

Abstract
There has been considerable interest in the generation of functional mesenchymal stromal cell (MSC) preparations from induced pluripotent stem cells (iPSCs) and this is now regarded as a potential source of unlimited, standardized, high‐quality cells for therapeutic applications in regenerative medicine. Although iMSCs meet minimal criteria for defining MSCs in terms of marker expression, there are substantial differences in terms of trilineage potential, specifically a marked reduction in chondrogenic and adipogenic propensity in iMSCs compared with bone marrow‐derived (BM) MSCs. To reveal the cellular basis underlying these differences, we conducted phenotypic, functional, and genetic comparisons between iMSCs and BM‐MSCs. We found that iMSCs express very high levels of both KDR and MSX2 compared with BM‐MSCs. In addition, BM‐MSCs had significantly higher levels of PDGFRα. These distinct gene expression profiles were maintained during culture expansion, suggesting that prepared iMSCs are more closely related to vascular progenitor cells (VPCs). Although VPCs can differentiate along the chondrogenic, osteogenic, and adipogenic pathways, they require different inductive conditions compared with BM‐MSCs. These observations suggest to us that iMSCs, based on current widely used preparation protocols, do not represent a true alternative to primary MSCs isolated from BM. Furthermore, this study highlights the fact that high levels of expression of typical MSC markers such as CD73, CD90, and CD105 are insufficient to distinguish MSCs from other mesodermal progenitors in differentiated induced pluripotent stem cell cultures. stem cells 2019;37:754–765
Keywords: Mesenchymal stromal cells, Induced pluripotent stem cells, iPSC‐derived mesenchymal stromal cells, Vascular progenitor cells
Significance Statement.
The generation of mesenchymal stromal cells (MSCs) from iMSCs has been proposed as an alternative strategy for obtaining unlimited sources of these cells of defined quality and capable of meeting increasing demands for research and therapeutic applications. However, there are distinctions between primary MSCs and iMSCs in terms of phenotype and differentiation. This study shows that iMSCs share phenotypic traits with bone marrow‐derived (BM)‐MSCs in terms of standard marker expression but shows marked differences in gene expression, especially in genes associated with vascular progenitor cells. These differences may explain the altered differentiation propensity of human iMSCs compared with primary MSCs.
Introduction
The discovery of induced pluripotent stem cells (iPSCs) has been a key breakthrough in stem cell biology and regenerative medicine. The properties of pluripotent differentiation and long‐term self‐renewal in vitro 1 exhibited by these cells are of great significance and potential value in developing new therapeutic strategies without encountering ethical obstacles associated with embryonic stem cells (ESCs). iPSCs have the same genetic background as the somatic cells from which they were derived, giving rise to the prospect of generating constantly renewable immunoprivileged cell populations for autologous cell therapy 2. Another opportunity of value is provided by the prospect of using iPSCs to uncover the origin and pathogenesis of human diseases 3, 4.
One aspect of iPSC technology that has drawn considerable interest has been the generation of functional mesenchymal stromal cell (MSC) preparations. This has arisen because of the strong interest in the research and therapeutic use of these cells. MSCs are multipotent cells which have been extensively tested as therapeutic agents for the treatment of degenerative diseases, autoimmune disorders and wound repair 5, 6. In addition, the cells can be used to model human diseases, for example, musculoskeletal disorders 7 and nonhematological mesenchymal cancers 8. MSCs are easily accessible in postnatal tissues, including bone marrow (BM), adipose tissue, and umbilical cord blood 9. However, problems with the inconsistency of supply, variable quality, and phenotypic changes associated with long‐term expansion have represented impediments to effective translation 10. To overcome these barriers, iPSCs, with the advantages mentioned above, have the potential to become a more standardized alternative to primary tissues for MSC production.
Human pluripotent stem cell‐derived MSCs (PSC‐MSCs) were first differentiated from ESCs, referred to ESC‐MSCs, using a strategy that involved differentiation of cells within the embryoid body (EB) 11. In recent years, protocols have also been developed for the generation of MSCs from human iPSCs. Commonly used protocols included: (a) transferring iPSCs to a suspension culture to form EBs followed by culturing on gelatin‐coated plates 12; (b) directly seeding dissociated iPSC colonies in precoated culture surfaces such as gelatin, collagen type 1, or synthetic polymers 13, 14, 15; or (c) simply replacing iPSC culture medium with MSC medium 16. Furthermore, inhibitors of kappaB kinase epsilon (IKKi), the TGF‐β/Activin type I receptor inhibitor (SB431542), Activin A, and BMP4 have been used to enhance early mesodermal induction in culture 17, 18. iPSC derivatives were termed iMSCs, because of their MSC‐like immunophenotype, plastic adherent ability, and apparent multipotent differentiation potential in chondro‐, osteo‐, and adipogenesis, therefore meeting the minimal criteria defined by the International Society for Cellular Therapy (ISCT standard) 19.
However, questions arise relating to the phenotypic fidelity of iMSCs because, irrespective of the derivation methods used, there are differences between these cells and primary MSCs. For instance, iMSCs generally have less capacity for adipogenic and chondrogenic differentiation. Adipocytes and chondrocytes differentiated from iMSCs often express significantly lower levels of lineage marker genes such as PPAR‐γ and ADIPOQ (adipogenesis) and ACAN and COL2A1 (chondrogenesis) than those derived from primary MSCs, 20, 21, 22, 23, 24, 25. Conversely, iMSCs are markedly efficient in osteogenesis based on the evaluation of matrix production and osteogenic marker expression 26, 27, 28, 29. The altered differentiation propensity may hinder the application of iMSCs in current research and therapeutic strategies such as those involving primary MSCs for disease modeling and tissue regeneration.
Previous hierarchical analysis of gene expression profiles (GEPs) suggested that both iMSCs and primary MSCs have the characteristics of mesodermal lineage but are clearly not identical. Gene clustering analysis showed that, irrespective of the differentiation methods used, iMSCs formed a cluster which was close to but separated from the primary MSC group 20. Moreover, Frobel et al. demonstrated the dissimilarity in DNA methylation patterns between the two cell types 21. However, the significance of the distinct GEPs between iMSCs and primary MSCs, and the possible relationship to differences in multipotency remain poorly understood.
To answer these questions, we compared the differentiation ability, immunophenotype, and GEPs between multiple iMSCs and BM‐MSC lines by looking at key genes representing different mesodermal stem cell populations. The phenotype, multipotency, and GEP of iMSCs in serial passages were also assessed to evaluate the impact of culture expansion. Our results showed that iMSCs demonstrated equivalent osteogenicity but less adipogenicity and chondrogencity when compared with BM‐MSCs. The GEPs of the two cell groups were significantly different and such distinction was maintained consistently during culture expansion, suggesting that both cell types represented different mesodermal progenitors and that iMSCs were, in fact, more similar to vascular progenitor cells (VPCs). Previous findings showed that even though the cell plasticity of VPCs endows them with capacities to undergo chondro‐, adipo‐, and osteogenesis, specific conditions are required to fully induce the lineage transformation of the cells, especially for chondrogenesis and adipogenesis 30, 31, 32, 33, 34. This may explain why iMSCs with VPC background showed an MSC‐like multipotent potential but displayed a functional inadequacy in a MSC preferred differentiation environment. In addition, a partially overlapped surface profile between VPCs and MSCs 35, 36 can make them indistinguishable by examining the markers selected in the ISCT standard. Taken together, our results suggested that it is insufficient to apply the ISCT standard alone to precisely define MSC populations derived from iPSCs. To reduce the risk of lineage misidentification, it is necessary to further validate the cell identity with various mesodermal progenitor markers.
Materials and Methods
Ethical Approvals
To isolate primary MSCs, BM was harvested from the iliac crest of three healthy donors with approval from both the NUI Galway Research Ethical and Galway University Hospitals Clinical Research Ethics Committees. Skin fibroblasts for generation of iPSC‐3 were obtained from a healthy donor with ethical approval by the University Hospital Galway research ethics committee.
Cell Lines
BM‐MSC lines from three healthy donors were isolated and cultured as previously described 37. iPSC‐1 and iPSC‐2 were kindly provided by Dr. Gareth Sullivan (University of Oslo) and Professor James Dutton (University of Minnesota), respectively. The conditions used for cell expansion and iPSC characterization protocols are included in Supporting Information.
Generation of iMSCs
The EB outgrowth method was applied as described previously with minor modifications. EBs were maintained in suspension culture for 8 days, followed by adherent culture on gelatin‐coated plates for outgrowth of differentiated cells. During this stage, EB formation medium was used which contained 80% knockout Dulbecco's modified Eagle's medium (KO‐DMEM, Invitrogen, Glasgow, UK), 20% fetal bovine serum (FBS; Sigma), 1 mM l‐glutamine (Invitrogen, Glasgow, UK), 14 μM β‐mercaptoethanol (Invitrogen, Glasgow, UK), and 1% nonessential amino acids (NEAA, Invitrogen, Glasgow, UK) 11. When outgrown cells formed confluent areas, they were passaged to new gelatin‐coated flasks and remaining EB clumps were removed using a 40 μm cell strainer. During this maintenance phase, cells were fed with medium comprising KO‐DMEM, 10% FBS, 1 mM l‐glutamine, and 1% NEAA 38. Finally, when the majority of the cells displayed an MSC‐like morphology, they were transferred to noncoated culture flasks with MSC culture medium (passage 0 [P0]). A continued homogeneous transition was achieved in each iMSC culture by eliminating nonadherent cells.
Proliferative Potential and Immunophenotypic Assessment
Cell growth rate was determined as cumulative population doubling (CPD) 39.
For immunophenotyping, flow cytometry analysis was performed using the human MSC analysis kit (BD Bioscience, San Diego, CA) according to the manufacturer's protocol. Individual antibodies against CD11b, CD19, CD45, CD34 (BD Bioscience, San Diego, CA), and human leukocyte antigen (HLA)‐DR (Thermo Fisher Scientific, MD) were also included in the analysis. Samples were analyzed on the BD FACS Canto II (BD Biosciences) and data analyzed using FlowJo 6.0.
Trilineage Differentiation
MSCs were subjected to chondro‐, adipo‐, and osteogenesis as described previously with minor modifications 37. Differentiated cells were analyzed by histological and immunocytochemical staining. The differentiation efficiency was also assessed by quantitation of related biomarkers. Detailed experimental procedures can be found in Supporting Information.
GEP Analysis
Real‐time quantitative polymerase chain reaction (qPCR) and semiquantitative PCR were performed to evaluate the GEP of the MSCs. Protocols for sample preparation and PCR analysis can be found in Supporting Information. Information relating to primers is given in Supporting Information Table S1.
Statistical Analysis
All values are presented as a mean ± SEM. Two‐tailed Student's t test was performed to detect the statistical differences, in gene expression, immunophenotype, and differentiation efficiency, between BM‐MSCs and iMSCs at the same passage. To detect differences in expression variation of the selected gene of the same cell type during culture expansion, One‐way analysis of variance followed by Bonferroni (compare all pairs of columns) correction were performed. p < .05 was considered statistically significant.
Results
Generation and Characterization of iPSC Lines
Three iPSC lines were included in this study, iPSC‐1, 2, and 3. All lines were derived from healthy donors using the standard retroviral reprogramming method 38. The pluripotent state of iPSC‐1 was validated previously 7 and characterization of iPSC‐2 and 3 included assessment of typical ESC‐like morphology (Supporting Information Fig. S1A), confirmation of a normal karyotype (Supporting Information Fig. S1B) and immunostaining to identify positive expression of key pluripotent markers OCT4, SOX2, NANOG, and TRA1‐60 (Supporting Information Fig. S1C). Furthermore, teratoma assay and EB formation were applied to examine the functional pluripotency of iPSC‐2 and 3, respectively. Histological analysis of teratoma sections derived from iPSC‐2 showed that the cells were able to generate different tissue types from three germ layers (Supporting Information Fig. S1D), whereas immunostaining of day 35 EBs confirmed pluripotency of iPSC‐3 with positive staining for α‐SMA (mesoderm), α‐Fetoprotein (endoderm), and β‐3‐tubulin (ectoderm; Fig. 1A–1E).
Figure 1.
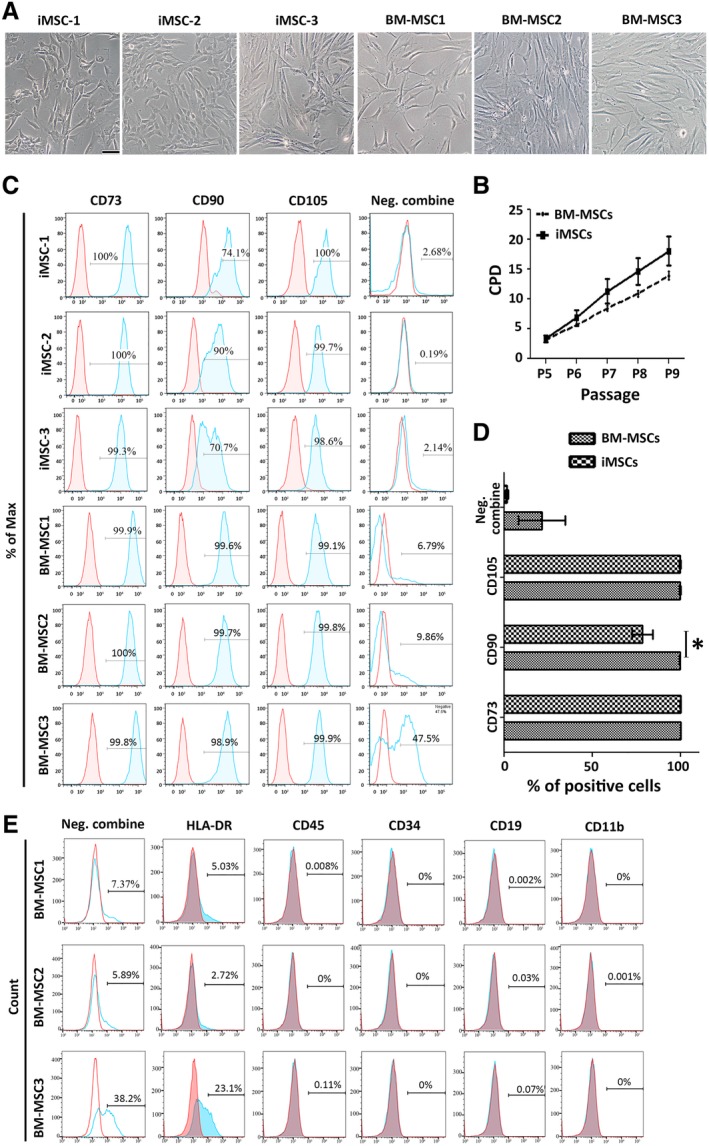
Preliminary characterization of induced mesenchymal stromal cells (iMSCs) and bone marrow (BM) derived‐MSCs. (A): Cellular morphology. Scale bar = 50 μm. (B): Proliferation capacity of each cell group compared based on the cumulative population doublings (CPD) (mean ± SEM). See Supporting Information Figure S2 for the growth rate of each line. (C–E): Surface profile analysis of the cells (P5) using flow cytometry. Red histograms represent isotype controls with the blue overlays representing each antigen; percentages of positive cells are shown within histograms. (C): Positive markers included CD90/73/105. The negative (Neg.) combine is a mixed antibody pool assessing CD45, CD34, CD19, CD11b, or HLA‐DR positive cells. (D): Summary of the immunophenotype of BM‐MSC and iMSC lines as detected by flow cytometry. Data represent mean ± SEM of n = 3; *, p < .05. (E): Individual antibody staining of hematopoietic populations to identify the positive signal shown in the Neg. combine group. See also Supporting Information Figure S3.
Phenotypic Comparison of iMSCs and BM‐MSCs
Since the EB outgrowth method is the most widely studied for the differentiation of iPSCs to MSCs 17, it was used in this study. In addition, three primary MSC lines were obtained from BM aspirates from the iliac crest of healthy donors. P5 was chosen as the earliest time point for analyzing iMSCs because earlier passage cells showed heterogeneous morphology and limited plastic adherence. The impact of serial passages on phenotype, differentiation and GEP was studied between P5 and P8 in both iMSCs and BM‐MSCs.
Preliminary analysis was carried out to compare the morphology, proliferative activity, and surface antigen expression between the two groups. Cell morphology was similar across all preparations (Fig. 1A) and there was no significant difference between iMSCs and BM‐MSCs in terms of their proliferative capacity (Supporting Information Fig. S2), both showing similar mean CPD (Fig. 1B). Immunophenotypic analysis confirmed that both cell groups expressed standard MSCs surface markers (CD73/90/105; Fig. 1C). Some preparations showed some positive expression of hematopoietic markers (Fig. 1C). When this was observed, we undertook a detailed analysis and found that it was specifically associated with HLA‐DR expression only and the cells were negative for other hematopoietic markers (CD11b/34/19/45; Fig. 1E; Supporting Information Fig. S3B, S3C). We concluded that the preparations did not contain hematopoietic progenitors and that the induction of HLA‐DR expression was a consequence of expansion in the FGF2‐containing medium, as described previously 40. We also noticed that iMSCs contained significantly less CD90+ cells than BM‐MSCs (p = .0238; Fig. 1D) and a bimodal pattern of CD90 expression was observed in iMSC preparations only (Fig. 1C). This appeared to be a consequence of changed CD90 expression in some cultures with passaging (Supporting Information Fig. S3A).
Functional Comparison of iMSCs and BM‐MSCs
The capacity for trilineage differentiation in both cell groups was assessed using standard differentiation conditions. Consistent with previous findings 20, 21, we observed that iMSCs demonstrated limited adipogenic differentiation. In adipocytes derived from BM‐MSCs, Oil Red O‐stained lipid droplets (LDs) were much larger compared with those from iMSCs (Fig. 2A). Consistently, the level of triacylglycerol (TAG) was significantly lower in iMSCs (20‐fold, p = .0056, Fig. 2B). Interestingly, the higher DNA content seen in differentiated iMSC cultures suggests potential differences in cell cycle regulation and proliferative activity between the two groups (Fig. 2C). The significantly lower ratio of TAG per DNA suggested a disrupted TAG synthesis in iMSCs compared with BM‐MSCs (40‐fold, p = .003, Fig. 2D). This difference remained in both early passage (P5; p = .006) and late (P8; p = .004) cultures (Fig. 2E), further indicating an incomplete adipogenic capacity of iMSCs.
Figure 2.
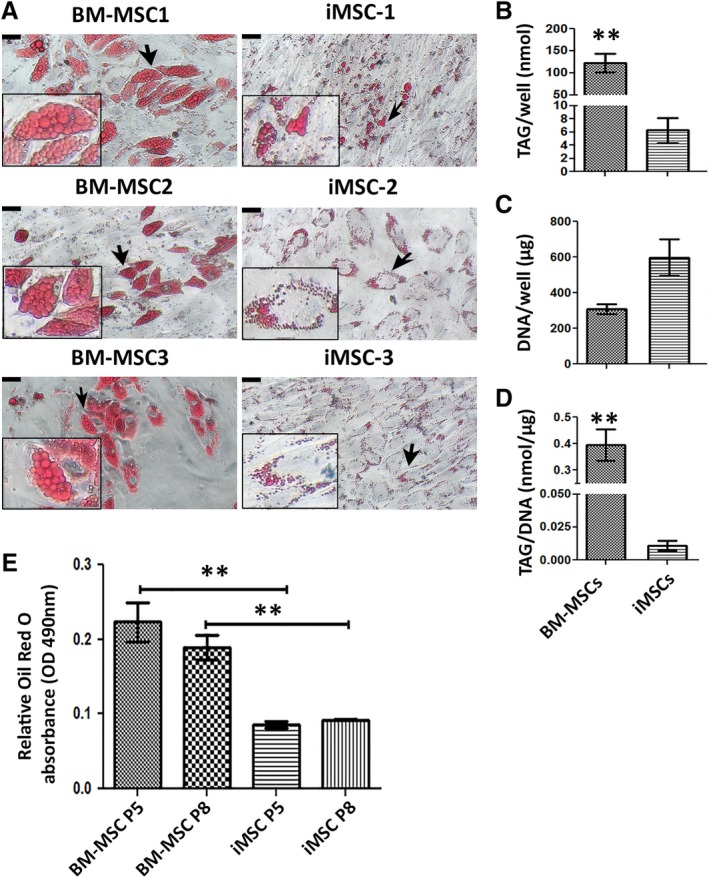
Adipogenic capacity of induced mesenchymal stromal cells (iMSCs) and bone marrow (BM) derived‐MSCs. (A): Morphologies of formed lipid droplets shown by staining with Oil Red O solution. Representative images of each cell line (P5) are shown. Insets are magnified images of the areas indicated by black arrows. Scale bar = 22.7 μm. (B–D): The amount of TAG and DNA and the ratio of TAG per DNA between the BM‐MSCs and iMSCs groups are shown. Data in each bar chart represents mean ± SEM of n = 3; **, p < .01. (E): Quantification of lipid elaborated by extraction of Oil Red O stain and measuring absorbance at O.D. 490 nm, n = 3 wells for each group. Results are mean ± SEM; **, p < .01.
Detailed analysis of chondrogenic potential revealed striking differences between iMSCs and BM‐MSCs. Toluidine blue staining showed an abundant glycosaminoglycan (GAG)‐rich extracellular matrix (ECM) in 35 days chondrogenic pellets of BM‐MSC and very limited GAG staining in iMSC pellets (Fig. 3A). Measurement of the accumulated GAG to DNA ratio (Fig. 3B) showed that BM‐MSCs produced 6.7‐fold higher GAG at P5 (p = .008) and 14‐fold higher at P8 (p = .1).
Figure 3.
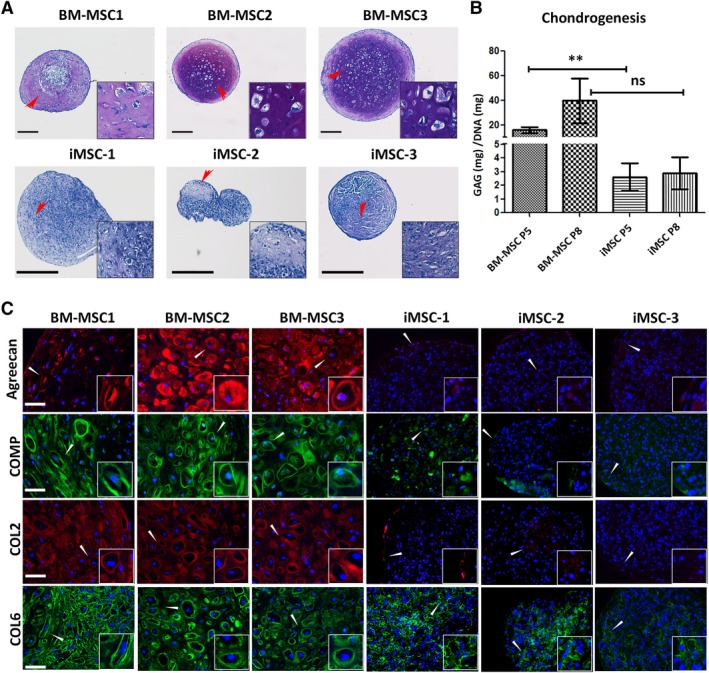
Comparison of chondrogenic pellets derived from induced mesenchymal stromal cells (iMSCs) and bone marrow (BM) derived‐MSCs. (A): Representative images of toluidine blue stained sections demonstrate the presence of GAG within the chondrogenic matrix of the pellets derived from MSCs at P5. Scale bar = 250 μm. (B): GAG quantification relative to DNA content of tested pellets. Data represents mean ± SEM of n = 3; **, p < .01. (C): Representative images of immunofluorescent staining for cartilaginous matrix components (aggrecan [red], COMP [green], COL2 [red], and COL6 [green]) on the pellets differentiated from P5 MSCs. Cell nuclei were stained with DAPI (blue). Scale bar = 50 μm. Insets displayed in (A) and (C) are magnified images of the areas indicated by the arrowheads.
Immunohistological analysis of specific ECM components underscored the differences in chondrogenic response between BM‐MSCs and iMSCs (Fig. 3C). This showed that collagen type II (COL2), collagen type VI (COL6), cartilage oligomeric matrix protein (COMP) and aggrecan were synthesized at a high level and distributed throughout the ECM in BM‐MSC derived pellets. However, aggrecan, COL2, and COMP, hallmarks of a cartilaginous phenotype, were absent or minimally expressed in iMSC pellets. COL6 was the only ECM component that appeared to be expressed at a high level in iMSC pellets and this was not uniformly distributed. Taken together, these results suggested a very limited chondrogenic proclivity of iMSCs.
In contrast, both iMSCs and BM‐MSCs demonstrated strong osteogenic capacity as measured by calcium deposition, osteocalcin synthesis, and assembly of a collagen type I (COL1)‐rich ECM. Osteocalcin was expressed abundantly in cell aggregates maintained in osteogenic medium for 35 days (Fig. 4A). The osteogenic cultures from both cell groups also showed similar abundant accumulation of calcium measured relative to total DNA content (Fig. 4B). In addition, analysis of COL1, the major collagen in bone 41, showed a richer accumulation in iMSC‐derived cultures compared with BM‐MSC‐derived cultures (Fig. 4C).
Figure 4.
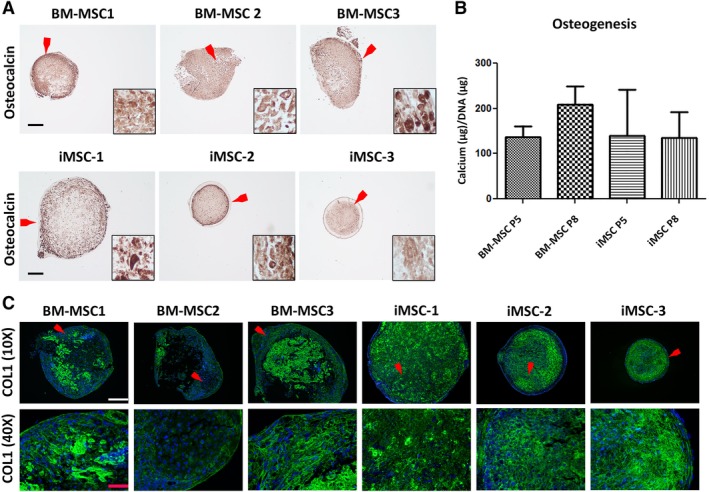
Comparison of osteogenic pellets derived from induced mesenchymal stromal cells (iMSCs) and bone marrow (BM) derived‐MSCs. (A): Representative images of immunohistochemical staining for detection of osteocalcin in pellets differentiated from P5 MSCs. Cell nuclei were stained with hematoxylin. Insets are magnified images of the areas indicated by the red arrows. Scale bar = 250 μm. (B): Calcium produced and standardized to DNA levels in osteogenic pellets derived from iMSCs and BM‐MSCs (P5 and P8) is shown. Data represents mean ± SEM of n = 3. (C): Representative immunohistochemistry demonstrates the expression and distribution of COL1 (green) in pellets derived from P5 MSCs (top row; white scale bar = 250 μm). Corresponding magnified images of COL1 staining from the areas indicated by the red arrowheads (bottom row; red scale bar = 50 μm).
iMSCs Were Derived Through Mesodermal Commitment
To interrogate the possibility that lack of trilineage differentiation capacity could be associated with the presence of heterogeneous mixtures of cells of nonmesodermal lineage, the expression of genes representing ectoderm and endoderm was assessed (Fig. 5). PCR analysis showed no expression of PAX6 and SOX1 indicating that cells of neuroectodermal lineages were not present in iMSCs and BM‐MSCs 42. Similarly, the absence of FOXA2 and CXCR4 indicated that no definitive endoderm was present. We did note, however, that the early regulators of endodermal differentiation, SOX17 and SOX7 43, were detected in iMSCs in P5 but absent in later passage cells (Fig. 5). These results suggested that differences in phenotype or differentiation potential between MSCs derived from iPSC and from BM could not be attributed to the presence of nonmesodermal lineage cells.
Figure 5.
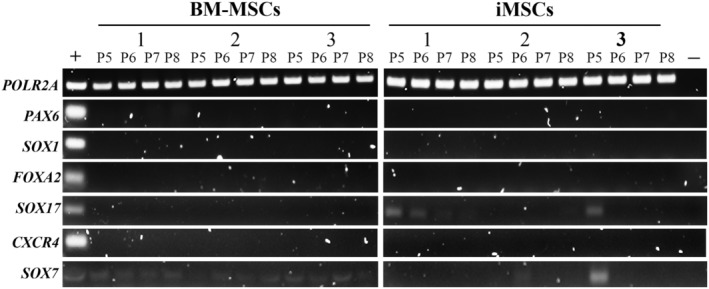
Ectodermic and endodermic gene profiles in induced mesenchymal stromal cells (iMSCs) and bone marrow‐derived MSCs by polymerase chain reaction (PCR) analysis. Cells were harvested from P5 to P8. Representative bands of the PCR products in 2% agarose gel electrophoresis are shown. “+” represents mixed RNA samples extracted from day 35 EBs derived from induced pluripotent stem cells as the positive control. “−” represents the negative control for each PCR reaction (nontemplate controls were performed for each PCR reaction).
We next considered whether iMSCs and BM‐MSCs differed in terms of early or late mesodermal commitment. Previous studies have suggested that iMSCs are less mature than BM‐MSCs and that this may influence differentiation capacity 44. To verify this, we selected a set of markers that would reveal the degree of mesodermal commitment for each group. We compared the expression levels of MIXL1, MESP2, and CDH1, all of which are involved in the regulation of early mesodermal commitment and differentiation. MIXL1 is mainly expressed in nascent mesoderm followed by downregulation at the point of commitment 45, 46. MESP2 is activated on initiation of segmentation in paraxial mesoderm (PM) 47. CDH1 is a marker of endomesoderm and its expression pattern is reduced during mesogenesis but maintained for endogenesis 48, 49.
There was no significant difference in expression of MIXL1 in iMSCs and BM‐MSCs over several passages in culture (from P5 to P8, Fig. 6A). BM‐MSCs showed higher expression of MESP2 compared with iMSCs across all passages. These differences were not significant except for P5 (p = .0068; Fig. 6B). This may suggest a PM origin of BM‐MSCs. In contrast to BM‐MSCs, iMSCs expressed significantly lower levels of CDH1 at P7 (20‐fold, p = .0221) and P8 (10‐fold, p = .0028; Fig. 6C). The marked change may be associated with maturation to a mesoderm state. Conversely, the high expression of both CDH1 and SOX7 in BM‐MSCs (Fig. 5) may infer the existence of an endodermal precursor which is in line with the potential of these cells to trans‐differentiate into endodermal lineages 50. Overall, a similarly low level of MIXL1 detected on both cell types implies their postmesodermal initiation state. Contrasting expression patterns of MESP2 indicate that iMSCs and BM‐MSCs may arise from different mesodermal subfractions.
Figure 6.
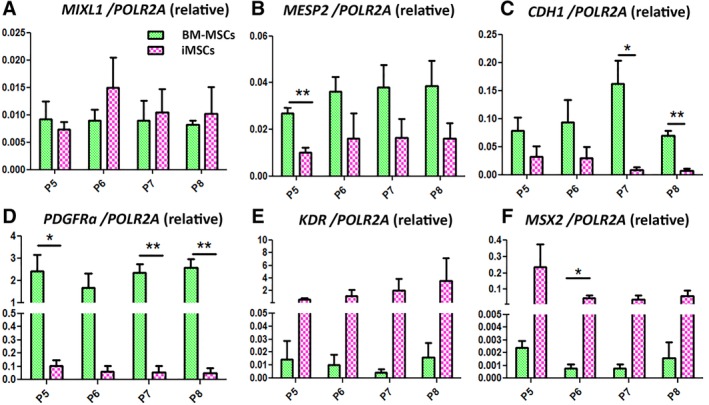
RT‐polymerase chain reaction analysis of mesodermal gene expression profiles between induced mesenchymal stromal cells (iMSCs) and bone marrow derived‐MSCs from P5 to P8. Data were normalized to POLR2A expression. Results are mean ± SEM. *, p < .05; **, p < .01.
iMSCs and BM‐MSCs Represent Distinct Mesodermal Progenitors
Next, we looked at the expression of genes that are widely used to distinguish subpopulations of mesodermal progenitors. We noted a dramatically higher level of PDGFRɑ expression by BM‐MSCs compared with iMSCs at all passages: P5 (24‐fold, p = .036), P6 (28‐fold, p = .0702), P7 (40‐fold, p = .0042), and P8 (55‐fold, p = .0028; Fig. 6D). A gradual diminution in the level of PDGFRɑ (39% over time) was also seen in iMSCs on passaging. Conversely, KDR expression was greater in iMSCs than in BM‐MSCs from 4‐fold (P5, p = .07) to 223‐fold (P8, p = .1; Fig. 6E). PDGFRα and KDR are restricted to PM and lateral plate mesoderm (LPM), respectively, during mesogenesis 51, 52. Recently, PDGFRα has emerged as a promising marker for isolation of primary MSCs from BM and heart 51. Overall, the very low level of PDGFRα in iMSCs suggests that MSC‐like cells are rare in iMSC cultures whereas the high expression of KDR suggests that iMSCs may originate in LPM.
In both human and mouse models, ESC‐derived KDR+ stem cells have been recognized as hemangioblasts, giving rise to hematopoietic lineages and VPCs 53. Based on flow cytometry analysis, the hematopoietic markers were expressed at extremely low levels in iMSC lines (Fig. 1; Supporting Information Fig. S3). Therefore, we speculate that KDR high iMSCs may be VPC‐like cells. The significantly stronger expression of MSX2 in iMSCs further supports this conjecture (Fig. 6F). Goupille et al. used nlacZ as a reporter for Msx2 expression in a transgenic mouse model and showed that this gene was expressed prominently in the vascular smooth muscle layer of peripheral arteries as well as in embryonic and adult aorta endothelium 54. Our results showed that the overall expression levels of MSX2 in iMSC group were 37.5‐ (P8) to 100‐fold (P5) higher than the levels in BM‐MSCs. Statistical significance was achieved by P6 (65.7‐fold, p = .046; Fig. 6F). In addition, the consistently high level of MSX2 in iMSCs may suggest a stable VPC phenotype 55. Furthermore, the unique GEP of iMSCs suggests a potential differentiation propensity to endothelial precursors and vascular smooth muscle progenitors 56.
Taken together, the results suggest that iMSCs are mature mesodermal progenitors with a VPC‐like background and that iMSCs and BM‐MSCs represent different mesodermal progenitor populations (Fig. 7). The proposal that iMSCs have a VPC‐like background provides a rationale for their functional divergence from BM‐MSCs.
Figure 7.
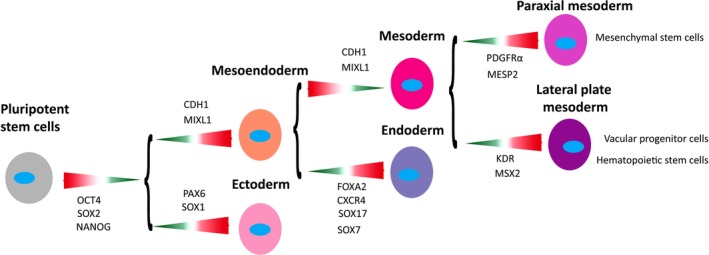
Schematic roadmap of lineage ontogenesis from pluripotent stem cells to different mesenchymal progenitors. Key genes associated with each developmental stage are shown. Lineage choice‐associated expression patterns are indicated by the colored arrows, where green with narrow end represents downregulation and red with open end upregulation.
Discussion
In this study, the iPSC derivatives were termed as iMSCs as the cells possessed the characteristics of MSCs as described by the ISCT standard (Figs. 1, 2, 3, 4). However, the GEP analysis performed argued that these two cell groups actually represented different mesodermal subfractions, where iMSCs were VPC‐like cells (PDGFRα low KDR high MSX2 high) and BM‐MSCs were a product of the mesengenic process (PDGFRα high KDR low MSX2 low; Fig. 6). The current understanding of VPCs provides the rationale for explaining inconsistencies between iMSCs and BM‐MSCS, that is, the compliance of iMSCs to the ISCT‐based cell surface phenotype and noncompliance with respect to differentiation capacity.
Firstly, flow cytometry analysis showed that iMSCs carried a BM‐MSC‐like surface profile (Fig. 1C). Previous findings proved that VPCs positively expressed not only CD73/90/105 but also CD44/166 and STRO‐1 57, 58, 59, 60, 61, 62. Due to the partially overlapping immunophenotype found, using these traditional MSC‐associated antigens contributes to an inability to discriminate iMSC with a VPC‐like background from the MSC population. Interestingly, the coexistence of CD90high and CD90low subsets was captured exclusively in iMSC lines (Fig. 1C). Of note, this immunophenotype was also observed in PSC‐MSC lines from other studies 13, 15, 29, 63, 64, 65. We found that continued expansion could induce a dynamic expression pattern of CD90 in the cells (Supporting Information Fig. S3A). Although Moraes et al. showed that depletion of CD90 in BM‐MSCs stimulated their osteogenic ability 66, this association was not found in iMSCs generated in our study (Fig. 4B). An alternate role of CD90 in adult cardiac vascular stem cells (CVSCs) was investigated by Gago‐Lopez et al., who reported that CD90− CVSCs expressed higher levels of genes involved in stemness when compared with CD90+ CVSCs 67. Whether a similar influence of CD90 variation can be found in iMSCs will need to be elucidated.
Secondly, both cell groups showed marked potential in trilineage differentiation. However, in contrast to BM‐MSCs, iMSCs were found to have significant osteogenic capacity; chondrogencity and adipogenicity were minimal (Figs. 2, 3, 4). This differentiation phenotype corresponds with the known fate plasticity of VPCs. It has been shown that VPCs display osteogenic capacity, which can be differentiated into osteoblasts in a primary MSC preferred condition, using β‐glycerol phosphate, ascorbic acid, and dexamethasone 30, 68, 69. However, for chondrogenesis, the cells require an additional treatment of BMP2, BMP4, or TGF‐β2 to complete a mesenchymal transition by interacting with members of the type I TGF‐β receptor 30, 32, 70, 71. In our study, TGF‐β3, as one of the commonest chondrogenic inducer for primary MSCs, was used for chondrogenesis of iMSCs. Although previous ligand‐receptor binding analysis demonstrated binding ability between TGF‐β3 and the type I receptor, this ligand has stronger binding preference and affinity to the type II receptor 72. Uniquely, CD105, a type III TGF‐β receptor, also binds TGF‐β3 with high affinity but fails to bind TGF‐β2, BMP2, and BMP4 73, 74. The distinct receptor preference of TGF‐β3 may explain the insufficient chondrogenesis of iMSCs. With reference to adipogenesis, Zhao et al. showed that tissue derived KDR+ CD31−CD34− cells, with characteristics of VPCs, were capable of adipogenesis 31, 33. Zimmerlin et al. compared the adipogenic capacity among vascular lineages and showed that the differentiation efficiency was low in CD31+endothelial lineages but high in CD146+smooth muscle cells or pericytes 34. These findings confirmed that VPCs have adipogenic potential but also implied that a pre‐VSMPs derivation of iMSCs may help to enhance their adipogenic efficiency. Taken together, the partially overlapped multipotency and disparate requirements for differentiation conditions between VPCs and MSCs provide the rationale to explain why iMSCs with a VPC‐like background showed the potential for trilineage differentiation but were dissimilar in differentiation efficiency when compared with BM‐MSCs.
GEP examination is a very important strategy for verifying cell type after differentiation of stem cells. In this study, redefining the lineage type of iMSCs based on their GEPs helped to explain the functional variation between the cells and BM‐MSCs. However, the number of publications including both GEP and functional comparison between PSC‐MSCs and primary MSCs is limited. Data reported previously indicated that PSC‐MSCs with functional inequivalence to primary MSCs commonly expressed high levels of genes associated with disparate mesodermal progenitors but exceedingly low levels of key MSC genes. Barbet et al. showed that in comparison to BM‐MSCs, ESC‐MSCs derived by a spontaneous differentiation method carried significantly higher levels of HAND1 (LPM marker) 75, NKX2‐5, and FLT4 (cardiovascular progenitor genes) 44, 76. In Billing et al.'s study, mesengenic derivation of ESCs directly in MSC maintenance medium led to the high expression of NKX2‐5 and FGF12 (a promoter of vascular smooth muscle commitment) 77, which were exclusively detected in the ESC‐MSCs 78. Diederichs and Tuan generated MSCs using these methods on iPSCs and demonstrated that KDR was significantly higher in the resultant cells than in BM‐MSCs. In addition, their data showed that the iPSC derivatives also expressed significantly higher MCAM, the elevation of which is associated with vascular smooth muscle transdifferentiation of primary MSCs 20, 79. Inefficient mesengenic induction was also seen even when the process was derived in a controlled manner. Mesengenesis of iPSCs by inhibiting TGF‐β/Activin/Nodal signaling with SB‐431542 resulted in a greater expression of VEGFR1 (FLT1), a marker of vascular endothelial cells in iMSCs than in BM‐MSCs 28, 80. Mesengenic induction using FGF2 and PDGF followed by cell sorting for a CD105+CD24− population also gave rise to a heterogeneous culture with greater expression of vascular endothelial markers (CL‐P1 81 and FAM43A 82) and neuroprogenitor markers (EFNB3 83, SLITRK5 84, and CKAP2L 85) in ESC‐MSCs than in BM‐MSCs 86. Moreover, irrespective of the derivation methods used, extremely low expression of MSC‐associated genes was often detected in these PSC‐MSCs, including IL6 20, 21, 28, BST1 21, 44, 86, SFRP4 21, 44, and VCAM1 21, 44, 86. The mismatches in GEPs between PSC‐MSCs and primary MSCs observed in other studies strongly support our speculation on the lack of efficiency of the commonly used differentiation methods in inducing the mesengenic specification of PSCs. Subsequently, this uncontrolled lineage commitment can lead to a generation of nonspecific mesodermal progenitor populations.
Conclusion
Our data clearly demonstrated that the ISCT standard is insufficient to precisely distinguish MSCs from other mesodermal progenitors. Distinct GEPs can be overlooked as a result of partially overlapping cellular characteristics between MSCs and other mesodermal progenitors. Inaccurate lineage identification as a result of incomplete characterization can compromise the ability to compare and contrast study outcomes and may be responsible for functional discrepancies of iMSC seen in other related studies. Additional examination of markers representing different mesodermal progenitors should be performed as a standard process for tracking differentiation and verifying the iMSC identity before used in future investigations.
On the other hand, to further validate the VPC background of iMSCs shown here, future analysis can be conducted to compare differentiation propensity, immunomodulatory activity and GEPs among iMSCs, BM‐MSCs, and tissue‐derived VPCs. It will also be of interest to investigate whether the cell origin of iPSCs can influence their mesengenic efficiency.
Author Contributions
M.X.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; G.S.: provision of study material, data analysis and interpretation, manuscript writing; M.M.: conception and design, data analysis and interpretation, manuscript writing; F.B.: conception and design, data analysis and interpretation, manuscript writing, final approval of the manuscript.
Disclosure of Potential Conflicts of Interest
M.M.'s spouse is a compensated advisor for Orbsen and has compensated shares in Osiris Therapeutics. F.B. is an uncompensated shareholder of Orbsen Therapeutics, Ltd. and Osiris Therapeutics, Inc. The other authors indicated no potential conflicts of interest.
Supporting information
Appendix S1: Supplementary Information
Figure S1 Characterization of iPSC‐2 and 3. (A) Representative cell morphology of iPSC colonies. Scale bar, 400 μm (B) G‐banding analysis confirmed normal karyotype of the cells. (C) Immunofluorescent staining demonstrated that iPSC positively expressed pluripotent markers (OCT4, SOX2, NANOG and TRA1‐60). White scale bar, 500 μm. (D) iPSC‐2 derived teratoma in vivo and hematoxylin and eosin‐stained teratoma sections. Detected tissue types included endoderm (glands), mesoderm (cartilage), and ectoderm (pigmented cells). Red scale bar, 200 mm. (E) EB formation was applied to induce spontaneous differentiation of iPSC‐3 in vitro. Representative images of immunofluorescent staining for germ layer markers on day 35 EB sections are shown, including α‐SMA (Mesoderm), AFP (Endoderm) and β‐3‐tublin (Ectoderm). Black scale bar, 400 μm and white scale bar, 500 μm.
Figure S2 Comparison of the growth rates of iMSC and BM‐MSC lines. The symbols embedded in each line indicate the time points of passage. See also Figure 1B.
Figure S3 Effects of serial passage on immunophenotyping of iMSCs and BM‐MSCs from P5 to P8. (A) Diagrams demonstrate the fraction of the cells expressing CD90, CD105, CD73 and Neg. combine on passaging. (B) Diagrams display the percentage of the cells positive for hematopoeitic markers during the expansion. (C) In iMSC group, an increased Neg. combine population was exclusively detected in iMSC‐3 from P5 to P8 (Fig. S3‐A). Flow cytometry analysis was conducted to specifically examine the hematopoeitic antigen expression profile of the cells at P8. Red histograms represent isotype controls with the blue overlays representing each antigen; percentages of positive cells are shown within histograms. See also Figure 1C and D.
Acknowledgments
This work was supported by Science Foundation Ireland (SFI 09/SRC.B1794) and the Irish Research Council (GOIPG/2014/96). This study has also received funding from the European Union's Seventh Framework Programme for Health under grant agreement number 223298. We thank Dr. Gareth Sullivan (Department of Molecular Medicine, Hybrid Technology Hub—Centre of Excellence, Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway & Oslo University Hospital) and Professor James R. Dutton (University of Minnesota) for providing the iPSCs and Dr. Linda Howard (National University of Ireland, Galway) for providing the skin fibroblast for generation of iPSC‐3. We acknowledge the help of Ms. Niamh Duffy, Regenerative Medicine Institute, NUI Galway for her help with cell culture. We also acknowledge the facilities and technical assistance of the Flow Cytometry Facility, the Centre for Microscopy & Imaging (www.imaging.nuigalway.ie).
Data Availability Statement:The data that support the findings of this study are available from the corresponding author upon reasonable request.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 2. Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol 2016;17:194–200. [DOI] [PubMed] [Google Scholar]
- 3. Suh W. A new era of disease modeling and drug discovery using induced pluripotent stem cells. Arch Pharm Res 2017;40:1–12. [DOI] [PubMed] [Google Scholar]
- 4. Sabapathy V, Kumar S. hiPSC‐derived iMSCs: NextGen MSCs as an advanced therapeutically active cell resource for regenerative medicine. J Cell Mol Med 2016;20:1571–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Munir H, McGettrick HM. Mesenchymal stem cell therapy for autoimmune disease: Risks and rewards. Stem Cells Dev 2015;24:2091–2100. [DOI] [PubMed] [Google Scholar]
- 6. Nakamura Y, Ishikawa H, Kawai K et al. Enhanced wound healing by topical administration of mesenchymal stem cells transfected with stromal cell‐derived factor‐1. Biomaterials 2013;34:9393–9400. [DOI] [PubMed] [Google Scholar]
- 7. Xu M, Stattin EL, Shaw G et al. Chondrocytes derived from mesenchymal stromal cells and induced pluripotent cells of patients with familial osteochondritis dissecans exhibit an endoplasmic reticulum stress response and defective matrix assembly. Stem Cells Translational Medicine 2016;5:1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garcia‐Castro J, Trigueros C, Madrenas J et al. Mesenchymal stem cells and their use as cell replacement therapy and disease modelling tool. J Cell Mol Med 2008;12:2552–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Williams AR, Hare JM. Mesenchymal stem cells: Biology, patho‐physiology, translational findings, and therapeutic implications for cardiac disease. Circ Res 2011;109:923–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zaim M, Karaman S, Cetin G et al. Donor age and long‐term culture affect differentiation and proliferation of human bone marrow mesenchymal stem cells. Ann Hematol 2012;91:1175–1186. [DOI] [PubMed] [Google Scholar]
- 11. Xu C, Jiang J, Sottile V et al. Immortalized fibroblast‐like cells derived from human embryonic stem cells support undifferentiated cell growth. Stem Cells 2004;22:972–980. [DOI] [PubMed] [Google Scholar]
- 12. Tang M, Chen W, Liu J et al. Human induced pluripotent stem cell‐derived mesenchymal stem cell seeding on calcium phosphate scaffold for bone regeneration. Tissue Eng Part A 2014;20:1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Villa‐Diaz LG, Brown SE, Liu Y et al. Derivation of mesenchymal stem cells from human induced pluripotent stem cells cultured on synthetic substrates. Stem Cells 2012;30:1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei H, Tan G, Manasi et al. One‐step derivation of cardiomyocytes and mesenchymal stem cells from human pluripotent stem cells. Stem Cell Res 2012;9:87–100. [DOI] [PubMed] [Google Scholar]
- 15. Hynes K, Menicanin D, Mrozik K et al. Generation of functional mesenchymal stem cells from different induced pluripotent stem cell lines. Stem Cells Dev 2014;23:1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou L, Luo Y, Chen M et al. A simple method for deriving functional MSCs and applied for osteogenesis in 3D scaffolds. Sci Rep 2013;3:2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luzzani CD, Miriuka SG. Pluripotent stem cells as a robust source of mesenchymal stem cells. Stem Cell Rev 2017;13:68–78. [DOI] [PubMed] [Google Scholar]
- 18. Deng P, Zhou C, Alvarez R et al. Inhibition of IKK/NF‐κB signaling enhances differentiation of mesenchymal stromal cells from human embryonic stem cells. Stem Cell Rep 2016;6:456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dominici M, Le Blanc K, Mueller I et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006;8:315–317. [DOI] [PubMed] [Google Scholar]
- 20. Diederichs S, Tuan RS. Functional comparison of human‐induced pluripotent stem cell‐derived mesenchymal cells and bone marrow‐derived mesenchymal stromal cells from the same donor. Stem Cells Dev 2014;23:1594–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frobel J, Hemeda H, Lenz M et al. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Rep 2014;3:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen YS, Pelekanos RA, Ellis RL et al. Small molecule mesengenic induction of human induced pluripotent stem cells to generate mesenchymal stem/stromal cells. Stem Cells Translational Medicine 2012;1:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang R, Zhou Y, Tan S et al. Mesenchymal stem cells derived from human induced pluripotent stem cells retain adequate osteogenicity and chondrogenicity but less adipogenicity. Stem Cell Res Ther 2015;6:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moslem M, Eberle I, Weber I et al. Mesenchymal stem/stromal cells derived from induced pluripotent stem cells support CD34(pos) hematopoietic stem cell propagation and suppress inflammatory reaction. Stem Cells Int 2015;2015:843058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vasko T, Frobel J, Lubberich R et al. iPSC‐derived mesenchymal stromal cells are less supportive than primary MSCs for co‐culture of hematopoietic progenitor cells. J Hematol Oncol 2016;9:43:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luzzani C, Neiman G, Garate X et al. A therapy‐grade protocol for differentiation of pluripotent stem cells into mesenchymal stem cells using platelet lysate as supplement. Stem Cell Res Ther 2015;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karlsson C, Emanuelsson K, Wessberg F et al. Human embryonic stem cell‐derived mesenchymal progenitors—Potential in regenerative medicine. Stem Cell Res 2009;3:39–50. [DOI] [PubMed] [Google Scholar]
- 28. Wang P, Liu X, Zhao L et al. Bone tissue engineering via human induced pluripotent, umbilical cord and bone marrow mesenchymal stem cells in rat cranium. Acta Biomater 2015;18:236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Y, Goldberg AJ, Dennis JE et al. One‐step derivation of mesenchymal stem cell (MSC)‐like cells from human pluripotent stem cells on a fibrillar collagen coating. PLoS One 2012;7:e33225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leszczynska A, O'Doherty A, Farrell E et al. Differentiation of vascular stem cells contributes to ectopic calcification of atherosclerotic plaque. Stem Cells 2016;34:913–923. [DOI] [PubMed] [Google Scholar]
- 31. Fang B, Liao L, Shi M et al. Multipotency of Flk1+CD34− progenitors derived from human fetal bone marrow. J Lab Clin Med 2004;143:230–240. [DOI] [PubMed] [Google Scholar]
- 32. Medici D, Olsen BR. Transformation of vascular endothelial cells into multipotent stem‐like cells: Role of the activin‐like kinase 2 receptor In: Hayat MA, ed. Stem Cells and Cancer Stem Cells, Volume 8: Therapeutic Applications in Disease and Injury. Dordrecht: Springer Netherlands, 2012:207–213. [Google Scholar]
- 33. Cao Y, Sun Z, Liao L et al. Human adipose tissue‐derived stem cells differentiate into endothelial cells in vitro and improve postnatal neovascularization in vivo. Biochem Biophys Res Commun 2005;332:370–379. [DOI] [PubMed] [Google Scholar]
- 34. Zimmerlin L, Donnenberg VS, Pfeifer ME et al. Stromal vascular progenitors in adult human adipose tissue. Cytometry A 2010;77:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hager G, Holnthoner W, Wolbank S et al. Three specific antigens to isolate endothelial progenitor cells from human liposuction material. Cytotherapy 2013;15:1426–1435. [DOI] [PubMed] [Google Scholar]
- 36. Lv FJ, Tuan RS, Cheung KM et al. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014;32:1408–1419. [DOI] [PubMed] [Google Scholar]
- 37. Murphy JM, Dixon K, Beck S et al. Reduced chondrogenic and adipogenic activity of mesenchymal stem cells from patients with advanced osteoarthritis. Arthritis Rheum 2002;46:704–713. [DOI] [PubMed] [Google Scholar]
- 38. Zhang J, Lian Q, Zhu G et al. A human iPSC model of Hutchinson Gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011;8:31–45. [DOI] [PubMed] [Google Scholar]
- 39. Bruckova L, Soukup T, Visek B et al. Proliferative potential and phenotypic analysis of long‐term cultivated human granulosa cells initiated by addition of follicular fluid. J Assist Reprod Genet 2011;28:939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bocelli‐Tyndall C, Zajac P, Di Maggio N et al. Fibroblast growth factor 2 and platelet‐derived growth factor, but not platelet lysate, induce proliferation‐dependent, functional class II major histocompatibility complex antigen in human mesenchymal stem cells. Arthritis Rheum 2010;62:3815–3825. [DOI] [PubMed] [Google Scholar]
- 41. Boskey AL. Bone composition: Relationship to bone fragility and antiosteoporotic drug effects. BoneKEy Rep 2013;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang X, Huang CT, Chen J et al. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 2010;7:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brown K, Legros S, Artus J et al. A comparative analysis of extra‐embryonic endoderm cell lines. PLoS One 2010;5:e12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barbet R, Peiffer I, Hatzfeld A et al. Comparison of gene expression in human embryonic stem cells, hESC‐derived mesenchymal stem cells and human mesenchymal stem cells. Stem Cells Int 2011;2011:368192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davis RP, Ng ES, Costa M et al. Targeting a GFP reporter gene to the MIXL1 locus of human embryonic stem cells identifies human primitive streak‐like cells and enables isolation of primitive hematopoietic precursors. Blood 2008;111:1876–1884. [DOI] [PubMed] [Google Scholar]
- 46. Ng ES, Azzola L, Sourris K et al. The primitive streak gene Mixl1 is required for efficient haematopoiesis and BMP4‐induced ventral mesoderm patterning in differentiating ES cells. Development 2005;132:873–884. [DOI] [PubMed] [Google Scholar]
- 47. Saga Y, Hata N, Koseki H et al. Mesp2: A novel mouse gene expressed in the presegmented mesoderm and essential for segmentation initiation. Genes Dev 1997;11:1827–1839. [DOI] [PubMed] [Google Scholar]
- 48. Burdsal CA, Damsky CH, Pedersen RA. The role of E‐cadherin and integrins in mesoderm differentiation and migration at the mammalian primitive streak. Development 1993;118:829–844. [DOI] [PubMed] [Google Scholar]
- 49. Tada S, Era T, Furusawa C et al. Characterization of mesendoderm: A diverging point of the definitive endoderm and mesoderm in embryonic stem cell differentiation culture. Development 2005;132:4363–4374. [DOI] [PubMed] [Google Scholar]
- 50. Azandeh S, Mohammad Gharravi A, Orazizadeh M et al. Improvement of mesenchymal stem cell differentiation into the endoderm lineage by four step sequential method in biocompatible biomaterial. BioImpacts 2016;6:9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Farahani RM, Xaymardan M. Platelet‐derived growth factor receptor alpha as a marker of mesenchymal stem cells in development and stem cell biology. Stem Cells Int 2015;2015:362753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamaguchi TP, Dumont DJ, Conlon RA et al. flk‐1, an flt‐related receptor tyrosine kinase is an early marker for endothelial cell precursors. Development 1993;118:489–498. [DOI] [PubMed] [Google Scholar]
- 53. Era T, Izumi N, Hayashi M et al. Multiple mesoderm subsets give rise to endothelial cells, whereas hematopoietic cells are differentiated only from a restricted subset in embryonic stem cell differentiation culture. Stem Cells 2008;26:401–411. [DOI] [PubMed] [Google Scholar]
- 54. Goupille O, Saint Cloment C, Lopes M et al. Msx1 and Msx2 are expressed in sub‐populations of vascular smooth muscle cells. Dev Dyn 2008;237:2187–2194. [DOI] [PubMed] [Google Scholar]
- 55. Cheng SL, Shao JS, Behrmann A et al. Dkk1 and MSX2‐Wnt7b signaling reciprocally regulate the endothelial‐mesenchymal transition in aortic endothelial cells. Arterioscler Thromb Vasc Biol 2013;33:1679–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Descamps B, Emanueli C. Vascular differentiation from embryonic stem cells: Novel technologies and therapeutic promises. Vascul Pharmacol 2012;56:267–279. [DOI] [PubMed] [Google Scholar]
- 57. Murakami Y, Hirata H, Miyamoto Y et al. Isolation of cardiac cells from E8.5 yolk sac by ALCAM (CD166) expression. Mech Dev 2007;124:830–839. [DOI] [PubMed] [Google Scholar]
- 58. Zhou L, Xia J, Qiu X et al. In vitro evaluation of endothelial progenitor cells from adipose tissue as potential angiogenic cell sources for bladder angiogenesis. PLoS One 2015;10:e0117644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alvero AB, Fu HH, Holmberg J et al. Stem‐like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells 2009;27:2405–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ditadi A, Sturgeon CM, Tober J et al. Human definitive haemogenic endothelium and arterial vascular endothelium represent distinct lineages. Nat Cell Biol 2015;17:580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pierelli L, Bonanno G, Rutella S et al. CD105 (endoglin) expression on hematopoietic stem/progenitor cells. Leuk Lymphoma 2001;42:1195–1206. [DOI] [PubMed] [Google Scholar]
- 62. Paprocka M, Krawczenko A, Dus D et al. CD133 positive progenitor endothelial cell lines from human cord blood. Cytometry A 2011;79:594–602. [DOI] [PubMed] [Google Scholar]
- 63. Kuhn LT, Liu Y, Boyd NL et al. Developmental‐like bone regeneration by human embryonic stem cell‐derived mesenchymal cells. Tissue Eng Part A 2014;20:365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yen ML, Hou CH, Peng KY et al. Efficient derivation and concise gene expression profiling of human embryonic stem cell‐derived mesenchymal progenitors (EMPs). Cell Transplant 2011;20:1529–1545. [DOI] [PubMed] [Google Scholar]
- 65. Li O, Tormin A, Sundberg B et al. Human embryonic stem cell‐derived mesenchymal stroma cells (hES‐MSCs) engraft in vivo and support hematopoiesis without suppressing immune function: Implications for off‐the shelf ES‐MSC therapies. PLoS One 2013;8:e55319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Moraes DA, Sibov TT, Pavon LF et al. A reduction in CD90 (THY‐1) expression results in increased differentiation of mesenchymal stromal cells. Stem Cell Res Ther 2016;7:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gago‐Lopez N, Awaji O, Zhang Y et al. THY‐1 receptor expression differentiates cardiosphere‐derived cells with divergent cardiogenic differentiation potential. Stem Cell Rep 2014;2:576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alves RD, Eijken M, van de Peppel J et al. Calcifying vascular smooth muscle cells and osteoblasts: Independent cell types exhibiting extracellular matrix and biomineralization‐related mimicries. BMC Genomics 2014;15:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aguilar‐Vazquez R, Carballo‐Molina OA, Collazo‐Navarrete O et al. Osteogenesis of human vascular endothelial cells in culture. Rev Invest Clin 2008;60:496–501. [PubMed] [Google Scholar]
- 70. Chaikuad A, Alfano I, Kerr G et al. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem 2012;287:36990–36998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. de Caestecker M. The transforming growth factor‐β superfamily of receptors. Cytokine Growth Factor Rev 2004;15:1–11. [DOI] [PubMed] [Google Scholar]
- 72. Radaev S, Zou Z, Huang T et al. Ternary complex of transforming growth factor‐beta1 reveals isoform‐specific ligand recognition and receptor recruitment in the superfamily. J Biol Chem 2010;285:14806–14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Castonguay R, Werner ED, Matthews RG et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J Biol Chem 2011;286:30034–30046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cheifetz S, Bellon T, Cales C et al. Endoglin is a component of the transforming growth factor‐beta receptor system in human endothelial cells. J Biol Chem 1992;267:19027–19030. [PubMed] [Google Scholar]
- 75. Onimaru K, Shoguchi E, Kuratani S et al. Development and evolution of the lateral plate mesoderm: Comparative analysis of amphioxus and lamprey with implications for the acquisition of paired fins. Dev Biol 2011;359:124–136. [DOI] [PubMed] [Google Scholar]
- 76. Nsair A, Schenke‐Layland K, Van Handel B et al. Characterization and therapeutic potential of induced pluripotent stem cell‐derived cardiovascular progenitor cells. PLoS One 2012;7:e45603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Song SH, Kim K, Jo EK et al. Fibroblast growth factor 12 is a novel regulator of vascular smooth muscle cell plasticity and fate. Arterioscler Thromb Vasc Biol 2016;36:1928–1936. [DOI] [PubMed] [Google Scholar]
- 78. Billing AM, Ben Hamidane H, Dib SS et al. Comprehensive transcriptomic and proteomic characterization of human mesenchymal stem cells reveals source specific cellular markers. Sci Rep 2016;6:21507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Espagnolle N, Guilloton F, Deschaseaux F et al. CD146 expression on mesenchymal stem cells is associated with their vascular smooth muscle commitment. J Cell Mol Med 2014;18:104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yamane A, Seetharam L, Yamaguchi S et al. A new communication system between hepatocytes and sinusoidal endothelial cells in liver through vascular endothelial growth factor and Flt tyrosine kinase receptor family (Flt‐1 and KDR/Flk‐1). Oncogene 1994;9:2683–2690. [PubMed] [Google Scholar]
- 81. Ohtani K, Suzuki Y, Eda S et al. The membrane‐type collectin CL‐P1 is a scavenger receptor on vascular endothelial cells. J Biol Chem 2001;276:44222–44228. [DOI] [PubMed] [Google Scholar]
- 82. Mariappan D, Niemann R, Gajewski M et al. Somitovasculin, a novel endothelial‐specific transcript involved in the vasculature development. Arterioscler Thromb Vasc Biol 2009;29:1823–1829. [DOI] [PubMed] [Google Scholar]
- 83. Laussu J, Audouard C, Kischel A et al. Eph/Ephrin signaling controls progenitor identities in the ventral spinal cord. Neural Dev 2017;12:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Aruga J, Yokota N, Mikoshiba K. Human SLITRK family genes: Genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 2003;315:87–94. [DOI] [PubMed] [Google Scholar]
- 85. Hussain MS, Battaglia A, Szczepanski S et al. Mutations in CKAP2L, the human homolog of the mouse Radmis gene, cause Filippi syndrome. Am J Hum Genet 2014;95:622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lian Q, Lye E, Suan Yeo K et al. Derivation of clinically compliant MSCs from CD105+, CD24− differentiated human ESCs. Stem Cells 2007;25:425–436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information
Figure S1 Characterization of iPSC‐2 and 3. (A) Representative cell morphology of iPSC colonies. Scale bar, 400 μm (B) G‐banding analysis confirmed normal karyotype of the cells. (C) Immunofluorescent staining demonstrated that iPSC positively expressed pluripotent markers (OCT4, SOX2, NANOG and TRA1‐60). White scale bar, 500 μm. (D) iPSC‐2 derived teratoma in vivo and hematoxylin and eosin‐stained teratoma sections. Detected tissue types included endoderm (glands), mesoderm (cartilage), and ectoderm (pigmented cells). Red scale bar, 200 mm. (E) EB formation was applied to induce spontaneous differentiation of iPSC‐3 in vitro. Representative images of immunofluorescent staining for germ layer markers on day 35 EB sections are shown, including α‐SMA (Mesoderm), AFP (Endoderm) and β‐3‐tublin (Ectoderm). Black scale bar, 400 μm and white scale bar, 500 μm.
Figure S2 Comparison of the growth rates of iMSC and BM‐MSC lines. The symbols embedded in each line indicate the time points of passage. See also Figure 1B.
Figure S3 Effects of serial passage on immunophenotyping of iMSCs and BM‐MSCs from P5 to P8. (A) Diagrams demonstrate the fraction of the cells expressing CD90, CD105, CD73 and Neg. combine on passaging. (B) Diagrams display the percentage of the cells positive for hematopoeitic markers during the expansion. (C) In iMSC group, an increased Neg. combine population was exclusively detected in iMSC‐3 from P5 to P8 (Fig. S3‐A). Flow cytometry analysis was conducted to specifically examine the hematopoeitic antigen expression profile of the cells at P8. Red histograms represent isotype controls with the blue overlays representing each antigen; percentages of positive cells are shown within histograms. See also Figure 1C and D.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.