

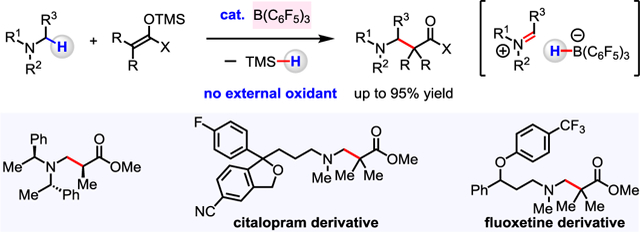
Abstract
An efficient method for the coupling of N-alkylamines with silicon enolates to generate β-amino carbonyl compounds is disclosed. These reactions proceed by activation of α-amino C–H bonds by B(C6F5)3, which likely generates a “frustrated” acid/base complex in the presence of large N-alkylamines. The transformation requires no external oxidant and releases hydrosilane as a by-product. The utility of this method is demonstrated in the late-stage functionalization of bioactive molecules such as citalopram, atomoxetine, and fluoxetine.
Graphical Abstract
Enolate nucleophiles are commonly employed in addition reactions to C=N bonds (i.e., Mannich-type processes), since this process reliably generates several classes of important β-amino carbonyl molecules.1–5 In these reactions, imine or iminium ion intermediates are either prepared in situ or in a separate operation through condensation of an amine and a carbonyl compound, α-fragmentation of iminium ion precursors, or oxidation of tertiary amines.1–5 The latter method (i.e., oxidative Mannich-type reactions: Figure 1A) employs organometallic catalysts (e.g., Ru, Rh, Cu, V complexes) and stoichiometric oxidants (e.g., t-BuOOH, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, O2) to convert N-alkylamines (I) to their corresponding iminium ions, which then react with various enolate equivalents (II).2,3 Such a process, while notable, requires external oxidants, and the scope of amines is largely confined to tetrahydroisoquinoline and N,N-dimethylaniline derivatives.2,3
Figure 1.
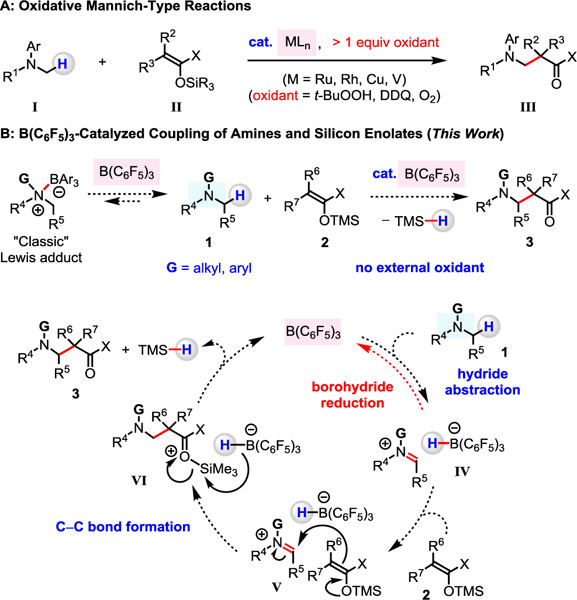
Coupling of N-Alkylamines and Silicon Enolates
We surmised that an attractive alternative to the oxidative Mannich-type reaction would entail rupture of an α-amino C–H bond of an N-alkylamine (1) by a boron-based Lewis acid (Figure 1B). Hydride abstraction from an N-alkylamine by organoborane compounds to furnish an iminium ion (IV) has been previously investigated.6–12 An important advantage of this approach is that an array of N-alkylamines (including those that lack the fused N-aryl groups) can be converted to IV without the use of external oxidant.6–10 Reaction of IV with an enol equivalent (2) would forge a C–C bond (V), subsequently releasing the β-amino carbonyl product 3, the Lewis acid catalyst, and a hydrosilane as an environmentally benign byproduct (VI). Here, we disclose the results of our studies regarding the realization of the above catalytic cycle.
To initiate our investigations, we needed an appropriate combination of acid catalyst and amine substrate that is capable of undergoing hydride transfer as opposed to forming a stable acid/base adduct. Accordingly, we considered pairing the strongly Lewis acidic B(C6F5)3 with a hindered amine (Scheme 1). We first probed the ability of B(C6F5)3 to convert a N,N-dimethylaniline to its derived iminium ion, which could then be trapped by a silyl ketene acetal (Scheme 1). Treatment of N,N-dimethylaniline (1a) and 1-methoxy-2-methyl-1-(trimethylsiloxy)propene (2a) with 20 mol % B(C6F5)3 afforded 3a in 31% yield (DCE, 22 °C). We reasoned that electron-donating N-aryl substituents might improve the efficiency of B(C6F5)3-catalyzed hydride abstraction by enhancing the hydride donor ability of the α-amino C–H bond and by stabilizing the resultant iminium ion intermediate. In the event, the reaction with electron-donating para-methoxy-substituted 1b afforded 3b in 25% yield. None of the desired product was observed with electron-deficient para-trifluoromethylphenyl-substituted 1c. With 3,5-di-tert-butyl-substituted 1d, the C–C bond forming product 3d was obtained in 38% yield.
Scheme 1.
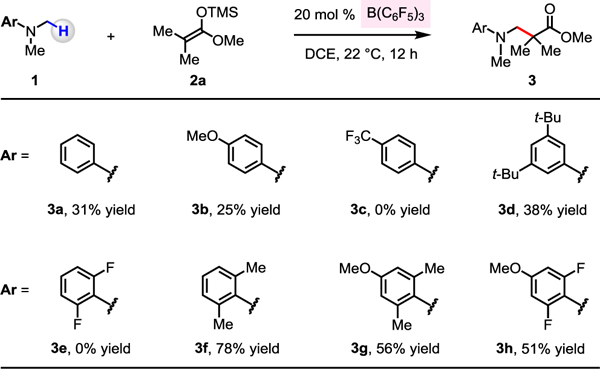
Evaluation of N-Aryl Substituents a,b
a Conditions: N,N-dimethylaniline (0.1 mmol), 1-methoxy-2-methyl-1-(trimethylsiloxy)propene (0.2 mmol), B(C6F5)3 (20 mol %), dichloroethane (0.25 mL), under N2, 22 °C, 12 h. b Yields were determined by 1H NMR analysis of unpurified product mixtures with mesitylene as the internal standard. See the Supporting Information for details.
To evaluate the effect of using more hindered amines, we tested a range of ortho-disubstituted anilines. Whereas 2,6-difluoro-N,N-dimethylaniline (1e) gave none of the Mannich-type product, reaction of larger and more electron-rich N,N,2,6-tetramethylaniline (1f) resulted in the formation of 3f in 78% yield, which marks a considerable improvement in efficiency. Encouraged by this finding, we studied the reaction with 4-methoxy-N,N,2,6-tetramethylaniline (1g), which gave 3g in 56% yield; the N-aryl substituent in 3g was removed under oxidative conditions.13 By using the less hindered and more electron-deficient 2,6-difluoro-4-methoxyphenyl-substituted 1h, we were able to obtain 3h in 51% yield.
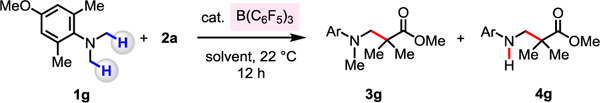
With the aim to further increase efficiency, we set out to identify the optimal conditions with the transformation that affords 3g, serving as the representative process (Table 1). There were no products formed without any B(C6F5)3 present (entry 2).
Table 1.
 | |||||
|---|---|---|---|---|---|
| entry | Lewis acid | catalyst loading (%) | solvent | yield (%) | |
| 3g | 4g | ||||
| 1 | B(C6F5)3 | 20 | DCE | 56 | 35 |
| 2 | none | 0 | DCE | 0 | 0 |
| 3 | B(C6F5)3 | 10 | DCE | 71 | 22 |
| 4 | B(C6F5)3 | 10 | Et2O | 83 | 17 |
| 5 | B(C6F5)3 | 5.0 | Et2O | 22 | <5 |
| 6 | B(C6F5)3 | 10 | THF | 38 | <5 |
| 7 | B(C6F5)3 | 10 | toluene | 75 | 21 |
| 8 | B(C6F5)3 | 10 | benzene | 81 | 16 |
| 9 | B(C6F5)3 | 5.0 | benzene | >95 | <5 |
| 10 | BF3•OEt2 | 10 | benzene | 0 | 0 |
| 11 | BPh3 | 10 | benzene | 0 | 0 |
Conditions: 4-methoxy-N,N,2,6-tetramethylaniline (0.1 mmol), 1-methoxy-2-methyl-1-(trimethylsiloxy)propene (0.2 mmol), Lewis acid, solvent (0.25 mL), under N2, 22 °C, 12 h.
Yields were determined by 1H NMR analysis of unpurified product mixtures with mesitylene as the internal standard. See the Supporting Information for details.
In some instances (entries 1 and 3–9), secondary amine 4g was also obtained, probably through the cleavage of the N–Me bond due to the reaction of in situ generated iminium ion with water. Among the N,N-dimethylanilines evaluated, only in the cases of ortho-dimethyl-substituted 1f and 1g were the latter type of byproducts formed. At lower catalyst loading (10 mol %) loss of the methyl unit was minimized and 3g was formed more selectively in 71% yield (entry 3). Next, we examined the effect of using ethereal solvents to investigate if these more polar solvents could facilitate the Mannich reaction which involves ionic intermediates. In diethyl ether, and with 10 mol % catalyst loading, 3g was formed in 83% yield, but with 5.0 mol % catalyst there was a considerable diminution in efficiency (22% yield, entries 4–5). With THF as the solvent, 3g was obtained in 38% yield (entry 6). Use of less polar aromatic hydrocarbons such as benzene and toluene as the solvent and with 10 mol % B(C6F5)3 led to the formation of 3g in 81% and 75% yield, respectively (entries 7–8). With benzene and 5.0 mol % B(C6F5)3 loading, 3g was obtained with high selectivity (<5% of 4g) and in >95% yield (entry 9). Use of less hindered BF3•OEt2 or less acidic BPh3 proved to be ineffective (entries 10–11), providing support for the hypothesis that acidic B(C6F5)3, along with sterically demanding and electron-rich N-alkylamines, represent the most effective catalyst/substrate combination.
A significant assortment of N,N-dialkylanilines may be used (Scheme 2). Reaction of N,N-dimethyl-substituted 1g afforded 3g in 80% yield after purification (5.0 mol % B(C6F5)3). With N-benzyl and N-cyclopropylmethyl-substituted substrates, 3i and 3j were isolated in 64% and 56% yield, respectively; thus, in these instances, α-amino methylene C–H bonds remained intact. α-Amino C–H bond of N-arylpyrrolidine 1k could be converted to a C–C bond by the use of 10 mol % B(C6F5)3, affording 3k in 90% yield. 1-(4-Methoxy-2,6-dimethylphenyl)pyrrolidin-3-ol (1l), possessing an unprotected hydroxyl group, furnished the desired products in their O-silylated forms 3l (31% yield, 4.0:1 trans:cis) and 3m (23% yield, >20:1 trans:cis).14 Reaction with α-methyl-substituted pyrrolidine delivered 3n in 70% yield and trans:cis of 7.0:1. When N-arylpiperidine 1o was used, 3o was isolated in 38% yield with the reaction being performed at 50 °C. A series of trialkyl-substituted amines that lack the fused N-aryl group were coupled efficiently with ((1-methoxyprop-1-en-1-yl)oxy)trimethylsilane (2b), leading to the formation of 5a−5d (88%–95% yield). Reaction of methyl (S)-3-(bis((S)-1-phenylethyl)amino)-2-methylpropanoate with 2b delivered 5d as a 1.6:1 mixture of diastereomers, which were separable through silica gel chromatography, allowing us to produce the β-amino esters in enantiomerically pure form (see the Supporting Information for details).
Scheme 2.
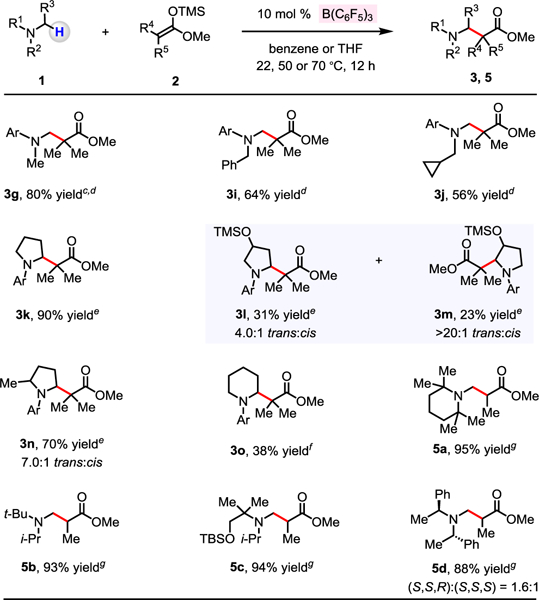
Evaluation of Different N-Alkylamines a,b
a Conditions: N,N-dialkylaniline (0.2 mmol), 1-methoxy-2-methyl-1-(trimethylsiloxy)propene (0.4 mmol), B(C6F5)3, solvent (0.5 mL), under N2, 22 °C, 12 h. b Yield of purified products. c B(C6F5)3 (5.0 mol %) was used. d Benzene (0.5 mL) was used. e THF (0.5 mL) was used. f B(C6F5)3 (10 mol %) and benzene (0.5 mL) were used and reaction was performed at 50 °C. g Benzene (0.5 mL) was used and reaction was performed at 70 °C. See the Supporting Information for details.
Next, we explored the range of silicon enolates (Scheme 3). Cyclopentyl- and cyclohexyl-substituted variants reacted with 1g to give 3p and 3q in 73% and 79% yield, respectively. The less sterically demanding ketene acetal (2b) afforded 3r in 66% yield. A broader range of ketene acetals proved to be applicable to reactions with N-arylpyrrolidine. α-Cycloalkylesters and methyl propionate could thus be installed, furnishing the corresponding 2-substituted pyrrolidine products 3s−3u in 60% to >95% yield. Nucleophilic partners derived from isopropyl isobutyrate and 3-methyldihydrofuran-2(3H)-one were found to be compatible, as indicated by efficient synthesis of 3v (90% yield) and 3w (70% yield, 1.3:1 dr). In addition to silyl ketene acetals, trimethyl((1-(methylthio)vinyl)oxy)silane could be used, as the transformation affording β-amino thioester 3x illustrates (52% yield).
Scheme 3.
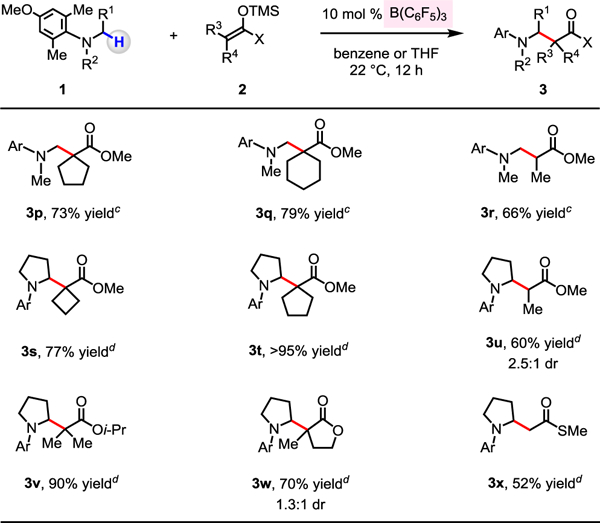
Evaluation of Various Silicon Enolates a,b
a Conditions: N,N-dialkylaniline (0.2 mmol), silicon enolate (0.4 mmol), B(C6F5)3 (10 mol %), solvent, under N2, 22 °C, 12 h. b Yield of purified products. c Benzene (0.5 mL) was used. d THF (0.5 mL) was used. See the Supporting Information for details.
The catalytic protocol is scalable, as highlighted by the 1.0 mmol synthesis of 3k, which was obtained in 95% yield by the use of 5.0 mol % B(C6F5)3 (Scheme 4A). The 4-methoxy-2,6-dimethylphenyl group of 3k could be readily removed under oxidative conditions to deliver 6k in 97% yield.13 This method is applicable in the late-stage functionalization of bioactive molecules containing an N-alkylamine moiety such as citalopram (antidepressant), atomoxetine (treatment for ADHD), and fluoxetine (antidepressant) (Scheme 4B). N-Methyl C–H bonds of these drug compounds were selectively converted to C–C bonds to afford a useful amount of β-amino carbonyl compounds 8a (23% yield, 0.30 g), 8b (25% yield, 92 mg), and 8c (27% yield, 113 mg).
Scheme 4.
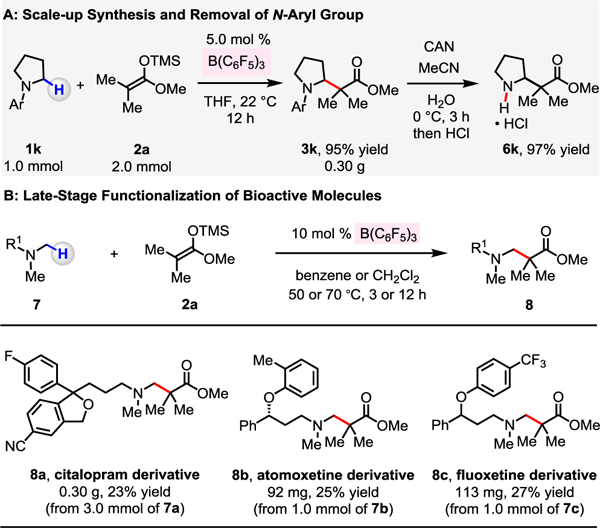
Scale-Up Synthesis and Late-Stage Functionalization of Bioactive Molecules
a See the Supporting Information for details.
In summary, we have developed a catalytic method for a B(C6F5)3-catalyzed C–C bond forming process that provides access to an assortment of β-amino carbonyl compounds. Acyclic as well as cyclic amines may be used as pro-electrophiles. On the basis of our mechanistic hypotheses, it should be possible to develop an enantioselective version of this C–C bond forming reaction through design of a chiral Lewis acid catalyst,15 and also to expand the scope of nucleophiles. Investigations along these lines are currently underway.
Supplementary Material
Acknowledgements.
We are grateful for financial support from the National Institutes of Health (GM-128695) and Boston College. We are thankful to Professor Amir H. Hoveyda (Boston College), Ms. Diana Fager (Boston College) and Mr. Richard Y. Liu (MIT) for helpful discussions.
Footnotes
The authors declare no competing financial interest.
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).For reviews on various types of Mannich-type reactions, see:; (a) Kobayashi S.; Ishitani H Catalytic Enantioselective Addition to Imines. Chem. Rev. 1999, 99, 1069–1094. [DOI] [PubMed] [Google Scholar]; (b) Córdova A The Direct Catalytic Asymmetric Mannich Reaction. Acc. Chem. Res. 2004, 37, 102–112. [DOI] [PubMed] [Google Scholar]; (c) Sodeoka M; Hamashima Y Development of Catalytic Enantioselective Reactions via Palladium Enolates as Key Intermediates. Bull. Chem. Soc. Jpn. 2005, 78, 941–956. [Google Scholar]; (d) Wenzel AG; Jacobsen EN in Enantioselective Synthesis of beta-Amino Acids, Juaristi E, Soloshonok V, Eds.; Wiley-VCH: New York, 2005; Chapter 4. [Google Scholar]; (e) Shibasaki M; Matsunaga S Metal/linked-BINOL complexes: Applications in direct catalytic asymmetric Mannich-type reactions. J. Organomet. Chem. 2006, 691, 2089–2100. [Google Scholar]; (f) Ting A; Schaus SE Organocatalytic Asymmetric Mannich Reactions: New Methodology, Catalyst Design, and Synthetic Applications. Eur. J. Org. Chem. 2007, 2007, 5797–5815. [Google Scholar]; (g) Verkade JMM; Hemert L. J. C.v..; Quaedflieg PJLM.; Rutjes FPJT Organocatalysed asymmetric Mannich Reaction. Chem. Soc. Rev. 2008, 37, 29–41. [DOI] [PubMed] [Google Scholar]; (h) Weiner B; Szymański W; Janssen DB; Minnaard AJ; Feringa BL Recent advances in the catalytic asymmetric synthesis of β-amino acids. Chem. Soc. Rev. 2010, 39, 1656–1691. [DOI] [PubMed] [Google Scholar]; (i) Karimi B; Enders D; Jafari E Recent Advances in Metal-Catalyzed Asymmetric Mannich Reactions. Synthesis 2013, 45, 2769–2812. [Google Scholar]; (j) Shirakawa S; Maruoka K Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem., Int. Ed. 2013, 52, 4312–4348. [DOI] [PubMed] [Google Scholar]
- (2).For selected examples of oxidative Mannich-type reactions, see:; (a) Catino AJ; Nichols JM; Nettles BJ; Doyle MP The Oxidative Mannich Reaction Catalyzed by Dirhodium Caprolactamate. J. Am. Chem. Soc. 2006, 128, 5648–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sureshkumar D; Sud A; Klussmann M Aerobic Oxidative Coupling of Tertiary Amines with Silyl Enolates and Ketene Acetals. Synlett 2009, 2009, 1558–1561. [Google Scholar]; (c) Chu L; Zhang X; Qing F-L CuBr-Catalyzed Oxidative Difluoromethylation of Tertiary Amines with Difluoroenol Silyl Ethers. Org. Lett. 2009, 11, 2197–2200. [DOI] [PubMed] [Google Scholar]; (d) Sud A; Sureshkumar D; Klussmann M Oxidative coupling of amines and ketones by combined vanadium- and organocatalysis. Chem. Commun. 2009, 3169–3171. [DOI] [PubMed] [Google Scholar]; (e) Rueping M; Vila C; Koenigs RM; Poscharny K; Fabry DC Dual catalysis: combining photoredox and Lewis base catalysis for direct Mannich reactions. Chem. Commun. 2011, 47, 2360–2362. [DOI] [PubMed] [Google Scholar]; (f) Boess E; Sureshkumar D; Sud A; Wirtz C; Farès C; Klussmann M Mechanistic Studies on a Cu-Catalyzed Aerobic Oxidative Coupling Reaction with N-Phenyl Tetrahydroisoquinoline: Structure of Intermediates and the Role of Methanol As a Solvent. J. Am. Chem. Soc. 2011, 133, 8106–8109. [DOI] [PubMed] [Google Scholar]; (g) Boess E; Schmitz C; Klussmann M A Comparative Mechanistic Study of Cu-Catalyzed Oxidative Coupling Reactions with N-Phenyltetrahydroisoquinoline. J. Am. Chem. Soc. 2012, 134, 5317–5325. [DOI] [PubMed] [Google Scholar]; (h) Ratnikov MO; Doyle MP Mechanistic Investigation of Oxidative Mannich Reaction with tert-Butyl Hydroperoxide. The Role of Transition Metal Salt. J. Am. Chem. Soc. 2013, 135, 1549–1557. [DOI] [PubMed] [Google Scholar]
- (3).For reviews of amine functionalization by catalytic oxidation of α-amino C–H bonds, see:; (a) Murahashi S; Zhang D. Ruthenium catalyzed biomimetic oxidation in organic synthesis inspired by cytochrome P-450. Chem. Soc. Rev. 2008, 37, 1490–1501. [DOI] [PubMed] [Google Scholar]; (b) Li C-J Cross-Dehydrogenative Coupling (CDC): Exploring C–C Bond Formations beyond Functional Group Transformations. Acc. Chem. Res. 2009, 42, 335–344. [DOI] [PubMed] [Google Scholar]; (c) Girard SA; Knauber T; Li C-J The Cross-Dehydrogenative Coupling of Csp3–H Bonds: A Versatile Strategy for C–C Bond Formations. Angew. Chem., Int. Ed. 2014, 53, 74–100. [DOI] [PubMed] [Google Scholar]
- (4).For recent advances of oxidant-free Mannich-type reactions through α-amino C–H functionalization, see:; (a) Chen W.; Seidel D The Redox-Mannich Reaction. Org. Lett. 2014, 16, 3158–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ma L; Seidel D Intramolecular Redox-Mannich Reactions: Facile Access to the Tetrahydroprotoberberine Core. Chem. Eur. J. 2015, 21, 12908–12913. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen W; Seidel D Redox-Annulation of Cyclic Amines and β‐Ketoaldehydes. Org. Lett. 2016, 18, 1024–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Chan JZ; Yao W; Hastings BT; Lok CK; Wasa M Direct Mannich-Type Reactions Promoted by Frustrated Lewis Acid/Bronsted Base Catalysts. Angew. Chem., Int. Ed. 2016, 55, 13877–13881. [DOI] [PubMed] [Google Scholar]; (b) Shang M; Cao M; Wang Q; Wasa M Enantioselective Direct Mannich-Type Reaction Catalyzed by Frustrated Lewis Acid/Brønsted Base Complexes. Angew. Chem., Int. Ed. 2017, 56, 13338–13341. [DOI] [PubMed] [Google Scholar]
- (6).For reviews of frustrated Lewis pair chemistry, see:; (a) Rokob TA.; Pápai I. Hydrogen Activation by Frustrated Lewis Pairs: Insights from Computational Studies. Top. Curr. Chem. 2013, 334, 157–211. [DOI] [PubMed] [Google Scholar]; (b) Ashley AE; O’Hare D Top. Curr. Chem. 2013, 334, 191–218. [DOI] [PubMed] [Google Scholar]; (c) Feng X; Du H Metal-free asymmetric hydrogenation and hydrosilylation catalyzed by frustrated Lewis pairs. Tetrahedron Lett. 2014, 55, 6959–6964. [Google Scholar]; (d) Stephan DW; Erker G Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem., Int. Ed. 2015, 54, 6400–6441. [DOI] [PubMed] [Google Scholar]; (e) Stephan DW Frustrated Lewis Pairs. J. Am. Chem. Soc. 2015, 137, 10018–10032. [DOI] [PubMed] [Google Scholar]; (f) Oestreich M; Hermeke J; Mohr J A unified survey of Si–H and H–H bond activation catalysed by electron-deficient boranes. Chem. Soc. Rev. 2015, 44, 2202–2220. [DOI] [PubMed] [Google Scholar]; (g) Stephan DW The broadening reach of frustrated Lewis pair chemistry. Science 2016, 354, aaf7229. [DOI] [PubMed] [Google Scholar]
- (7).For organoborane-mediated hydride abstraction from N-alkylamines, see:; (a) Millot N.; Santini CC.; Fenet B.; Basset JM. Formation and Characterization of Zwitterionic Stereoisomers from the Reaction of B(C6F5)3 and NEt2Ph: (E)- and (Z)- [EtPhN+=CHCH2–B–(C6F5)3]. Eur. J. Inorg. Chem. 2002, 3328–3335. [Google Scholar]; (b) Focante F; Mercandelli P; Sironi A; Resconi L Complexes of tris(pentafluorophenyl)boron with nitrogen-containing compounds: Synthesis, reactivity and metallocene activation. Coord. Chem. Rev. 2006, 250, 170–188. [Google Scholar]; (c) Farrell JM; Heiden ZM; Stephan DW Metal-Free Transfer Hydrogenation Catalysis by B(C6F5)3. Organometallics 2011, 30, 4497–4500. [Google Scholar]; (d) Schwendemann S; Fröhlich R; Kehr G; Erker G Intramolecular frustrated N/B lewis pairs by enamine hydroboration. Chem. Sci. 2011, 2, 1842–1849. [Google Scholar]; (e) Chen J; Chen EX-Y Reactivity of Amine/E(C6F5)3 (E = B, Al) Lewis Pairs toward Linear and Cyclic Acrylic Monomers: Hydrogenation vs. Polymerization. Molecules 2015, 20, 9575–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chen G-Q; Kehr G; Daniliuc CG; Bursch M; Grimme S; Erker G Intermolecular Redox-Neutral Amine C–H Functionalization Induced by the Strong Boron Lewis Acid B(C6F5)3 in the Frustrated Lewis Pair Regime. Chem. Eur. J. 2017, 23, 4723–4729. [DOI] [PubMed] [Google Scholar]
- (8).For B(C6F5)3-catalyzed dehydrogenation of N-heterocycles, see:; (a) Maier AFG.; Tussing S.; Schneider T.; Flörke U.; Qu A-W.; Grimme S.; Paradies J. Frustrated Lewis Pair Catalyzed Dehydrogenative Oxidation of Indolines and Other Heterocycles. Angew. Chem., Int. Ed. 2016, 55, 12219–12223. [DOI] [PubMed] [Google Scholar]; (b) Kojima M; Kanai M Tris(pentafluorophenyl)borane-catalyzed Acceptorless Dehydrogenation of N-Heterocycles. Angew. Chem., Int. Ed. 2016, 55, 12224–12227. [DOI] [PubMed] [Google Scholar]
- (9).For organoborane-catalyzed allylic C–H abstraction, see:; (a) Chatterjee I.; Oestreich M. B(C6F5)3-Catalyzed Transfer Hydrogenation of Imines and Related Heteroarenes Using Cyclohexa-1,4-dienes as a Dihydrogen Source. Angew. Chem., Int. Ed. 2015, 54, 1965–1968. [DOI] [PubMed] [Google Scholar]; (b) Chatterjee I; Qu Z-W; Grimme S; Oestreich MB (C6F5)3¬-Catalyzed Transfer of Dihydrogen from One Unsaturated Hydrocarbon to Another. Angew. Chem., Int. Ed. 2015, 54, 12158–12162. [DOI] [PubMed] [Google Scholar]; (c) Keess S; Oestreich M Cyclohexa-1,4-dienes in transition-metal-free ionic transfer processes. Chem. Sci. 2017, 8, 4688–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shang M; Chan JZ; Cao M; Chang Y; Wang Q; Cook B; Torker S; Wasa M C–H Functionalization of Amines via Alkene-Derived Nucleophiles through Cooperative Action of Chiral and Achiral Lewis Acid Catalysts: Applications in Enantioselective Synthesis. J. Am. Chem. Soc. 2018, 140, 10593–10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For reviews regarding α-amino C–H functionalization, see:; (a) Campos KR. Direct sp3 C–H bond activation adjacent to nitrogen in heterocycles. Chem. Soc. Rev. 2007, 36, 1069–1084. [DOI] [PubMed] [Google Scholar]; (b) Davies HML; Manning JR Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chiral Amine Synthesis: Methods, Developments and Applications; Nugent TC., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2010. [Google Scholar]; (d) Mitchell EA; Peschiulli A; Lefevre N; Meerpoel L; Maes BUW Direct α-functionalization of saturated cyclic amines. Chem. Eur. J. 2012, 18, 10092–10142. [DOI] [PubMed] [Google Scholar]; (e) Girard SA; Knauber T; Li C-J The cross-dehydrogenative coupling of Csp3–H bonds: A versatile strategy for C–C bond formations. Angew. Chem., Int. Ed. 2014, 53, 74–100. [DOI] [PubMed] [Google Scholar]; (f) Haibach MC; Seidel D C–H Bond functionalization through intramolecular hydride transfer. Angew. Chem., Int. Ed. 2014, 53, 5010–5036. [DOI] [PubMed] [Google Scholar]
- (12).For representative examples of α-amino C–H bond functionalization, see:; (a) Davies HML; Hansen T; Hopper DW; Panaro SA Highly Regio-, Diastereo-, and Enantioselective C–H Insertions of Methyl Aryldiazoacetates into Cyclic N-Boc-Protected Amines. Asymmetric Synthesis of Novel C2-Symmetric Amines and threo-Methylphenidate. J. Am. Chem. Soc. 1999, 121, 6509–6510. [Google Scholar]; (b) Li Z; Li C-J CuBr-Catalyzed Efficient Alkynylation of sp3 C–H Bonds Adjacent to a Nitrogen Atom. J. Am. Chem. Soc, 2004, 126, 11810–11811. [DOI] [PubMed] [Google Scholar]; (c) DiRocco DA; Rovis T Catalytic Asymmetric α-Acylation of Tertiary Amines Mediated by a Dual Catalysis Mode: N-Heterocyclic Carbene and Photoredox Catalysis. J. Am. Chem. Soc. 2012, 134, 8094–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cordier CJ; Lundgren RJ; Fu GC Enantioconvergent Cross-Coupling of Racemic Alkylmetal Reagents with Unactivated Secondary Alkyl Electrophiles: Catalytic Asymmetric Negishi α-Alkylations of N-Boc-pyrrolidine. J. Am. Chem. Soc. 2013, 135, 10946–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bergonzini G; Schindler CS; Wallentin C-J; Jacobsen EN; Stephenson CRJ Photoredox activation and anion binding catalysis in the dual catalytic enantioselective synthesis of β-amino esters. Chem. Sci. 2014, 5, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Espelt LR; McPherson IS; Wiensch EM; Yoon TP Enantioselective conjugate additions of α-amino radicals via cooperative photoredox and Lewis acid catalysis. J. Am. Chem. Soc. 2015, 137, 2452–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Osverger TJ; Rogness DC; Kohrt JT; Stepan AF; White MC Oxidative diversification of amino acids and peptides by small-molecule iron catalysis. Nature 2016, 537, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Le C; Liang Y; Evans RW; Li X; MacMillan DWC Selective sp3 C–H alkylation via polarity-match-based cross-coupling. Nature 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Jain P; Verma P; Xia G; Yu J-Q Enantioselective amine α-functionalization via palladium-catalysed C–H arylation of thioamides. Nat. Chem. 2017, 9, 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Chen W; Ma L; Paul A; Seidel D Direct α-C–H bond functionalization of unprotected cyclic amines. Nat. Chem. 2018, 10, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) McManus JB; Onuska NPR; Nicewicz DA Generation and alkylation of α-carbamyl radicals via organic photoredox catalysis. J. Am. Chem. Soc. 2018, 140, 9056–9060. [DOI] [PubMed] [Google Scholar]; (l) Ye J; Kalvet I; Schoenebeck F; Rovis T Direct α-alkylation of primary aliphatic amines enabled by CO2 and electrostatics. Nat. Chem. 2018, 10, 1037–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jurberg ID; Peng B; Wöstefeld E; Wasserloos M; Maulide N Intramolecular Redox-Triggered C–H Functionalization. Angew. Chem., Int. Ed. 2012, 51, 1950–1953. [DOI] [PubMed] [Google Scholar]
- (14).After isolation by silica gel column chromatography, 3l and 3m were treated with an aqueous solution of 4 N HCl and characterized as their corresponding non-silylated compounds. See the Supporting Information for details.
- (15).Süsse L; Hermeke J; Oestreich M The Asymmetric Piers Hydrosilylation. J. Am. Chem. Soc. 2016, 138, 6940–6943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.