

Abstract
Precise regulation of the glial cell cycle is essential during nervous system development and in response to injury, whereas disruption of cell cycle control is associated with malignant glial tumors and other nervous system diseases. The Ras signaling pathway plays a central role in regulating the mammalian cell cycle, and uncontrolled Ras signaling has been implicated in a wide range of human cancers, including malignant gliomas. Recent studies in glia have demonstrated that activation of Ras can either induce or inhibit proliferation through complex interactions among downstream signaling pathways impinging on cell cycle regulatory proteins. Studies in Schwann cells have begun to delineate the pathways by which Ras regulates the cell cycle in normal and pathological glia, and have identified promising targets for therapeutic intervention in the treatment of PNS and CNS malignant glial tumors. NEUROSCIENTIST 8(2):93–97, 2002
Keywords: Ras, Glioma, Neurofibromatosis-type 1 (NF1), Schwann cell, Cyclin-dependent kinase inhibitor, Tumor suppressor protein
Malignant gliomas are considered one of the deadliest forms of human cancer for which there is no known cure or satisfactory treatment. Gliomas, the most common type of primary brain tumors, arise from astrocytes, oligodendrocytes, or their precursors; can become malignant; and form highly invasive tumors that infiltrate the brain (see Maher and others 2001 for review). In the PNS, Schwann cells (SCs) can form malignant peripheral nerve sheath tumors, or Schwannomas. Although there has been rapid progress in cancer cell biology and genetics, most of the therapeutic approaches used to successfully treat extraneural solid tumors have proven ineffective in the treatment of malignant glial tumors. Insights into the cellular and molecular mechanisms that regulate normal glial proliferation could lead to the development of novel therapeutic strategies for blocking malignant glial growth.
The Ras signaling pathway is significantly overactivated in high-grade gliomas (see Bredel and Pollack 1999 for review) and malignant peripheral glial tumors (Cichowski and Jacks 2001). Ras is a 21 kDa G protein that transduces mitogenic and environmental signals from tyrosine kinase membrane receptors and other intracellular pathways to the nucleus (Satoh and others 1992). Recent studies in PNS glia have shown that Ras can either promote cell cycle progression or induce cell cycle arrest depending upon the level and duration of activation and the ensuing balance of positive and negative signals that regulate cell cycle proteins. Primary rodent SCs have proven to be a useful model system to study the interactions between Ras and the cell cycle. They can be cultured from peripheral nerves for extended periods, and abnormal activation of Ras through the loss of the GTP-ase activating protein, neurofibromin, has been implicated in the formation of peripheral nerve tumors in patients with neurofibromatosis type 1 (NF1). NF1 is a common inherited disease characterized by multiple benign neurofibromas. These tumors can progress to malignant peripheral nerve sheath tumors (MPNSTs), and NF1 patients are also predisposed to brain and optic tract gliomas as well as extraneural forms of cancer (Korf 2000; Cichowski and Jacks 2001). This update article will focus on the newly defined signaling pathways regulating the cell cycle through Ras in normal and malignant glia, with an emphasis on the positive and negative regulators of SC growth.
Regulation of Nuclear Cell Cycle Proteins in Glia
Major progress has been made in understanding the signals and proteins involved in the regulation of the cell cycle in oligodendrocyte progenitor cells (OPCs), astrocytes, and more recently SCs (Tikoo and others 1997; 2000; Ghiani and Gallo 2001). Cell proliferation is regulated by coordinated interactions between multiple proteins at distinct phases of the cell cycle. The three principal players are the cyclins (D, E, A, and B), cyclin-dependent kinases (Cdks), and cyclin-dependent kinase inhibitors (CKIs) (Fig. 1). Growth factors and axonal mitogens stimulate quiescent glial cells to enter the G1 phase of the cell cycle by inducing expression of the D-type cyclins. These cyclins subsequently assemble with their catalytic partners, the cyclin-dependent kinases (Cdk4 or Cdk6). Cyclin D-Cdk complexes are required for G1 progression and serve to couple cell cycle machinery with extracellular signals (see Sherr and Roberts 1999 for review).
Fig. 1.
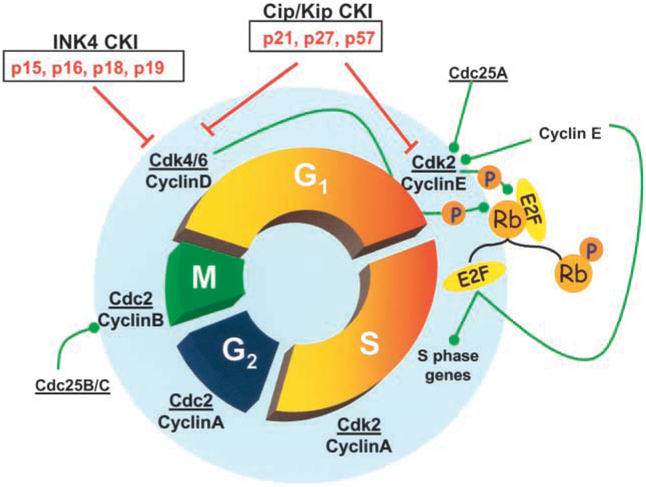
Glial cell proliferation is regulated by coordinated interactions between multiple proteins at distinct phases of the cell cycle. The cyclins (D, E, A, and B) assemble with their catalytic partners, the cyclin-dependent kinases (Cdk2, 4, 6, and cdc2), which are positively regulated by cyclin-cdk activating kinases (not shown), and via the serine/threonine phosphatase, cdc25. Negative regulation is mediated cyclin-dependent kinase inhibitors (CKIs), from the INK4 or Cip/Kip family, which bind to cyclin/cdk complexes. Progression to S phase requires the phosphorylation (inactivation) of retinoblastoma protein (pRb) by cyclinE-cdk2, which permits the E2F family of transcription factors (E2F) to regulate key S phase gene expression.
The sequential activation of different Cdks coordinates the orderly progression through the cell cycle, and the activity of Cdks is regulated by several mechanisms. Activation of Cdks occurs via phosphorylation of cyclin-CDK complexes by cyclin- Cdk activating kinases (CAKs) (Fisher and Morgan 1994), and via the serine/threonine phosphatase cdc25 (Morgan 1997). Negative regulation is mediated by two families of CKIs, the Cip/Kip family (p21, p27, p57) or Ink family (p15, p16, p18, p19) (Sherr and Roberts 1999) (Fig. 1). All of these proteins bind and inhibit specific cyclin-Cdk complexes, giving an additional level of control of the cell cycle.
Transition from G1 into S phase (DNA synthesis phase) is a critical cell cycle restriction point in glial cells. Progression to S phase in SCs and OPCs requires formation and activation of cyclinE-Cdk2 complexes (Tikoo and others 2000; Ghiani and Gallo 2001) and the subsequent phosphorylation (inactivation) of the retinoblastoma protein (pRb). Phosphorylation of Rb leads to the release of the E2F family of transcription factors, which are key regulators of S phase gene expression (Mulligan and Jacks 1998) (Fig. 1). CKIs can prevent entry into S phase by binding to G1 cyclin-Cdk complexes. Both p21 and p27 levels are known to increase in OPCs either during permanent cell cycle withdrawal and differentiation or during reversible cell cycle arrest in G1 in response to neuronal signals (Casaccia-Bonnefil and others 1999; Ghiani and others 1999). Although induction of CKIs has been shown to regulate cell cycle arrest during differentiation, the contribution of the Ras pathway in these developmental events is not clear.
Positive Regulation of Glial Cell Cycle by Ras
Ras can positively regulate cell cycle progression in glia during development and following nerve injury. Mitogen-dependent induction of the Cyclin D gene is dependent on the Ras/Raf/ERK pathway (Lavoie and others 1996). Growth factor and axonal signals, such as β neuregulin, induce SC proliferation through activation of the PI3 kinase and the mitogen-activated protein kinase (MAPK) pathways (Kim and others 1997; Maurel and Salzer 2000). In vitro growth factors such as platelet-derived growth factor (PDGF) or basic fibroblast growth factor are ineffective mitogens for purified SCs without the synergistic action of agents that elevate cAMP levels (i.e., forskolin). Until recently, the mechanisms for the cooperative interaction between the cAMP-dependent protein kinase A (PKA) and Ras pathways were poorly understood. It has been hypothesized that cAMP functions as a mitogen by stimulating the expression of growth factor receptors in SCs. This is supported by data showing that PDGF receptor expression is significantly up-regulated by cAMP in vitro (Weinmaster and Lemke 1990).
A recent study by Kim and others (2001) provides compelling evidence for an alternative mechanism. Rather than acting at the level of growth factor receptors, cAMP acts downstream and functions to regulate cyclin D1 expression during the G1 phase of the cell cycle. Loss-of-function studies have demonstrated that cyclin D1 is necessary for growth of mature SCs in vitro and in vivo (Kim and others 2000). By using an inducible retroviral vector, it was demonstrated that ectopic expression of cyclin D1 in SCs eliminated the requirement for cAMP in PDGF-treated cultures. It was further shown that elevated cAMP levels initiated and sustained G1 progression in SCs primed with PDGF by maintaining cyclin D1 expression (Fig. 2A). Collectively, these results demonstrate that cyclin D1 is a critical downstream target of Ras in SCs and that coactivation of the PKA pathway is required for sustaining growth-factor-induced cell cycle progression. Interestingly, in most mammalian cells including CNS glia, the cAMP-PKA pathway inhibits proliferation and induces differentiation through inactivation of the Ras/Raf/MAPK pathway. This reversed relationship between cAMP and proliferation further illustrates the complexity of the interactions between the Ras pathways and the cell cycle, even among different glial cell types.
Fig. 2.
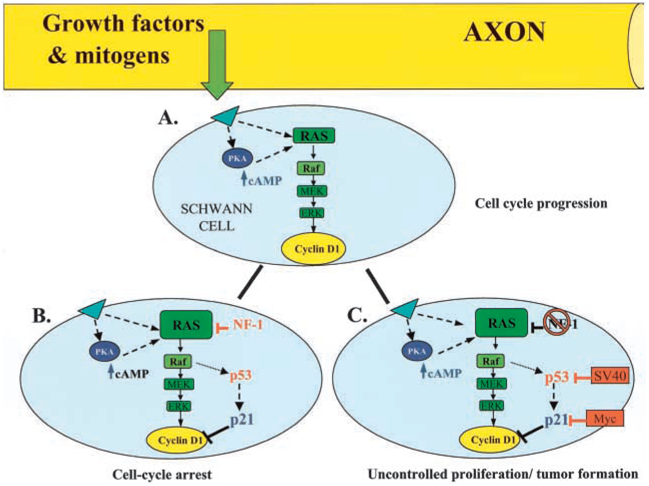
Model for Ras-dependent regulation of the Schwann cell cycle. A, Transient activation of Ras by axonal mitogens induces cyclin D1 expression through activation of the Ras/Raf/ MAPK pathway. Coactivation of the PKA-cAMP pathway is required for sustaining cyclin D1 levels and cell cycle progression in Schwann cells.B, Robust or sustained Ras activity can induce cell cycle arrest via the p53-dependent induction of the CKI, p21CIP. Cell cycle arrest is also mediated via the inhibitory actions of tumor suppressor proteins, such as the NF1 protein, neurofibromin.C, Uncontrolled proliferation and tumor formation occurs when these negative growth controls are antagonized by oncogenes, such as SV40 or Myc, or mutations in tumor suppressor proteins.
Negative Regulation of Glial Cell Cycle by the Ras Pathway
Although activation of the Ras signaling pathways is generally considered proliferative, recent research has demonstrated that sustained or strong activation of the Ras/Raf/ MAPK pathway can, in fact, inhibit proliferation (Sewing and others 1997). Cell cycle arrest must occur during development before glial cells terminally differentiate or initiate myelination. In the adult nervous system, cell cycle arrest may serve to control glial cell number or act as a protective mechanism against transformation and tumor formation. SCs have proven to be an excellent model system to study the molecular mechanisms of Ras-dependent inhibition of cell proliferation. Cell cycle arrest can occur through two principle mechanisms: Ras-dependent induction of CKIs or the action of tumor suppressor proteins (Fig. 2B).
Cell Cycle Arrest via Ras- Dependent Induction of CDKIs
Ras-dependent cell cycle arrest occurs during senescence (Wynford-Thomas 1997), a process that cells enter at the end of their proliferative life span. In many human cells, senescence is the result of a mitotic counting mechanism related to the incremental shortening of telomeres (the ends of chromosomes) with successive divisions (Bodnar and others 1998). A telomere-independent cell cycle arrest has been observed via Ras-dependent induction of CKIs in cultured rodent cells, which maintain constant telomere length (Sherr and DePinho 2000). Recent in vitro studies have suggested that glial cells may differ from cells such as fibroblasts in that Ras-induced G1 arrest does not necessarily induce replicative senescence, as expected. Activation of the Ras/Raf/MAPK pathway in primary rat SCs and fibroblasts induces a cell cycle arrest through a p53-dependent induction of the CKI, p21cip1 (Lloyd and others 1997; Serrano and others 1997) (Fig. 2B). p53 is an important tumor suppressor protein, which induces p21 expression and cell cycle arrest following stress or DNA damage (Agami and Bernards 2000; Walworth 2000). Interestingly, unlike fibroblasts, this Raf-stimulated G1 arrest did not induce a flattened, senescent-like cell morphology in SCs (Lloyd and others 1997).
Two groups recently challenged the idea that cultured somatic cells have a limited replicate life span (Mathon and others 2001; Tang and others 2001). In one study, the proliferative capacity of primary SCs after continual passaging in vitro was tested and compared to that of fibroblasts obtained from the same nerve (Mathon and others 2001). Unlike fibroblasts, which predictably entered replicative senescence after ~4 passages in vitro, SCs continued to divide at a constant rate through extensive passaging (> 50) and failed to undergo replicative senescence unless this was induced by cell cycle arresting agents such as amphidocolin. In addition, SCs retained all of the checkpoints (such as CKIs and p53) often lost during immortalization or transformation. Similar findings were observed in cultured rat oligodendrocyte precursor cells (Tang and others 2001). These results demonstrate that although cell cycle arrest could be induced under certain conditions, rodent glial cells do not necessarily have a limited proliferative lifespan, as previously thought. Taken together, these results suggest that cell cycle arrest in normal glial cells may function as a protective response to inappropriate environmental signals acting through Ras-dependent induction of CKIs.
Cell Cycle Arrest via Tumor Suppressor Proteins
Tumor suppressor genes can also induce Ras-dependent growth arrest, thus protecting cells from malignant transformation either by acting directly on the Ras pathway or by the regulation of key cell cycle proteins. The tumor suppressor protein p53 is an example of the latter. Activation of p53 can induce expression of the CKI, p21 as discussed earlier. The NF1 protein, neurofibromin, functions as a GTPase activating protein (GAP), which directly inhibits Ras by increasing the conversion of Ras-GTP to Ras-GDP in vivo and in vitro (Martin and others 1990; Xu and others 1990). As described previously, elevated cAMP initiates G1 progression in SCs by elevating and maintaining cyclin D1 levels (Kim and others 2001). It has been recently demonstrated that NF1 may function as a tumor suppressor by antagonizing the accumulation of cAMP and sustained cyclin D1 expression in SCs. SCs from NF1-deficient mice have increased Ras activation and elevated cAMP levels, and this agonistic action of cAMP and subsequent sustained increase in cyclin D1 could contribute to tumor formation (Kim and others 2001). The mechanism by which the loss of NF1 increases cAMP levels remains unclear.
Ras-Dependent Deregulation of the Cell Cycle in Malignant Glia
Uncontrolled Ras signaling has been implicated in many forms of cancer including CNS gliomas (see Bredel and Pollack 1999 for review). The oncogenic transformation of normal glia can occur when inappropriate Ras signaling antagonizes the normal cell cycle controls. It is now known that transforming Ras requires the presence of cooperating oncogenes or the loss of tumor suppressor genes (Lloyd and others 1997; Lloyd 1998). It has been proposed that interactions between oncogenes and the Ras pathway offset the balance between Ras and the CDKIs and mediate the transformation of benign tumors into malignant tumors or gliomas. For example, oncogenic Myc can overcome a p53-dependent cell cycle arrest by antagonizing p21 induction (Hermeking and others 1995). Similarly, viral oncogenes such as simian virus 40 (SV40LT) can antagonize the inhibitory effects of CDKIs and tumor supressor genes such as p53 (Lloyd and others 1997; Lloyd 1998) (Fig. 1C).
Loss of tumor suppressor genes such as NF1 or p53 can also lead to cellular transformation. The majority of NF1 tumors are benign, but in a small percentage of cases, neurofibromas can develop into malignant Schwannomas or MPNSTs (Cichowski and Jacks 2001; Korf 2000). Studies of SCs with mutant NF1 have demonstrated that NF1-deficient SCs are invasive and angiogenic, similar to SCs isolated from human neurofibromas. These events were enhanced in NF1 null SCs, which SCs can be induced to hyperproliferate by agents that elevate cAMP levels, as discussed previously (Kim and others 1997).
What mechanisms underlie the transformation from benign to maligant peripheral tumors? Mutations of both copies of the NF1 gene have been observed in MPNSTs, but this is also seen in benign neurofibromas (Colman and others 1995). It has been proposed that second-hit mutations are required for progression to malignancy (Cichowski and Jacks 2001). Loss of the p53 gene is frequently seen in MPNSTs from patients with NF1 (Legius and others 1994), and there is evidence to support a definitive role for p53 in the transformation of neurofibromas to MPNSTs. The NF1 and p53 genes are closely linked on chromosome 11, and recently double knockout mouse models were developed that carried cis or trans mutations for both genes (Cichowski and others 1999; Vogel and others 1999). These groups reported that cells carrying mutations for both genes on the same chromosome developed MPNSTs at high frequency whereas NF1 and p53 trans mutations developed benign tumors resembling those seen in heterozygous NF1 or p53 animals. These findings suggest that the cooperative loss in both genes contributes to the formation of malignant PNS tumors.
Therapeutic Approaches to the Treatment of NF-1 Tumors and Glioma
Pharmacological inhibition of Ras and related G proteins is being used as a therapeutic strategy for blocking malignant glial cell growth. Growing evidence for the role of Ras signaling in NF1 peripheral tumors has made it a promising target for novel anticancer therapies. Inhibition of the Ras pathway in NF1 patients either by using neutralizing antibodies to Ras or by increasing Ras-GAP activity results in tumor cell reversion (De Clue and others 1992). Inhibitors of farnesyl protein transferase (FTI), an enzyme that promotes the attachment of Ras to the cell membrane, significantly reduced hyperproliferation of NF1-null SCs (Kim and others 1997). This agent is currently being tested in clinical trials and could have therapeutic benefits to NF1 patients. There have also been a series of papers recently, reporting similar findings using FTIs and other Ras pathway inhibitors in the treatment of CNS gliomas (Bredel and others 1998; Pollack and others 1999; Feldkamp and others 2001).
Future Directions
The field of glioma research is expanding rapidly and changing directions, as researchers are beginning to take a more integrative approach to understanding this deadly disease. New studies are bridging the fields of developmental neuroscience, cell biology, genetics, and neuro-oncology to understand the cellular and molecular mechanisms involved in the development of malignant glial tumors. Glioma derive from many glial cell types, and they involve diverse genetic alterations and clinical features. This complexity has contributed to the lack of a successful therapeutic treatment. Despite this complexity, Ras plays a common role in many gliomas, and although rarely mutated, the majority of malignant gliomas involve some form of uncontrolled Ras signaling. Studies in PNS glia have yielded important insights into the Ras-dependent mechanisms regulating the cell cycle in normal and malignant glia. Future research in understanding the extracellular signals and converging signal transduction pathways regulating the cell cycle of CNS and PNS glia will bring us closer to developing successful pharmacological and genetic therapies to combat glioma.
Acknowledgments
We thank Dr. Philip Lee for critically reviewing the manuscript, and Dr. Shibeshih Belachew for helpful discussions, and for contributing to Figure 1. Beth Stevens is concurrently enrolled in the Neuroscience and Cognitive Science Program at the University of Maryland, College Park.
References
- Agami R, Bernards R. 2000. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell 102:55–66. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, and others. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279:349. [DOI] [PubMed] [Google Scholar]
- Bredel M, Pollack IF. 1999. The p21-Ras signal transduction pathway and growth regulation in human high-grade gliomas. Brain Res Rev 29:232–49. [DOI] [PubMed] [Google Scholar]
- Bredel M, Pollack IF, Freund JM, Hamilton AD, Sebti SM. 1998. Inhibition of Ras and related G-proteins as a therapeutic strategy for blocking malignant glioma growth. Neurosurgery 43:124–31. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Hardy RJ, Teng KK, Levine JM, Koff A, Chao MV. 1999. Loss of p27Kip1 function results in increased proliferative capacity of oligodendrocyte progenitors but unaltered timing of differentiation. Development 126:4027–37. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Jacks T. 2001. NF1 tumor suppressor gene function: narrowing the GAP. Cell 104:593–604. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, and others. 1999. Mouse models of tumor development in Neurofibromatosis type 1. Science 286:2172–6. [DOI] [PubMed] [Google Scholar]
- Colman SD, Williams CA, Wallace MR. 1995. Benign neurofibromas in type 1 neurofibromatosis (NF1) show somatic deletions of the NF1 gene. Nat Genet 11:90–2. [DOI] [PubMed] [Google Scholar]
- De Clue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, and others. 1992. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in type 1 neurofibromatosis. Cell 69:265–73. [DOI] [PubMed] [Google Scholar]
- Feldkamp MM, Lau N, Roncari L, Guha A. 2001. Isotype-specific Ras GTP-levels predict the efficacy of farnesyl transferase inhibitors against human astrocytomas regardless of Ras mutational status. Cancer Res 61:4425–31. [PubMed] [Google Scholar]
- Fisher RP, Morgan DO. 1994. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 78:713–24. [DOI] [PubMed] [Google Scholar]
- Ghiani CA, Gallo V. 2001. Inhibition of CyclinE-CDK2 complex formation and activity is associated with cell cycle arrest and withdrawal in oligodendrocyte progenitor cells. J Neurosci 21:1274–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiani CA, Yuan X, Eisen AM, Knutson PL, DePinho RA, McBain CJ, and others. 1999. Voltage-activated K+ channels and membrane depolarization regulate accumulation of the cyclin-dependent kinase inhibitors p27 (Kip1) and p21(CIP1) in glial progenitor cells. J Neurosci 19:5380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Funk JO, Reichert M, Ellwart JW, Eick D. 1995. Abrogation of p53-induced cell cycle arrest by c-Myc: evidence for an inhibitor of p21WAF1/ CIP1/SDI1. Oncogene 11:1409–15. [PubMed] [Google Scholar]
- Kim HA, Ling B, Ratner N. 1997. NF1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol Cell Biol 17:862–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HA, Pomeroy SL, Whoriskey W, Pawlitzky I, Benowitz LI, Sicinski P, and others. 2000. A developmentally regulated switch directs regenerative growth of Schwann cells through cyclin D1. Neuron 26:405–16. [DOI] [PubMed] [Google Scholar]
- Kim HA, Ratner N, Roberts TM, Stiles CD. 2001. Schwann cell proliferative responses to cAMP and NF1 are mediated by cyclin D1. J Neurosci 21:1110–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf BR. 2000. Malignancy in neurofibromatosis type 1. The Oncologist 5:477–85. [DOI] [PubMed] [Google Scholar]
- Lavoie JN, L’Allenmuin G, Brunet A, Muller R, Pouyssegur J. 1996. Cyclin D1 expression is regulated positively by the P42/P44 MAPK and negatively by the P38/HOG MAPK pathway. J Biol Chem 271:608–16. [DOI] [PubMed] [Google Scholar]
- Legius EH, Dierick R, Wu BK, Hall P, Marynen JJ, Cassiman JJ, and others. 1994. P53 mutations are frequent in malignant NF1 tumors. Gene Chrom Cancer 10:250–5. [DOI] [PubMed] [Google Scholar]
- Lloyd AC. 1998. Ras versus cyclin-dependent kinase inhibitors. Curr Opin Genet Dev 8:43–8. [DOI] [PubMed] [Google Scholar]
- Lloyd AC, Obermuller S, Staddon, Barth CF, McMahon M, Land H. 1997. Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes Dev 11:663–77. [DOI] [PubMed] [Google Scholar]
- Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Lois DN, Cavenee WK, and others. 2001. Malignant glioma: genetics and biology of a grave matter. Genes Dev 15:1311–33. [DOI] [PubMed] [Google Scholar]
- Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, and others. 1990. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63:843–9. [DOI] [PubMed] [Google Scholar]
- Mathon NF, Malcolm DS, Harrisingh MC, Cheng L, Lloyd AC. 2001. Lack of replicative senescence in normal rodent glia. Science 291:872–5. [DOI] [PubMed] [Google Scholar]
- Maurel P, Salzer J. 2000. Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination require PI 3-kinase activity. J Neurosci 201:4635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO. 1997. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 13:261–91. [DOI] [PubMed] [Google Scholar]
- Mulligan G, Jacks T. 1998. The retinoblastomas gene family: cousins with overlapping interests. Trends Genet 14:223–9. [DOI] [PubMed] [Google Scholar]
- Pollack IF, Bredel M, Erff M, Hamilton AD, Sebti SM. 1999. Inhibition of Ras and related guanosine triphosphate-dependent proteins as a therapeutic strategy for blocking malignant glioma growth: II—preclinical studies in a nude mouse model. Neurosurgery 45:1208–14. [DOI] [PubMed] [Google Scholar]
- Satoh T, Nakafuka Y, Kaziro. 1992. Function of Ras as a molecular switch in signal transduction. J Biol Chem 267:24149–52. [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrah ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593–602. [DOI] [PubMed] [Google Scholar]
- Sewing A, Wiseman B, Lloyd AC, Land H. 1997. High intensity Raf signal causes cell cycle arrest mediated by p21. Mol Cell Biol 17:5588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, DePinho RA. 2000. Cellular senescence: mitotic clock or culture shock? Cell 102:407–10. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13:501–12. [DOI] [PubMed] [Google Scholar]
- Tang DG, Tokumoto M, Apperly JA, Lloyd AC, Raff MC. 2001. Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science 291:868–70. [DOI] [PubMed] [Google Scholar]
- Tikoo R, Casaccia-Bonnefil P, Chao M, Koff A. 1997. Changes in cyclin-dependent kinase 2 and p27kip1 accompany glial cell differentiation of central glia. J Biol Chem 272:442–7. [DOI] [PubMed] [Google Scholar]
- Tikoo R, Zanzanni G, Shiffman D, Salzer J, Chao M. 2000. Cell cycle control of Schwann cell proliferation: role of cyclin-dependent kinase-2. J Neurosci 20:4627–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parda LF. 1999. Mouse tumor model for neurofibromatosis type 1. Science 286:2176–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NC. 2000. Cell-cycle checkpoint kinases: checking in on the cell cycle. Curr Opin Cell Biol 12:697–704. [DOI] [PubMed] [Google Scholar]
- Weinmaster G, Lemke G. 1990. Cell-specific cyclic AMP-mediated induction of the PDGF receptor. EMBO J 9:915–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynford-Thomas D. 1997. Proliferative lifespan checkpoints: cell-type specificity and influence on tumor biology. Eur J Cancer 33:716–26. [DOI] [PubMed] [Google Scholar]
- Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, and others. 1990. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants in S. cerevisiae. Cell 63:835–41. [DOI] [PubMed] [Google Scholar]