1·4)** | 0·7 (0·5, 2·0)** | 0·8 (0·5, 1·9)** | |
| Gal3BP/sCD166 (μg/ml) | Alive | 1·2 (0·8, 2·0) | 1·3 (0·7, 2·4) | 1·0 (0·9, 2·0) |
| Dead | 2·0 (1·7, 3·1)** | 2·4 (1·7, 4·1)** | 2·7 (1·9, 3·4)* | |
| CatS (ng/ml) | Alive | 53 (47, 61) | 56 (50, 61) | 55 (50, 59) |
| Dead | 55 (51, 60) | 56 (38, 62) | 60 (54, 67)** | |
| CD147 (ng/ml) | Alive | 9·1 (7·3, 10·2) | 9·4 (7·4, 11·1) | 8·9 (7·4, 10·3) |
| Dead | 8·7 (7·5, 10·0) | 9·9 (8·2, 11·7) | 10·1 (8·1, 12·4) | |
| Monocyte/macrophage activation | ||||
| CCL18/PARC (ng/ml) | Alive | 65 (55, 88) | 79 (58, 102) | 78 (68, 96) |
| Dead | 102 (78, 115)* | 103 (78, 134)** | 115 (85, 155)** | |
| sCD163 (ng/ml) | Alive | 421 (317, 690) | 516 (353, 645) | 562 (317, 699) |
| Dead | 650 (494, 831) | 824 (595, 1066)** | 865 (557, 1145)** | |
| Alcam/sCD166 (ng/ml) | Alive | 102 (92, 121) | 107 (92, 112) | 105 (97, 121) |
| Dead | 101 (84, 115) | 107 (89, 121) | 102 (89, 131) |
P < 0·05;
P < 0·01 comparing non‐survivors and survivors.
CRP = C‐reactive protein; sTNF‐R1 = soluble tumor necrosis factor receptors; PTX3 = pentraxin‐related protein/TNF‐inducible gene 14 protein (TSG‐14); OPG = osteoprotegerin; vWF = Von Willebrand factor; ePCR = endothelial cell protein C receptor; CXCL = chemokine ligand; DLL1 = delta‐like protein 1; ECM = extracellular matrix; GDF15 = growth differentiation factor 15; Gal3BP = galectin‐3 binding‐protein/CYT‐MAA/K90/Mac‐2BP; CatS = cathepsin S; PARC = pulmonary and activation‐regulated chemokine.
Baseline levels of inflammatory markers in relation to disease severity
Of the six selected inflammatory markers, several were associated with different measures of disease severity, as shown in Table 3. First, significantly higher levels of GDF15, endostatin, PARC and CRP were observed in patients with more organs affected. Secondly, GDF15, Gal3BP and CRP were significantly higher in patients with stage M1c, reflecting a higher tumor burden. Similarly, OPG, GDF15, PARC and CRP levels were significantly higher in those with higher LDH as a marker of tissue damage, above the upper limit of normal individuals.
Table 3.
Serum levels quartiles of selected markers at start of treatment in relation to disease severity
| Organs involved | M stage | LDH | ||||
|---|---|---|---|---|---|---|
| OPG (ng/ml) | ≤2 | 4·0 (3·4–4·6) | M1a/b | 3·7 (2·9–4·7) | Norm | 4·0 (3·4–4·6) |
| >2 | 4·9 (4·2–5·7) | M1c | 4·6 (4·1–5·2) | >ULN | 5·0 (4·3–6·0)* | |
| GDF15 (ng/ml) | ≤2 | 0·5 (0·3–0·7) | M1a/b | 0·4 (0·2–0·7) | Norm | 0·4 (0·3–0·7) |
| >2 | 1·0 (0·7–1·5)* | M1c | 0·8 (0·6–1·1)* | >ULN | 1·1 (0·7–1·8)** | |
| Endostatin (ng/ml) | ≤2 | 139 (127–151) | M1a/b | 135 (118–155) | Norm | 142 (130–154) |
| >2 | 162 (148–178)* | M1c | 154 (143–166) | >ULN | 161 (146–178) | |
| PARC (ng/ml) | ≤2 | 75 (66–86) | M1a/b | 74 (60–92) | Norm | 76 (67–85) |
| >2 | 99 (87–113)** | M1c | 90 (80–100) | >ULN | 103 (90–119)** | |
| Gal3BP (µg/ml) | ≤2 | 1·7 (1·3–2·2) | M1a/b | 1·1 (0·8–1·6) | Norm | 1·5 (1·2–1·9) |
| >2 | 1·8 (1·4–2·3) | M1c | 1·9 (1·6–2·3)* | >ULN | 2·1 (1·6–2·7) | |
| CRP (µg/ml) | ≤2 | 1·1 (0·7–1·7) | M1a/b | 0·8 (0·4–1·5) | Norm | 1·1 (0·7–1·7) |
| >2 | 2·1 (1·3–3·4)* | M1c | 1·8 (1·3–2·6)* | >ULN | 2·3 (1·4–3·8)* |
Data are given as back‐transformed estimated marginal means adjusted for age and sex. > ULN = above upper level of normal; LDH = lactate dehydrogenase; M stage = metastatic stage (AJCC cancer staging manual); OPG = osteoprotegerin; GDF15 = growth differentiation factor 15; PARC = pulmonary and activation‐regulated chemokine; Gal3BP = galectin‐3 binding‐protein/CYT‐MAA/K90/Mac‐2BP; CRP = C‐reactive protein.
P < 0·05;
P < 0·01 comparing non‐survivors and survivors.
Predictors of outcome
We next evaluated the accuracy of the selected markers in predicting disease outcome using ROC analysis. As shown in Fig. 1a, all the selected markers at baseline gave good discrimination between survivors and non‐survivors, with AUC between 0·70 and 0·76. Further, all markers, except Gal3BP, displayed a symmetrical area indicating that median levels could be a reasonable cut‐off. Kaplan–Meier survival curves for all‐cause mortality, using the median to dichotomize the data, confirmed that higher levels of each marker were associated with poor prognosis. We next evaluated the prognostic value of the selected markers in multivariable Cox‐regression models. Figure 2a shows the univariate and multivariable analysis of clinical markers, indicating that these markers were significantly associated with all‐cause mortality, and also when put together in a multivariable model. These markers of disease severity were used in subsequent adjustment strategies. Figure 2b shows the univariate and multivariable analysis (i.e. adjusted for LDH levels, M‐stage and number of organs affected) for each of the suggested biomarkers. All markers were associated with all‐cause mortality in univariate analysis, with hazard ratios (HR) between 1·7 and 2·2. The association between survival and GDF15, OPG and PARC were markedly attenuated by multivariable adjustment and were no longer significantly associated with death. Endostatin and Gal3BP were also modified by adjustment but remained significantly associated with all‐cause death. Figure 2c shows a Cox regression model with a stepwise selection of the best clinical and biochemical variables and indicates that endostatin and LDH are the strongest predictors, followed by M‐stage.
Figure 1.
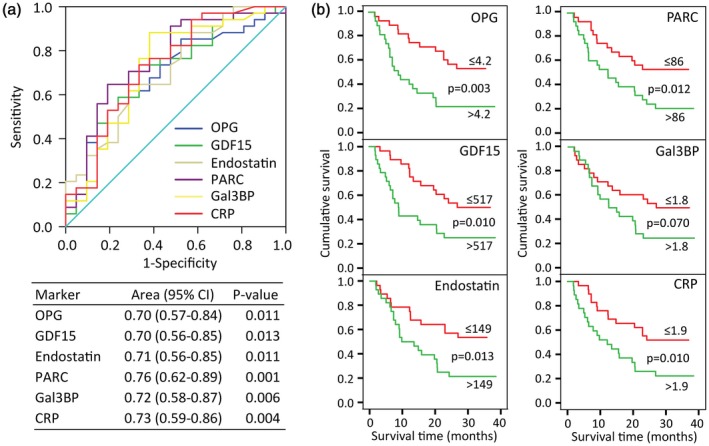
Survival analysis of inflammatory biomarkers during ipilimumab therapy. (a) Receiver operating curves (ROC) and diagnostic accuracy for all‐cause mortality. (b) Kaplan–Meier curves for all‐cause mortality by dichotomized levels of inflammatory markers.
Figure 2.
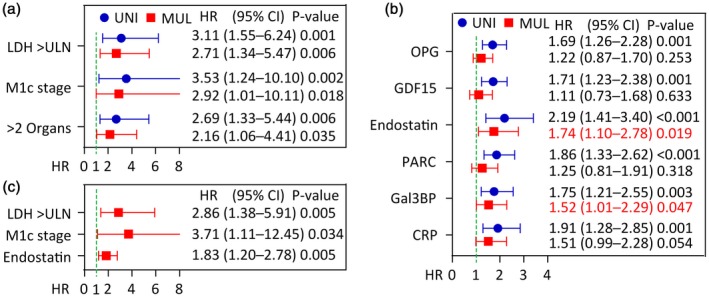
Univariate (blue circle) and multivariable (red square) Cox regression of clinical and inflammatory biomarkers as predictors of all‐cause mortality during ipilimumab therapy. (a) Clinical predictors. (b) Inflammatory biomarkers alone and after adjustment with LDH, M stage, and organs affected. (c). Stepwise model of the strongest predictors including both clinical and inflammatory markers. Hazard ratios (HR) for inflammatory markers are expressed as log‐transformed per standard deviation change in marker.
Discussion
Ipilimumab has improved survival in subgroups of patients with MM, but no biomarker is currently available for predicting treatment benefit. The present study was initiated to evaluate if biomarkers reflecting inflammation, vascular inflammation, angiogenesis and ECM remodeling could predict prognosis in patients treated with ipilimumab. Several markers reflecting different pathways correlated with indices of disease severity were consistently elevated in non‐survivors after initiation of treatment. In particular, high levels of Gal3BP and endostatin were also independently associated with poor survival in adjusted analysis, and deserve further attention as possible prognostic biomarkers in this population.
Whereas ipilimumab enhances the immune response against tumor, persistent low‐grade local and systematic immune response and inflammation could also promote tumor progression and death of patients with MM 24, 25. Several studies have revealed an association between leukocyte counts, conventional markers such as CRP and LDH and clinical outcome in MM 20, 21, 22, 23, 25, 26, 27, 28, 29, 30, 31. The present study supports and extends these findings by showing that biomarkers that reflect a range of inflammatory‐related pathways, including vascular inflammation, ECM remodeling/fibrosis and angiogenesis, correlate with different indices of disease severity, and are consistently up‐regulated in non‐survivors after initiation of ipilimumab. Thus, low or normal levels of OPG, GDF15, endostatin, PARC, Gal3BP and CRP in MM patients have the potential to identify patients who may benefit from such therapy. Indeed, comparing these biomarkers in multivariable analysis revealed that two of these markers, i.e. endostatin and Gal3BP, were independently associated with poor prognosis, suggesting that these markers could reflect distinct processes related to MM progression that is not necessarily reversed by ipilimumab therapy.
Endostatin is a 20‐kDa C‐terminal fragment from type XVIII collagen, generally found in epithelial and endothelial vascular basement membranes, and is an endogenous inhibitor of angiogenesis 32, 33. Endostatin blocks the proliferation and organization of endothelial cells into new blood vessels and inhibits angiogenesis and growth of both primary tumors and secondary metastasis 34, 35, 36, 37, 38. Interestingly, endostatin seems to be predominantly linked to angiogenesis arriving from pathogenic sources such as tumors, while processes such as wound healing are not affected 39, and endostatin has been suggested as adjuvant therapy in various malignancies. High expression of endostatin in tumor tissue may also indicate poor prognosis, probably reflecting substantial tumor burden and active stromal remodeling 40. Thus, the association between high endostatin levels, the number of organs affected and poor outcome in our study could potentially reflect an enhanced compensatory response to limit angiogenesis as well as ECM remodeling. Our finding is supported by previous studies demonstrating that high circulating endostatin levels are associated with poor outcome or an aggressive phenotype in a variety of malignancies 40, 41, 42, 43, 44. It is also possible that endostatin could counteract the effect of ipilimumab by attenuating the infiltration of activated T cells into the tumor, but this hypothesis needs to be proved in future studies.
Galectins are a family of beta‐galactoside‐binding proteins implicated in modulating cell–cell and cell–matrix interactions, angiogenesis and apoptosis 45. Galectin‐3‐binding protein is a glycoprotein expressed in normal cells, but elevated circulating levels have been demonstrated in multiple cancers 46, 47, 48, 49, 50, 51, 52 including melanoma 53. The role of Gal3BP in cancer prognosis seems equivocal with both negative and positive influences on the prognosis of various cancers, but high levels are mainly associated with shorter survival, disease progression 54, 55 and reduced response to chemotherapy. Our finding of consistently increased Gal3BP following immunotherapy supports a previous study showing an association between high levels and clinical outcome in melanoma patients 45, 54, but so far there are no data on Gal3BP or endostatin as biomarkers of ipilimumab therapy. Mechanisms underlying over‐expression of Gal3BP in cancer is not fully understood, but its association with multiple forms of cancer could be related to the multidomain nature of the protein and ability to bind different ligands in different tumor tissues.
As this was not a placebo‐controlled trial, it is hard to conclude if the association of high levels of endostatin and Gal3BP was a marker of treatment efficiency or related to disease progression independently of therapy. At present, we can only suggest that high levels of these markers reflect disease progression during ipilimumab therapy. However, the mechanisms for this association seem to differ between the two markers. Thus, as endostatin has been shown to have anti‐angiogenetic properties with potential attenuating effects on carcinogenesis, the high levels may reflect counteracting mechanisms in a tumor with a large pro‐angiogenetic potential and therefore poor prognosis. Conversely, Gal3BP may reflect activation of pathways that promote maladaptive ECM remodeling which could enhance tumor progression. Indeed, a recent study in melanoma shows that the combination of ipilimumab with the vascular endothelial growth factor (VEGF)‐A blocking antibody bevacizumab induced increased antibody response to galectin‐3, and this response was associated with improved prognosis 56. No such antibody response was seen during PD‐1 inhibition.
To the best of our knowledge, endostatin or GAL3BP have not been reported as markers for treatment efficacy during immunotherapy in melanoma or other cancers. However, a galectin 3 inhibitor, in combination with ipilimumab or PD‐1 inhibition, is being currently evaluated for the treatment of patients with metastatic melanoma 57 underscoring that galectin 3 or its marker Gal3BP (as well as endostatin) could be part of a panel of biomarkers that predicts the outcome for immunotherapy in melanoma. Our data suggest endostatin and Gal3BP as potential prognostic biomarkers for ipilimumab in MM. The study does not provide mechanistic insight into the relationship between biomarkers and induced responses, but it is tempting to speculate that the correlation with disease severity and consistent up‐regulation of multiple inflammatory‐related markers in non‐survivors could reflect that these patients have progressed too far to respond to ipilimumab. Analysis of pretreatment blood samples could enable the identification of possible biomarkers that could help in making treatment decisions in patients with MM. However, the number of included patients is small, and the results need to be confirmed in a larger study. Moreover, short‐term observation, as in the present study (2 years), will ‘select’ for biomarkers that predict short‐term progression rather than a favorable clinical outcome. Accordingly, larger studies with long‐term follow‐up are needed to evaluate these molecules as potential biomarkers for long‐term survival following ipilimumab therapy. The strength is, however, that this is a prospective study and the data are derived from a ‘real‐world’ patient cohort, and the selected candidates (i.e. endostatin and Gal3BP) should be further investigated in MM during various forms of therapeutic approaches.
Disclosures
All the authors have no conflicts of interest to disclose.
Author contributions
M. N. and T. U. contributed to design, acquiring and analysis of the data, statistical calculations as well as writing the manuscript. E. A., T. K. G. and P. B. contributed to obtaining and analysis of the data and revision of the manuscript. K. D. J. was Principal Investigator for the IPI4 study, contributed to acquiring the data and revision of the manuscript. S. A. and K. T. H. designed the protocol for the IPI4 study, contributed to the revision of the manuscript and S. A. obtained the grants for the IPI4 study. A. Y., B. H., P. A., K. A. T. and G. M. M. contributed to the design, acquiring and analysis of the data and revision of the manuscript. All authors approved the manuscript for submission.
Acknowledgements
Thanks to Peder Braadland, Kjetil Boye, Helene Hartvedt Grytli and Abhilash D. Pandya for statistical assistance and technical support. The IPI4 national Phase IV study was financed by the Department of Health, Oslo, Norway (NCT0268196/EudraCT 2013‐002408‐2015). Antibody analyses were supported by The Cancer Clinic, Oslo University Hospital through the Metaflammation Strategic Focus Area. This paper is part of the PhD thesis of M. N., that is supported by the Bodil and Magnes Foundation.
References
- 1. Balch CM, Gershenwald JE, Soong SJ et al Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009; 27:6199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hodi FS, O’Day SJ, McDermott DF et al Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garbe C, Peris K, Hauschild A et al Diagnosis and treatment of melanoma. European consensus‐based interdisciplinary guideline – update 2016. Eur J Cancer 2016; 63:201–17. [DOI] [PubMed] [Google Scholar]
- 4. Maio M, Grob JJ, Aamdal S et al Five‐year survival rates for treatment‐naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a Phase III trial. J Clin Oncol 2015; 33:1191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schadendorf D, Hodi FS, Robert C et al Pooled analysis of long‐term survival data from Phase II and Phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015; 33:1889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robert C, Schachter J, Long GV et al Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015; 372:2521–32. [DOI] [PubMed] [Google Scholar]
- 7. Robert C, Long GV, Brady B et al Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–30. [DOI] [PubMed] [Google Scholar]
- 8. Larkin J, Chiarion‐Sileni V, Gonzalez R et al Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lebbe C, Weber JS, Maio M et al Survival follow‐up and ipilimumab retreatment of patients with advanced melanoma who received ipilimumab in prior Phase II studies. Ann Oncol 2014; 25:2277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kahler KC, Hassel JC, Heinzerling L et al Management of side effects of immune checkpoint blockade by anti‐CTLA‐4 and anti‐PD‐1 antibodies in metastatic melanoma. J Dtsch Dermatol Ges 2016; 14:662–81. [DOI] [PubMed] [Google Scholar]
- 11. Gnjatic S, Bronte V, Brunet LR et al Identifying baseline immune‐related biomarkers to predict clinical outcome of immunotherapy. J Immunother Cancer 2017; 5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6:392–401. [DOI] [PubMed] [Google Scholar]
- 13. Zhong JH, Huang DH, Chen ZY. Prognostic role of systemic immune‐inflammation index in solid tumors: a systematic review and meta‐analysis. Oncotarget 2017; 8:75381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sund M, Kalluri R. Tumor stroma derived biomarkers in cancer. Cancer Metastasis Rev 2009; 28:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016; 16:275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hopkins AM, Rowland A, Kichenadasse G et al Predicting response and toxicity to immune checkpoint inhibitors using routinely available blood and clinical markers. Br J Cancer 2017; 117:913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jacquelot N, Roberti MP, Enot DP et al Predictors of responses to immune checkpoint blockade in advanced melanoma. Nat Commun 2017; 8:592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ribatti D, Annese T, Longo V. Angiogenesis and melanoma. Cancers (Basel) 2010; 2:114–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vajaitu C, Draghici CC, Solomon I et al The central role of inflammation associated with checkpoint inhibitor treatments. J Immunol Res 2018; 2018:4625472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martens A, Wistuba‐Hamprecht K, Geukes Foppen M et al Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res 2016; 22:2908–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Delyon J, Mateus C, Lefeuvre D et al Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol 2013; 24:1697–703. [DOI] [PubMed] [Google Scholar]
- 22. Martens A, Wistuba‐Hamprecht K, Yuan J et al Increases in absolute lymphocytes and circulating CD4+ and CD8+ T cells are associated with positive clinical outcome of melanoma patients treated with ipilimumab. Clin Cancer Res 2016; 22:4848–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Simeone E, Gentilcore G, Giannarelli D et al Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother 2014; 63:675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–74. [DOI] [PubMed] [Google Scholar]
- 25. Roxburgh CS, McMillan DC. Role of systemic inflammatory response in predicting survival in patients with primary operable cancer. Future Oncol 2010; 6:149–63. [DOI] [PubMed] [Google Scholar]
- 26. Bjoern J, Juul Nitschke N, Zeeberg Iversen T, Schmidt H, Fode K, Svane IM. Immunological correlates of treatment and response in stage IV malignant melanoma patients treated with Ipilimumab. Oncoimmunology 2016; 5:e1100788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gebhardt C, Sevko A, Jiang H et al Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res 2015; 21:5453–9. [DOI] [PubMed] [Google Scholar]
- 28. Khoja L, Atenafu EG, Templeton A et al The full blood count as a biomarker of outcome and toxicity in ipilimumab‐treated cutaneous metastatic melanoma. Cancer Med 2016; 5:2792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iero M, Valenti R, Huber V et al Tumour‐released exosomes and their implications in cancer immunity. Cell Death Differ 2008; 15:80–8. [DOI] [PubMed] [Google Scholar]
- 30. Schaefer C, Butterfield LH, Lee S et al Function but not phenotype of melanoma peptide‐specific CD8(+) T cells correlate with survival in a multiepitope peptide vaccine trial (ECOG 1696). Int J Cancer 2012; 131:874–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer 2015; 15:321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Reilly MS, Boehm T, Shing Y et al Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997; 88:277–85. [DOI] [PubMed] [Google Scholar]
- 33. Ferreras M, Felbor U, Lenhard T, Olsen BR, Delaisse J. Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett 2000; 486:247–51. [DOI] [PubMed] [Google Scholar]
- 34. Schuch G, Heymach JV, Nomi M et al Endostatin inhibits the vascular endothelial growth factor‐induced mobilization of endothelial progenitor cells. Cancer Res 2003; 63:8345–50. [PubMed] [Google Scholar]
- 35. Hajitou A, Grignet C, Devy L et al The antitumoral effect of endostatin and angiostatin is associated with a down‐regulation of vascular endothelial growth factor expression in tumor cells. FASEB J 2002; 16:1802–4. [DOI] [PubMed] [Google Scholar]
- 36. Kim YM, Jang JW, Lee OH et al Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res 2000; 60:5410–3. [PubMed] [Google Scholar]
- 37. Kim YM, Hwang S, Kim YM et al Endostatin blocks vascular endothelial growth factor‐mediated signaling via direct interaction with KDR/Flk‐1. J Biol Chem 2002; 277:27872–9. [DOI] [PubMed] [Google Scholar]
- 38. Abdollahi A, Hahnfeldt P, Maercker C et al Endostatin’s antiangiogenic signaling network. Mol Cell 2004; 13:649–63. [DOI] [PubMed] [Google Scholar]
- 39. Berger AC, Feldman AL, Gnant MF et al The angiogenesis inhibitor, endostatin, does not affect murine cutaneous wound healing. J Surg Res 2000; 91:26–31. [DOI] [PubMed] [Google Scholar]
- 40. Szarvas T, Laszlo V, Vom Dorp F et al Serum endostatin levels correlate with enhanced extracellular matrix degradation and poor patients' prognosis in bladder cancer. Int J Cancer 2012; 130:2922–9. [DOI] [PubMed] [Google Scholar]
- 41. Kantola T, Vayrynen JP, Klintrup K et al Serum endostatin levels are elevated in colorectal cancer and correlate with invasion and systemic inflammatory markers. Br J Cancer 2014; 111:1605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohlund D, Ardnor B, Oman M, Naredi P, Sund M. Expression pattern and circulating levels of endostatin in patients with pancreas cancer. Int J Cancer 2008; 122:2805–10. [DOI] [PubMed] [Google Scholar]
- 43. Li M, Liu F, Sun P et al Correlations between serum levels of vascular endothelial growth factor and endostatin with clinical pathological characteristics of patients with gastrointestinal cancers. Hepatogastroenterology 2012; 59:1865–8. [DOI] [PubMed] [Google Scholar]
- 44. Xu CJ, Song JF, Su YX, Liu XL. Expression of b‐FGF and endostatin and their clinical significance in human osteosarcoma. Orthop Surg 2010; 2:291–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grassadonia A, Tinari N, Iurisci I et al 90K (Mac‐2 BP) and galectins in tumor progression and metastasis. Glycoconj J 2004; 19:551–6. [DOI] [PubMed] [Google Scholar]
- 46. Iacobelli S, Sismondi P, Giai M et al Prognostic value of a novel circulating serum 90K antigen in breast cancer. Br J Cancer 1994; 69:172–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Strizzi L, Muraro R, Vianale G et al Expression of glycoprotein 90K in human malignant pleural mesothelioma: correlation with patient survival. J Pathol 2002; 197:218–23. [DOI] [PubMed] [Google Scholar]
- 48. Marchetti A, Tinari N, Buttitta F et al Expression of 90K (Mac‐2 BP) correlates with distant metastasis and predicts survival in stage I non‐small cell lung cancer patients. Cancer Res 2002; 62:2535–9. [PubMed] [Google Scholar]
- 49. Iacovazzi PA, Guerra V, Elba S, Sportelli F, Manghisi OG, Correale M. Are 90K/MAC‐2BP serum levels correlated with poor prognosis in HCC patients? Preliminary results. Int J Biol Markers 2003; 18:222–6. [DOI] [PubMed] [Google Scholar]
- 50. Fornarini B, D'Ambrosio C, Natoli C, Tinari N, Silingardi V, Iacobelli S. Adhesion to 90K (Mac‐2 BP) as a mechanism for lymphoma drug resistance in vivo. Blood 2000; 96:3282–5. [PubMed] [Google Scholar]
- 51. Gentiloni N, Caradonna P, Costamagna G et al Pancreatic juice 90K and serum CA 19–9 combined determination can discriminate between pancreatic cancer and chronic pancreatitis. Am J Gastroenterol 1995; 90:1069–72. [PubMed] [Google Scholar]
- 52. Piccolo E, Tinari N, D’Addario D et al Prognostic relevance of LGALS3BP in human colorectal carcinoma. J Transl Med 2015; 13:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cesinaro AM, Natoli C, Grassadonia A, Tinari N, Iacobelli S, Trentini GP. Expression of the 90K tumor‐associated protein in benign and malignant melanocytic lesions. J Invest Dermatol 2002; 119:187–90. [DOI] [PubMed] [Google Scholar]
- 54. Reynolds SR, Vergilis IJ, Szarek M, Ferrone S, Bystryn JC. Cytoplasmic melanoma‐associated antigen (CYT‐MAA) serum level in patients with melanoma: a potential marker of response to immunotherapy? Int J Cancer 2006; 119:157–61. [DOI] [PubMed] [Google Scholar]
- 55. Vergilis IJ, Szarek M, Ferrone S, Reynolds SR. Presence and prognostic significance of melanoma‐associated antigens CYT‐MAA and HMW‐MAA in serum of patients with melanoma. J Invest Dermatol 2005; 125:526–31. [DOI] [PubMed] [Google Scholar]
- 56. Wu X, Giobbie‐Hurder A, Connolly EM et al Anti‐CTLA‐4 based therapy elicits humoral immunity to galectin‐3 in patients with metastatic melanoma. Oncoimmunology 2018; 7:e1440930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Farhad M, Rolig AS, Redmond WL. The role of Galectin‐3 in modulating tumor growth and immunosuppression within the tumor microenvironment. Oncoimmunology 2018; 7:e1434467. [DOI] [PMC free article] [PubMed] [Google Scholar]