

Summary
Structural maintenance of chromosomes (SMC) complexes shape the genomes of virtually all organisms but how they function remains incompletely understood. Recent studies in bacteria and eukaryotes have led to a unifying model in which these ring-shaped ATPases act along contiguous DNA segments processively enlarging DNA loops. In support of this model, single-molecule imaging experiments indicate that Saccharomyces cerevisiae condensin complexes can extrude DNA loops in an ATP hydrolysis dependent manner in vitro. Here, using time-resolved chromosome conformation capture (Hi-C) we investigate the interplay between ATPase activity of the Bacillus subtilis SMC complex and loop formation in vivo. We show that point mutants in the SMC nucleotide binding domain that impair but do not eliminate ATPase activity not only exhibit delays in de novo loop formation but also have reduced rates of processive loop enlargement. These data provide in vivo evidence that SMC complexes function as loop extruders.
Keywords: SMC, ParB, condensin, cohesin, loop extrusion, TAD
Introduction
SMC complexes are present in most eubacteria and all eukaryotes where they play central roles in a wide array of DNA transactions including the compaction and resolution of sister chromatids in mitosis and the formation of topologically associating domains (TADs) in interphase (Hirano, 2016; Uhlmann, 2016). These broadly conserved complexes are composed of two SMC subunits, a kleisin family member, and accessory factors (Haering and Gruber, 2016). SMC proteins contain a long (40–50 nm) antiparallel coiled coil. At one end of the coiled coil the N- and C-terminal domains form an ATP-binding “head” domain that resembles those found in ATP-binding cassette (ABC) transporters. The hinge domain at the other end interacts with the hinge of a second SMC protein forming a V-shaped dimer. The kleisin subunit bridges the two nucleotide binding domains generating a large tripartite ring (Fig. 1A).
Figure 1. Identification of SMC mutants with decreased ATPase activity.
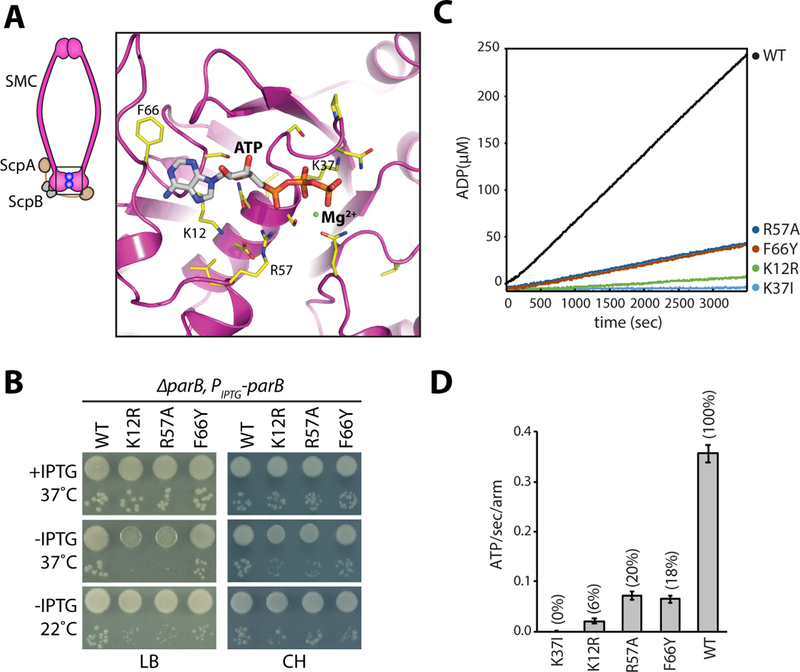
A, Homology model of the ATPase domain of B. subtilis SMC. Only one of the two composite ATP binding domains is shown. Amino acids that were mutated are indicated (yellow). A schematic of the condensin complex with its head domains (boxed) bound to ATP (blue balls) is shown on the left. B, Growth of wild-type and the relevant mutants in the presence and absence of ParB on agar plates. The 10−2 and 10−5 dilutions are shown. Under Hi-C assay conditions (22˚C no IPTG and 37˚C with IPTG in CH medium), the mutants grow similar to wild-type. C, NADH-coupled ATPase activity assay for the indicated mutants. D, Bar graph showing ATPase activities. Error bars show the standard deviation of four replicates. See also Figures S1, S2, Tables S1 and S2.
Studies in a number of organisms suggest that these ring-shaped assemblies are topologically loaded onto DNA and then redistribute along the chromosome arms (Cuylen and Haering, 2011; Ercan et al., 2009; Gligoris et al., 2014; Hu et al., 2011; Minnen et al., 2016; Schmidt et al., 2009; Wilhelm et al., 2015). ATP-dependent head engagement is critical for association of the SMC complex with its DNA loading site (Hu et al., 2011; Minnen et al., 2016), while hydrolysis is required for topological entrapment and release from this site. It has been suggested that the rings then passively slide along the DNA buffeted by RNA polymerase complexes engaged in transcription elongation (Davidson et al., 2016; Ocampo-Hafalla and Uhlmann, 2011). Alternatively, it has been proposed that cycles of ATP hydrolysis by SMC’s ATPase domains provide the motive force for DNA transport (Alipour and Marko, 2012; Minnen et al., 2016). This translocation activity has been invoked to explain how SMC complexes structure genomes. In this model, tethered rings (or a single ring embracing a small loop) load onto DNA and translocate away from this site processively enlarging a DNA loop (Alipour and Marko, 2012; Dekker and Mirny, 2016; Fudenberg et al., 2016; Goloborodko et al., 2016; Kimura et al., 1999; Nasmyth, 2001; Naumova et al., 2013; Nichols and Corces, 2015; Sanborn et al., 2015). Loop extrusion by SMC complexes loaded along chromosome arms provides a simple solution for how condensin complexes linearly compact and resolve sister chromatids in mitosis and how cohesin complexes, aided by the insulator protein CTCF, generate TADs during interphase.
Recent single-molecule experiments using purified condensin complexes from Saccharomyces cerevisiae provide biochemical support for the loop-extrusion model (Eeftens et al., 2017; Ganji et al., 2018; Keenholtz et al., 2017; Terakawa et al., 2017). These studies demonstrated that condensin complexes can translocate along DNA and generate DNA loops in an ATP hydrolysis dependent manner. It has also recently been reported that expression of Walker A and Walker B Smc3 mutants in mouse lymphoid cells reduced but did not eliminate mutant cohesin complexes at CTCF anchoring sites (Vian et al., 2018). These results were interpreted as indicative of ATP-dependent translocation in vivo with residual ATPase activity of the mutant complexes leading to reduced DNA translocation.
Here, using a new assay to monitor SMC dynamics (Wang et al., 2017), we sought to rigorously investigate whether the ATPase activity of the SMC complex from the bacterium Bacillus subtilis powers DNA transport in vivo. The B. subtilis SMC condensin complex is composed of an SMC homodimer, a kleisin subunit ScpA, and the kite family accessory protein ScpB (Palecek and Gruber, 2015). This ring-shaped complex is similar in size, architecture, and inter-subunit contacts to its eukaryotic counterparts (Burmann et al., 2013; Diebold-Durand et al., 2017; Hirano, 2016; Kamada et al., 2013). The B. subtilis SMC complex is recruited to the origin of replication by the broadly conserved partitioning protein ParB bound to centromeric parS sites (Gruber and Errington, 2009; Sullivan et al., 2009). ATP binding by SMC is required for its interaction with the ParB-parS nucleoprotein complex and ATP hydrolysis is necessary for topological entrapment and release from the parS site (Minnen et al., 2016; Wilhelm et al., 2015). Once loaded, time-resolved ChIP-seq and Hi-C experiments indicate that SMC complexes travel ~2 Mb to the replication terminus at rates >50 kb/min while tethering the left and right chromosome arms together (Fig. S1A) (Marbouty et al., 2015; Wang et al., 2017; Wang et al., 2015). Directed movement is independent of transcription and replication (Wang et al., 2017; Wang et al., 2015) indicating that another mechanism is responsible for active transport.
Here we show that point mutants in the SMC head domain that impair but do not eliminate ATPase activity not only exhibit delays in the initiation of DNA juxtaposition but also have reduced rates of chromosome arm alignment, providing in vivo evidence that ATPase activity is required for both SMC complex loading (Minnen et al., 2016) and DNA translocation.
Results
We modeled the SMC nucleotide binding domain from B. subtilis using the crystal structure of the SMC head domain from Pyrococcus furiosus bound to ATP (Lammens et al., 2004) and the apo-structure from B. subtilis (Burmann et al., 2013) and identified 17 residues that were predicted to help position or stabilize the amino acids involved in ATP binding and hydrolysis (Fig. 1A, Fig. S1B). Amino acid substitutions were introduced into the endogenous smc gene in B. subtilis and their impact on function was assessed based on colony size in the presence and absence of the ParB loader (Fig. S1C). Synthetic interactions between hypomorphic smc alleles and null mutations in parB have been reported previously (Gruber and Errington, 2009; Wang et al., 2014).
Among the 21 mutants tested, 7 phenocopied the smc null and could only form colonies at 22˚C on LB medium in the presence of ParB and were inviable in its absence (Fig. S1C, D). Among these mutants were G1092S in the signature motif and D1124E in the D-loop. These mutations, when introduced into S. cerevisiae smc1 in the presence of wild-type smc3 were found to be hypomorphic, impairing distinct activities of the SMC cohesin complex (Camdere et al., 2015; Elbatsh et al., 2016). The stronger phenotype observed here is likely due to the fact that B. subtilis SMC forms a homodimer and therefore both subunits in the complex contain these amino acid substitutions. Four of the SMC mutants (including Q143A) had intermediate phenotypes. These grew slower than wild-type in the presence of ParB at 37˚C and failed to grow in its absence (Fig. S1C, E). The remaining 11 mutants formed colonies in the presence of ParB that were indistinguishable from wild-type. In the absence of ParB, four of these (K12R, D42A, R57A, and R57K) were unable to form colonies; one (F66Y) had a small-colony phenotype; and five (including D42E) resembled wild-type (Fig. 1B, Fig. S1C, E). When grown on defined rich (Casein Hydrolysate, CH) medium at 37˚C, SMC(K12R), SMC(R57A), and SMC(F66Y) mutants lacking ParB formed smaller colonies than the matched wild-type control (Fig. 1B).
We purified 9 mutants that encompassed the full range of phenotypes described above, including a canonical Walker A motif mutant (K37I) that does not bind ATP (Hirano et al., 2001), and wild-type B. subtilis SMC from E. coli as SMC-His6 fusions (Fig. S2). As anticipated, the mutants that phenocopied the smc null (K37I in the Walker A motif, G1092S in the signature motif, D1124E in the D-loop, and N33A) (Fig. S1C, D) had virtually undetectable ATPase activity in vitro (Fig. S2B, C). One of the mutants that grew slowly in the presence of ParB (Q143A) (Fig. S1C, E) had <1% of the ATPase activity of wild-type. Finally, three of the hypomorphic mutants (K12R, R57A, and F66Y) with relatively mild growth defects in the absence of ParB had reduced ATPase activity ranging from 6–20% of wild-type (Fig. 1C, D and S2). A previous study found a similar reduction in ATPase activity for the R57A mutant (Hirano and Hirano, 2006). Similar reductions were observed in the presence of single and double stranded DNA (Fig. S2E) (Hirano and Hirano, 2006).
To investigate whether a decrease in condensin’s ATPase activity impacts DNA translocation and loop formation, we analyzed the rate of DNA juxtaposition by time-resolved Hi-C. In this assay, strains harbor an IPTG-inducible allele of the condensin loader ParB and a single origin-proximal parS site. Upon addition of IPTG, the extent of juxtaposition of the left and right arm is monitored in a Hi-C time course. Importantly, our previous ChIP-seq analysis indicates that all three condensin subunits are specifically enriched along the juxtaposed region. Furthermore, time-resolved ChIP-seq experiments using an ectopic parS site inserted at −94˚ indicate that SMC travels along the chromosome arms at rates comparable to DNA juxtaposition (Wang et al., 2017). Here, we use rates of juxtaposition as a proxy for condensin movement because when condensin is loaded at the origin, ChIP-seq does not provide sufficient resolution of SMC enrichment for rate determination (Wang et al., 2017). Wild-type SMC and the three mild hypomorphic mutant strains were grown in defined rich (CH) medium in the absence of inducer at 22˚C to ensure similar growth rates (Fig. 1B). Cultures were then treated with IPTG, shifted to 37˚C, and samples were collected for Hi-C at 5- or 10-minute intervals. Importantly, upon addition of IPTG all four strains grew at indistinguishable rates (τ=34 min) and had similar levels of SMC, ScpA, ScpB, and ParB (Fig. 2D). As observed previously (Wang et al., 2017), in cells with wild-type SMC, 20 minutes after ParB induction, ~685 kb of DNA on either side of the origin-proximal parS site were juxtaposed (Fig. 2A). 5 minutes later, ~930 kb were aligned. Analysis of the complete Hi-C time course revealed that the left and right chromosome arms zipped up at a rate of 50±2 kb/min (Fig. 2C, Fig. S3).
Figure 2. SMC ATPase mutants have reduced rates of DNA juxtaposition.

A, Normalized Hi-C interaction maps displaying contact frequencies for pairs of 10 kb bins. The strain (BWX4077) contains wild-type smc, a single parS site at −1˚ and an IPTG-inducible parB allele. Maps show interaction frequencies before and after addition of IPTG. Time after induction is indicated above the maps. Axes present genome positions in degrees and are oriented with the replication origin at the center. The rate of DNA juxtaposition is indicated on the right. See Figure S3 for analysis. The scale bar depicts Hi-C interaction scores for all contact maps presented in this study. B, Hi-C time course of strains containing smc point mutations, K12R (BWX4149), R57A (BWX4078) and F66Y (BWX4152). C, Extent of juxtaposition over time averaged for the left and right arms in the indicated strains. Extrapolation to the abscissa shows the relative lag before juxtaposition begins. See Figure S3 for details. D, Immunoblot analysis of the same strains presented in A and B in the presence of IPTG. The levels of SMC variants, ScpA, ScpB and ParB are similar in all four strains. For the ScpA blot, the bottom band is ScpA and the top band is non-specific. SigA controls for loading. See also Figures S3, S4, S5 and Table S1.
Consistent with the idea ATP hydrolysis is required for topological loading of the condensin ring (Wilhelm et al., 2015), all three SMC mutants displayed a lag prior to the appearance of juxtaposed DNA flanking the origin (Fig. 2B, C, Fig. S3). The mild SMC(F66Y) mutant had a 4-minute delay compared to wild-type while the stronger mutants (K12R and R57A) were delayed by ~10 minutes. Importantly, after the lag, the left and right arms processively zipped up in all three mutants. Analysis of the rates of DNA juxtaposition revealed that the mild F66Y mutant zipped up the arms at 41±2 kb/min, while K12R and R57A had rates of 30±3 and 33±1 kb/min, respectively. These rates of DNA juxtaposition are 18–40% slower than wild-type and were reproducible in an independent Hi-C time course (Fig. S4). The reduced rates of condensin-mediated juxtaposition did not directly correlate with the decrease in ATPase activity of the SMC mutants and we suspect that this is due to the limitations of the in vitro assays available for the B. subtilis SMC complexes (Hirano and Hirano, 2004). However, we note that the lag times in the initiation of juxtaposition, which likely reflect the requirement for ATP hydrolysis for topological loading in vivo, correlated with the slower rates of movement down the arms.
Importantly, the SMC mutant (Q143A) that had a more severe growth defect (Fig. S1C, E) and <1% of the ATPase activity of wild-type (Fig. S2B, C) failed to juxtapose the chromosome arms even after 40 min of ParB induction (Fig. S5C). By contrast, one of the mutants (D42E) with no discernable growth phenotype (Fig. S1C, E) and 82% of the ATPase activity of wild-type (Fig. S2B, C) zipped up the two arms in a manner similar to wild-type (Fig. S5C). Finally, a strain in which SMC itself was under IPTG-inducible control juxtaposed the left and right arms without a lag and at rates similar to wild-type upon addition of inducer (Fig. S5A, B). That the juxtaposition was similar to wild-type in this strain indicates that any DNA entanglements that might have arisen during growth prior to SMC induction in this strain and prior to ParB induction in SMC mutants studied here (Gruber et al., 2014; Wang et al., 2014) did not impair condensin movement after their production.
Discussion
These data extend the analysis of mammalian cohesin in lymphoid cells (Vian et al., 2018) and complement the single-molecule studies of the S. cerevisiae condensin complex (Eeftens et al., 2017; Ganji et al., 2018; Keenholtz et al., 2017; Terakawa et al., 2017) providing in vivo support for the model that SMC complexes translocate along chromosomes powered by ATP hydrolysis and could therefore structure genomes by loop extrusion. Intriguingly, the in vitro rate of DNA extrusion determined for S. cerevisiae condensin (up to 1.5 kb/sec) (Ganji et al., 2018) is similar to the rate of loop formation we observe in vivo (1.67 kb/sec, 0.83 kb/sec per chromosome arm). Furthermore, the ensemble rates of ATP hydrolysis for B. subtilis SMC and the S. cerevisiae condensin complex are within a factor of two of each other. Thus, these rates may represent a biological optimum for loop extrusion efficiency.
While we attribute the reduced translocation rates of the SMC hypomorphic mutants to their decreased ATPase activity, other possibilities could account for our findings. For example, it is possible that the mutations affected some other aspect of the complex that hindered its movement along the chromosome arms. We consider this unlikely because we obtained similar results with three distinct mutations in the nucleotide binding domain. It is also possible that the mutants affected the on- or off-rates of the complex on DNA. While formally possible, we note that previous studies (Tran et al., 2017; Wang et al., 2017) indicate that condensin-dependent DNA juxtaposition initiates at parS suggesting that productive loading at other positions along the chromosome is negligible. An alternative model is that the number of SMC complexes loaded onto the DNA influences the rate of DNA translocation (Terakawa et al., 2017), and mutations that reduce the rate of loading and therefore the number of SMC complexes indirectly affect the rate of DNA translocation. This model predicts that as more and more condensin complexes are loaded onto the chromosome the rate of DNA transport will increase. However, we observed that the rates of DNA juxtaposition were constant as a function of time in all strains tested despite continuous loading of condensins at the parS site and their increasing numbers on the DNA (Wang et al., 2017). These considerations lead us to conclude that our data are most consistent with a model in which cycles of ATP hydrolysis by the SMC head domains are required for DNA transport (Badrinarayanan et al., 2012).
Structural and biophysical studies on the B. subtilis condensin indicate that the complex undergoes a large structural transition from a ring- to rod-like conformation during its ATPase cycle (Soh et al., 2015). More recent studies have led to the proposal that DNA translocation is mediated by stepwise loop formation involving a peristalsis-like mechanism in which two regions of DNA along the duplex separately interact with the head and hinge of the (ATP-bound) ring-shaped complex (Burmann et al., 2017; Diebold-Durand et al., 2017). The ring-to-rod transition triggered by ATP hydrolysis and nucleotide release drives the DNA bound at the hinge toward the head. The two regions of DNA that constitute the base of a loop are then captured in a meta-chamber created by the kleisin subunit and the SMC head domains (Kschonsak et al., 2017). The re-binding of ATP leads to ring opening and capture of a new chromosomal loop, likely facilitated by conformational fluctuations of the DNA (Lawrimore et al., 2017; Marko et al., 2018). ATP hydrolysis drives this second captured loop into the meta-chamber where it merges with one held in this compartment (Marko et al., 2018). In this way cycles of ATP hydrolysis expand the loop in a stepwise manner. Consistent with this model, the in vitro single-molecule studies on S. cerevisiae condensin have shown that the complex can extrude a loop by anchoring DNA and reeling the duplex in from one side (Ganji et al., 2018). We note that studies in B. subtilis and C. crescentus (Tran et al., 2017; Wang et al., 2017) indicate that bacterial condensin complexes loaded at the origin generate a symmetric loop as they translocate down the chromosome arms. These data raise the possibility that paired SMC complexes might be the functional unit for loop extrusion (Diebold-Durand et al., 2017). Future studies in vitro and in vivo will be required to establish how these novel DNA translocases travel along the chromosome while accommodating all the DNA transactions encountered along their paths.
STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-SMC polyclonal rabbit antibody | (Lindow et al., 2002) | N/A |
| Anti-ParB polyclonal rabbit antibody | (Lin et al., 1997) | N/A |
| Anti-ScpA polyclonal rabbit antibody, affinity purified | (Wang et al., 2017) | N/A |
| Anti-ScpA polyclonal rabbit antibody, affinity purified | (Wang et al., 2017) | N/A |
| Anti-SigA polyclonal rabbit antibody | (Fujita, 2000) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Benzonase | EMD | Cat # 7046 |
| Ni-NTA agarose | Qiagen | Cat # 30230 |
| HiTrap Q HP | GE Healthcare | Cat # 17115301 |
| Bradford Assay Kit | ThermoFisher | Cat # 23236 |
| ATP | Dot Scientific | Cat # DSA30030–5 |
| NADH | Acros Organic | Cat # 271100010 |
| Phosphoenolypyruvate | Tokyo Chemical Industry | Cat # P0256 |
| Rabbit Pyruvate Kinase | Roche Diagnostics | Cat # 10109045001 |
| Lactate Dehydrogenase | Sigma, Darmstadt, Germany | Cat # L2500–25KU |
| Formaldehyde 37% | Sigma | Cat # F8775 |
| Ready-Lyse Lysozyme | Epicentre | Cat # R1802M |
| HindIII | NEB | Cat # R0104M |
| Klenow | NEB | Cat # M0210L |
| Biotin-14-dATP | ThermoFisher | Cat # 19524016 |
| T4 DNA ligase | NEB | Cat # M0202M |
| Proteinase K | NEB | Cat # P8107S |
| T4 DNA Polymerase | NEB | Cat # M0203L |
| Critical Commercial Assays | ||
| ATPase/GTPase Activity Assay Kit | Sigma | Cat # MAK113 |
| NEBNext Ultra II DNA Library Prep Kit | NEB | Cat # E7645S |
| Deposited Data | ||
| Raw and analyzed Hi-C data | This paper | GEO: GSE95137 |
| Original Data | Mendeley Data | http://dx.doi.org/10.17632/tygwp234gr.1 |
| Experimental Models: Organisms/Strains | ||
| Bacillus subtilis strains, see Table S1 | ||
| Oligonucleotides | ||
| See Table S3 | ||
| Recombinant DNA | ||
| See Table S2 | ||
| Software and Algorithms | ||
| MATLAB 8.5 (R2015) | (Wang et al., 2017) | https://www.mathworks.com/ |
| hiclib | (Imakaev et al., 2012) | https://bitbucket.org/mirnylab/hiclib |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, David Rudner (rudner@hms.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacillus subtilis Strains and Growth
Bacillus subtilis strains were derived from the prototrophic strain PY79 (Youngman et al., 1983). Strains, plasmids and oligonucleotides are listed in Tables S1–S3. For the Hi-C time courses, cells were grown overnight (for 12–14 h) in defined rich (Casein Hydrolysate, CH) medium (Harwood and Cutting, 1990) in the absence of IPTG at 22˚C. The following morning, logarithmically growing cultures were diluted into fresh CH medium to an OD600 of 0.075 and incubated with aeration at 22˚C for ~2 h until an OD600 reached 0.15. IPTG was added to a final concentration of 1 mM and the flasks were placed at 37˚C. Samples were taken before or after induction at specified time points.
METHOD DETAILS
Homology Modeling
A structure of B. subtilis SMC has been reported (PDB ID 3ZGX)(Burmann et al., 2013), but this structure lacks a bound nucleotide and is poorly ordered in the ATPase domain, offering limited guidance for the design of mutants. However, high resolution structural data are available for Pyrococcus furiosus SMC in complex with ATP (PDB ID 1XEX) (Lammens et al., 2004). To enable rational design of mutants in the B. subtilis protein, we employed a multiple template homology modeling method using the two structures. Briefly, the two template structures were aligned to each other by secondary structure matching in Coot (Emsley and Cowtan, 2004). A sequence alignment was constructed on the basis of the aligned structures and was used for multi-template modeling in MODELLER (Webb and Sali, 2016). Residues 199 – 1195 are located far from the ATPase domain, and were omitted from the modeling. A bound ATP and Mg2+ ion were included during modeling as rigid bodies, positioned based on the P. furiosus SMC structure. A total of 20 models were created and the top scoring model was used as the basis for the design of mutagenesis experiments and the preparation of Figure 1A and Figure S1B.
Protein Purification
SMC-His6 wild-type and mutants were expressed in E. coli BL21 DE3 pLysS. Cells were grown in LB at 37˚C to an OD600 of 0.4 and induced with 0.5 mM IPTG and harvested after 2 hours. All subsequent manipulations were carried out at 4˚C. 250 ml of cells were harvested by centrifugation and resuspended in 20 ml lysis buffer (50 mM Hepes Na+ pH 7.6, 75 mM NaCl). A crude extract was prepared by freeze-thawing the cells followed by the addition of 5 mM β-Mercaptoethanol, 1 mM PMSF and 1 mM MgCl2, 28 U/ml Benzonase (EMD 7046) and Lysozyme (0.5 mg/ml), and then sonication. The lysates were clarified at 100,000 g for 1 hour. To the soluble fraction, NaCl and Imidazole were added to achieve a final concentration of 300 mM and 10 mM respectively. The soluble fraction was then loaded onto a 3 ml Ni2+-NTA agarose (Qiagen) column equilibrated with Buffer I (50 mM Hepes Na+ pH 7.6, 300 mM NaCl, 5 mM β-Mercaptoethanol, 10mM Imidazole). Bound proteins were washed with Buffer II (20 mM Hepes Na+ pH 7.6, 300 mM NaCl, 5 mM β-Mercaptoethanol, 50 mM Imidazole, 10% glycerol) followed by elution in Buffer II containing 500 mM Imidazole. Peak fractions were pooled and dialyzed with three changes into Buffer A (20 mM Hepes K+ pH7.6, 50 mM KCl, 1mM EDTA, 10% glycerol, 5 mM β-Mercaptoethanol). The dialysate was loaded onto 2 tandem 1ml HiTrap Q column on a FPLC (AKTA, GE Healthcare Life Sciences), washed with Buffer A and eluted with a linear KCl gradient. Peak fractions were pooled, dialyzed into Buffer A, aliquoted and flash frozen in liquid nitrogen. Protein concentrations were determined using the Bradford reagent from Thermo Scientific.
ATPase Activity Assays
Steady-state ATPase activity was monitored using the NADH-coupled assay (De La Cruz et al., 2000; Hass et al., 1961; Imamura et al., 1966; Trentham et al., 1972). Purified SMC wild-type and mutant proteins were diluted to 400 nM in reaction buffer (20 mM Hepes K+ pH7.6, 50 mM KCl, 1mM EDTA, 10% glycerol, 5 mM β-Mercaptoethanol). In a separate tube, a 2x NADH cocktail was prepared, which contained reaction buffer, 2 mM ATP, 4 mM MgCl2, 0.8 mM NADH (Acros Organic), 1 mM phosphoenolypyruvate (Tokyo Chemical Industry), 10 U/mL rabbit pyruvate kinase (Roche Diagnostics), 16 U/mL lactate dehydrogenase (Sigma, Darmstadt, Germany). 40 μL of the diluted proteins were mixed with 40 μL of 2x NADH cocktail. After mixing, 30 μL of each reaction was transferred into a 384-well plate in duplicate. The plate was pulse centrifuged at 1000 g and placed in a microplate spectrophotometer (BioTek, Winooski, VT) where NADH oxidation was monitored at 340 nm over time. The amount of NADH oxidized (equivalent to the amount of ADP produced) was calculated via a NADH standard curve. ADP product formation was plotted over time and fitted to a line. The obtained rates were divided by the final SMC concentration. The experiment was performed four times using aliquots of proteins from the same protein preparation.
For comparison, a malachite green assay was used for a subset of the mutants. SMC (WT), SMC (K37I), SMC (K12R), SMC (R57A) and SMC (F66Y), were separately purified and subjected to a malachite green assay using Sigma-Aldrich ATPase/GTPase Activity Assay Kit (MAK113), in a buffer containing 20 mM Hepes K+ pH 7.6, 50 mM KCl, 2 mM MgCl2 and 1 mM ATP. The assays were performed at 37°C for 30 min in triplicate. To ensure the reactions were in the linear range of the assay, 50 nM wild-type SMC, 200 nM SMC (R57A) and SMC (F66Y), and 400 nM SMC (K12R) and SMC (K37I) were used. The ATPase rates were divided by the protein concentration. For the 5 mutants tested, the ATPase rates measured using NADH-coupled assay were ~25% lower than using the malachite green assay (Figure S2C, D). However and importantly, the relative rates of the mutants compared to the wild-type were very similar between the two assays.
Hi-C
The detailed Hi-C procedure was previously described in Wang et al, 2015 (Wang et al., 2015). Briefly, cells were crosslinked with 3% formaldehyde at room temperature for 30 min, quenched with 125 mM glycine for 5 min. 5×107 cells were used for each Hi-C reaction. Cells were lysed using Ready-Lyse Lysozyme (Epicentre, R1802M) followed by 0.5% SDS treatment. Solubilized chromatin was digested with HindIII in the presence of 1% Triton for 2 h at 37˚C. The cleaved ends were filled in with Klenow and Biotin-14-dATP, dGTP, dCTP, dTTP. The products were then ligated with T4 DNA ligase overnight at 16˚C. Crosslinking was reversed at 65˚C overnight in the presence proteinase K. The DNA was then extracted twice with phenol/chloroform/isoamylalcohol (25:24:1) (PCI), precipitated with ethanol, and resuspended in 20 μL of QIAGEN EB buffer. Biotin from non-ligated ends was removed using T4 polymerase (4 h at 20˚C) followed by extraction with PCI. The DNA was then sheared by sonication in 10s on - 10s off cycles for 12 min with 60% amplitude using a Qsonica Q800 water bath sonicator. The sheared DNA was used for library preparation with the NEBNext Ultra II DNA Library Prep kit (E7645S) according to the manufacturer’s instructions for end repair, adapter ligation, and size selection. Biotinylated DNA fragments were purified using 10 μL streptavidin beads. 5 μL DNA-bound beads were used for PCR in a 50 μL reaction for 14 cycles. PCR products were purified using Ampure beads and sequenced using Illumina NextSeq 500.
Generation of Hi-C contact maps
Paired-end sequencing reads were mapped to the genome of B. subtilis PY79 (NCBI Reference Sequence NC_022898.1) using the same pipeline described previously (Wang et al., 2017; Wang et al., 2015). The B. subtilis PY79 genome was first divided into 404 10-kb bins. The interaction matrices were generated, and normalized using an iterative normalization procedure, implemented using the hiclib library for Python (https://bitbucket.org/mirnylab/hiclib) (Imakaev et al., 2012; Le et al., 2013; Wang et al., 2015). Subsequent analysis and visualization were done using R scripts or MATLAB 8.5 (R2015) (MathWorks, Natick, MA). For presentation, the circular genome was linearized at the replication terminus to generate origin-centered contact maps.
Mapping endpoints of DNA juxtaposition on Hi-C maps
The endpoints of DNA juxtaposition on Hi-C maps (Fig. 2A, B) were determined as in Wang et al., 2017 (Wang et al., 2017). Essentially, a three-step process was used. First, the mean and standard deviation (σ) of contact probabilities for each Hi-C map were computed. To minimize biases to the estimates of the mean and standard deviation (σ) from contributions arising from chromosome features like condensin-dependent interactions, we used robust statistics: as an estimate of the mean, we calculated the median; and as an estimate of the standard deviation, we calculated the median absolute deviation and multiplied it by 1.4826 (Rousseeuw and Croux, 1993). The contact probabilities equal to 0.2x5x, 0.50x, 0.75x, 1.00x, or 1.25x standard deviations (σ) above the mean were set as the threshold values. Next, interaction probabilities above or below the threshold were assigned a value of 1 or 0 respectively, generating an enrichment map with a binary profile showing points with a Hi-C score above the specified threshold (Figure S3, lower panels points in light green and yellow). Lastly, a point-connecting algorithm was employed to identify the largest contiguous region of enrichment (Figure S3, lower panels, highlighted in yellow) after applying a mask of 30–35 pixels (300–350 kb) to remove contributions arising from the primary diagonal that represents short-range interactions along the chromosome arms.
Lastly, to avoid missing entire sections of connected segments, especially at higher threshold values, we chose to connect neighbors separated by as many as 15 unconnected bins. This was performed by the “imclose()” function in MATLAB 8.5 (R2015) using a diamond structuring element. From this contiguous region, the coordinates of the maximum excursion away from the parS site were obtained (white dotted lines), and labeled on the Hi-C contact maps (Figure S3, upper panels, blue dotted lines). For consistency throughout the study, the measurements using a threshold of 0.50xσ (0.50 standard deviation above the mean) were reported in the main text.
Calculating rates of DNA juxtaposition
The rates of DNA juxtaposition were calculated from a linear fit to the position-versus-time plots, where position was the endpoint of DNA juxtaposition determined as described above. The reported rates and their errors were calculated from the slope of the line-of-best-fit and the standard error of the regression, respectively. To more accurately determine the rates, the early time points when the juxtaposition endpoints were close to the primary diagonal and late time points when the two arms appear fully juxtaposed were omitted from the analysis (Figure S3).
Immunoblot analysis
Immunoblot analysis was performed as described previously (Wang et al., 2015). Cells were collected and resuspended in lysis buffer (20 mM Tris pH 7.0, 1 mM EDTA, 10 mM MgCl2, 1 mg/ml lysozyme, 10 μg/ml DNase I, 100 μg/ml RNase A, with protease inhibitors: 1 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml pepstatin) to a final OD600 of 10 for equivalent loading. The cells were incubated at 37˚C for 10 min followed by the addition of an equal volume of sodium dodecyl sulfate (SDS) sample buffer (0.25 M Tris pH 6.8, 4% SDS, 20% glycerol, 10 mM EDTA) containing 10% β-Mercaptoethanol. Samples were heated for 5 min at 80˚C prior to loading. Proteins were separated by SDS-PAGE on 10% (SMC and SigA) or 15% (ParB, ScpA, and ScpB) polyacrylamide gels, electroblotted onto Immobilon-P membranes (Millipore) and blocked in 5% nonfat milk in phosphate-buffered saline (PBS) with 0.5% Tween 20. The blocked membranes were probed with anti-ParB (1:5,000) (Lin et al., 1997), anti-SMC (1:5,000) (Lindow et al., 2002), anti-SigA (1:10,000) (Fujita, 2000), anti-ScpA (1:10,000) (Wang et al., 2017), or anti-ScpB (1:10,000) (Wang et al., 2017) diluted into 3% BSA in 1x PBS with 0.05% Tween 20. Primary antibodies were detected using horseradish peroxidase-conjugated goat anti-rabbit IgG (BioRad) and Western Lightening Plus chemiluminescence reagent as described by the manufacturer (Perkin Elmer). The signal was captured using Bio-Rad ChemiDoc XRS.
Plasmid construction
pWX722 [yvbJ::Pspank (optRBS) spo0J (ΔparS) (cat)] was constructed by inserting spo0J (ΔparS) containing an optimized ribosomal binding site (optRBS) (amplified from pWX599 (Wang et al., 2015) using oWX1668 and oWX999 and digested with XmaI and NheI) into pER134 between XmaI and NheI. pER134 [yvbJ::Pspank (cat)] is an ectopic integration vector with an IPTG-inducible promoter (D.Z.R. unpublished).
pWX740 [smc(K12R)-(his)6 (kan)], pWX741 [smc(R57A)-(his)6 (kan)], pWX742 [smc(F66Y)-(his)6 (kan)], pWX743 [smc(K37I)-(his)6 (kan)], pWX758 [smc(N33A)-(his)6 (kan)], pWX759 [smc(D42E)-(his)6 (kan)], pWX760 [smc(Q143A)-(his)6 (kan)], pWX762 [smc(G1092S)-(his)6 (kan)], pWX763 [smc(D1124E)-(his)6 (kan)] were generated using site-directed mutagenesis from template pKM309 [smc-(his)6 (kan)] (Sullivan et al., 2009), with oligos oWX1732 and 1733 (for pWX740), oWX1736 and 1737 (for pWX741), oWX1738 and 1739 (for pWX742), oWX1744 and 1745 (for pWX743), oWX1625 and 1626 (for pWX758), oWX1666 and 1667 (for pWX759), oWX1634 and 1635 (for pWX760), oWX1792 and 1793 (for pWX762), oWX1794 and 1795 (for pWX763), . The smc gene was sequenced using oWX848, oWX1194, oWX1195, oWX1196, oWX1746 and oWX1747.
Strain construction
BWX3976: smc (WT) loxP-kan-loxP.
In the wild-type cells, a loxP-flanked kan cassette was inserted upstream of the smc gene using isothermal assembly. The isothermal assembly reaction contained 3 PCR products: 1) a region upstream of smc (amplified from wild-type genomic DNA using oWX1620 and oWX1621); 2) loxP-kan-loxP cassette (amplified from pWX470 using universal primers oWX438 and oWX439) and 3) a region containing the 5’ end of smc (amplified from wild-type genomic DNA using primers oWX1622 and oWX822). The insertion was verified using oWX1623 and oWX1624. The smc gene was sequenced using oWX523, oWX848, oWX1194, oWX1195 and oWX1196 and oWX1624. The genomic DNA of this strain (BWX3976) was transformed into BWX4070, which harbors a single parS at −1˚ and an IPTG-inducible parB, to build BWX4077.
BWX3990: smc (R57A) loxP-kan-loxP.
The smc(R57A) mutation was constructed by transforming the product of an isothermal assembly reaction into wild-type cells. The isothermal assembly reaction contained 2 PCR products: 1) a region upstream of smc and loxP-kan-loxP cassette (amplified from BWX3976 genomic DNA using oWX1620 and oWX1631) and 2) a downstream region (amplified from BWX3976 using oWX1633 and oWX822). The smc gene was sequenced using oWX523, oWX848, oWX1194, oWX1195 and oWX1196 and oWX1624. The genomic DNA of this strain (BWX3990) was transformed into BWX4070, which harbors a single parS at −1˚ and an IPTG-inducible parB, to build BWX4078. The smc (R57A) mutation in the final strain was sequence-confirmed using oWX1624.
BWX4129: smc (K12R) loxP-kan-loxP.
The smc(K12R) mutation was constructed and verified using the same method, except that the 2 PCR products used for isothermal assembly were: 1) the region upstream of smc and loxP-kan-loxP cassette (amplified from BWX3976 genomic DNA using oWX1620 and oWX1696) and 2) the downstream region (amplified from BWX3976 using oWX1661 and oWX822).
BWX4137: smc (F66Y) loxP-kan-loxP.
The smc(F66Y) mutation was constructed and verified using the same method, except that the 2 PCR products used for isothermal assembly were: 1) the region upstream of smc and loxP-kan-loxP cassette (amplified from BWX3976 genomic DNA using oWX1620 and oWX1702) and 2) the downstream region (amplified from BWX3976 using oWX1703 and oWX822).
Quantification and Statistical Analysis
For the NADH-coupled ATPase assay, absorbance at 340 nm was measured every 10 seconds for 60 min. The extent of NADH oxidized (equivalent to the amount of ADP produced) was calculated using an NADH standard curve. ADP product formation was plotted over time and fitted to a line. The obtained rates were divided by the input SMC concentration. The experiment was performed four times using aliquots of proteins from the same purification. The data were averaged and the standard deviation was calculated.
For the malachite green ATPase assay, absorbance at 620 nm was measured 30 min after the start of the reaction. The experiment was performed three times using aliquots of proteins from the same purification. The data were averaged and the standard deviation was calculated.
Supplementary Material
Acknowledgements
Hi-C data were deposited in the Gene Expression Omnibus (accession no. GSE95137). We thank members of the Wang, Rudner and Bernhardt labs and J. Loparo for stimulating discussions; S. Zheng for help with FPLC purification; A. Grossman for antibodies; M. Greenberg for generously sharing the NextSeq and A. Alegrado for help with sample loading. Support for this work comes from the National Institute of Health grants GM086466 and GM073831 (D.Z.R.), start-up funds from Indiana University (X.W.), the Vallee Foundation (A.C.K.), the National Science Foundation grants CHE1412499 (M.G.O.) and MCB1614514 (J.C.C.), and a Natural Sciences and Engineering Research Council of Canada Fellowship (H.B.B).
Footnotes
Supplemental Information
Supplemental information includes five figures and three tables and can be found with this article online.
Declaration of Interests
The authors declare no competing interests.
Data and Software Availability
Protein gels and immunoblot analyses are available at Mendeley data http://dx.doi.org/10.17632/tygwp234gr.1. The accession number for Hi-C data (raw and analyzed) is GEO: GSE95137. The scripts for the Hi-C analyses are available upon request.
References
- Alipour E, and Marko JF (2012). Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res 40, 11202–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badrinarayanan A, Reyes-Lamothe R, Uphoff S, Leake MC, and Sherratt DJ (2012). In vivo architecture and action of bacterial structural maintenance of chromosome proteins. Science 338, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmann F, Basfeld A, Vazquez Nunez R, Diebold-Durand ML, Wilhelm L, and Gruber S (2017). Tuned SMC Arms Drive Chromosomal Loading of Prokaryotic Condensin. Mol Cell 65, 861–872 e869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmann F, Shin HC, Basquin J, Soh YM, Gimenez-Oya V, Kim YG, Oh BH, and Gruber S (2013). An asymmetric SMC-kleisin bridge in prokaryotic condensin. Nat Struct Mol Biol 20, 371–379. [DOI] [PubMed] [Google Scholar]
- Camdere G, Guacci V, Stricklin J, and Koshland D (2015). The ATPases of cohesin interface with regulators to modulate cohesin-mediated DNA tethering. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuylen S, and Haering CH (2011). Deciphering condensin action during chromosome segregation. Trends Cell Biol 21, 552–559. [DOI] [PubMed] [Google Scholar]
- Davidson IF, Goetz D, Zaczek MP, Molodtsov MI, Huis In ‘t Veld PJ, Weissmann F, Litos G, Cisneros DA, Ocampo-Hafalla M, Ladurner R, et al. (2016). Rapid movement and transcriptional re-localization of human cohesin on DNA. EMBO J 35, 2671–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cruz EM, Sweeney HL, and Ostap EM (2000). ADP inhibition of myosin V ATPase activity. Biophys J 79, 1524–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, and Mirny L (2016). The 3D Genome as Moderator of Chromosomal Communication. Cell 164, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold-Durand ML, Lee H, Ruiz Avila LB, Noh H, Shin HC, Im H, Bock FP, Burmann F, Durand A, Basfeld A, et al. (2017). Structure of Full-Length SMC and Rearrangements Required for Chromosome Organization. Mol Cell 67, 334–347 e335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeftens JM, Bisht S, Kerssemakers J, Kschonsak M, Haering CH, and Dekker C (2017). Real-time detection of condensin-driven DNA compaction reveals a multistep binding mechanism. EMBO J 36, 3448–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbatsh AMO, Haarhuis JHI, Petela N, Chapard C, Fish A, Celie PH, Stadnik M, Ristic D, Wyman C, Medema RH, et al. (2016). Cohesin Releases DNA through Asymmetric ATPase-Driven Ring Opening. Mol Cell 61, 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Ercan S, Dick LL, and Lieb JD (2009). The C. elegans dosage compensation complex propagates dynamically and independently of X chromosome sequence. Curr Biol 19, 1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell reports 15, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M (2000). Temporal and selective association of multiple sigma factors with RNA polymerase during sporulation in Bacillus subtilis. Genes Cells 5, 79–88. [DOI] [PubMed] [Google Scholar]
- Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, and Dekker C (2018). Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligoris TG, Scheinost JC, Burmann F, Petela N, Chan KL, Uluocak P, Beckouet F, Gruber S, Nasmyth K, and Lowe J (2014). Closing the cohesin ring: structure and function of its Smc3-kleisin interface. Science 346, 963–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloborodko A, Imakaev MV, Marko JF, and Mirny L (2016). Compaction and segregation of sister chromatids via active loop extrusion. eLife 5, e14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber S, and Errington J (2009). Recruitment of condensin to replication origin regions by ParB/SpoOJ promotes chromosome segregation in B. subtilis. Cell 137, 685–696. [DOI] [PubMed] [Google Scholar]
- Gruber S, Veening JW, Bach J, Blettinger M, Bramkamp M, and Errington J (2014). Interlinked Sister Chromosomes Arise in the Absence of Condensin during Fast Replication in B. subtilis. Curr Biol 24, 293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haering CH, and Gruber S (2016). SnapShot: SMC Protein Complexes Part I. Cell 164, 326–326 e321. [DOI] [PubMed] [Google Scholar]
- Harwood CR, and Cutting SM (1990). Molecular Biological Methods for Bacillus (Wiley; ). [Google Scholar]
- Hass LF, Boyer PD, and Reynard AM (1961). Studies on possible phosphoryl enzyme formation in catalysis by hexokinase, pyruvate kinase, and glucose 6-phosphatase. J Biol Chem 236, 2284–2291. [PubMed] [Google Scholar]
- Hirano M, Anderson DE, Erickson HP, and Hirano T (2001). Bimodal activation of SMC ATPase by intra- and inter-molecular interactions. EMBO J 20, 3238–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M, and Hirano T (2004). Positive and negative regulation of SMC-DNA interactions by ATP and accessory proteins. EMBO J 23, 2664–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M, and Hirano T (2006). Opening closed arms: long-distance activation of SMC ATPase by hinge-DNA interactions. Mol Cell 21, 175–186. [DOI] [PubMed] [Google Scholar]
- Hirano T (2016). Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 164, 847–857. [DOI] [PubMed] [Google Scholar]
- Hu B, Itoh T, Mishra A, Katoh Y, Chan KL, Upcher W, Godlee C, Roig MB, Shirahige K, and Nasmyth K (2011). ATP hydrolysis is required for relocating cohesin from sites occupied by its Scc2/4 loading complex. Curr Biol 21, 12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Tada M, and Tonomura Y (1966). The pre-steady state of the myosin--adenosine triphosphate system. IV. Liberation of ADP from the myosin--ATP system and effects of modifiers on the phosphorylation of myosin. J Biochem 59, 280–289. [PubMed] [Google Scholar]
- Kamada K, Miyata M, and Hirano T (2013). Molecular basis of SMC ATPase activation: role of internal structural changes of the regulatory subcomplex ScpAB. Structure 21, 581–594. [DOI] [PubMed] [Google Scholar]
- Keenholtz RA, Dhanaraman T, Palou R, Yu J, D’Amours D, and Marko JF (2017). Oligomerization and ATP stimulate condensin-mediated DNA compaction. Sci Rep 7, 14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Rybenkov VV, Crisona NJ, Hirano T, and Cozzarelli NR (1999). 13S condensin actively reconfigures DNA by introducing global positive writhe: implications for chromosome condensation. Cell 98, 239–248. [DOI] [PubMed] [Google Scholar]
- Kschonsak M, Merkel F, Bisht S, Metz J, Rybin V, Hassler M, and Haering CH (2017). Structural Basis for a Safety-Belt Mechanism That Anchors Condensin to Chromosomes. Cell 171, 588–600 e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammens A, Schele A, and Hopfner KP (2004). Structural biochemistry of ATP-driven dimerization and DNA-stimulated activation of SMC ATPases. Curr Biol 14, 1778–1782. [DOI] [PubMed] [Google Scholar]
- Lawrimore J, Friedman B, Doshi A, and Bloom K (2017). RotoStep: A Chromosome Dynamics Simulator Reveals Mechanisms of Loop Extrusion. Cold Spring Harb Symp Quant Biol. [DOI] [PubMed] [Google Scholar]
- Le TB, Imakaev MV, Mirny LA, and Laub MT (2013). High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342, 731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DC, Levin PA, and Grossman AD (1997). Bipolar localization of a chromosome partition protein in Bacillus subtilis. Proc Natl Acad Sci U S A 94, 4721–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindow JC, Kuwano M, Moriya S, and Grossman AD (2002). Subcellular localization of the Bacillus subtilis structural maintenance of chromosomes (SMC) protein. Mol Microbiol 46, 997–1009. [DOI] [PubMed] [Google Scholar]
- Marbouty M, Le Gall A, Cattoni DI, Cournac A, Koh A, Fiche JB, Mozziconacci J, Murray H, Koszul R, and Nollmann M (2015). Condensin- and Replication-Mediated Bacterial Chromosome Folding and Origin Condensation Revealed by Hi-C and Super-resolution Imaging. Mol Cell 59, 588–602. [DOI] [PubMed] [Google Scholar]
- Marko JF, De Los Rios P, Barducci A, and Gruber S (2018). DNA-segment-capture model for loop extrusion by structural maintenance of chromosome (SMC) protein complexes. bioRxiv 325373 10.1101/325373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnen A, Burmann F, Wilhelm L, Anchimiuk A, Diebold-Durand ML, and Gruber S (2016). Control of Smc Coiled Coil Architecture by the ATPase Heads Facilitates Targeting to Chromosomal ParB/parS and Release onto Flanking DNA. Cell reports 14, 2003–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K (2001). Disseminating the genome: joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu Rev Genet 35, 673–745. [DOI] [PubMed] [Google Scholar]
- Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, and Dekker J (2013). Organization of the mitotic chromosome. Science 342, 948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols MH, and Corces VG (2015). A CTCF Code for 3D Genome Architecture. Cell 162, 703–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo-Hafalla MT, and Uhlmann F (2011). Cohesin loading and sliding. Journal of cell science 124, 685–691. [DOI] [PubMed] [Google Scholar]
- Palecek JJ, and Gruber S (2015). Kite Proteins: a Superfamily of SMC/Kleisin Partners Conserved Across Bacteria, Archaea, and Eukaryotes. Structure 23, 2183–2190. [DOI] [PubMed] [Google Scholar]
- Rousseeuw PJ, and Croux C (1993). Alternatives to the Median Absolute Deviation. Journal of the American Statistical Association 88, 1273–1283. [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112, E6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CK, Brookes N, and Uhlmann F (2009). Conserved features of cohesin binding along fission yeast chromosomes. Genome biology 10, R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh YM, Burmann F, Shin HC, Oda T, Jin KS, Toseland CP, Kim C, Lee H, Kim SJ, Kong MS, et al. (2015). Molecular basis for SMC rod formation and its dissolution upon DNA binding. Mol Cell 57, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan NL, Marquis KA, and Rudner DZ (2009). Recruitment of SMC by ParB-parS organizes the origin region and promotes efficient chromosome segregation. Cell 137, 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terakawa T, Bisht S, Eeftens JM, Dekker C, Haering CH, and Greene EC (2017). The condensin complex is a mechanochemical motor that translocates along DNA. Science 358, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NT, Laub MT, and Le TBK (2017). SMC Progressively Aligns Chromosomal Arms in Caulobacter crescentus but Is Antagonized by Convergent Transcription. Cell reports 20, 2057–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trentham DR, Bardsley RG, Eccleston JF, and Weeds AG (1972). Elementary processes of the magnesium ion-dependent adenosine triphosphatase activity of heavy meromyosin. A transient kinetic approach to the study of kinases and adenosine triphosphatases and a colorimetric inorganic phosphate assay in situ. The Biochemical journal 126, 635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F (2016). SMC complexes: from DNA to chromosomes. Nat Rev Mol Cell Biol 17, 399–412. [DOI] [PubMed] [Google Scholar]
- Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, Huang SC, El Khattabi L, Dose M, Pruett N, et al. (2018). The Energetics and Physiological Impact of Cohesin Extrusion. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Brandao HB, Le TB, Laub MT, and Rudner DZ (2017). Bacillus subtilis SMC complexes juxtapose chromosome arms as they travel from origin to terminus. Science 355, 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Le TB, Lajoie BR, Dekker J, Laub MT, and Rudner DZ (2015). Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev 29, 1661–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tang OW, Riley EP, and Rudner DZ (2014). The SMC condensin complex is required for origin segregation in Bacillus subtilis. Curr Biol 24, 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B, and Sali A (2016). Comparative Protein Structure Modeling Using MODELLER. Current protocols in protein science 86, 2 9 1–2 9 37. [DOI] [PubMed] [Google Scholar]
- Wilhelm L, Bürmann F, Minnen A, Shin H-C, Toseland CP, Oh B-H, and Gruber S (2015). SMC condensin entraps chromosomal DNA by an ATP hydrolysis dependent loading mechanism in Bacillus subtilis. eLife 10, 7554/eLife.06659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman PJ, Perkins JB, and Losick R (1983). Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proc Natl Acad Sci U S A 80, 2305–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.