

Abstract
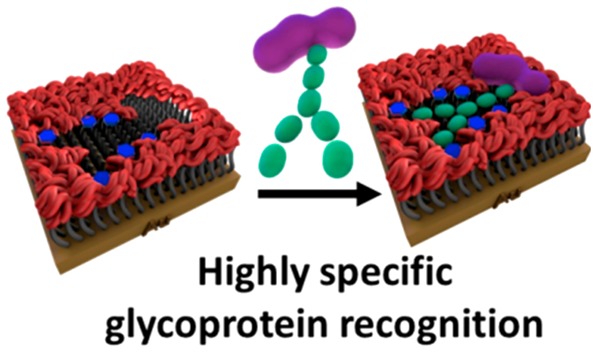
Since glycoproteins have become increasingly recognized as key players in a wide variety of disease processes, there is an increasing need for advanced affinity materials for highly selective glycoprotein binding. Herein, for the first time, a surface-initiated controlled radical polymerization is integrated with supramolecular templating and molecular imprinting to yield highly reproducible synthetic recognition sites on surfaces with dissociation constants (KD) in the low micromolar range for target glycoproteins and minimal binding to nontarget glycoproteins. Importantly, it is shown that the synthetic strategy has a remarkable ability to distinguish the glycosylated and nonglycosylated forms of the same glycoprotein, with a >5-fold difference in binding affinity. The precise control over the polymer film thickness and positioning of multiple carbohydrate receptors plays a crucial role in achieving an enhanced affinity and selectivity. The generated functional materials of unprecedented glycoprotein recognition performance open up a wealth of opportunities in the biotechnological and biomedical fields.
Keywords: glycoprotein recognition, selectivity, molecular imprinting, radical polymerization, ATRP
Introduction
Glycoproteins make up the majority of human proteins, with many having a close association with disease progression.1−3 Therefore, the recognition and quantification of glycoproteins are of paramount importance for a variety of research purposes and practical applications, including life sciences and medical research, clinical diagnostics, medical devices, and imaging.4−7 However, major challenges remain regarding their specific recognition and in particular how the structural features of both the protein and glycan parts of the glycoprotein can be simultaneously recognized to maximize selectivity.8,9
Antibodies raised against glycoproteins are typically specific for the protein units,10 which are recognized irrespective of the presence or not of particular glycan structures. On the other hand, lectins act as affinity tools for glycans, even if they are limited in terms of their ability to recognize a full glycan structure,11 but are not able to provide structural information about the protein unit of the glycoprotein. Considering these limitations in biological recognition materials, there is considerable motivation and opportunities to develop synthetic materials capable of highly specific molecular recognition for such challenging macromolecular targets.
Molecular imprinting (MI),12,13 which involves a process of template-induced formation of molecular cavities featuring recognition sites in a material, emerges as a very important concept to meet such a major challenge. In MI, the synthetic receptors used to construct the recognizing sites into the imprinted cavity play a key role in promoting target affinity and selectivity via the formation of multiple interactions, including covalent bonding, hydrogen bonding, electrostatic, hydrophobic, and van der Waals interactions.14−16 By virtue of their ability to covalently and reversibly bind with diols of carbohydrates,17−20 boronic acid (BA) entities emerged as front-runners to act as synthetic receptors for glycoprotein recognition at imprinted cavities.
Earlier attempts to prepare synthetic recognition systems for glycoproteins on material surfaces using MI and BA-based carbohydrate receptors left significant room for improvement in terms of meeting the requirements of high selectivity and high affinity.21−24 To date, two main MI pathways have been adopted. One strategy is based on glycoprotein immobilization on a BA-terminated monolayer, upon which a polymer network is formed around the template glycoprotein.21−23 Since the carbohydrate receptors are arranged in a monolayer fashion, glycan binding to the boronic acid-terminated monolayers occurs randomly. The strategy can, thus, potentially promote nonspecific binding from other glycoproteins. The other strategy relies on an initial glycoprotein/BA complex formation, followed by fixation of the complex on the surface and construction of a molecular scaffold around the glycoprotein template.24 Although this is an attractive method to form specific glycoprotein glycan binding, the drawback is that the spatial arrangement of the carbohydrate receptors occurs in a two-dimensional display.24 Thus, while these strategies yielded surfaces with important glycoprotein binding properties, the glycoprotein selectivity is limited by the degree of molecular manipulation that can be achieved to create a well-defined carbohydrate receptor pattern that is complementary to the glycoprotein template.
In the context of creating highly selective imprinted glycoprotein cavities on surfaces, strategies that are amenable to a three-dimensional (3D) display of the carbohydrate receptors are particularly appealing since they can more closely create a sterically and chemically complementary cavity to the natural 3D glycan structure. With this proviso in mind, herein, we demonstrate a surface imprinting methodology that meets these criteria by relying on the precomplexation of BA-based carbohydrate receptors with the target glycoprotein and atom transfer radical polymerization (ATRP), which enables precise control over the surface macromolecular structure and functionality (Figure 1).25
Figure 1.
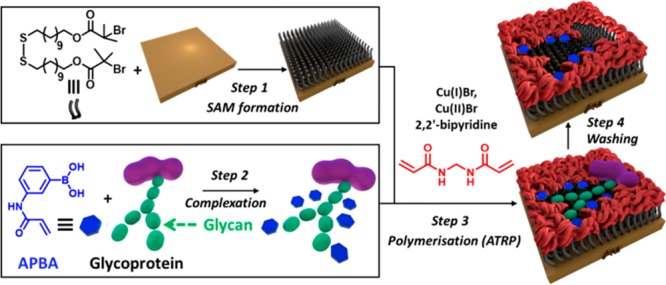
A schematic (not to scale) illustration of the molecular imprinting process involving precomplexation of BA-based carbohydrate receptors with the target glycoprotein and surface-initiated ATRP polymerization to form highly selective molecular cavities on surfaces for glycoprotein recognition.
Our modular strategy involves four main steps: (1) functionalization of a gold surface with an ATRP initiator-terminated SAM; (2) formation of a high order BA/glycoprotein complex using protein compatible conditions; (3) surface-initiated highly controlled ATRP polymerization in the presence of the preformed BA/glycoprotein complexes to create glycoprotein glycan-specific 3D interaction sites within ultrathin, imprinted polymer films; (4) following formation of the well-controlled molecular cavities, the glycoprotein template can be easily removed by washing with an elution buffer due to the reversible nature of the BA/diol interactions.24,26 This strategy provides the ability to achieve surface binding sites, which are complementary not only to the glycoprotein template in their size and shape but also to the very specific orientation and sugar sequence of the glycoprotein glycan. This latter recognition mode is facilitated by the initial generation of a BA/glycoprotein complex (step 2), in which the spatial arrangement of the multiple BA receptors in the complex is preserved upon surface incorporation via surface-initiated ATRP polymerization (step 3). Because of the characteristic feature of a glycoprotein glycan, i.e., one glycan bears multiple hydroxyl groups, multiple BAs anchored at appropriate positions in the recognition cavities interact synergistically with a glycoprotein glycan to promote affinity and selectivity. RNase B, which comprises a single glycosylated site with five diverse high-mannose glycans, is chosen as our model glycoprotein template to form the imprinted molecular cavities. The nonglycosylated RNase form, RNase A, and two highly glycosylated glycoproteins, α1-acid glycoprotein and horseradish peroxidase, are employed as controls to demonstrate selectivity. The chosen control proteins provide a range of sizes, charges, and percentages of glycosylation (Table S1) to give a representative selection of different properties that can contribute to the binding response.
Results and Discussion
The ATRP initiator-terminated SAMs were formed using a synthesized disulfide functionalized with tert-butyl bromide ATRP initiators, 11,11′-dithiobis[1-(2-bromo-2-methylpropionyloxy)undecane] (11-DTMBD) (see the Supporting Information for details on the synthesis of 11-DTMBD and characterization). The 11-DTMBD SAMs were created by first cleaning gold substrates using piranha solution for 10 min, following which they were immersed in a 1 mM ethanolic solution of 11-DTMBD for 24 h. In order to confirm the formation of a high-quality 11-DTMBD monolayer, contact angle, ellipsometry, and X-ray photoelectron spectroscopy (XPS) analysis were performed. The 11-DTMBD SAM has an advancing contact angle of 73.0 ± 1.8° and receding contact angle of 61.8 ± 2.1°, agreeing well with the presence of a tert-butyl bromide-terminated monolayer.27 The contact angle hysteresis (the difference between the advancing and receding angles) of 11.2° indicates the presence of a packed monolayer. The ellipsometric thickness observed for the 11-DTMBD derived SAMs is 1.7 ± 0.1 nm, which is less than the theoretical molecular length of the 11-DTMBD moiety as determined from ChemDraw3D, i.e., 1.9 nm. The difference between molecular length and SAM thickness is attributed to the tilt angle of the SAM molecules.28,29
XPS was employed to study the elemental composition of the 11-DTMBD SAM. As anticipated, the high-resolution scans confirmed the presence of all of the expected elements, showing signals from S (2p), C (1s), O (1s), and Br (3d) (Figure 2).
Figure 2.

High-resolution XPS spectra of (a) S (2p), (b) C (1s), (c) O (1s), and (d) Br (3d) of the 11-DTMBD SAM.
The S (2p) spectra reveals two doublet peaks, with the doublet peak at a lower binding energy, indicating that the sulfur is chemisorbed on the gold surface.30,31 The second doublet, weaker and at a higher binding energy, is ascribed to the S—H bonds, indicating the presence of some unbound sulfur.31 No oxidized sulfur is observed, as demonstrated by the absence of sulfur peaks above 166 eV. An analysis of the C (1s) spectra shows four carbon environments consistent with C—C, C—Br, C—O, and C=O. Furthermore, the C=O bond was observed in the O (1s) spectra at a binding energy of 533.3 eV alongside the C—O bond at 531.9 eV, which further indicates that the end groups of the 11-DTMBD molecule are present. This observation is further supported by the high-resolution spectra of the Br (3d) that shows the presence of the 3d5/2 and 3d3/2 peaks consistent with the C—Br bond observed in the C (1s) spectra. Collectively, the contact angle, ellipsometry, and XPS data are used to assess SAM quality and to demonstrate the formation of a well-packed 11-DTMBD SAM.
After the formation of a well-ordered ATRP initiator-terminated SAM, attention was turned toward the generation of very thin polymer layers. The surface-initiated ATRP reactions from the 11-DTMBD SAMs were undertaken using ethyl 2-bromoisobutyrate as the sacrificial initiator, N,N′-methylenebis(acrylamide) (MEBA) as the cross-linking monomer, 2,2′-bipyridyl as the ligand, and an optimized 1:1.5 ratio of Cu(I)Br/Cu(II)Br as the catalyst. Figure 3 shows the thickness of the poly(MEBA) surface as a function of time. The film thickness increases with good reproducibility over the first 30 min and then levels off with thickness values around 8 nm. Polymerization ceases likely as a result of different processes, such as bimolecular reaction,32 catalytic radical termination,33 or migration–termination.34 The trend in thickness growth over time is dependent on the ATRP conditions, which affect the polymerization rate and cessation of polymerization.35−37 These results demonstrate that this system provides elegant control over the thicknesses of the polymer layer and thus can be used to reliably grow surface-confined ultrathin molecularly imprinted polymer (MIP) films.
Figure 3.
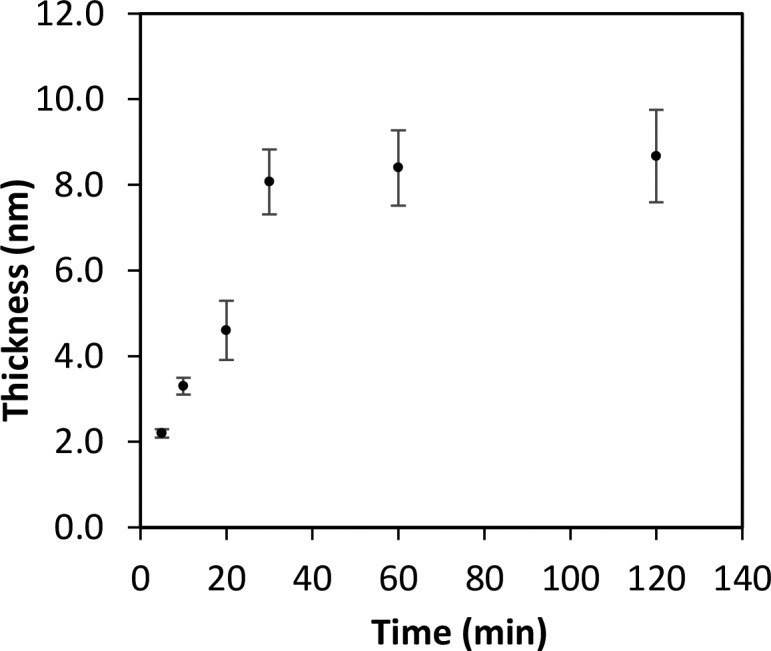
Kinetic study to monitor the rate of thickness growth of MEBA over 2 h from a tert-butyl bromide initiator-terminated surface using ATRP.
Molecular imprinting polymerization was performed by using RNase B as the glycoprotein template, wherein BA derivatives were first complexed with RNase B prior to the imprinting process. The BA derivative (3-acrylamidophenyl boronic acid, APBA) contains an acrylamide moiety for cross-linking with the tert-butyl bromide ATRP initiator-terminated surface and the MEBA cross-linking monomer, thus allowing for the 3D spatially controlled BA grafting within the imprinted cavities. Complexation of RNase B with APBA was carried out using 75% (v/v) 10× PBS and 25% (v/v) MeOH at pH 8.6. The chosen alkaline pH promotes the formation of stable cyclic esters between BAs and the diols of the RNase B,26 which comprises high-mannose glycans. If other carbohydrate residues, such as sialic acid, are also present on the glycan, the pH needs to be carefully considered. Unlike other carbohydrates (e.g., mannose, glucose, galactose), the binding between BAs and sialic acid is favored at an acidic to physiological pH.38
The MeOH was introduced to circumvent the low aqueous solubility of APBA. In order to establish that MeOH would not adversely affect the native structure of the protein, circular dichroism (CD) studies of RNase B were performed in different percentages of MeOH (i.e., 0–50%). As illustrated in Figure S1, all of the CDs show a similar spectral shape, indicating that MeOH does not cause any conformational changes to the tertiary structure of the glycoprotein.
The complexation and subsequent imprinting were carried out using different RNase B/APBA mole ratios, namely, 1:10, 1:15, and 1:20. Molecular imprinting was performed by the concurrent polymerization of the RNase B/APBA complex and MEBA monomer for 20 min. The different ratios of the RNase B template to APBA receptors under investigation (1:10, 1:15, and 1:20) allowed tuning the density of BAs in the imprinted cavities, which in turn can have a strong influence on dictating the binding affinity and selectivity of the RNase B-imprinted surface. Binding studies on the molecularly imprinted surfaces as well as on nonimprinted surfaces (i.e., the same protocol as that for imprinting, including the presence of BAs, but without the RNase B as a template) were conducted by surface plasmon resonance (SPR) spectroscopy. Briefly, each protein was injected across the imprinted and nonimprinted surfaces at a range of concentrations (i.e., 1.6–50 μM) and the responses at equilibrium (Req) measured. Following each injection, the surface was regenerated by an acidic wash to remove the protein.
As illustrated in Figure 4, all of the imprinted surfaces preferentially captured RNase B over the control proteins at all of the concentrations tested. For each concentration, 2 measurements from 3 individual chips were taken (n = 6), from which the average and standard deviation values were then calculated. The small standard deviation associated with RNase B binding into the imprinted surfaces illustrates that the imprinting method can provide reproducible synthetic recognition sites on surfaces. Furthermore, as shown in Table 1, the imprinted surfaces prepared from the 1:10 RNase B/APBA ratio provided the highest affinity and selectivity for RNase B, with an impressive 4.5-fold selectivity over its nonglycosylated homologue, and a >5.3-fold selectivity over other glycosylated proteins. These selectivity values are higher than those obtained for other glycoprotein imprinting systems,23,24 thus highlighting the efficacy of the approach to fabricate imprinted cavities with sugar recognition properties. The degree of selectivity is better at a 1:10 RNase B/APBA ratio than at 1:15 and 1:20 ratios mostly due to the fact that the RNase B binding affinity is slightly higher for the 1:10 ratio. A possible explanation for a small decrease in affinity to a higher APBA ratio could be steric hindrance caused by neighboring APBA moieties that hamper the diol-BA binding.
Figure 4.
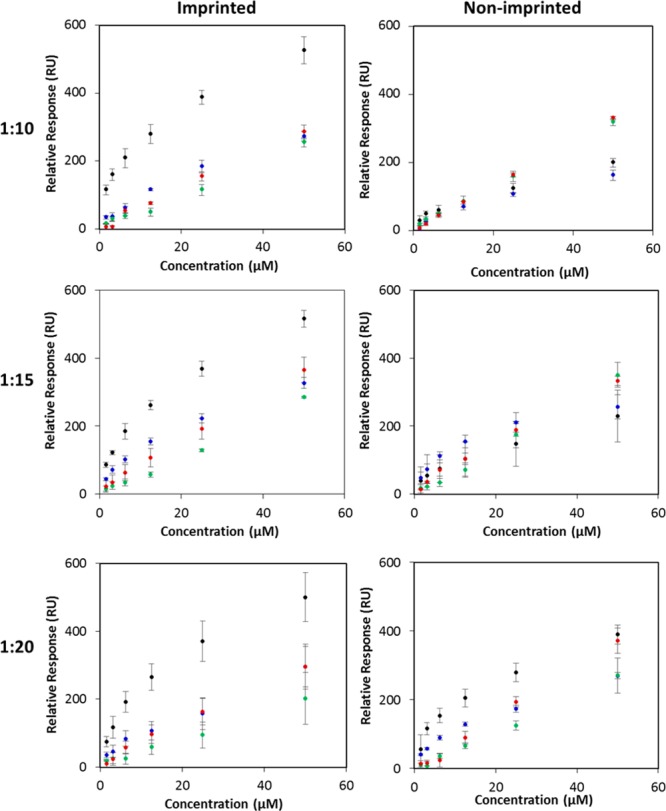
Effect of the RNase B/APBA ratio (1:10, 1:15, and 1:20) and protein concentration on the Req of imprinted poly(MEBA)-APBA surfaces and nonimprinted poly(MEBA)-APBA surfaces to RNase B (black), RNase A (blue), α1-acid glycoprotein (green), and HRP (red).
Table 1. KD for Each of the Proteins for the Imprinted Poly(MEBA)-APBA Surfaces Formed Using RNase B/APBA Ratiosa.
|
KD (μM) |
|||
|---|---|---|---|
| RNase B/APBA (1:10) | RNase B/APBA (1:15) | RNase B/APBA (1:20) | |
| RNase B | 14.2 ± 2.1 | 19.7 ± 2.1 | 23.0 ± 2.6 |
| RNase A | 63.3 ± 7.2 | 40.8 ± 3.7 | 60.6 ± 6.0 |
| α1-acid glycoprotein | 105.4 ± 11.6 | 135 ± 11.2 | 217.3 ± 20.0 |
| HRP | 75.3 ± 8.5 | 69.9 ± 6.1 | 90.5 ± 8.6 |
| selectivity (KDA/KDB) | 4.5 | 2.1 | 2.6 |
The selectivity values of the imprinted poly(MEBA)-APBA surfaces for RNase B with respect to RNase A are shown at the bottom of the table.
These results provide evidence that the contribution to the binding affinity from imprinted cavities arose from shape-matching and functional interactions. Since RNase B and its nonglycan containing analogue (RNase A) have similar dimensions and isoelectric points (Table S1) but very distinct binding affinities (Table 1), it is reasonable to suggest that not as much shape-matching but RNase B-BA interactions predominantly determine the overall binding strength of the imprinted surfaces toward RNase B. Remarkably, despite the high percentage of glycosylation on the two other glycoproteins (45% glycosylation for α1-acid glycoprotein and 21% glycosylation for HRP) that could enable their glycans to interact with the boronic acid moieties, weak affinities were obtained. This behavior might be a result of a combination of unmatched shape and unmatched spatial arrangement of the boron monomers on the cavity. The low, similar nonspecific binding observed on the nonimprinted surfaces for all of the proteins investigated provide further evidence that the imprinting is responsible for the observed selectivity. Overall, these findings suggest that the remarkable recognition specificity of the imprinted surfaces toward RNase B can be attributed to the precise control exerted by ATRP over the RNase B/APBA complex immobilization that results in multiple BAs being anchored in appropriate positions in the recognition cavity to interact specifically with the target RNase B glycoprotein.
In order to gain insights on the effect of the layer thickness on the binding properties of the imprinted cavities, the imprinted surfaces described above that were produced with a 20 min polymerization time and the optimum 1:10 RNase B/APBA ratio were compared with those produced with shorter (10 min) and longer times (60 min). Figure 5a reports the ellipsometry results for the three imprinted and nonimprinted polymerization times. An increase in thickness of the imprinted surfaces is clearly observed with an increase in polymerization time, in which the values are significantly higher than those obtained for the nonimprinted surfaces. These results highlight that the inclusion of the template protein is affecting the polymerization process.39 A plausible explanation might be that the presence of the protein, which imparts the polymeric growth around the template, lowers the termination rate constant, resulting in the formation of a thicker layer.40
Figure 5.
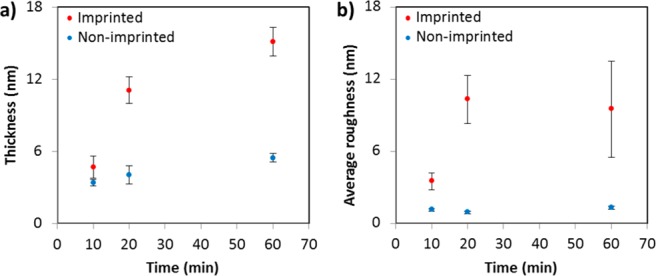
(a) Ellipsometry film thickness and (b) AFM average roughness changes of the imprinted and nonimprinted poly(MEBA)-APBA surfaces as a function of the polymerization time.
Apart from differences in thickness, the imprinted and nonimprinted surfaces are morphologically different as observed by atomic force microscopy (AFM) (Figure S2). The presence of the template protein during imprinting produced less smooth surfaces and more pronounced three-dimensional structures due to the impressions of the protein formed within the polymer matrices. The sequential increase in the average surface roughness values (Ra) and the variation of these values for the imprinted surfaces (Figure 5b) are in agreement with the ellipsometry thickness trends obtained.
The binding properties for the 10 and 60 min polymerized imprinted and nonimprinted surfaces are summarized in Figure 6 and Table 2, and it can be compared with those obtained for 20 min that are illustrated in Figure 4 and Table 1. For the 10 min polymerized surfaces, the responses of both surfaces to any of the proteins tested were relatively low and neither the imprinted nor nonimprinted surfaces showed any discernible selectivity for any one protein over another. As shown in Tables 1 and 2, the 10 min polymerized imprinted surfaces exhibit a much lower affinity toward RNase B than the 20 min polymerized imprinted surfaces. Furthermore, the errors associated with the binding affinities are significantly higher, compared with the values obtained for 20 and 60 min, indicating that the imprinting using shorter polymerization times results in poorly controlled and unreproducible imprinted recognition sites.
Figure 6.
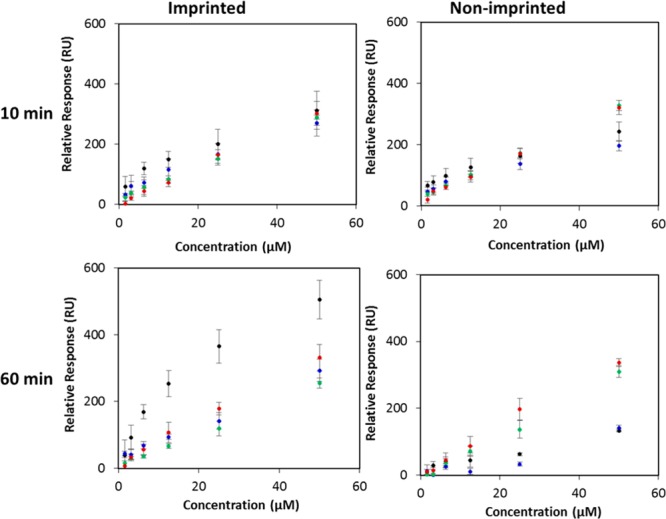
Effect of polimerization time (10 or 60 min) and protein concentration on the Req of imprinted poly(MEBA)-APBA surfaces and nonimprinted poly(MEBA)-APBA surfaces to RNase B (black), RNase A (blue), α1-acid glycoprotein (green), and HRP (red). A 1:10 RNase B/APBA ratio was used.
Table 2. KD for Each of the Proteins for the Imprinted Poly(MEBA)-APBA Surfaces Formed Using 1:10 RNase B/APBA Ratio and Either 10 or 60 min Polymerization Timea.
|
KD (μM) |
||
|---|---|---|
| polymerization (10 min) | polymerization (60 min) | |
| RNase B | 85.3 ± 31.9 | 21.5 ± 4.6 |
| RNase A | 145.5 ± 52.9 | 61.6 ± 11.3 |
| α1-acid glycoprotein | 186.3 ± 67.1 | 123.3 ± 21.6 |
| HRP | 237.7 ± 85.0 | 77.4 ± 14.0 |
| selectivity (KDA/KDB) | 1.7 | 2.9 |
The selectivity values of the imprinted poly(MEBA)-APBA surfaces for RNase B with respect to RNase A are shown at the bottom of the table.
The RNase B glycoprotein has dimensions of 3.8 nm × 2.8 nm × 2.2 nm, and the thickness obtained for the imprinted surfaces using 10 min polymerization was 4.7 ± 0.9 nm. While the dimensions of RNase B are closer to the film thickness, the results suggest that the polymer layer is too thin to generate enough complementary BA spatial arrangement for RNase B and/or enough depth for shape-matching.
In comparison with the 20 min imprinted surfaces, the 60 min surfaces likewise demonstrated that imprinting within this polymer matrix is possible. As was observed for the 20 min nonimprinted surfaces, no selectivity for any particular protein including RNase B is apparent from the 60 min nonimprinted surfaces. However, the imprinted surfaces exhibited selectivity for RNase B over all of the control proteins. As with the 20 min imprinted surfaces, the sequential increase in the response of the 60 min imprinted surfaces to increasing concentrations of RNase B is observed, which indicates that cavities well-matched to the structure and functionality of RNase B have been formed within the polymer. Nevertheless, the 60 min imprinted surfaces achieve less homogeneous recognition across different samples and also show a lower selectivity for RNase B. The selectivity of the imprint for RNase B over RNase A remained at 2.9-fold. Furthermore, the 60 min imprinted surfaces exhibit a slightly lower affinity for RNase B in comparison with the 20 min imprinted surfaces (KD of 21.5 μM vs 14.2 μM). The loss of selectivity and affinity can be attributed to the thicker polymer layer of the 60 min imprinted surface that can potentially lead to imprinted sites being “buried” deep within the polymer layer, hindering their accessibility and recognition properties. These findings establish that the thickness of the imprinting layer is critical for the binding properties of the imprinted surface, with 20 min polymerization time providing optimum conditions to endow the surface-confined cavities with specific glycoprotein recognition properties.
Conclusions
The generation of affinity tools that specifically recognizes and binds glycoproteins remains a critical bottleneck in biomedical research and diagnostic test development. In this work, a whole new strategy was developed to tailor synthetic materials that possess highly selective molecular recognition properties with affinity in the low micromolar range for glycoproteins. The unprecedented performance relies on the recognition capabilities of the surface-confined imprinted cavities that are ultimately dictated by the well-defined pattern of multiple BAs within the cavity that are sterically complementary to the unique molecular structure of the glycan on the target glycoprotein. This step is initially controlled by the stoichiometric ratio of glycoprotein target to BA moieties during complexation, in which, for RNase B, a 1:10 BA/RNase B ratio yielded imprinted surfaces with superior binding properties when compared with higher molar ratios of BA. An excess of carbohydrate receptors in the cavity can be detrimental to the overall affinity and selectivity to the target glycoprotein. In addition, we showed that the thickness of the recognition layer influences the affinity and selectivity of the resulting imprinted surface for its template, providing an additional parameter for tuning the properties of the imprinted surfaces.
This study provides an unprecedented strategy, which combines supramolecular assembly and well-controlled surface-confined ATRP polymerization, to create robust and highly reproducible template-induced glycoprotein recognition sites on material surfaces. These advanced recognition materials are of great interest in many biomedical applications, in which they are deemed particularly well-suited for achieving highly specific biosensing and bioanalytical platforms for biomedical research, glycoprotein analysis, clinical diagnosis, and cancer detection. For instance, many clinical biomarkers in cancer are glycoproteins, such as CEA in colorectal cancer, CA125 in ovarian cancer, HER2 in breast cancer, PSA in prostate cancer, and α-fetoprotein in liver cancer.41,42 In this regard, the developed glycoprotein recognition platform provides a highly valuable tool for cancer detection.
Acknowledgments
The authors acknowledge financial support of this work by the EPSRC (EP/K027263/1) and ERC (consolidator grant 614787).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsabm.9b00289.
Materials, methods, and results from circular dichroism and atomic force microscopy, and characteristics of the proteins investigated by SPR (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hwang H.; Zhang J.; Chung K. A.; Leverenz J. B.; Zabetian C. P.; Peskind E. R.; Jankovic J.; Su Z.; Hancock A. M.; Pan C.; Montine T. J.; Pan S.; Nutt J.; Albin R.; Gearing M.; Beyer R. P.; Shi M.; Zhang J. Glycoproteomics in neurodegenerative diseases. Mass Spectrom. Rev. 2009, 29, 79–125. 10.1002/mas.20221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J. B.; Marth J. D. A genetic approach to Mammalian glycan function. Annu. Rev. Biochem. 2003, 72, 643–691. 10.1146/annurev.biochem.72.121801.161809. [DOI] [PubMed] [Google Scholar]
- Adamczyk B.; Tharmalingam T.; Rudd P. M. Glycans as cancer biomarkers. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820, 1347–1353. 10.1016/j.bbagen.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Gilgunn S.; Conroy P. J.; Saldova R.; Rudd P. M.; O’Kennedy R. J. Aberrant PSA Glycosylation-A Sweet Predictor of Prostate Cancer. Nat. Rev. Urol. 2013, 10, 99–107. 10.1038/nrurol.2012.258. [DOI] [PubMed] [Google Scholar]
- Hudak J. E.; Bertozzi C. R. Glycotherapy: New Advances Inspire a Reemergence of Glycans in Medicine. Chem. Biol. 2014, 21, 16–37. 10.1016/j.chembiol.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanctot P. M.; Gage F. H.; Varki A. P. The Glycans of Stem Cells. Curr. Opin. Chem. Biol. 2007, 11, 373–380. 10.1016/j.cbpa.2007.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos-Dos-Santos A.; Oliveira I. A.; Lucena M. C.; Mantuano N. R.; Whelan S. A.; Dias W. B.; Todeschini A. R. Biosynthetic Machinery Involved in Aberrant Glycosylation: Promising Targets for Developing of Drugs Against Cancer. Front. Oncol. 2015, 5, 138. 10.3389/fonc.2015.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. S.; Shah P.; Eshghi S. T.; Yang W. M.; Trikannad N.; Yang S.; Chen L. J.; Aiyetan P.; Hoti N.; Zhang Z.; Chan D. W.; Zhang H. Comprehensive Analysis of Protein Glycosylation by Solid-Phase Extraction of N-Linked Glycans and Glycosite-Containing Peptides. Nat. Biotechnol. 2016, 34, 84–88. 10.1038/nbt.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H. P.; Suttapitugsakul S.; Sun F. X.; Wu R. H. Mass Spectrometry-Based Chemical and Enzymatic Methods for Global Analysis of Protein Glycosylation. Acc. Chem. Res. 2018, 51, 1796–1806. 10.1021/acs.accounts.8b00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner E.; Flanagan N.; Gildersleeve J. C. Perspectives on Anti-Glycan Antibodies Gleaned from Development of a Community Resource Database. ACS Chem. Biol. 2016, 11, 1773–1783. 10.1021/acschembio.6b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Luo S.; Zhang B. The Use of Lectin Microarray for Assessing Glycosylation of Therapeutic Proteins. mAbs 2016, 8, 524–535. 10.1080/19420862.2016.1149662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. X.; Xu S. F.; Li J. H. Recent Advances in Molecular Imprinting Technology: Current Status, Challenges and Highlighted Applications. Chem. Soc. Rev. 2011, 40, 2922–2942. 10.1039/c0cs00084a. [DOI] [PubMed] [Google Scholar]
- Haupt K. Biomaterials Plastic Antibodies. Nat. Mater. 2010, 9, 612–614. 10.1038/nmat2818. [DOI] [PubMed] [Google Scholar]
- Mahon C. S.; Fulton D. A. Mimicking Nature with Synthetic Macromolecules Capable of Recognition. Nat. Chem. 2014, 6, 665–672. 10.1038/nchem.1994. [DOI] [PubMed] [Google Scholar]
- Mourao C. A.; Bokeloh F.; Xu J. J.; Prost E.; Duma L.; Merlier F.; Bueno S. M. A.; Haupt K.; Bui B. T. S. Dual-Oriented Solid-Phase Molecular Imprinting: Toward Selective Artificial Receptors for Recognition of Nucleotides in Water. Macromolecules 2017, 50, 7484–7490. 10.1021/acs.macromol.7b01782. [DOI] [Google Scholar]
- Kitayama Y.; Yoshikawa K.; Takeuchi T. Post-Cross-Linked Molecular Imprinting with Functional Polymers as a Universal Building Block for Artificial Polymeric Receptors. Macromolecules 2017, 50, 7526–7534. 10.1021/acs.macromol.7b01233. [DOI] [Google Scholar]
- Stephenson-Brown A.; Wang H. C.; Iqbal P.; Preece J. A.; Long Y. T.; Fossey J. S.; James T. D.; Mendes P. M. Glucose Selective Surface Plasmon Resonance-Based Bis-Boronic Acid Sensor. Analyst 2013, 138, 7140–7145. 10.1039/c3an01233f. [DOI] [PubMed] [Google Scholar]
- D’Hooge F.; Elfeky S. A.; Flower S. E.; Pascu S. I.; Jenkins A. T. A.; van den Elsen J. M. H.; James T. D.; Fossey J. S. Biotinylated Boronic Acid Fluorophore Conjugates: Quencher Elimination Strategy for Imaging and Saccharide Detection. RSC Adv. 2012, 2, 3274–3280. 10.1039/c2ra00542e. [DOI] [Google Scholar]
- Stephenson-Brown A.; Yong S.; Mansor M. H.; Hussein Z.; Yip N. C.; Mendes P. M.; Fossey J. S.; Rawson F. J. Electronic Communication of Cells with a Surface Mediated by Boronic Acid Saccharide Interactions. Chem. Commun. 2015, 51, 17213–17216. 10.1039/C5CC04311E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. C.; Zhou H.; Chen B. Q.; Mendes P. M.; Fossey J. S.; James T. D.; Long Y. T. A Bis-Boronic Acid Modified Electrode for the Sensitive and Selective Determination of Glucose Concentrations. Analyst 2013, 138, 7146–7151. 10.1039/c3an01234d. [DOI] [PubMed] [Google Scholar]
- Huang J.; Wu Y. N.; Cong J. J.; Luo J.; Liu X. Y. Selective and Sensitive Glycoprotein Detection via a Biomimetic Electrochemical Sensor Based on Surface Molecular Imprinting and Boronate-Modified Reduced Graphene Oxide. Sens. Actuators, B 2018, 259, 1–9. 10.1016/j.snb.2017.12.049. [DOI] [Google Scholar]
- Luo J.; Huang J.; Cong J. J.; Wei W.; Liu X. Y. Double Recognition and Selective Extraction of Glycoprotein Based on the Molecular Imprinted Graphene Oxide and Boronate Affinity. ACS Appl. Mater. Interfaces 2017, 9, 7735–7744. 10.1021/acsami.6b14733. [DOI] [PubMed] [Google Scholar]
- Wang S. S.; Ye J.; Bie Z. J.; Liu Z. Affinity-Tunable Specific Recognition of Glycoproteins via Boronate Affinity-Based Controllable Oriented Surface Imprinting. Chem. Sci. 2014, 5, 1135–1140. 10.1039/c3sc52986j. [DOI] [Google Scholar]
- Stephenson-Brown A.; Acton A. L.; Preece J. A.; Fossey J. S.; Mendes P. M. Selective glycoprotein detection through covalent templating and allosteric click-imprinting. Chem. Sci. 2015, 6, 5114–5119. 10.1039/C5SC02031J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X.; Li X.; Husson S. M. Surface Molecular Imprinting by Atom Transfer Radical Polymerization. Biomacromolecules 2005, 6, 1113–1121. 10.1021/bm049311i. [DOI] [PubMed] [Google Scholar]
- Stephenson-Brown A.; Wang H. C.; Iqbal P.; Preece J. A.; Long Y.; Fossey J. S.; James T. D.; Mendes P. M. Glucose Selective Surface Plasmon Resonance-Based Bis-Boronic Acid Sensor. Analyst 2013, 138, 7140–7145. 10.1039/c3an01233f. [DOI] [PubMed] [Google Scholar]
- Bain C. D.; Troughton E. B.; Tao Y. T.; Evall J.; Whitesides G. M.; Nuzzo R. G. Formation of Monolayer Films by the Spontaneous Assembly of Organic Thiols from Solution onto Gold. J. Am. Chem. Soc. 1989, 111, 321–335. 10.1021/ja00183a049. [DOI] [Google Scholar]
- Ulman A. Formation and Structure of Self-Assembled Monolayers. Chem. Rev. 1996, 96, 1533–1554. 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]
- Zhang H. L.; Zhang J.; Li H. Y.; Liu Z. F.; Li H. L. Structural Investigation of a New Series of Azobenzene-Containing Self-Assembled Monolayers on Gold. Mater. Sci. Eng., C 1999, 8–9, 179–185. 10.1016/S0928-4931(99)00020-X. [DOI] [Google Scholar]
- Weidner T.; Bretthauer F.; Ballav N.; Motschmann H.; Orendi H.; Bruhn C.; Siemeling U.; Zharnikov M. Correlation Between the Molecular Structure and Photoresponse in Aliphatic Self-Assembled Monolayers with Azobenzene Tailgroups. Langmuir 2008, 24, 11691–11700. 10.1021/la802454w. [DOI] [PubMed] [Google Scholar]
- Castner D. G.; Hinds K.; Grainger D. W. X-ray Photoelectron Spectroscopy Sulfur 2p Study of Organic Thiol and Disulfide Binding Interactions with Gold Surfaces. Langmuir 1996, 12, 5083–5086. 10.1021/la960465w. [DOI] [Google Scholar]
- Gopireddy D.; Husson S. M. Macromolecules (Washington, DC, U. S.) 2002, 35, 4218–4221. 10.1021/ma012254q. [DOI] [Google Scholar]
- Ribelli T. G.; Augustine K. F.; Fantin M.; Krys P.; Poli R.; Matyjaszewski K. Macromolecules 2017, 50, 7920–7929. 10.1021/acs.macromol.7b01552. [DOI] [Google Scholar]
- Zhou D.; Gao X.; Wang W.-J; Zhu S. Macromolecules 2012, 45, 1198–1207. 10.1021/ma202640x. [DOI] [Google Scholar]
- Barbey R.; Lavanant L.; Paripovic D.; Schuewer N.; Sugnaux C.; Tugulu S.; Klok H.-A. Polymer Brushes via Surface-Initiated Controlled Radical Polymerization: Synthesis, Characterization, Properties, and Applications. Chem. Rev. 2009, 109, 5437–5527. 10.1021/cr900045a. [DOI] [PubMed] [Google Scholar]
- Huang W. X.; Baker G. L.; Bruening M. L. Controlled Synthesis of Cross-Linked Ultrathin Polymer Films by Using Surface-Initiated Atom Transfer Radical Polymerization. Angew. Chem., Int. Ed. 2001, 40, 1510–1512. . [DOI] [PubMed] [Google Scholar]
- Jiang J. S.; Zhang Y.; Guo X. Z.; Zhang H. Q. Ambient Temperature Synthesis of Narrow or Monodisperse, Highly Cross-Linked, and “Living” Polymer Microspheres by Atom Transfer Radical Precipitation Polymerization. RSC Adv. 2012, 2, 5651–5662. 10.1039/c2ra01249a. [DOI] [Google Scholar]
- Otsuka H.; Uchimura E.; Koshino H.; Okano T.; Kataoka K. Anomalous Binding Profile of Phenylboronic Acid with N-Acetylneuraminic Acid (Neu5Ac) in Aqueous Solution with Varying pH. J. Am. Chem. Soc. 2003, 125, 3493–3502. 10.1021/ja021303r. [DOI] [PubMed] [Google Scholar]
- Polowinski S. Template Polymerisation and Co-polymerisation. Prog. Polym. Sci. 2002, 27, 537–577. 10.1016/S0079-6700(01)00035-1. [DOI] [Google Scholar]
- Tan Y. Y.; van Ekenstein G. O. R. A. A Generalized Kinetic-Model for Radical-Initiated Template Polymerizations in Dilute Template Systems. Macromolecules 1991, 24, 1641–1647. 10.1021/ma00007a029. [DOI] [Google Scholar]
- Pinho S. S.; Reis C. A. Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- Munkley J.; Elliott D. J. Hallmarks of glycosylation in cancer. Oncotarget 2016, 7, 35478–35489. 10.18632/oncotarget.8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.