

A novel antibiotic combination of the oral cephalosporin ceftibuten (CTB) and the β-lactamase inhibitor clavulanate (CLA) is currently in development for urinary tract infections, including those caused by extended-spectrum-β-lactamase (ESBL)-producing organisms. This study aimed to identify the pharmacodynamic index and magnitude of this index for CLA, when combined with a fixed CTB exposure (∼59% free time above the CTB-CLA MIC) against ESBL-producing Escherichia coli and Klebsiella pneumoniae (CTB-CLA MICs of 0.25/0.125 to 1/0.5 μg/ml) using the in vitro chemostat model.
KEYWORDS: beta-lactam/beta-lactamase inhibitor, cephalosporin, extended-spectrum beta-lactamase, urinary tract infection
ABSTRACT
A novel antibiotic combination of the oral cephalosporin ceftibuten (CTB) and the β-lactamase inhibitor clavulanate (CLA) is currently in development for urinary tract infections, including those caused by extended-spectrum-β-lactamase (ESBL)-producing organisms. This study aimed to identify the pharmacodynamic index and magnitude of this index for CLA, when combined with a fixed CTB exposure (∼59% free time above the CTB-CLA MIC) against ESBL-producing Escherichia coli and Klebsiella pneumoniae (CTB-CLA MICs of 0.25/0.125 to 1/0.5 μg/ml) using the in vitro chemostat model. Dose fractionation studies identified the time that free CLA concentrations remained above a threshold concentration (fT>threshold) to be the best pharmacodynamic index (R2 = 0.85) compared with the free area under the curve (AUC)/threshold ratio (R2 = 0.62) and free maximum concentration/threshold ratio (R2 = 0.37). For E. coli isolates, stasis and 1-log10 CFU reductions were achieved at 30.9 and 47.9% fT>CTB concentrations of the 2:1 CTB-CLA MIC (fT>MIC here), respectively. For K. pneumoniae isolates, stasis and 1-log10 CFU reductions were achieved at 51.9 and 92.0% fT>MIC, respectively. These data inform exposure requirements for CLA combined with CTB for optimizing pharmacodynamics against Enterobacteriaceae and should be useful in designing dosage regimens for this combination antibiotic.
INTRODUCTION
Urinary tract infections comprise a substantial portion of emergency department visits and hospital admissions annually in the United States (1, 2). When considering empirical antibiotic therapy for infections originating from urine, coverage of Escherichia coli and Klebsiella species is essential, as these two Gram-negative enterics account for upwards of 80% of identified bacterial causes (2, 3). Antimicrobial resistance among Gram-negative bacteria has become a global threat, and uropathogens are not exempt from this danger. The primary mechanism of resistance in these pathogens is the production of extended-spectrum β-lactamases (ESBLs), often in conjunction with fluoroquinolone and aminoglycoside resistance determinants on the same plasmid, thereby defining these strains as multidrug resistant. Recent surveillance studies in the United States have observed ESBL rates within urinary E. coli and Klebsiella species isolates of 8 to 18% (3, 4). Notably, ESBL rates can be significantly higher in other countries (3, 5, 6).
Treatment options for complicated urinary tract infections (cUTIs) caused by ESBL-producing bacteria are limited; therefore, intravenous carbapenems are frequently prescribed as the agents of choice. Oral antibiotic options, particularly for ESBL cUTI, are even more elusive; fluoroquinolone and trimethoprim-sulfamethoxazole resistance within E. coli is substantial, most tetracyclines do not adequately reach the site of infection, and oral fosfomycin and nitrofurantoin are approved only for uncomplicated cystitis in the United States (6, 7). New oral options for the treatment of cUTI caused by ESBL-producing bacteria would be a welcome addition to our anti-infective armamentarium.
A novel combination of the oral cephalosporin ceftibuten (CTB) and the β-lactam-based β-lactamase inhibitor clavulanate (CLA) is being developed for the treatment of cUTIs, including those caused by ESBL-producing Enterobacteriaceae. Although much is known about the pharmacodynamics of cephalosporin antibiotics (8), little is known about the exposure-response relationships of the β-lactamase components, specifically clavulanate (9, 10). The objectives of the present study were to determine ceftibuten pharmacodynamics alone and then clavulanate pharmacodynamics when combined with ceftibuten against select E. coli and Klebsiella pneumoniae isolates using the in vitro chemostat pharmacodynamic model. These data should be useful in guiding the selection of an optimal dosage of ceftibuten and clavulanate for further study.
RESULTS
Isolate MICs.
The results of the in vitro susceptibility studies for each of the selected isolates included in the study are presented in Table 1, together with the corresponding ESBL enzymatic profiles. The combination of CTB-CLA (using clavulanate concentrations precisely 0.5 times that of the ceftibuten concentration at each dilution) resulted in potentiated MICs that were at least 64-fold lower than those of ceftibuten alone against the four ESBL isolates; the MIC against the wild-type isolate was the same as that of ceftibuten alone. As expected, clavulanate alone had poor activity against these strains.
TABLE 1.
Modal MICs for ceftibuten, clavulanate, and ceftibuten-clavulanate against the Enterobacteriaceae isolates used in this studya
| Isolate | ESBL type(s) | Ceftibuten MIC (μg/ml) | Clavulanate MIC (μg/ml) | CTB-CLA MICb (μg/ml) |
|---|---|---|---|---|
| EC25922 | None | 0.25 | 32 | 0.25/0.125 |
| EC477 | CTX-M-55 | >64 | 32 | 1/0.5 |
| EC478 | CTX-M-15, TEM | >64 | 32 | 0.5/0.25 |
| KP547 | CTX-M-15, SHV-12 | >64 | 16 | 0.25/0.125 |
| KP548 | CTX-M-14, SHV-12 | 16 | 32 | 0.25/0.125 |
ESBL, extended-spectrum β-lactamase; EC, E. coli; KP, Klebsiella pneumoniae.
For CTB-CLA, the MIC is listed as the concentration of ceftibuten/concentration of clavulanate.
Ceftibuten and clavulanate concentrations.
Observed versus predicted concentrations for ceftibuten (n = 608 observations) and clavulanate (n = 997 observations) throughout all experiments are provided in Fig. 1. For both drugs, concentrations achieved in the in vitro chemostat models were accurate and unbiased, as implied by the R2 value, y intercept, and slope from linear regression analyses. For the experiments involving the ESBL-producing strains where no clavulanate was added to the chemostat model, it was difficult to maintain ceftibuten concentrations, presumably due to hydrolysis of the β-lactam ring; regardless, ceftibuten-alone exposures against ESBL-producing stains were intended to be low (i.e., 0% free time above the MIC [fT>MIC]), and these low exposures were realized. The median ceftibuten fT>CTB-CLA MIC when no clavulanate was present in the model was 39% (range, 23 to 48%). Observed ceftibuten and clavulanate concentrations in each chemostat model fit a one-compartment model well (data not shown) and were used for determination of precise pharmacodynamic exposure in each model.
FIG 1.
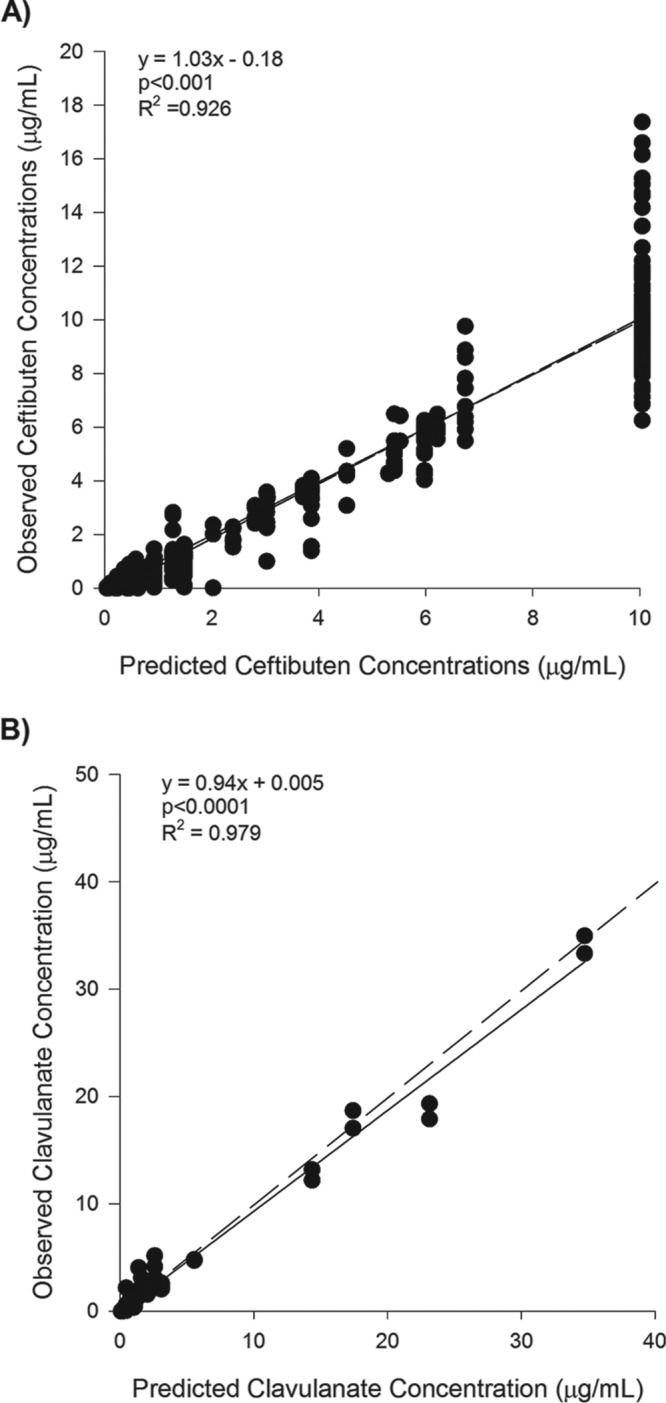
Observed versus predicted concentrations of ceftibuten (A) and clavulanate (B) in the in vitro chemostat model. Black dots are observed concentrations. Black lines are the regression line of best fit. The dashed line (which is superimposed for ceftibuten) is the line of unity.
Ceftibuten pharmacodynamics.
Ceftibuten pharmacodynamics in the absence of clavulanate were characterized against wild-type E. coli ATCC 25922 (EC25922). It was assumed that like all other β-lactam antibiotics, the ceftibuten free time above the MIC (fT>MIC) was the pharmacodynamic index; therefore, these studies focused on identifying the fT>MIC threshold associated with stasis and 1-log10 reductions in CFU at 24 h. The time-kill results of various ceftibuten fT>MIC exposures are provided in Fig. S1 in the supplemental material. Figure 2 demonstrates the maximal-effect (Emax) model describing the relationship between ceftibuten exposure and CFU reductions. Stasis and 1-log10 CFU reductions were observed at 54% and 59% fT>MIC, respectively.
FIG 2.
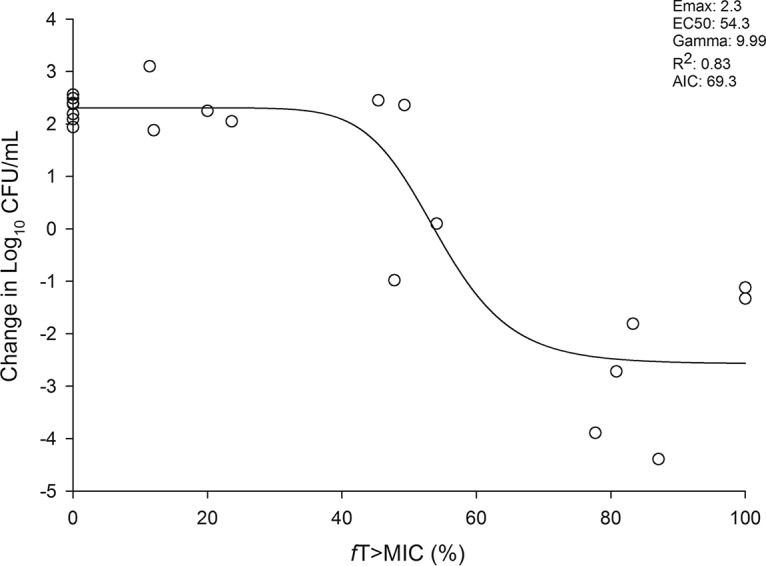
Reduction in E. coli 25922 (wild type) CFU per milliliter versus observed ceftibuten fT>MIC exposures. Each symbol represents an individual observed exposure in a chemostat model. The line drawn through the data points represents the line of best fit based on the sigmoid Emax (maximal-effect) formula. EC50, fT>MIC associated with 50% of the maximal effect; Gamma, slope or Hill coefficient; AIC, Akaike information criterion.
Clavulanate dose fractionation studies.
EC477, an E. coli isolate harboring a CTX-M-55 ESBL (ceftibuten MIC > 64 μg/ml; CTB-CLA MIC = 1/0.5 μg/ml), was used for clavulanate dose fractionation studies. During these experiments, ceftibuten was delivered to the chemostat models to achieve ∼59% fT>MIC using the ceftibuten MIC of the CTB-CLA combination; thus, the correct clavulanate exposure should return ceftibuten killing to an ∼1-log10 CFU reduction. The time-kill results for various clavulanate exposures against this strain are provided in Fig. S2 in the supplemental material. Emax model exposure-response curves for the pharmacodynamic parameters of the ratio of the free maximum concentration to a threshold concentration (fCmax/threshold), ratio of the free area under the curve to a threshold concentration (fAUC/threshold), and free time that the clavulanate concentration remains above a threshold (fT>threshold) are presented in Fig. 3. A concentration of 1 μg/ml (i.e., the ceftibuten concentration of the CTB-CLA MIC for EC477) was arbitrarily applied as the threshold for initial assessment. The pharmacodynamic index that was best associated with the ability for clavulanate exposures to return ceftibuten to a bactericidal antibiotic was fT>threshold (R2 = 0.85; Akaike information criterion value [AIC] = 58.6), followed by the fAUC/threshold ratio (R2 = 0.62; AIC = 99.5) and the fCmax/threshold ratio (R2 = 0.37; AIC value = 111.5). Of note, testing of different threshold values changes the R2 for the fT>threshold index (Table 2) but does not alter the R2 fits for the fCmax/threshold or fAUC/threshold ratio.
FIG 3.

Emax model fits of clavulanate dose fractionation studies (combined with ceftibuten to achieve 59% fT>CTB-CLA MIC) against EC477, containing a CTX-M-55 ESBL. The threshold concentration was set at 1 μg/ml, the ceftibuten concentration of the CTB-CLA MIC, during these analyses. Each symbol represents an individual observed exposure in a chemostat model. The line drawn through the data points represents the line of best fit based on the sigmoid Emax (maximal-effect) formula. EC50, fT>MIC associated with 50% of the maximal effect; Gamma, slope or Hill coefficient; AIC, Akaike information criterion.
TABLE 2.
Interisolate summary of clavulanate fT>threshold Emax model fits along with stasis and 1-log CFU reduction exposure requirementsa
| Isolate | CTB-CLA MICb (μg/ml) | fT>threshold (μg/ml) | R2 | AIC | % fT>CTB to achieve stasis | % fT>CTB to achieve 1-log10 CFU reduction |
|---|---|---|---|---|---|---|
| EC477 | 1/0.5 | >2 | 0.67 | 95.3 | 24.9 | 27.7 |
| >1 | 0.85 | 58.6 | 31.0 | 35.9 | ||
| >0.5 | 0.83 | 80.1 | 36.3 | 41.0 | ||
| >0.25 | 0.83 | 80.1 | 42.4 | 47.9 | ||
| >0.125 | 0.83 | 80.3 | 48.6 | 54.9 | ||
| EC478 | 0.5/0.25 | >1 | 0.46 | 46.7 | 8.1 | 21.5 |
| >0.5 | 0.86 | 30.5 | 22.7 | 44.0 | ||
| >0.25 | 0.85 | 30.9 | 32.0 | 54.4 | ||
| >0.125 | 0.84 | 32.1 | 40.2 | 63.7 | ||
| KP547 | 0.25/0.125 | >1 | 0.07 | 49.3 | 23.5 | NA |
| >0.5 | 0.80 | 30.7 | 66.1 | 85.7 | ||
| >0.25 | 0.79 | 31.8 | 74.6 | 91.0 | ||
| >0.125 | 0.74 | 34.1 | 80.2 | 96.7 | ||
| >0.06 | 0.70 | 35.6 | 85.0 | 100.0 | ||
| KP548 | 0.25/0.125 | >1 | 0.78 | 35.0 | 5.0 | NA |
| >0.5 | 0.90 | 24.4 | 19.4 | 30.9 | ||
| >0.25 | 0.91 | 23.4 | 29.4 | 47.7 | ||
| >0.125 | 0.91 | 23.1 | 39.4 | 66.0 | ||
| >0.06 | 0.91 | 23.0 | 49.9 | 84.4 | ||
Ceftibuten was coadministered to achieve 59% fT>MIC (i.e., ceftibuten concentration in the CTB-CLA MIC [the median observed exposure was 57% fT>MIC]) during all experiments. EC, E. coli; KP, Klebsiella pneumoniae; AIC, Akaike information criterion; NA, not applicable.
For CTB-CLA, the MIC is listed as the concentration of ceftibuten/concentration of clavulanate.
Clavulanate pharmacodynamic magnitude determination.
Four ESBL-producing strains (EC477, EC478, KP547, and KP548) were used to determine the magnitude of clavulanate fT>threshold required for stasis and 1-log10 CFU reductions. When coadministered with clavulanate, observed ceftibuten exposures during these experiments ranged between 50% and 66% fT>MIC (median, 57% fT>MIC). Time-kill results are available in Fig. S3 to S5 in the supplemental material. Table 2 demonstrates the Emax model fits (R2 and AIC) and resulting fT>threshold values for stasis and 1-log10 CFU reductions for each organism assessed using different threshold values. Based on the highest R2 and lowest AIC for EC477, EC478, KP547, and KP548, the best-fit models were defined by fT>1 μg/ml, fT>0.5 μg/ml, fT>0.5 μg/ml, and fT>0.06 μg/ml, respectively. Notably, the best fits for the individual E. coli isolates were both threshold concentrations equal to the ceftibuten concentration in the CTB-CLA MIC. Individual best fits for the two K. pneumoniae isolates varied but were very similar across threshold values of between 0.125 and 0.5 μg/ml. Because of these differences, E. coli and K. pneumoniae were modeled separately to arrive at final threshold concentrations for each genus. The thresholds that best fit these E. coli (R2 = 0.77; AIC = 139.65) and K. pneumoniae (R2 = 0.77; AIC = 74.92) isolates were fT>MIC (i.e., ceftibuten concentration in the CTB-CLA MIC) (Fig. 4), which were excellent fits for composite organism curves. For E. coli isolates, stasis and 1-log10 CFU reductions were achieved at 30.9 and 47.9% fT>MIC, respectively. For K. pneumoniae isolates, stasis and 1-log10 CFU reductions were achieved at 51.9 and 92.0% fT>MIC, respectively.
FIG 4.

Relationship between the change in log10 CFU and the clavulanate fT>threshold by bacterial genus. (A) Composite Emax (maximal-effect) model of best fit for EC477 and EC478, which found the threshold concentration to be the value of the ceftibuten concentration in the CTB-CLA MIC; (B) composite Emax model of best fit for KP547 and KP548, which found the threshold concentration to be the value of the ceftibuten concentration in the CTB-CLA MIC. Each symbol represents an individual observed exposure in a chemostat model, where the different colors designate individual isolates. The line drawn through the data points represents the line of best fit based on the sigmoid Emax formula. EC50, fT>MIC associated with 50% of the maximal effect; Gamma, slope or Hill coefficient; AIC, Akaike information criterion.
DISCUSSION
The pharmacodynamic behavior of β-lactamase inhibitors has not been well established (11). Recent pharmacodynamic studies have begun to identify the exposure-response relationships of the β-lactamase inhibitor component required to restore activity of the β-lactam (9, 10, 12–14). Notably, the pharmacodynamic index may be different for these inhibitors; fT>threshold has been identified for tazobactam in combination with ceftolozane, cefepime, and piperacillin, with different threshold concentrations as a fraction of the MIC identified dependent on the β-lactam backbone. In contrast, the fAUC/threshold ratio was the index for CB-618 and vaborbactam (11). When combined with aztreonam, avibactam correlated with a free time above a fixed concentration of 2 to 2.5 μg/ml, regardless of the MIC (9). In the present study, we sought to use the in vitro chemostat model to identify the pharmacodynamic index for clavulanate and the magnitude of this index that restores ceftibuten bactericidal activity against select ESBL-producing Enterobacteriaceae.
Ceftibuten alone was observed to require 54% to 59% fT>MIC for stasis and 1-log10 CFU reductions, respectively, against a wild-type E. coli strain. These exposure requirements are consistent with those for other cephalosporin antibiotics against Gram-negative bacteria (8, 15–17). Therefore, for all subsequent clavulanate dose fractionation and magnitude-of-index experiments, the goal was to maintain ceftibuten exposures at 59% fT>MIC based on the corresponding CTB-CLA MIC for each isolate. A range of exposures was achieved throughout the experiments (50 to 66% fT>MIC), with a median of 57% fT>MIC, which is in line with exposures needed to achieve between stasis and a 1-log10 CFU reduction, assuming that all included isolates would behave like EC25922. That said, the variability in CFU reductions observed during the clavulanate-containing experiments could be due to different exposure-response relationships for the ESBL-producing isolates or, if our assumptions were correct, to the steep ceftibuten response-exposure curve (Fig. 2), where the difference between 50 and 66% fT>MIC could result in stasis to >1-log10 CFU reductions at 24 h. Other in vitro pharmacodynamic studies assessed exposure-response relationships of the inhibitor using a human-simulated concentration profile for the β-lactam component based on the commonly used dosing regimen (10, 12–14). A different approach to ceftibuten dosing was applied in this study because alternative dosing regimens might be advantageous when combining ceftibuten with a β-lactamase inhibitor. Currently, there is no specific threshold reduction guidance for antibiotics being developed for complicated urinary tract infections, including acute pyelonephritis. However, for antibiotic development in pneumonia (community acquired and hospital acquired), regulatory approval was predicted by a high probability of achieving exposures needed for stasis to 1-log10 CFU reductions (18).
During dose fractionation studies using an ESBL-producing E. coli isolate (EC477), clavulanate fT>threshold was identified as the pharmacodynamic index that best described the return of a ceftibuten microbiological response (R2 values of 0.83, versus 0.62 for the fAUC/threshold ratio and 0.37 for the fCmax/threshold ratio) (Fig. 3). A threshold of 1 μg/ml, which was the ceftibuten concentration in the CTB-CLA-potentiated MIC for this isolate, was applied during these analyses. However, it is worth noting that various threshold concentrations were tested (Table 2), and applying concentrations of between 0.125 and 2 μg/ml did not alter the result of the best pharmacodynamic driver. The R2 value for the fAUC/threshold and fCmax/threshold ratios, due to the nature of both indexes being ratios, does not change with different threshold values. To the best of our knowledge, the present study is the first to investigate and clarify the pharmacodynamic driver for clavulanate against Enterobacteriaceae, despite several decades of vast clinical use of this companion drug. A follow-up murine thigh infection model by our group, also reported in this issue (19), likewise identified fT>threshold to be the pharmacodynamic parameter for clavulanate. These observations are consistent with those for other β-lactamase inhibitors, such as avibactam and tazobactam, where CFU reduction was associated with fT>threshold (9, 12, 13).
The specific threshold and magnitude required for stasis and a 1-log10 CFU reduction were explored against two ESBL-producing E. coli and two K. pneumoniae isolates with CTB-CLA MICs ranging from 0.25/0.125 to 1/0.5 μg/ml. One advantage of the in vitro chemostat model is the opportunity for multiple sampling throughout the experiment, which allows precise estimates of pharmacodynamic exposure and permits full characterization of each isolate individually. When each isolate was fit to the sigmoidal Emax models, various threshold concentrations resulted in “best fits” based on R2 and AIC (Table 2). For the two E. coli isolates, fT>MIC (using the ceftibuten concentration of the CTB-CLA MIC) best correlated with CFU reductions. For K. pneumoniae isolates, the best model fits were observed using concentrations of 0.5 μg/ml (1 dilution above the MIC) and 0.06 μg/ml (2 dilutions below the MIC) for KP547 and KP548, respectively. As a result of observed differences between E. coli and K. pneumoniae, composite curves were modeled for each genus (Fig. 4). The CTB-CLA MIC remained the best threshold for E. coli and was also identified as the best threshold for both K. pneumoniae isolates using the composite analysis.
These results are in contrast to those of murine thigh infection studies, which, when using a composite curve of 11 E. coli and K. pneumoniae isolates, observed a fixed threshold of 0.5 μg/ml (19). As noted above, various thresholds have been identified for the other β-lactamase inhibitors, where fT>threshold was the pharmacodynamic index. For example, avibactam, when combined with ceftazidime, was associated with an fT>threshold of 1 μg/ml regardless of the MIC in the neutropenic thigh infection model consistently against a number of Pseudomonas aeruginosa isolates (20), while fixed thresholds between 0.25 and 0.5 μg/ml were identified in hollow-fiber studies against Enterobacteriaceae (21). In contrast, in combination with aztreonam, critical thresholds were identified at fixed concentrations of 2 to 2.5 μg/ml (9). Tazobactam thresholds were dependent on the type and level of β-lactamase expression in the isolates but ranged from 0.05 to 0.25 μg/ml in one experiment with ceftolozane (12) and from 0.25 to 2 μg/ml with piperacillin (13) or were equal to 0.5× MIC of the organism in a third experiment (22). The different thresholds identified for clavulanate between the in vitro and the murine thigh infection models could be due to the different isolates included, the different ceftibuten regimens coadministered (i.e., human simulated in the in vivo trial), or some combination; however, we argue the results between studies are more similar than not. While a fixed threshold of 0.5 μg/ml was not the best fit in the present study, it provided reasonable R2 values for individual isolates (Table 2) and resulted in an R2 value of 0.73 for a composite fit of E. coli and K. pneumoniae together (data not shown).
Based on these composite analyses, stasis and 1-log10 CFU reductions were achieved against E. coli with clavulanate fT>MIC of 30.9 and 47.9%, respectively. In contrast, higher fT>MIC values were required for the K. pneumoniae isolates; stasis and 1-log10 CFU reductions were achieved at 51.9 and 92.0% fT>MIC, respectively. The greater clavulanate requirements for Klebsiella spp. was a result of observed CFU reductions with KP547. This isolate had the same CTB-CLA MIC as KP548 and largely the same ESBL profile (CTX-M-15 and SHV-12 versus CTX-M-14 and SHV-12); however, it required significantly greater exposure to achieve stasis and 1-log10 CFU reductions. We speculate that this could be due to enzyme transcription levels within the isolate, as observed with tazobactam in previous studies (12, 22). Alternatively, this could have little to do with clavulanate and could be related to ceftibuten requirements. A murine intra-abdominal infection experiment found ceftibuten to require greater time above the MIC (2.2 h) for K. pneumoniae than for E. coli (1.6 h) to save 100% of the animals (23).
Limitations of this experiment include the limited number of Enterobacteriaceae (two ESBL-producing E. coli and two ESBL-producing K. pneumoniae) isolates studied. Isolate-to-isolate variability in ceftibuten %fT>MIC requirements combined with enzyme expression differences could lead to different clavulanate exposure requirements than identified here, as is exemplified by the murine data (19). Second, the in vitro chemostat model has no immune system, and thus, the contributions of such to both ceftibuten and clavulanate exposure requirements still need to be determined. Finally, since the exposure requirements are referenced to the ceftibuten concentration of the potentiated MIC, these observations assume that the 2:1 ceftibuten-clavulanate MIC is, in and of itself, optimized. Should a reduced ratio of ceftibuten to clavulanate reduce the MIC further, this could lead to different threshold values or exposure requirements. However, the 4 ESBL-producing strains used here demonstrated potentiated MICs that were similar to or within 1 to 2 dilutions of the ceftibuten MIC against the wild-type strain (EC25922), suggesting that, at least against these strains, this ratio is achieving maximal or near-maximal ceftibuten MIC reduction.
These data demonstrate that clavulanate fT>threshold was the pharmacodynamic index associated with the ability to restore ceftibuten stasis and 1-log CFU reductions against ESBL-producing E. coli and K. pneumoniae. For these isolates, the threshold that best correlated with these observations was the CTB-CLA MIC. These data can be helpful in designing dosage regimens for both ceftibuten and clavulanate for phase 1 to 3 clinical studies.
MATERIALS AND METHODS
Antimicrobial agents.
Ceftibuten (Aurobindo Pharma Ltd., Telangana, India) and clavulanate (Shandong New Time Pharmaceutical Co., Sandong, China) powders were prepared according to the manufacturers’ instructions. A ceftibuten stock solution with a concentration of 2 mg/ml was prepared using a ceftibuten standard powder with a potency of 86.58%, dissolved in 100% dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO). This resulted in a final concentration of DMSO in the chemostat models of no more than 0.5%. A clavulanate stock solution with a concentration of 1.68 mg/ml was prepared using a clavulanate standard powder with a potency of 42.1%, dissolved in 20 mM NH4-acetate. Tazobactam (Sigma-Aldrich, St. Louis, MO) powder was dissolved in 20 mM NH4-acetate, according to the sponsor’s instructions, to achieve a solution containing 20 mM tazobactam. Avibactam (Chem Shuttle, Hayward, CA) powder was dissolved in 0.01 M phosphate-buffered saline to achieve a solution containing 10 mM avibactam. Tazobactam and avibactam solutions were added to the pharmacokinetic sample cryovials to avoid postcollection degradation of ceftibuten and the analytic assay internal standard, amoxicillin, due to the ESBLs present.
Isolates.
Five Enterobacteriaceae isolates were included in this study. All isolates were provided by Achaogen, Inc. The isolates included the following: one wild-type E. coli isolate, two E. coli isolates producing ESBLs, and two Klebsiella pneumoniae isolates producing ESBLs.
Susceptibility studies.
MIC studies were conducted by broth microdilution in triplicate at the Center for Anti-Infective Research and Development (Hartford, CT) to determine the modal MIC. MIC trays were prepared for ceftibuten alone, clavulanate alone, and the combination of ceftibuten and clavulanate. Concentrations of ceftibuten (in DMSO) in the trays ranged from ≤0.063 to ≥64 μg/ml. For clavulanate trays, standard clavulanate powder was dissolved in phosphate buffer at a pH of 6 (0.1 mol/liter), and the range of concentrations in the tray was ≤0.03125 to ≥32 μg/ml. For combined ceftibuten-clavulanate trays, standard powders of ceftibuten and clavulanate were used, dissolved in DMSO and phosphate buffer, respectively, and applied in a 2:1 ratio to the tray, thus providing an MIC range of ≤0.063/0.312 to ≥64/32 μg/ml.
In vitro pharmacodynamic model.
A one-compartment in vitro chemostat model was employed for all experiments. The 300-ml glass chemostats were filled with cation-adjusted Mueller-Hinton II broth (CAMHB) (20 to 25 mg/liter calcium, 10 to 12.5 mg/liter magnesium; Becton, Dickinson and Company, Sparks, MD) and placed in a water bath at 35°C. Magnetic stirrers were used for consistent mixing of the contents for each model during the experiment. The starting inoculum was 106 CFU/ml. Once inoculated, the bacteria were allowed to enter log-phase growth over 30 min before exposure to the antibiotics. For each drug exposure-bacterium combination, the experiment was conducted over 24 h and consisted of a drug-free control and two experimental (i.e., drug-containing) replicate models. Data from the two replicate models were treated as independent exposures given that there is some expected variability in obtained concentrations due to small inconsistencies in volume flows between the chemostat models. A peristaltic pump (Masterflex L/S model 7524-40; Cole-Parmer, Vernon Hills, IL) was set to infuse CAMHB into the models at a desired elimination rate for ceftibuten and clavulanate. Broth samples were collected from the chemostat throughout the experiment to determine ceftibuten and clavulanate free-drug concentrations and bacterial density. Antibiotic carryover was minimized by serial dilution of samples. Bacterial density was determined after 18 to 24 h of incubation at 37°C. The lower limit of detection for bacterial density in this model was 1.7 log10 CFU/ml.
Ceftibuten fT>MIC determination.
A wild-type E. coli strain (EC25922) was used for ceftibuten fT>MIC determination. Exposures that target 10, 20, 40, 60, 80, and 100% fT>MIC over 24 h were dosed into the chemostat model, targeting a ceftibuten peak of 10 μg/ml to simulate clinically achievable free maximum ceftibuten concentrations in humans after a 400-mg dose (24). Notably, this peak concentration would be maintained throughout all ceftibuten-containing experiments, regardless of the fAUC/MIC or fT/MIC ratio targeted. The control models served to define 0% fT>MIC results. The change in log10 CFU per milliliter at 24 h was the primary endpoint for the exposure-response relationship. The magnitudes of fT>MIC required for stasis and 1-log10 reductions were estimated with an inhibitory sigmoidal Emax model in Phoenix WinNonlin version 6.3 (Pharsight Corp., Mountain View, CA).
Clavulanate dose fractionation studies.
EC477, producing a CTX-M-55 ESBL, was selected for clavulanate dose fractionation studies. A ceftibuten exposure required for a ∼1-log10 CFU reduction against the MIC of EC477 was simulated in the chemostat models. The ceftibuten peak was again targeted at 10 μg/ml, and each ceftibuten dose was administered every 8 h to target a specific fT>CTB concentration of the CTB-CLA MIC. Reference tested clavulanate exposures (based on the 24-h AUC after 125 mg every 8 to 24 h [25]) were initially defined as follows for dose fractionation studies. Low and high fAUCs were 3.75 μg · h/ml and 15 μg · h/ml, approximately equivalent to those of clavulanate at 125 mg every 24 h (q24h) and at 125 mg q8h, respectively, with 30% protein binding. Clavulanate was dosed every 24, 12, or 6 h to achieve a range of fCmax, fAUC0–24, and fT>threshold exposures. Additional clavulanate exposures, representing supraphysiological fAUCs, were defined as 30, 36, 48, 60, and 72 μg · h/ml. These exposures were required for dose fractionation studies because lower doses, even when fractionated every 6 h, could not maintain high fT>1 μg/ml using the flow rates achievable in the in vitro model.
Experiments with ceftibuten alone served as zero exposure for clavulanate. Bacterial density was determined at 0, 1, 6, 12, 18, and 24 h for each experiment, and both ceftibuten and clavulanate concentrations were confirmed. The change in log10 CFU per milliliter at 24 h was plotted against pharmacodynamic exposures and analyzed using an inhibitory sigmoidal Emax model in Phoenix WinNonlin. The following exposure relationships were assessed: fCmax/threshold ratio, fAUC0–24/threshold ratio, and %fT>threshold. The final pharmacodynamic index was selected based on R2, AIC, and visual inspection of plots.
Clavulanate pharmacodynamic magnitude determination.
EC477, EC478, KP547, and KP548 were used for clavulanate pharmacodynamic magnitude determination. Ceftibuten exposures were again fixed as described above to achieve an approximately 1-log10 CFU/ml reduction using the isolate-specific CTB concentration of the CTB-CLA MIC. Clavulanate exposures ranging from 0% to 100% fT>threshold were investigated, based on results from the dose fractionation studies. The change in log10 CFU per milliliter at 24 h was plotted against the pharmacodynamic index and analyzed using an inhibitory sigmoidal Emax model in Phoenix WinNonlin. For each isolate, exposures that resulted in stasis and 1-log10 CFU reductions were calculated. Thresholds investigated varied by 1 to 3 dilutions above and below the CTB-CLA MIC for each isolate. The final magnitude was selected based on R2, AIC, and visual inspection of plots.
Ceftibuten and clavulanate concentration determination.
Broth samples collected for drug concentration determinations were immediately frozen and stored at −80°C until shipped for analysis. Ceftibuten and clavulanate concentrations were analyzed by Achaogen, Inc., and Intertek Pharmaceutical Services (San Diego, CA) by high-performance liquid chromatography. Inter- and intraday coefficients of variation were less than 10% for ceftibuten and clavulanate quality controls. The lower limits of detection for both ceftibuten and clavulanate were 0.01 μg/ml.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by Achaogen, Inc., South San Francisco, CA. J.L.K. and D.P.N. have received research funding from Achaogen, Inc., and are members of the speakers’ bureau. The other authors report no conflicts of interest.
We acknowledge the assistance of Jennifer Tabor-Rennie, Kimelyn Greenwood, Debora Santini, and Sara Robinson from the Center for Anti-Infective Research and Development, Hartford Hospital, for their assistance with the in vitro model experiments. We acknowledge John Cremin from Achaogen, Inc., for determination of ceftibuten and clavulanate concentrations in broth.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00144-19.
REFERENCES
- 1.Barber AE, Norton JP, Spivak AM, Mulvey MA. 2013. Urinary tract infections: current and emerging management strategies. Clin Infect Dis 57:719–724. doi: 10.1093/cid/cit284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. 2015. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol 13:269–284. doi: 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sader HS, Flamm RK, Jones RN. 2014. Frequency of occurrence and antimicrobial susceptibility of Gram-negative bacteremia isolates in patients with urinary tract infection: results from United States and European hospitals (2009-2011). J Chemother 26:133–138. doi: 10.1179/1973947813Y.0000000121. [DOI] [PubMed] [Google Scholar]
- 4.Lob SH, Nicolle LE, Hoban DJ, Kazmierczak KM, Badal RE, Sahm DF. 2016. Susceptibility patterns and ESBL rates of Escherichia coli from urinary tract infections in Canada and the United States, SMART 2010-2014. Diagn Microbiol Infect Dis 85:459–465. doi: 10.1016/j.diagmicrobio.2016.04.022. [DOI] [PubMed] [Google Scholar]
- 5.Ponce-de-Leon A, Rodríguez-Noriega E, Morfín-Otero R, Cornejo-Juárez DP, Tinoco JC, Martínez-Gamboa A, Gaona-Tapia CJ, Guerrero-Almeida ML, Martin-Onraët A, Vallejo Cervantes JL, Sifuentes-Osornio J. 2018. Antimicrobial susceptibility of gram-negative bacilli isolated from intra-abdominal and urinary-tract infections in Mexico from 2009 to 2015: results from the Study for Monitoring Antimicrobial Resistance Trends (SMART). PLoS One 13:e0198621. doi: 10.1371/journal.pone.0198621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazzariol A, Bazaj A, Cornaglia G. 2017. Multi-drug-resistant Gram-negative bacteria causing urinary tract infections: a review. J Chemother 29:2–9. doi: 10.1080/1120009X.2017.1380395. [DOI] [PubMed] [Google Scholar]
- 7.Zhanel GG, Zhanel MA, Karlowsky JA. 2018. Oral fosfomycin for the treatment of acute and chronic bacterial prostatitis caused by multidrug-resistant Escherichia coli. Can J Infect Dis Med Microbiol 2018:1404813. doi: 10.1155/2018/1404813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turnidge JD. 1998. The pharmacodynamics of beta-lactams. Clin Infect Dis 27:10–22. doi: 10.1086/514622. [DOI] [PubMed] [Google Scholar]
- 9.Singh R, Kim A, Tanudra MA, Harris JJ, McLaughlin RE, Patey S, O’Donnell JP, Bradford PA, Eakin AE. 2015. Pharmacokinetics/pharmacodynamics of a beta-lactam and beta-lactamase inhibitor combination: a novel approach for aztreonam/avibactam. J Antimicrob Chemother 70:2618–2626. doi: 10.1093/jac/dkv132. [DOI] [PubMed] [Google Scholar]
- 10.VanScoy BD, Tenero D, Turner S, Livermore DM, McCauley J, Conde H, Bhavnani SM, Rubino CM, Ambrose PG. 2017. Pharmacokinetics-pharmacodynamics of tazobactam in combination with cefepime in an in vitro infection model. Antimicrob Agents Chemother 61:e01052-17. doi: 10.1128/AAC.01052-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ambrose PG, Lomovskaya O, Griffith DC, Dudley MN, VanScoy B. 2017. Beta-lactamase inhibitors: what you really need to know. Curr Opin Pharmacol 36:86–93. doi: 10.1016/j.coph.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 12.VanScoy B, Mendes RE, Nicasio AM, Castanheira M, Bulik CC, Okusanya OO, Bhavnani SM, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2013. Pharmacokinetics-pharmacodynamics of tazobactam in combination with ceftolozane in an in vitro infection model. Antimicrob Agents Chemother 57:2809–2814. doi: 10.1128/AAC.02513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicasio AM, VanScoy BD, Mendes RE, Castanheira M, Bulik CC, Okusanya OO, Bhavnani SM, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2016. Pharmacokinetics-pharmacodynamics of tazobactam in combination with piperacillin in an in vitro infection model. Antimicrob Agents Chemother 60:2075–2080. doi: 10.1128/AAC.02747-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.VanScoy BD, Trang M, McCauley J, Conde H, Bhavnani SM, Friedrich LV, Alexander DC, Ambrose PG. 2016. Pharmacokinetics-pharmacodynamics of a novel beta-lactamase inhibitor, CB-618, in combination with meropenem in an in vitro infection model. Antimicrob Agents Chemother 60:3891–3896. doi: 10.1128/AAC.02943-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craig WA. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26:1–10; quiz, 11–12. doi: 10.1086/516284. [DOI] [PubMed] [Google Scholar]
- 16.Maglio D, Ong C, Banevicius MA, Geng Q, Nightingale CH, Nicolau DP. 2004. Determination of the in vivo pharmacodynamic profile of cefepime against extended-spectrum-beta-lactamase-producing Escherichia coli at various inocula. Antimicrob Agents Chemother 48:1941–1947. doi: 10.1128/AAC.48.6.1941-1947.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacVane SH, Kuti JL, Nicolau DP. 2014. Clinical pharmacodynamics of antipseudomonal cephalosporins in patients with ventilator-associated pneumonia. Antimicrob Agents Chemother 58:1359–1364. doi: 10.1128/AAC.01463-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bulik CC, Bhavnani SM, Hammel JP, Forrest A, Dudley MN, Ellis-Grosse EJ, Drusano GL, Ambrose PG. 2013. Evaluation of the probability of regulatory approval based on pre-clinical pharmacokinetic/pharmacodynamic target attainment for community-acquired bacterial (CABP) and hospital-acquired bacterial pneumonia (HABP), abstr A-295. Abstr 53rd Intersci Conf Antimicrob Agents Chemother. American Society for Microbiology, Washington, DC. [Google Scholar]
- 19.Abdelraouf K, Stainton SM, Nicolau DP. 2019. In vivo pharmacodynamic profile of ceftibuten-clavulanate combination against extended-spectrum-β-lactamase-producing Enterobacteriaceae in the murine thigh infection model. Antimicrob Agents Chemother 63:e00145-19. doi: 10.1128/AAC.00145-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berkhout J, Melchers MJ, van Mil AC, Seyedmousavi S, Lagarde CM, Schuck VJ, Nichols WW, Mouton JW. 2016. Pharmacodynamics of ceftazidime and avibactam in neutropenic mice with thigh or lung infection. Antimicrob Agents Chemother 60:368–375. doi: 10.1128/AAC.01269-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coleman K, Levasseur P, Girard AM, Borgonovi M, Miossec C, Merdjan H, Drusano G, Shlaes D, Nichols WW. 2014. Activities of ceftazidime and avibactam against beta-lactamase-producing Enterobacteriaceae in a hollow-fiber pharmacodynamic model. Antimicrob Agents Chemother 58:3366–3372. doi: 10.1128/AAC.00080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanscoy B, Mendes RE, McCauley J, Bhavnani SM, Bulik CC, Okusanya OO, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2013. Pharmacological basis of beta-lactamase inhibitor therapeutics: tazobactam in combination with ceftolozane. Antimicrob Agents Chemother 57:5924–5930. doi: 10.1128/AAC.00656-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Onyeji CO, Nicolau DP, Nightingale CH, Quintiliani R. 1994. Optimal times above MICs of ceftibuten and cefaclor in experimental intra-abdominal infections. Antimicrob Agents Chemother 38:1112–1117. doi: 10.1128/AAC.38.5.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pernix Therapeutics. 2010. Cedax (ceftibuten dihydrate capsule) prescribing information. Pernix Therapeutics, LLC, Gonzales, LA. [Google Scholar]
- 25.Vree TB, Dammers E, Exler PS. 2003. Identical pattern of highly variable absorption of clavulanic acid from four different oral formulations of co-amoxiclav in healthy subjects. J Antimicrob Chemother 51:373–378. doi: 10.1093/jac/dkg082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.