

Summary
Regulatory T (Treg) cells represent an essential component of peripheral tolerance. Given their potently immunosuppressive functions that is orchestrated by the lineage‐defining transcription factor forkhead box protein 3 (FoxP3), clinical modulation of these cells in autoimmunity and cancer is a promising therapeutic target. However, recent evidence in mice and humans indicates that Treg cells represent a phenotypically and functionally heterogeneic population. Indeed, both suppressive and non‐suppressive Treg cells exist in human blood that are otherwise indistinguishable from one another using classical Treg cell markers such as CD25 and FoxP3. Moreover, murine Treg cells display a degree of plasticity through which they acquire the trafficking pathways needed to home to tissues containing target effector T (Teff) cells. However, this plasticity can also result in Treg cell lineage instability and acquisition of proinflammatory Teff cell functions. Consequently, these dysfunctional CD4+FoxP3+ T cells in human and mouse may fail to maintain peripheral tolerance and instead support immunopathology. The mechanisms driving human Treg cell dysfunction are largely undefined, and obscured by the scarcity of reliable immunophenotypical markers and the disregard paid to Treg cell antigen‐specificity in functional assays. Here, we review the mechanisms controlling the stability of the FoxP3+ Treg cell lineage phenotype. Particular attention will be paid to the developmental and functional heterogeneity of human Treg cells, and how abrogating these mechanisms can lead to lineage instability and Treg cell dysfunction in diseases like immunodysregulation polyendocrinopathy enteropathy X‐linked (IPEX) syndrome, type 1 diabetes, rheumatoid arthritis and cancer.
Keywords: antigen specificity, cell therapy, human immunology, regulatory T cells, regulatory T cell dysfunction
Introduction
Regulatory T (Treg) cells are essential mediators of peripheral tolerance to self and non‐self‐antigens 1. Treg cells achieve this immunoregulatory control through multiple suppressive mechanisms that inhibit cells of innate immunity, antigen‐presenting cell (APC) functions, as well as adaptive B, CD4+ or CD8+ effector T (Teff) cell responses 2. Pioneering experiments identified these potently immunosuppressive cells as CD4+CD25+ T cells as the transfer of CD25‐depleted splenocytes into lymphopenic mice conferred multi‐organ autoimmunity 3. Homologous CD4+CD25high human counterparts were identified shortly thereafter 4, 5, 6, 7, 8. In 2003, forkhead box P3 (FoxP3) was identified as the lineage‐defining transcription factor of Treg cells. Indeed, gene deletion or abrogation of its functions caused severe inflammatory autoimmune disorders in mice and humans by abolishing Treg cell development 9, 10, 11.
FoxP3+ Treg cells can be categorized into two ontogenic categories: thymic‐derived/natural Treg (tTreg) cells and peripheral/induced Treg (pTreg) cells. The former develop within the thymus from single‐positive CD4+ thymocytes following a moderate‐ to high‐avidity T cell receptor (TCR) engagement with self‐antigens on major histocompatibility complex (MHC)‐II molecules by medullary thymic epithelial cells 12. The latter arise in the periphery from naïve, CD4+ conventional T (Tconv) cells that are antigen‐activated in the presence of FoxP3‐inducing cytokines [transforming growth factor (TGF)‐β, interleukin (IL)‐2], dietary constituents (retinoic acid) or drugs (glucocorticoids, rapamycin) 13. Both Treg cell types play central roles in the global immunoregulatory potential in hosts. Alterations in their development, homeostasis or function may predispose to a variety of disease conditions including allergy, autoimmunity, graft rejection, cancer and response to immunotherapies.
Current research is focused on developing novel therapies to enhance endogenous Treg cell functions in vivo with cytokines and small drugs, use ex‐vivo manipulated Treg cells in autologous adoptive transfers to promote immunoregulation in settings of autoimmunity, and induce antigen‐specific Treg cells to strengthen tolerance in allergic inflammation 14. However, Treg cells represent a phenotypically and functionally diverse array of cell subsets with differing effector functions and fates in circulation and tissues 15, 16. Here, we provide an overview of the factors and mechanisms influencing the development and heterogeneity of Treg cells in human health and disease.
FoxP3, the master regulator of Treg cell lineage commitment
FoxP3, a 431 amino acid forkhead winged helix family transcriptional regulator, is the master transcription factor driving the genetic programming of Treg cells 17. Natural or experimental mutations of the foxp3 gene result in congenital deficiencies in Treg cell development and function. These lead to spontaneous, multi‐organ, immune pathology in scurfy mice and humans with immunodysregulation polyendocrinopathy enteropathy X‐linked (IPEX) syndrome 9, 10, 11.
FoxP3 acts primarily as a transcriptional repressor of key genes involved in T cell activation and effector functions, including proliferation and synthesis of proinflammatory cytokines [e.g. IL‐2, IL‐4, IL‐17A and interferon (IFN)‐γ], all the while endowing the cell with potent suppressive functions 18, 19. Sustained expression of FoxP3 in Treg cells is required for lineage commitment and stability, and several key mechanisms including cytokine signaling, epigenetic control of the foxp3 locus and interactions of FoxP3 with other proteins, contribute to the regulation of FoxP3 expression and, consequently, maintenance of peripheral tolerance (Fig. 1).
Figure 1.
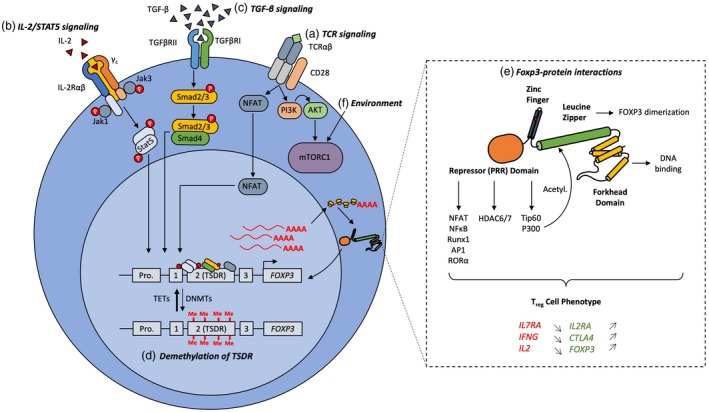
Mechanisms preserving the stability of the regulatory T cell (Treg) phenotype. Treg cell lineage stability is reliant on the strength of forkhead box protein 3 (FoxP3) expression. There are several mechanisms in place to ensure robust FoxP3 expression in Treg cells. A, T cell receptor (TCR) signaling leads to nuclear factor of activated T cells (NFAT) binding to the CNS2 region of the foxp3 locus for transactivation of gene expression. B, Constitutive High level of CD25 expression, the interleukin (IL)‐2 receptor α, on the Treg cell surface confers a high sensitivity to IL‐2 in the environment. IL‐2 signaling through Janus kinase (Jak)1 and Jak3 result in signal transducer and activator of transcription (STAT‐5) phosphorylation and dimerization and subsequent translocation into the nucleus. Phosphorylated (p)STAT‐5 binding to the conserved non‐coding DNA sequence (CNS)2 drives FoxP3 expression. C, Transforming growth factor (TGF)‐β signaling through TGF‐βRI and TGF‐βRII result in Smad2/3 phosphorylation, association with the transcription Smad4 and the translocation of the complex into the nucleus. Smad2/3/4 bind to the foxp3 promoter and drive FoxP3 expression. In the presence of TCR signaling, TGF‐β‐driven FoxP3 expression in naïve CD4+ conventional T (Tconv) results in induced (i)Treg/peripheral (p)Treg induction. D, To enable transcription factor binding to the foxp3 locus enhancer region, certain sites are specifically demethylated in Treg cells. In the CNS2 enhancer region, this is referred to as the Treg‐specific demethylated region (TSDR). Demethylation of the TSDR is mediated by the 10–11 translocation (Tet) family demethylases Tet1 and Tet2. DNA methyl transferases (Dnmt) such as Dnmt1 methylate the TSDR and destabilize Foxp3 expression. E, Once FoxP3 is expressed, it heterodimerizes and can associate with many different binding partners (~700), including transcription factors, histone deacetylases and histone acetyl transferases. Binding to these proteins are necessary for transcriptional repression of various genes (il7ra, ifng, il2) and activation of others (il2ra, ctla4, foxp3). F, Significant focus has been devoted to studying the environmental signals controlling FoxP3 expression. Phosphoinositide‐3‐kinase–protein kinase B (PI3K‐Akt) signaling downstream TCR and CD28 signaling is needed for transient mammalian target of rapamycin complex 1 (mTORC1) activation and consolidation of the Treg cell phenotype. However, chronic activation of mTORC1 (e.g. through environmental signals such as glutamine) result in sustained mTORC1 activation and therefore deregulation of Treg cells. Thus, mTORC1 inhibitors (e.g. rapamycin) are used in the in‐vitro expansion of Treg cells.
Cytokine control of FoxP3+ Treg cell homeostasis
IL‐2 is necessary for global Treg cell homeostasis by promoting their development, survival and function in the thymus and periphery 20, 21, 22. IL‐2 activates the signal transducer and activator of transcription (STAT)‐5, which binds to several sites on the foxp3 promoter to enhance FoxP3 expression and thus establish the Treg cell genetic program. A defining feature of Treg cells, unlike other T cell subsets, is their constitutive expression of CD25, the α chain of the heterotrimeric high‐affinity IL‐2R. Indeed, Treg cells have a higher sensitivity to IL‐2 signaling than Teff cells due to preferential binding of IL‐2 through high expression of CD25 and higher activity of PP1 and PP2A phosphatases which modulate IL‐2 signaling 23. Defects in IL‐2 signaling (e.g. mutations in CD25) can give rise to IPEX‐like autoimmunity as a consequence of Treg cell dysfunction 24.
TGF‐β is another essential cytokine promoting the development of Treg cells. In conjunction with TCR stimulation, TGF‐β mediates the conversion of CD4+ FoxP3− naïve Tconv cells into iTreg/pTreg cells (in‐vitro/in‐vivo) 25. TGF‐β signaling activates Smad2/3 transcription factors, which bind directly to enhancer regions of the foxp3 locus to promote FoxP3 expression in these pTreg cells 25, 26. TGF‐β signaling is also critical for tTreg cell development and function. Although a CD4+ T cell‐specific deletion of the TGF‐β receptor II (TGF‐βRII) in mice had no defect in Treg cell frequencies, mice nevertheless developed multi‐organ T cell‐mediated autoimmunity, suggesting a defect in immunoregulation 27, 28. Moreover, a specific knock‐out of the TGF‐β receptor I (TGF‐βRI) in murine T cells curtailed tTreg cell generation within the first week of life of neonatal mice, supporting a requirement for TGF‐β in tTreg cell development 29. Additionally, intrathymic injection of precursor CD4−CD8− thymocytes from cd4C re tgfbr1 fl/fl mice into syngeneic wild‐type hosts failed to yield tTreg cells 30. Thus, global Treg cell development is dependent on TGF‐β signalling.
Epigenetic regulation of FoxP3 expression
Enzymatic demethylation of specific cytosine–phosphate–guanine (CpG) motifs within the intronic enhancer evolutionary conserved non‐coding sequence (CNS) 2, also called the Treg‐specific demethylated region (TSDR), is another critical mechanism maintaining FoxP3 expression in Treg cells and is a feature that is absent in Tconv cells 31, 32, 33. Demethylation of these critical non‐coding sequences opens up the foxp3 enhancer region to large multi‐molecular complexes containing FoxP3, c‐Rel, nuclear factor of activated T cells (NFAT), STAT‐5, runt‐related transcription factor 1‐core‐binding factor (Runx1‐CBF)β, cAMP responsive element‐binding/activating transcription factor (Creb/ATF) and ETS proto‐oncogene 1 (Ets1). These multi‐molecular complexes bring the CNS2 into contact with the foxp3 promoter, thereby driving FoxP3 expression. Indeed, it was recently demonstrated that demethylation of the CNS2 resulted in enhanced Treg cell lineage stability by increasing Treg cell sensitivity to IL‐2 through greater STAT‐5 occupancy of the foxp3 enhancer 34. Accordingly, recruitment of the DNA methylase Dnmt1 to the foxp3 promoter and its subsequent methylation severely impaired the maintenance of FoxP3. Demethylation of CNS2 in Treg cells is mediated by the 10–11 translocation (Tet) family of demethylases, which are themselves recruited to the foxp3 locus by STAT‐5 35. Deletion of Tet1 and Tet2 in murine Treg cells yields severe autoimmunity as a consequence of poor Treg cell lineage commitment.
CNS1 is also important for Treg cell lineage commitment. Deletion of CNS1, which contains a binding site for Smad2/3 upstream of CNS2, abrogates pTreg cell development in mice and leads to inflammation at mucosal sites 36. Moreover, the newly identified CNS0 was also shown to be necessary for Treg cell stability 37. CNS0 is bound by the genomic organizer Satb1 in FoxP3− tTreg cell thymic precursors and alters chromatin accessibility in order to orchestrate the changes needed for future FoxP3 expression and Treg cell development in mice.
FoxP3 binding‐partners and Treg cell stability
FoxP3 imparts its functions by directly or indirectly binding to ~ 700 transcriptional promoters, either repressing or activating the target genes 38, 39. However, the transcriptional control of most genes inherent to the Treg cell transcriptional program is achieved by the indirect binding of FoxP3 through other molecular factors. For example, complexes between FoxP3 and NFAT, nuclear factor (NF)‐κB, acute myeloid leukemia 1 (AML1)/Runx1, activator protein 1 (AP1) and retinoic acid orphan receptor (ROR)α transcription factors are known to impair the NFAT/AP1 transcriptional program that leads to il2 expression in Tconv cells, thereby maintaining anergy in Treg cells 1. To identify other FoxP3 binding partners, Rudra et al. expressed the prokaryotic biotin ligase, BirA, and FoxP3 with an N‐terminal BirA ligase biotinylation site in a murine T cell hybridoma line to purify protein complexes containing FoxP3 and identified 361 binding partners by mass spectrometry 40. Along with FoxP3, these binding partners formed a complex transcriptional program with multiple positive and negative feedback loops consolidating the Treg cell transcriptional program. Hence, FoxP3 is necessary but not sufficient for maintaining the Treg cell phenotype 40, 41.
Key binding partners of FoxP3 include several histone deacetylases (HDAC) such as HDAC1, 7 and 9, and histone acetyltransferases (HAT), such as the Tat‐interacting protein 60 (TIP60) and p300 42, 43, 44. Interactions with these proteins are necessary for transcriptional silencing of target genes as well as for protein modifications that enhance FoxP3 function. For example, acetylation of FoxP3 upon complexing with p300 allows it to evade proteasomal degradation, which constitutes a post‐translational mechanism of FoxP3 stability 43. Furthermore, we have recently shown that abrogating the FoxP3–Tat‐interacting protein 60 (TIP60) interaction, a defect caused by the germline foxp3 A384T IPEX mutation, relieves human Treg cell suppressive capacity while retaining major aspects of the Treg cell phenotype including the repression of proliferation and inflammatory cytokines 45. Restoring this interaction using a TIP60 allosteric modifier rescued FoxP3 functions and Treg cell suppressive capacity in vitro and in vivo. Thus, several mechanisms maintain FoxP3 stability and consequently establish the requisite transcriptional program to ensure FoxP3+ Treg cell functional development.
Functional heterogeneity among human FoxP3+ Treg cells
Despite FoxP3 ensuring a robust immunosuppressive phenotype, significant functional heterogeneity exists among human FoxP3+ Treg cells. We previously developed a single‐cell strategy to examine the phenotypical and functional heterogeneity of human CD4+CD25highCD127low Treg cells relative to FoxP3 expression from blood of healthy individuals 16. CD4+CD25high/bright Treg cells, albeit highly enriched in suppressive FoxP3+ T cells, harbour a pool of bona fide FoxP3+ Treg cells with compromised suppressive function, despite the maintenance of hallmark phenotypic, epigenetic and transcriptional features of Treg cells. These FoxP3+ Treg cells with compromised suppressive function also produce proinflammatory cytokines such as IL‐2, IL‐17 and IFN‐γ following polyclonal activation 16. Whether this heterogeneity relates to FoxP3+ Treg cell subsets that have acquired unique effector functions or Treg cells losing their phenotype remains to be determined. These non‐suppressive CD25high FoxP3+ cells in healthy peripheral blood may be involved in the onset of autoimmune or inflammatory states, and further characterization is needed to understand more clearly their roles in health and disease.
We recently identified Helios, an Ikaros family transcription factor, to be preferentially expressed on suppressive Treg cells as opposed to non‐suppressive Treg cells in human blood during immune quiescence and in disease 46. Although, Helios was initially proposed as a marker for differentiating between tTreg and iTreg cells, there are no data supporting this definition in humans. We further demonstrated that co‐expression of the surface receptors T cell immunoreceptor with immunoglobulin (Ig) and ITIM domains (TIGIT) and Fc receptor‐like 3 (FcRL3) identified most Helios+ Treg cells in the periphery and were absent from Teff cells at the steady state and following TCR stimulation 46.
Antigen‐specific Treg cell functions
Heterogeneity in Treg cell function also lies in TCR specificities that drive their developmental ontogeny, peripheral homeostasis and effector functions. As stated earlier, tTreg and pTreg cells differ in their ontogenies. Consequently, tTreg cells possess a diverse, self‐restricted TCR repertoire distinct from Tconv cells, whereas pTreg cells retain the antigen specificities of their naïve Tconv precursors. In‐vitro evidence shows that tTreg cell suppressive function is TCR activation‐dependent, and although antigen non‐specific suppression has been described, antigen‐specific signals are largely viewed as necessary for optimal tTreg cell suppressive functions within the periphery 47, 48, 49. For example, pancreatic autoantigen‐specific Treg cells and myelin basic protein (MBP)‐specific Treg cells preferentially prevent disease in mouse models of type‐1 diabetes (T1D) and multiple sclerosis, respectively 50, 51, 52. Nevertheless, naïve (CD25lowCD45RA+) non‐antigen‐experienced tTreg cell populations are critical for homeostasis of the global tTreg cell pool 53. Expressing high levels of the prosurvival molecule Bcl‐2 and relying on IL‐7 signaling in the periphery, naïve tTreg cells are rapidly proliferative and readily differentiate into potently suppressive memory tTreg cells upon TCR engagement by their cognate self‐antigen 53, 54.
Allergy represents the best‐studied area regarding the functional consequences of pTreg cell antigen specificity. Barrier tissues are constantly exposed to a myriad of environmental, dietary and commensal microbial antigens requiring homeostatic immune control mediated by the local Treg or Teff cell antigen‐specific repertoires. Consequently, they are particularly specialized sites for the de‐novo generation of pTreg cells in order to ensure immune homeostasis and maintain tolerance 55, 56. A break of Treg cell‐mediated tolerance often results in allergic inflammation at these sites. Specialized APCs, such as alveolar macrophages in the lungs and CD103+ dendritic cells (DCs) in the intestinal mucosa, favour pTreg cell differentiation via a TGF‐β‐ or retinoic acid‐dependent process 57, 58, 59. In the lungs, the TCR specificities from pTreg cells are often restricted to common aeroantigens (e.g. house dust mite, plant pollen, birch and Aspergillus fumigatus), and dampen allergic inflammation 60. Bacher et al. recently developed an antigen‐reactive T cell enrichment technology to study aeroantigen‐specific Treg cell function in adults 60. The technique relied on the detection and isolation from peripheral blood mononuclear cells (PBMC) of rare antigen‐specific Treg and Teff cells by the use of 4‐1BB (CD137) or CD40L (CD154) expression, respectively 60. The authors showed that Treg cells specific to common aeroantigens were enriched in allergic adults and existed at greater numbers than aeroantigen‐specific Teff cells, thus leading to the hypothesis that aeroantigen‐specific Treg cells suppressed Teff cell expansion. In other barrier tissues such as the gut, pTreg cells maintain tolerance towards the diversity of commensal microorganisms and food antigens to which the immune system is continuously being exposed 55, 56.
Antigen‐specific Treg cells have also been implicated to play a role in autoimmunity and cancer. Ooi et al. recently demonstrated that Treg cells with a TCR self‐restriction towards the type IV collagen α chain (peptides 135–145) presented specifically on human leukocyte antigen D‐related (HLA‐DR1) provided dominant resistance to Goodpasture’s syndrome, a rapidly progressive and fatal autoimmune disease of the kidneys and lungs, in humanized mice 48. In rheumatoid arthritis (RA), TCR‐sequencing of synovial CD14−CD4+CD25highCD127− Treg cells yielded a few expanded clonotypes 61. These Treg cells were enriched in suppressive and activated (HLA‐DR+) Treg cell subsets, thereby demonstrating a functional relevance for Treg cell antigen‐specificity in RA. In cancer, Treg cells play a key role in promoting immune‐evasion by cancer cells. For example, increased frequencies of Treg cells specific to NY‐ESO‐1, an immunogenic tumor antigen, are observed in peripheral blood of melanoma patients 62, and their frequency correlates with metastatic potential and poor prognosis.
Functional plasticity, or instability, of Treg cells
We and others have shown that adoptive transfer of FoxP3+ Treg cells into lymphopenic mice resulted in a loss of FoxP3 expression, loss of suppressive activity and consequential acquisition of inflammatory characteristics 15. Experiments conducted in foxp3 GFPCre Rosa26‐loxP‐stop‐loxP‐YFP fate mapping mice showed that a significant proportion of Treg cells that expressed FoxP3 at one point in time (YFP+) lost FoxP3 expression (GFP−) in homeostatic settings 15. These YFP+GFP− cells, termed exFoxP3, were phenotypically similar to memory Teff cells and produced proinflammatory cytokines. It is unclear whether exTreg cells originate from bona fide committed tTreg cells, from an uncommitted CD25low subset found therein, or if they represent Teff cells that have not completely converted into FoxP3+ pTreg cells 63. Whether Treg cell plasticity occurs in the normal pathophysiology of human disease is currently unknown.
Extrinsic factors promoting the instability of Treg cell function
Cytokine signaling, repeated antigen exposure and methylation patterns within the foxp3 promoter are key factors enabling the stability of Treg cell function. IL‐6 is a proinflammatory cytokine known to counteract IL‐2 signaling through STAT‐3 dimers which occupy STAT‐5‐binding sites of the foxp3 locus, thereby attenuating FoxP3 expression 22. Moreover, IL‐6 in the presence of TGF‐β can favour the development of Th17 cells over pTreg from naïve CD4+ T cells in mice and humans 64. Whether Treg cells can themselves convert into Th17 cells, thereby demonstrating true plasticity, remains unknown in both humans and mice. The nature and strength of antigenic stimulation also influences Treg stability. Development of tTreg cells in the mouse thymus depends on strong TCR stimulation leading to demethylation of the TSDR and subsequent binding of STAT‐5 to the foxp3 locus 65. In contrast, induction of murine pTreg cells is improved when TCR stimulation is weaker and co‐stimulation is reduced 66. In humans, repeated TCR stimulation of CD4+CD25highCD127low Treg cells attenuated, and even completely abrogated, FoxP3 expression while increasing production of proinflammatory cytokines 67, although the exFoxP3 cells identified here may have originated from contaminating Teff cells (see Conclusion).
Functional adaptation of Treg cells
Local inflammatory signals can drive CD4+ T cells to undergo functional plasticity to acquire specialized effector functions in inflammatory sites. Growing evidence indicates that FoxP3+ Treg cells can also acquire tissue‐specific adaptations that promote their homing to inflammatory sites for the control of immune responses driven by various Teff cell lineages. Treg cells achieve this by up‐regulating the transcription factors of other T cell lineages in the presence of specific polarizing conditions, namely T‐bet (Th1), interferon regulator factor 4 (IRF4) and GATA binding protein 3 (GATA3) (Th2) and STAT‐3 and RORγt (Th17) 68, 69, 70, 71. For example, expression of CCR6 (CD196), a chemokine receptor characteristic of Th17 cells, driven by STAT‐3 expression in Treg cells, is thought to dampen Th17 cell‐mediated inflammation and tissue pathology in crescentic glomerulonephritis, a potent inflammatory kidney disease 72. However, the co‐expression of secondary transcription factors may also represent a transition where Treg cells convert into Teff cells as the acquired transcription factors may endow Treg cells with the ability to produce proinflammatory cytokines. Indeed, human and mouse RORγt+ Treg cells obtained through in‐vitro Th17 polarizing conditions produce IL‐17A and demonstrate attenuated suppressive capacities despite intact FoxP3 expression 71, 73. Whether these cells can completely convert into FoxP3− Teff cells or whether this plasticity is reversible remains to be determined. Moreover, whether this occurs in‐vivo in humans is completely unknown.
Treg cell dysfunction in human disease
How Treg cell dysfunction occurs and impacts the outcome of human disease is an important question. Here, we will highlight a few key mechanisms leading to Treg cell dysfunction in human disease: (i) genetic defects, (ii) abrogation of Treg cell‐promoting signals, (iii) presence of Treg cell destabilizing factors and (iv) co‐opting Treg cell suppressive function.
Congenital Treg cell defects: IPEX syndrome, the human ‘FoxP3 knock‐out’
The most extreme example of Treg cell dysregulation occurs in IPEX syndrome, a sex‐linked congenital disease that is frequently fatal within infancy. It is largely caused by single loss‐of‐function germline point mutations in the Foxp3 locus that abrogate Treg cell function to different degrees 9. More than 60 such mutations have been identified with a clinical spectrum whose severity is dependent on the nature of the mutation and the protein domain affected 74. Mutations in the N‐terminal proline‐rich repressor (PRR) domain often result in improper Treg cell suppressive function and production of proinflammatory cytokines (e.g. E70H and T108M) 75. Moreover, IPEX mutations on the leucine zipper domain (e.g. ∆E251) prevent FoxP3 from exerting transcriptional control of target genes and are often associated with severe clinical manifestations of autoimmunity. However, most IPEX cases are caused by mutations in the forkhead (FKH) domain of FoxP3, which either prevent DNA‐binding (e.g. R397W and I363V) or interactions with other proteins that aid in orchestrating the Treg transcriptional program (e.g. A384T) 45, 75. Importantly, the presence of a mutation does not necessarily imply diminished FoxP3 protein expression levels (e.g. F373A 76), Treg numbers (e.g. A384T 45) or even loss of Treg suppressive functions (e.g. R347H). Clearly, the IPEX case demonstrates that, simple enumeration of Treg cells is often not sufficient to make claims about Treg cell function in vivo.
Developmental and homeostatic Treg cell dysfunction in autoimmunity
The IL‐2/IL‐2R pathway is necessary for the thymic development and peripheral homeostasis of Treg cells, and congenital or homeostatic disruption of key components of this pathway can provoke Treg dysregulation and give rise to a spectrum of diseases ranging from IPEX‐like autoimmunity 24 to type 1 diabetes (T1D).
Implications for T1D pathogenesis
Mouse studies show that a hallmark of diabetes onset is the apoptosis of pancreatic islet Treg cells, alongside decreased CD25 and Bcl‐2 expression, suggesting local IL‐2 deprivation 77. In some T1D cohorts, patients have decreased IL‐2 production 78, and diminished CD25 expression within FoxP3+ Treg cells 79. Long et al. found that IL‐2 sensitivity of CD4+CD25+ Treg cells was decreased in diabetic patients compared to healthy controls 80. However, this observation was not seen by other studies 23, or only reproduced when looking at subjects bearing T1D‐predisposing mutations on genes of the IL‐2 pathway (IL2RA and PTPN2) 81, 82. Indeed, the IL2RA susceptibility haplotype was associated with decreased sensitivity to low doses of IL‐2 in vitro, diminished suppressive function, lower CD25 expression and lower levels of FoxP3 expression by Helios+ Treg cells under limiting conditions of IL‐2 (Fig. 2A) 81. There is still no consensus on whether Treg cell frequencies correlate with T1D onset 81, 83. Furthermore, some have argued that Teff cells from diabetic subjects are resistant to suppression by Treg cells 84, possibly through a STAT‐3‐dependent mechanism 85.
Figure 2.
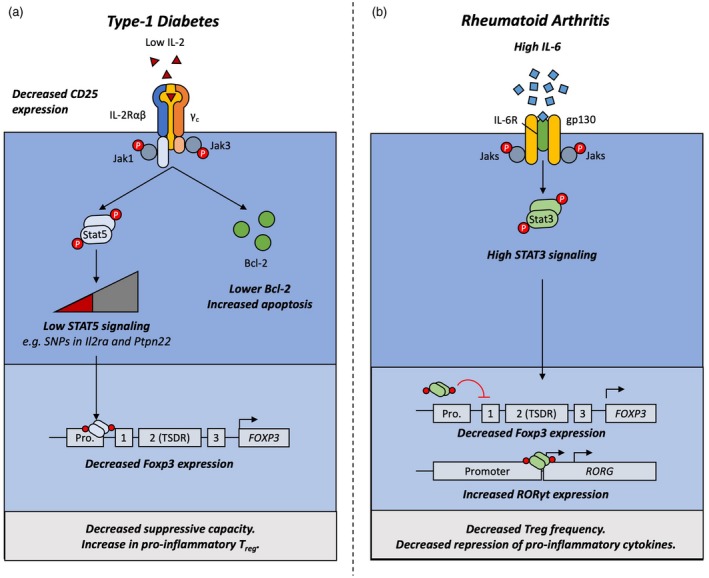
Mechanisms driving Treg cell dysfunction in type‐1 diabetes and rheumatoid arthritis. Forkhead box protein 3 (FoxP3) expression is destabilized by extrinsic factors in type‐1 diabetes and rheumatoid arthritis. A, Local deprivation in interleukin (IL)‐2 and diminished sensitivity to IL‐2 increases susceptibility to apoptosis through diminished B‐cell lymphoma 2 (Bcl‐2) production. Furthermore, lack of this positive signal reduces phosphorylated signal transducer and activator of transcription (pSTAT)‐5 activation and occupancy of the foxp3 promoter, leading to diminished FoxP3 expression. As a result, regulatory T cells (Treg) have a lower suppressive capacity in vitro and can start secreting proinflammatory cytokines. B, High levels of IL‐6 in the inflammatory pannus of rheumatoid arthritis patients trigger STAT‐3 signaling through the IL‐6 receptor (IL‐6R). STAT‐3 occupies the STAT‐5‐binding sites on the foxp3 locus, which attenuates FoxP3 expression. Furthermore, STAT‐3 binding to the rorc promoter enhances retinoic acid orphan receptor (ROR)γt expression, the T helper type 17 (Th17) master transcription factor. As a result, Th17 cells develop preferentially over Treg cells during disease flares.
In‐vitro, as stated earlier, diminished FoxP3 expression is correlated with loss of a suppressive phenotype and secretion of proinflammatory cytokines 67. Indeed, patients with either new‐onset 86 or established diabetes 87 have an increased frequency of proinflammatory, cytokine‐secreting, less suppressive Treg cells in their peripheral blood. However, it is as yet unknown if this dysfunction is linked causally to poor IL‐2 signaling or if it is a consequence of islet inflammation. Altogether, these results indicate that susceptibility mutations in genes of the IL‐2 pathway impair Treg cell sensitivity to IL‐2 signals, leading to diminished pSTAT‐5 levels and reduced expression levels of FoxP3. This dysregulation could ultimately lead to defective suppression by Treg cells.
Translational relevance for immunotherapy
Defects in IL‐2 signaling preferentially tamper with Treg cell homeostasis. Therefore, Low‐dose IL‐2 therapy could specifically stimulate the Treg compartment and re‐establish immune homeostasis in diseases such as T1D, graft‐versus‐host disease (GVHD) 88 and hepatitis C virus (HCV)‐induced vasculitis 89. The DF‐IL‐2 trial showed low‐dose aldesleukin [recombinant human (rh)IL‐2] increased Treg cell frequency in a dose‐dependent manner 90. In the DILT1D trial, a single dose of aldesleukin improved Treg suppressive function in vitro 91. In Alopecia areata, an autoimmune disease targeting hair follicles, hair regrowth upon treatment with rhIL‐2 was associated with the recruitment of CD4+CD25+FoxP3+ cells at the site of lesion and persisted for 2 months after the end of treatment, suggesting that Treg cells require a threshold of IL‐2R activation to acquire migratory capacity 92.
Finding a therapeutic window that allows for specific activation of Treg cells over other immune cell subsets will be key to the successful development of low‐dose IL‐2 therapy. As harmful cell types such as NK cells, CD4+ and CD8+ Teff cells constitutively express the intermediate‐affinity IL‐2R, the potential for accelerating the course of the disease exists. Indeed, Todd et al reported that for all doses tested at day 1 post‐administration, the plasma concentrations of aldesleukin was higher than the activation threshold of NK cells and activated memory Teff cells 91. To circumvent these issues, one approach is the engineering of IL‐2 superagonists to improve durability and selectivity through increased affinity, prolonged half‐life and lower doses. One example is IL‐2/CD25 fusion proteins, where IL‐2 is bound to CD25 by a non‐cleavable linker to increase the persistence of IL‐2 and reduce binding to the intermediate‐affinity IL‐2R 93. Another investigated compound is IL‐2‐anti‐IL‐2 monoclonal antibody immune complexes (IL‐2IC) 94, 95, 96, where IL‐2 is bound to the IL‐2IC antibody such that the CD25‐binding epitope is exposed and the CD122 (IL‐2Rβ)‐binding epitope is blocked (e.g. clone JES6‐1), preferentially inducing the expansion of Treg cells over Tconv cells. Moreover, a human IL‐2‐anti‐IL‐2 monoclonal antibody (F5111.2) immune complex was generated to preferentially enhance human Treg cell proliferation in humanized mice, and successfully used to ameliorate autoimmunity and GVHD in non‐obese diabetic (NOD) mice 97. Finally, Sockolosky et al. devised a strategy to selectively stimulate engineered T cells in the context of T cell therapy. The infused T cells express a mutant ortho‐IL‐2Rβ receptor that signals through the native STAT‐5 pathway but does not bind to wild‐type IL‐2. Instead, these receptors bind ortho‐IL‐2, an engineered cytokine–receptor complex that acts as an agonist of ortho‐IL‐2Rβ but not of any form of the wild‐type IL‐2R 98. They applied this strategy for the expansion of tumor‐reactive CD8+ cytotoxic T lymphocytes (CTL) in a mouse model of melanoma. However, a similar approach could be conceived to expand engineered ortho‐IL‐2Rβ Treg cells and enhance the efficacy of Treg cell therapy.
Inflammation‐mediated destabilization of Treg cell function
Proinflammatory cytokines such as IL‐6 and TNF‐α can interfere with the stability of FoxP3 expression in Treg cells, alter the Treg/Teff balance locally or systemically and ultimately provoke a loss of peripheral tolerance. Indeed, IL‐6 and TNF‐α are readily over‐expressed in a number of autoimmune and chronic inflammatory conditions and prompts us to consider its relevance as a target of immunotherapy in autoimmunity.
The case for rheumatoid arthritis (RA)
The inflammatory environment of the synovial pannus in RA represents an obvious setting where cytokine‐mediated Treg cell dysfunction may occur. High levels of IL‐6 may inhibit Treg cell homeostasis and function and enhance the development of proinflammatory T cell subsets. One evidence in support of IL‐6‐mediated dysfunction of Treg cells is the preferential development of Th17 cells over Treg cells in the periphery of RA patients 99, 100. This reciprocal regulation can be explained as, in the presence of TGF‐β, IL‐6 enhances the expression of RORγt through STAT‐3 while repressing FoxP3 expression (Fig. 2B).
Another proinflammatory cytokine affecting Treg cell function is TNF‐α, which signals via the TNF‐RII receptor, and can subsequently down‐regulate FoxP3 expression and alter Treg cell suppressive function 101. TNF‐α may also impair Treg cell function by altering the formation of the immunological synapse between APCs and Treg cells. Here, PKCθ plays a role in the integration of TCR and CD28 signals in Teff cells upstream of NF‐κB. Contrary to Tconv cells, PKCθ is sequestered from the immunological synapse in Treg cells. TNF‐α promotes the recruitment of PKCθ to the TCR in Treg cells, and through downstream Akt signalling inhibits their suppressive capacity (Fig. 2B). Consistently, in‐vitro administration of a PKCθ inhibitor on Treg cells from RA patients enhanced their suppressive function 102.
However, studies evaluating the function of Treg cells in RA reveal inconsistent findings. While in new‐onset patients or during disease flares the frequency of Treg cells in circulation is diminished 103, Treg cell frequencies are normal in patients with clinically managed disease. However, a defect in repression, and not in suppressive function, of proinflammatory cytokines was reported in Treg cells in RA 104, linked to defective CTLA‐4 expression 105. Furthermore, Van Amelsfort et al. reported that synovial Treg cells from RA patients have a very activated phenotype and that synovial Teff cells are resistant to suppression 106. Indeed, in addition to dysregulating Treg cells, IL‐6 and TNF‐α signaling render Teff cells resistant to suppression via a protein kinase B (PKB)‐dependent mechanism 107.
Translational relevance for immunotherapy
IL‐6 and TNF also contribute to systemic symptoms such as fever and asthenia, and fuel tissue damage through recruitment of neutrophils to the inflamed joints and the differentiation of osteoclasts through the NF‐κB pathway. Consequently, antibodies directed against these cytokines were developed to block their signaling. Anti‐TNF pharmacological agents such as etanercept, adalimumab and infliximab are a major part of the therapeutic arsenal available to treat RA. Anti‐TNF treatment induces an increase in circulating Treg cells in responding patients, which correlates with a decrease in the titres of C‐reactive protein, a biomarker of inflammation 108. Tocilizumab, an IL‐6R inhibitor, has been authorized as a second line of treatment for RA patients, after failure of methotrexate or anti‐TNFs. As observed with anti‐TNF drugs, the clinical benefits of tocilizumab treatment are accompanied by an increase in the frequency of peripheral Treg cells after 6 months of treatment 101.
Co‐opting Treg cell suppressive function in cancer
Cancer immunity provides a setting where increased Treg cell suppression contributes to cancer onset, progression and metastasis. To sustain their growth and gain the potential to metastasize, tumors develop a variety of tumor‐induced immunosuppressive mechanisms to escape anti‐tumor immunity, such as the expression or secretion of anti‐inflammatory mediators [indoleamine 2,3‐dioxygenase (IDO), IL‐10, TGF‐β], and the recruitment of a wide variety of suppressive leukocytes such as Treg cells, myeloid‐derived suppressor cells (MDSCs), tolerogenic DCs and tumor‐associated macrophages (TAMs).
Establishment of a Treg cell niche in the tumor microenvironment
Tumor cells contribute to the establishment of a Treg cell niche by expressing IDO, an enzyme involved in tryptophan degradation. Its expression diminishes tryptophan availability and produces metabolites that induce T cell apoptosis 109 while promoting increased Treg cell frequencies in the tumor infiltrate (Fig. 3). Reprogramming of Treg cells into Th17 cells has been shown to promote early anti‐tumor CTL responses 110. However, IDO inhibits this process by suppressing IL‐6 secretion in DCs and through the GCN2 kinase pathway in FoxP3+ cells. Finally, IDO expression also silences the mTORC2/Akt pathway, thus stabilizing the Treg cell lineage inside the tumor microenvironment 111.
Figure 3.
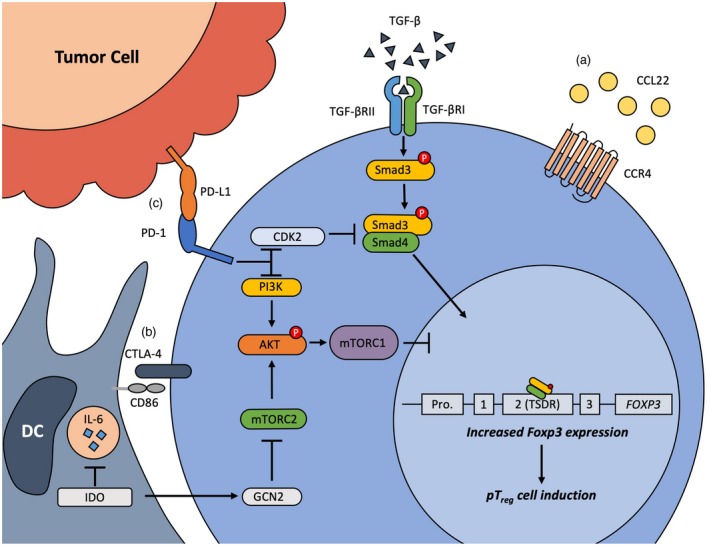
Mechanisms promoting regulatory T cell (Treg) development and immunosuppression in the tumor microenvironment. A, Treg cells are recruited to the tumor through chemokine attraction. B, Interaction of Treg cells with antigen‐presenting cells (APCs) through cytotoxic T lymphocyte antigen (CTLA)‐4 deprive T effector (Teff) cells of co‐stimulatory signals, polarizes dendritic cells (DCs) towards a tolerogenic phenotype and induces the expression of indoleamine 2,3‐dioxygenase (IDO), which catabolizes metabolites, thereby inducing Teff cell apoptosis. Furthermore, it inhibits the reprogramming of Treg cells into Th17 cells by suppressing interleukin (IL)‐6 secretion and promotes Treg cell lineage stability by inhibiting the transient mammalian target of rapamycin complex 1/protein kinase B (mTORC2/Akt) pathway. C, Tumor cells express programmed cell death ligand 1 (PD‐L1), which binds programmed cell death 1 (PD‐1) at the surface of Treg cells. The PD‐1 pathway stabilizes forkhead box protein 3 (FoxP3) expression by inhibiting the phosphoinositide‐3‐kinase (PI3K)/Akt pathway and synergizes with transforming growth factor (TGF)‐β by diminishing the level of Smad3 necessary to promote the conversion of naïve CD4+ T cells into peripheral (p)Treg cells, while inducing Teff cell exhaustion.
Treg cells play an essential role in establishing immunosuppression in the tumor microenvironment. Indeed, depletion of Treg cells with an anti‐CD25 monoclonal antibody (mAb) leads to tumor clearance in murine syngeneic tumor models 112. Treg cells infiltrate several types of human cancers, including melanoma, breast, pancreas and liver 113. Higher frequencies of tumor‐infiltrating Treg cells have been correlated with worse prognosis and metastatic potential. In the example of ovarian cancer, a high frequency of infiltrating Treg cells and a decreased CTL to Treg cell ratio have both been associated with reduced survival of patients 114, 115, consistent with Treg cells suppressing anti‐tumoral immune response. Tumor‐infiltrating Treg cells also display a very activated phenotype and have high levels of expression of immune checkpoint molecules such as CTLA‐4 (CD152), programmed cell death 1 (PD‐1) (CD279) 116 and TIGIT 117, and activation markers such as GITR 118. These data point towards enhanced activity of Treg cells in the tumor microenvironment contributing to immune evasion.
Translational relevance for immunotherapy
Immune check point inhibitors were developed following the rationale that these biologics compete with the binding of natural ligands to the target co‐inhibitory receptor, thus alleviating Tconv and CTL inhibitory signaling. Numerous molecules targeting these pathways have been developed successfully for the treatment of many cancers, such as anti‐CTLA‐4 mAbs (ipilimumab, tremelimumab) and anti‐PD‐1 mAbs (nivolumab, pembrolizumab).
Studies now show that the protective effect of some of these biologics was through impairment of Treg cell function. Since it is abolished in FcγR–/– mice, the protective effect of anti‐CTLA‐4 is very likely mediated by antibody‐dependent cell‐mediated cytotoxicity (ADCC) 119. Indeed, depletion of Treg cells in a mouse model of melanoma was dependent on the presence of FcγR‐expressing macrophages infiltrating the tumor 120. In melanoma patients, ADCC assays showed depletion of Treg cells through the interaction of ipilimumab and FcγRIIIA+ monocytes. Furthermore, responders to ipilimumab treatment have increased proportions of macrophages in their tumors 121, and treatment efficacy is correlated with decreasing frequencies of Treg cells in tumors 122. In contrast, tremelimumab, another anti‐CTLA‐4 mAb, likely functions without ADCC of Treg cells as it suppresses Treg cell function without affecting cell numbers 123. Therefore, research is needed to assess the impact of check point inhibitors on the functional dynamics of Treg cell subsets in blood and in particular within the tumor microenvironment.
For example, the role of CTLA‐4 in Treg cell suppressive function is well established. CTLA‐4 acts in a cell‐intrinsic manner by competing with CD28 for its shared ligands. It also acts on APCs by inducing IDO expression 124 and reducing surface expression of CD80 and CD86 through endocytosis and down‐regulating transcription of their mRNA 125. This leads to the emergence of tolerogenic DCs and limits the availability of co‐stimulatory ligands to Teff cells.
Conversely, PD‐1 is also highly expressed on Treg cells, but its role is not well identified. In melanoma patients, in‐vitro treatment with nivolumab down‐regulated FoxP3 expression in sorted Treg cells 126 and inhibited Treg cell suppressive function 127. However, PD‐1–/– mice have a similar frequency of circulating Treg cells to their wild‐type counterparts, and do not display a diminished suppressive capacity 128. Nevertheless, PD‐1–/– CD4+ T cells have a diminished capacity to differentiate into pTreg cells when transferred into lymphopenic hosts 128. Indeed, the PD‐L1/PD‐1 pathway plays a role in the development of pTreg cells by synergizing with TGF‐β signaling through Smad3 to promote the conversion of naïve T cells into iTreg cells. Furthermore, PD‐1 signaling inhibits the PI3K/Akt pathway, which is known to destabilize FoxP3 expression 129. Taken together, these results suggest that PD‐1 does not necessarily play a role in Treg cell suppressive function but regulates their homeostasis and stability, thus contributing to the regulation of the Treg/Teff cell balance.
Conclusion and future perspectives
Despite the enormous efforts by the immunological community to characterize the molecular and cellular basis of Treg cell development and function in health and disease, several knowledge gaps remain in this area which future research must address. Various mechanisms ensure a robust and sustained immunosuppressive phenotype in Treg cells. However, there is significant functional and phenotypical heterogeneity that remains to be captured in human disease. One of the future priorities relate to the urgent need for novel strategies to monitor Treg cell function in human blood more effectively, and in particular in tissues, in states of health and disease. Part of the problem lies with the phenotypical markers used to distinguish human Treg cells and subsets found therein from a variety of Tconv cells. Both CD25 and FoxP3, the quintessential markers of Treg cells, are up‐regulated on Tconv cells upon TCR stimulation, and can only be used in situations of immune quiescence, not states of immune activation or inflammation. Other markers have been proposed to overcome this issue. For example, Treg cells are often defined as CD127low given the inverse correlation between CD127 and FoxP3 expression 130, 131. Nevertheless, this gating strategy fails to eliminate activated Teff cells and excludes a large proportion of bona fide Treg cells. More recently described Treg cell markers include HLA‐DR, CTLA‐4, CCR6, GARP, CD15s, CD39, CD49d, CD147, TNFRII, GITR and LAP (Table 1) 132. Nonetheless, these markers are readily modulated on the surface of Treg and Tconv cells consequently impeding proper discrimination and their use for downstream functional and phenotypical studies. We recently demonstrated that TIGIT and FcRL3 are reliable and specific markers for identifying and sorting Helios+ Treg cells, and given the stable suppressive phenotype of these cells, we envision that sorted TIGIT+FcRL3+CD25highCD127low cells can be isolated and clinically manipulated for therapeutic use 46.
Table 1.
List of human Treg cell markers
| Location | Marker | Treg | Teff |
|---|---|---|---|
| Surface | CD25 (IL‐2Rα) | High expression on most cells | Up‐regulated with activation |
| CD127 (IL‐7Rα) | Low/negative expression | Down‐regulated with activation | |
| TIGIT1 | High expression correlating with Helios | Up‐regulated with activation | |
| CD307c (FcRL3)1 | High expression on Helios+ cells | Low/negative expression | |
| HLA‐DR | Expression on terminally differentiated cells | Up‐regulated with activation | |
| CD15s (Sialyl Lewis x) | Up‐regulated with activation | Weakly up‐regulated with activation | |
| GARP | High expression on activated cells | Not expressed | |
| CD39 | Up‐regulated with activation | Not expressed | |
| CD49d | Down‐regulated with activation | Highly expressed | |
| CD120b (TNF‐RII) | High expression | Up‐regulated with activation | |
| CD357 (GITR, TNF‐RSF18) | High expression | Up‐regulated with activation | |
| LAP | High expression on activated cells | Not expressed | |
| CD147 (Basigin/Emmprin) | Constitutive expression | Up‐regulated with activation | |
| CCR6 | Expression on memory Treg cells only | Expressed on Th17 cells and with activation | |
| Intracellular | FoxP3 | High expression | Transient, high up‐regulation with activation |
| Helios | High expression | Not expressed | |
| CD152 (CTLA‐4) | High intracellular expression | Not expressed intracellularly |
When TIGIT and FcRL3 are used in conjunction, they capture most Helios+ memory Treg cells, which have a stably immunosuppressive phenotype.
IL = interleukin; TIGIT = T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine‐based inhibition motif (ITIM) domains; FcRL3 = Fc receptor‐like protein 3; GARP = glycoprotein‐A repetitions predominant protein; HLA‐DR = human leukocyte antigen D‐related; TNF = tumor necrosis factor; GITR = glucocorticoid‐induced TNF‐R‐related protein; CTLA‐4 = cytotoxic T lymphocyte antigen; Th17 = T helper type 17; Treg = regulatory T cell; Teff = effector T.
Another issue that has thus far prevented a proper definition of Treg cell function in human disease is the lack of discrimination of antigen‐specific responses, particularly in conventional, in‐vitro polyclonal suppression assays. These methods mask the important effects of antigen‐specific Treg cells that may serve different physiological roles than other Treg cell subsets (e.g. aeroantigen‐specific Treg cells maintaining tolerance to airborne allergens, but not necessarily counteracting inflammation following pulmonary viral infections). Novel methodologies are emerging now to understand more clearly the antigen‐specific effects of Treg cells in human disease. MHC‐II tetramer technology, which has largely been ineffective at reliably isolating rare CD4+ TCR specificities, is being vastly improved through adjustments such as barcoding to improve the number of epitopes studied, or dual staining of identically loaded tetramers with different fluorochromes, in conjunction with surface markers such as CD137 to improve specificity and isolate antigen‐stimulated Treg cells by flow cytometry. Ultimately, these important advancements will enable us to monitor Treg cell functions in unprecedented depth, thereby enhancing our understanding of Treg cells in human health and disease.
Finally, several hurdles affect the therapeutic potential of Treg cells in disease. Novel strategies will need to be developed to overcome the limitations related to survival and cell persistence in vivo, stability of the Treg cell functional phenotype and selective engagement or repression of antigen‐specific responses in defined disease settings. In this regard, a better understanding of the genetic factors [single nucleotide polymorphisms (SNPs) or epigenetic], mechanisms of transcriptional regulation (splice isoforms, miRNAs and transcription factor activity) and post‐translational modifications (phosphorylation, acetylation or ubiquitination) influencing FoxP3 gene and protein expression or activity will be required to further modulate the function of endogenous, or adoptively transferred expanded Treg cells in therapy.
Disclosures
There are no competing interests.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Regulatory T cells: exploring mechanisms for future therapies. Clinical and Experimental Immunology 2019, 197: 11–13.
From stability to dynamics: understanding molecular mechanisms of regulatory T cells through Foxp3 transcriptional dynamics. Clinical and Experimental Immunology 2019, 197: 14–23.
The role of FOXP3+ regulatory T cells in human autoimmune and inflammatory diseases. Clinical and Experimental Immunology 2019, 197: 24–35.
Methods to manufacture regulatory T cells for cell therapy. Clinical and Experimental Immunology 2019, 197: 52–63.
References
- 1. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008; 133:775–87. [DOI] [PubMed] [Google Scholar]
- 2. Sakaguchi S, Wing K, Onishi Y, Prieto‐Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol 2009; 21:1105–11. [DOI] [PubMed] [Google Scholar]
- 3. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self‐tolerance maintained by activated T cells expressing IL‐2 receptor alpha‐chains (CD25). Breakdown of a single mechanism of self‐tolerance causes various autoimmune diseases. J Immunol 1995; 155:1151. [PubMed] [Google Scholar]
- 4. Ng WF, Duggan PJ, Ponchel F et al Human CD4(+)CD25(+) cells: a naturally occurring population of regulatory T cells. Blood 2001; 98:2736. [DOI] [PubMed] [Google Scholar]
- 5. Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human Cd4(+)Cd25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med 2001; 193:1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of Cd4(+)Cd25(+) T cells with regulatory properties from human blood. J Exp Med 2001; 193:1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stephens Leigh A, Mottet C, Mason D, Powrie F. Human CD4+CD25+ thymocytes and peripheral T cells have immune suppressive activity in vitro . Euro J Immunol 2001; 31:1247–54. [DOI] [PubMed] [Google Scholar]
- 8. Baecher‐Allan C, Brown JA, Freeman GJ, Hafler DA. CD4(+)CD25(+) regulatory cells in human peripheral blood. J Immunol 2001; 167:1245. [DOI] [PubMed] [Google Scholar]
- 9. Brunkow ME, Jeffery EW, Hjerrild KA et al Disruption of a new forkhead/winged‐helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 2001; 27:68. [DOI] [PubMed] [Google Scholar]
- 10. Wildin RS, Ramsdell F, Peake J et al X‐linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 2001; 27:18. [DOI] [PubMed] [Google Scholar]
- 11. Bennett CL, Christie J, Ramsdell F et al The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001; 27:20. [DOI] [PubMed] [Google Scholar]
- 12. Hsieh C‐S, Lee H‐M, Lio C‐WJ. Selection of regulatory T cells in the thymus. Nat Rev Immunol 2012; 12:157. [DOI] [PubMed] [Google Scholar]
- 13. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Ann Rev Immunol 2012; 30:733–58. [DOI] [PubMed] [Google Scholar]
- 14. Miyara M, Ito Y, Sakaguchi S. TREG‐cell therapies for autoimmune rheumatic diseases. Nat Rev Rheumatol 2014; 10:543. [DOI] [PubMed] [Google Scholar]
- 15. Zhou X, Bailey‐Bucktrout S, Jeker LT et al Foxp3 instability leads to the generation of pathogenic memory T cells in vivo. Nat Immunol 2009; 10:1000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. d’Hennezel E, Yurchenko E, Sgouroudis E, Hay V, Piccirillo CA. Single‐cell analysis of the human T regulatory population uncovers functional heterogeneity and instability within FOXP3+ cells. J Immunol 2011; 186:6788–97. [DOI] [PubMed] [Google Scholar]
- 17. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4:330. [DOI] [PubMed] [Google Scholar]
- 18. Komatsu N, Mariotti‐Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3(+) T cells: a committed regulatory T‐cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci 2009; 106:1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duarte João H, Zelenay S, Bergman M‐L, Martins Ana C, Demengeot J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur J Immunol 2009; 39:948–55. [DOI] [PubMed] [Google Scholar]
- 20. Zorn E, Nelson EA, Mohseni M et al IL‐2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT‐dependent mechanism and induces the expansion of these cells in vivo. Blood 2006; 108:1571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL‐2 receptor β‐dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol 2007; 178:280. [DOI] [PubMed] [Google Scholar]
- 22. Yao Z, Kanno Y, Kerenyi M et al Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood 2007; 109:4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu A, Snowhite I, Vendrame F et al Selective IL‐2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low‐dose IL‐2 therapy in type 1 diabetes. Diabetes 2015; 64:2172–83. [DOI] [PubMed] [Google Scholar]
- 24. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X‐linked‐like syndrome, and defective IL‐10 expression from CD4 lymphocytes. J Allergy Clin Immunol 2007; 119:482–7. [DOI] [PubMed] [Google Scholar]
- 25. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol 2007; 9:194. [DOI] [PubMed] [Google Scholar]
- 26. Ruan Q, Kameswaran V, Tone Y et al Development of Foxp3+ regulatory T cells is driven by the c‐Rel enhanceosome. Immunity 2009; 31:932–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early‐onset autoimmunity in mice with the T cell‐specific targeting of transforming growth factor‐β receptor. Immunity 2006; 25:441–54. [DOI] [PubMed] [Google Scholar]
- 28. Li MO, Sanjabi S, Flavell RA. Transforming growth factor‐β controls development, homeostasis, and tolerance of T cells by regulatory T cell‐dependent and ‐independent mechanisms. Immunity 2006; 25:455–71. [DOI] [PubMed] [Google Scholar]
- 29. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF‐β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol 2008; 9:632. [DOI] [PubMed] [Google Scholar]
- 30. Konkel JE, Jin W, Abbatiello B, Grainger JR, Chen W. Thymocyte apoptosis drives the intrathymic generation of regulatory T cells. Proc Natl Acad Sci 2014; 111:E465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim H‐P, Leonard WJ. CREB/ATF‐dependent T cell receptor–induced FoxP3 gene expression: a role for DNA methylation. J Exp Med 2007; 204:1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Janson PCJ, Winerdal ME, Marits P, Thörn M, Ohlsson R, Winqvist O. FOXP3 promoter demethylation reveals the committed Treg population in humans. PLOS ONE 2008; 3:e1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T‐cell lineage? Nat Rev Immunol 2009; 9:83. [DOI] [PubMed] [Google Scholar]
- 34. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014; 158:749–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang R, Qu C, Zhou Y et al Hydrogen sulfide promotes Tet1‐ and Tet2‐mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity 2015; 43:251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Josefowicz SZ, Niec RE, Kim HY et al Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012; 482:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kitagawa Y, Ohkura N, Kidani Y et al Guidance of regulatory T cell development by Satb1‐dependent super‐enhancer establishment. Nat Immunol 2016; 18:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marson A, Kretschmer K, Frampton GM et al Foxp3 occupancy and regulation of key target genes during T‐cell stimulation. Nature 2007; 445:931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zheng Y, Josefowicz SZ, Kas A, Chu T‐T, Gavin MA, Rudensky AY. Genome‐wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 2007; 445:936. [DOI] [PubMed] [Google Scholar]
- 40. Rudra D, deRoos P, Chaudhry A et al Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol 2012; 13:1010–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fu W, Ergun A, Lu T et al A multiple redundant genetic switch locks in the transcriptional signature of T regulatory cells. Nat Immunol 2012; 13:972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li B, Samanta A, Song X et al FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci 2007; 104:4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Loosdregt J, Vercoulen Y, Guichelaar T et al Regulation of Treg functionality by acetylation‐mediated Foxp3 protein stabilization. Blood 2010; 115:965. [DOI] [PubMed] [Google Scholar]
- 44. Xiao Y, Li B, Zhou Z, Hancock WW, Zhang H, Greene MI. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr Opin Immunol 2010; 22:583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bin Dhuban K, d’Hennezel E, Nagai Y et al Suppression by human FOXP3+ regulatory T cells requires FOXP3‐TIP60 interactions. Sci Immunol 2017; 2:eaai9297. [DOI] [PubMed] [Google Scholar]
- 46. Bin Dhuban K, d’Hennezel E, Nashi E et al Coexpression of TIGIT and FCRL3 identifies helios+ human memory regulatory T cells. J Immunol 2015; 194:3687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schmidt AM, Lu W, Sindhava VJ et al Regulatory T cells require TCR signaling for their suppressive function. J Immunol (Balt) 1950; 194:4362–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ooi JD, Petersen J, Tan YH et al Dominant protection from HLA‐linked autoimmunity by antigen‐specific regulatory T cells. Nature 2017; 545:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoeppli RE, MacDonald KG, Levings MK, Cook L. How antigen specificity directs regulatory T‐cell function: self, foreign and engineered specificity. HLA 2016; 88:3–13. [DOI] [PubMed] [Google Scholar]
- 50. Tonkin DR, He J, Barbour G, Haskins K. Regulatory T cells prevent transfer of type 1 diabetes in NOD mice only when their antigen is present in vivo . J Immunol 2008; 181:4516. [DOI] [PubMed] [Google Scholar]
- 51. Kim YC, Zhang A‐H, Yoon J et al Engineered MBP‐specific human Tregs ameliorate MOG‐induced EAE through IL‐2‐triggered inhibition of effector T cells. J Autoimmunity 2018; 92:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hori S, Haury M, Coutinho A, Demengeot J. Specificity requirements for selection and effector functions of CD25+4+ regulatory T cells in anti‐myelin basic protein T cell receptor transgenic mice. Proc Natl Acad Sci 2002; 99:8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miyara M, Yoshioka Y, Kitoh A et al Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009; 30:899–911. [DOI] [PubMed] [Google Scholar]
- 54. Silva SL, Albuquerque AS, Serra‐Caetano A et al Human naïve regulatory T‐cells feature high steady‐state turnover and are maintained by IL‐7. Oncotarget 2016; 7:12163–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lathrop SK, Bloom SM, Rao SM et al Peripheral education of the immune system by colonic commensal microbiota. Nature 2011; 478:250–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luu M, Steinhoff U, Visekruna A. Functional heterogeneity of gut‐resident regulatory T cells. Clin Trans Immunol 2017; 6:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Soroosh P, Doherty TA, Duan W et al Lung‐resident tissue macrophages generate Foxp3(+) regulatory T cells and promote airway tolerance. J Exp Med 2013; 210:775–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Coombes JL, Siddiqui KRR, Arancibia‐Cárcamo CV et al A functionally specialized population of mucosal CD103(+) DCs induces Foxp3(+) regulatory T cells via a TGF‐β– and retinoic acid‐dependent mechanism. J Exp Med 2007; 204:1757–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Benson MJ, Pino‐Lagos K, Rosemblatt M, Noelle RJ. All‐trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co‐stimulation. J Exp Med 2007; 204:1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bacher P, Heinrich F, Stervbo U et al Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell 2016; 167:1067–1078.e1016. [DOI] [PubMed] [Google Scholar]
- 61. Rossetti M, Spreafico R, Consolaro A et al TCR repertoire sequencing identifies synovial Treg cell clonotypes in the bloodstream during active inflammation in human arthritis. Ann Rheumatic Diseases 2017; 76:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vence L, Palucka AK, Fay JW et al Circulating tumor antigen‐specific regulatory T cells in patients with metastatic melanoma. Proc Natl Acad Sci 2007; 104:20884–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Miyao T, Floess S, Setoguchi R et al Plasticity of Foxp3+ T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 2012; 36:262–75. [DOI] [PubMed] [Google Scholar]
- 64. Bettelli E, Carrier Y, Gao W et al Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006; 441:235. [DOI] [PubMed] [Google Scholar]
- 65. Burchill MA, Yang J, Vang KB et al Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity 2008; 28:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Semple K, Nguyen A, Yu Y, Wang H, Anasetti C, Yu X‐Z. Strong CD28 costimulation suppresses induction of regulatory T cells from naive precursors through Lck signaling. Blood 2011; 117:3096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hoffmann P, Boeld TJ, Eder R et al Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Euro J Immunol 2009; 39:1088–97. [DOI] [PubMed] [Google Scholar]
- 68. Koch MA, Tucker‐Heard GS, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T‐bet controls regulatory T cell homeostasis and function during type‐1 inflammation. Nat Immunol 2009; 10:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zheng Y, Chaudhry A, Kas A et al Regulatory T‐cell suppressor program co‐opts transcription factor IRF4 to control T(H)2 responses. Nature 2009; 458:351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chaudhry A, Rudra D, Treuting P et al CD4(+) regulatory T cells control Th17 responses in a Stat3‐dependent manner. Science 2009; 326:986–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kluger MA, Meyer MC, Nosko A et al RORγt(+)Foxp3(+) cells are an independent bifunctional regulatory T cell lineage and mediate crescentic GN. J Am Soc Nephrol 2016; 27:454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kluger MA, Luig M, Wegscheid C et al Stat3 programs Th17‐specific regulatory T cells to control GN. J Am Soc Nephrol 2014; 25:1291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ayyoub M, Deknuydt F, Raimbaud I et al Human memory FOXP3(+) Tregs secrete IL‐17 ex vivo and constitutively express the T(H)17 lineage‐specific transcription factor RORγt. Proc Natl Acad Sci U S A 2009; 106:8635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. d’Hennezel E, Bin Dhuban K, Torgerson T, Piccirillo C. The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet 2012; 49:291–302. [DOI] [PubMed] [Google Scholar]
- 75. Bin Dhuban K, Piccirillo CA. The immunological and genetic basis of immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome. Curr Opin Allergy Clin Immunol 2015; 15:525–32. [DOI] [PubMed] [Google Scholar]
- 76. Bacchetta R, Passerini L, Gambineri E et al Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest 2006; 116:1713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tang Q, Adams JY, Penaranda C et al Central role of defective interleukin‐2 production in the triggering of islet autoimmune destruction. Immunity 2008; 28:687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Roncarolo MG, Zoppo M, Bacchetta R et al Interleukin‐2 production and interleukin‐2 receptor expression in children with newly diagnosed diabetes. Clin Immunol Immunopathol 1988; 49:53–62. [DOI] [PubMed] [Google Scholar]
- 79. Parackova Z, Kayserova J, Danova K et al T regulatory LYMPHOCYTES in type 1 diabetes: impaired CD25 expression and IL‐2 induced STAT5 phosphorylation in pediatric patients. Autoimmunity 2016; 49:523–31. [DOI] [PubMed] [Google Scholar]
- 80. Long SA, Cerosaletti K, Bollyky PL et al Defects in IL‐2R signaling contribute to diminished maintenance of FOXP3 expression in CD4+CD25+ regulatory T‐cells of type 1 diabetic subjects. Diabetes 2010; 59:407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Garg G, Tyler JR, Yang JHM et al Type 1 diabetes‐associated IL2RA variation lowers IL‐2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol 2012; 188:4644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yang JHM, Cutler AJ, Ferreira RC et al Natural variation in interleukin‐2 sensitivity influences regulatory T‐cell frequency and function in individuals with long‐standing type 1 diabetes. Diabetes 2015; 64:3891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hull CM, Peakman M, Tree TIM. Regulatory T cell dysfunction in type 1 diabetes: what’s broken and how can we fix it? Diabetologia 2017; 60:1839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol 2008; 181:7350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ihantola EL, Viisanen T, Gazali AM et al Effector T cell resistance to suppression and STAT3 signaling during the development of human type 1 diabetes. J Immunol 2018; 201:1144–53. [DOI] [PubMed] [Google Scholar]
- 86. Marwaha AK, Crome SQ, Panagiotopoulos C et al Cutting edge: Increased IL‐17‐secreting T cells in children with new‐onset type 1 diabetes. J Immunol 2010; 185:3814–8. [DOI] [PubMed] [Google Scholar]
- 87. McClymont SA, Putnam AL, Lee MR et al Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol 2011; 186:3918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Koreth J, Matsuoka K, Kim HT et al Interleukin‐2 and regulatory T cells in graft‐versus‐host disease. N Engl J Med 2011; 365:2055–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Saadoun D, Rosenzwajg M, Joly F et al Regulatory T‐cell responses to low‐dose interleukin‐2 in HCV‐induced vasculitis. N Engl J Med 2011; 365:2067–77. [DOI] [PubMed] [Google Scholar]
- 90. Rosenzwajg M, Churlaud G, Mallone R et al Low‐dose interleukin‐2 fosters a dose‐dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun 2015; 58:48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Todd JA, Evangelou M, Cutler AJ et al Regulatory T cell responses in participants with type 1 diabetes after a single dose of interleukin‐2: a non‐randomised, open label, Adaptive Dose‐Finding Trial. PLoS Med 2016; 13:e1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Castela E, Le Duff F, Butori C et al Effects of low‐dose recombinant interleukin 2 to promote T‐regulatory cells in alopecia areata. JAMA Dermatol 2014; 150:748–51. [DOI] [PubMed] [Google Scholar]
- 93. Ward NC, Yu A, Moro A et al IL‐2/CD25: a long‐acting fusion protein that promotes immune tolerance by selectively targeting the IL‐2 receptor on regulatory T cells. J Immunol 2018; 201:2579–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yokoyama Y, Iwasaki T, Kitano S et al IL‐2–anti‐IL‐2 monoclonal antibody immune complexes inhibit collagen‐induced arthritis by augmenting regulatory T cell functions. J Immunol 2018; 201:1899. [DOI] [PubMed] [Google Scholar]
- 95. Spangler JB, Tomala J, Luca VC et al Antibodies to interleukin‐2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity 2015; 42:815–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Spangler JB, Trotta E, Tomala J et al Engineering a single‐agent cytokine/antibody fusion that selectively expands regulatory T cells for autoimmune disease therapy. J Immunol 2018; 201:2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Trotta E, Bessette PH, Silveria SL et al A human anti‐IL‐2 antibody that potentiates regulatory T cells by a structure‐based mechanism. Nat Med 2018; 24:1005–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sockolosky JT, Trotta E, Parisi G et al Selective targeting of engineered T cells using orthogonal IL‐2 cytokine‐receptor complexes. Science 2018; 359:1037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Samson M, Audia S, Janikashvili N et al Brief report: inhibition of interleukin‐6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum 2012; 64:2499–503. [DOI] [PubMed] [Google Scholar]
- 100. Pesce B, Soto L, Sabugo F et al Effect of interleukin‐6 receptor blockade on the balance between regulatory T cells and T helper type 17 cells in rheumatoid arthritis patients. Clin Exp Immunol 2013; 171:237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Valencia X, Stephens G, Goldbach‐Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T‐regulatory cells. Blood 2006; 108:253–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zanin‐Zhorov A, Ding Y, Kumari S et al Protein kinase C‐θ mediates negative feedback on regulatory T cell function. Science 2010; 328:372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lawson CA, Brown AK, Bejarano V et al Early rheumatoid arthritis is associated with a deficit in the CD4+CD25high regulatory T cell population in peripheral blood. Rheumatology (Oxf) 2006; 45:1210–7. [DOI] [PubMed] [Google Scholar]
- 104. Ehrenstein MR, Evans JG, Singh A et al Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti‐TNFα therapy. J Exp Med 2004; 200:277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Flores‐Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA‐4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci USA 2008; 105:19396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum 2004; 50:2775–85. [DOI] [PubMed] [Google Scholar]
- 107. Wehrens EJ, Mijnheer G, Duurland CL et al Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c‐AKT hyperactivation in effector cells. Blood 2011; 118:3538–48. [DOI] [PubMed] [Google Scholar]
- 108. Nadkarni S, Mauri C, Ehrenstein MR. Anti‐TNF‐alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF‐beta. J Exp Med 2007; 204:33–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Godin‐Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3‐dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res 2011; 17:6985–91. [DOI] [PubMed] [Google Scholar]
- 110. Sharma MD, Hou DY, Baban B et al Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross‐presentation and CD8(+) T cell priming in naive mice. Immunity 2010; 33:942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter‐regulation, and tolerance. Trends Immunol 2016; 37:193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti‐CD25 (interleukin‐2 receptor alpha) monoclonal antibody. Cancer Res 1999; 59:3128–33. [PubMed] [Google Scholar]
- 113. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer 2010; 127:759–67. [DOI] [PubMed] [Google Scholar]
- 114. Curiel TJ, Coukos G, Zou L et al Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942–9. [DOI] [PubMed] [Google Scholar]
- 115. Sato E, Olson SH, Ahn J et al Intraepithelial CD8+ tumor‐infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci 2005; 102:18538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Syed Khaja AS, Toor SM, El Salhat H et al Preferential accumulation of regulatory T cells with highly immunosuppressive characteristics in breast tumor microenvironment. Oncotarget 2017; 8:33159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. De Simone M, Arrigoni A, Rossetti G et al Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor‐infiltrating T regulatory cells. Immunity 2016; 45:1135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Krausz LT, Fischer‐Fodor E, Major ZZ, Fetica B. GITR‐expressing regulatory T‐cell subsets are increased in tumor‐positive lymph nodes from advanced breast cancer patients as compared to tumor‐negative lymph nodes. Int J Immunopathol Pharmacol 2012; 25:59–66. [DOI] [PubMed] [Google Scholar]
- 119. Bulliard Y, Jolicoeur R, Windman M et al Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor‐targeting antibodies. J Exp Med 2013; 210:1685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Simpson TR, Li F, Montalvo‐Ortiz W et al Fc‐dependent depletion of tumor‐infiltrating regulatory T cells co‐defines the efficacy of anti–CTLA‐4 therapy against melanoma. J Exp Med 2013; 210:1695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Romano E, Kusio‐Kobialka M, Foukas PG et al Ipilimumab‐dependent cell‐mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A 2015; 112:6140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hodi FS, Butler M, Oble DA et al Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte‐associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci 2008; 105:3005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Khan S, Burt DJ, Ralph C, Thistlethwaite FC, Hawkins RE, Elkord E. Tremelimumab (anti‐CTLA4) mediates immune responses mainly by direct activation of T effector cells rather than by affecting T regulatory cells. Clin Immunol (Orlando) 2011; 138:85–96. [DOI] [PubMed] [Google Scholar]
- 124. Fallarino F, Grohmann U, Hwang KW et al Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol 2003; 4:1206–12. [DOI] [PubMed] [Google Scholar]
- 125. Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down‐regulate co‐stimulatory molecules on antigen‐presenting cells. Eur J Immunol 2000; 30:1538–43. [DOI] [PubMed] [Google Scholar]
- 126. Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen‐specific CTL by CD4+CD25Hi regulatory T cells. Int Immunol 2009; 21:1065–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wang C, Thudium KB, Han M et al In vitro characterization of the anti‐PD‐1 antibody nivolumab, BMS‐936558, and in vivo toxicology in non‐human primates. Cancer Immunol Res 2014; 2:846–56. [DOI] [PubMed] [Google Scholar]
- 128. Chen X, Fosco D, Kline DE et al PD‐1 regulates extrathymic regulatory T‐cell differentiation. Eur J Immunol 2014; 44:2603–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Francisco LM, Salinas VH, Brown KE et al PD‐L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 2009; 206:3015–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Liu W, Putnam AL, Xu‐yu Z et al CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 2006; 203:1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yu N, Li X, Song W, et al CD4+CD25+CD127low/− T cells: a more specific Treg population in human peripheral blood. Inflammation 2012; 35:1773–80. [DOI] [PubMed] [Google Scholar]
- 132. Miyara M, Chader D, Sage E et al Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3high regulatory T cells in humans. Proc Natl Acad Sci USA 2015; 112:7225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]