

Abstract
Background
Disease modeling with patient-derived induced pluripotent stem cells (iPSCs) is a powerful tool for elucidating the mechanisms underlying disease pathogenesis and developing safe and effective treatments. Patient peripheral blood (PB) cells are used for iPSC generation in many cases since they can be collected with minimum invasiveness. To derive iPSCs that lack immunoreceptor gene rearrangements, hematopoietic stem and progenitor cells (HSPCs) are often targeted as the reprogramming source. However, the current protocols generally require HSPC mobilization and/or ex vivo expansion owing to their sparsity at the steady state and low reprogramming efficiencies, making the overall procedure costly, laborious, and time-consuming.
Methods
We have established a highly efficient method for generating iPSCs from non-mobilized PB-derived CD34+ HSPCs. The source PB mononuclear cells were obtained from 1 healthy donor and 15 patients and were kept frozen until the scheduled iPSC generation. CD34+ HSPC enrichment was done using immunomagnetic beads, with no ex vivo expansion culture. To reprogram the CD34+-rich cells to pluripotency, the Sendai virus vector SeVdp-302L was used to transfer four transcription factors: KLF4, OCT4, SOX2, and c-MYC. In this iPSC generation series, the reprogramming efficiencies, success rates of iPSC line establishment, and progression time were recorded. After generating the iPSC frozen stocks, the cell recovery and their residual transgenes, karyotypes, T cell receptor gene rearrangement, pluripotency markers, and differentiation capability were examined.
Results
We succeeded in establishing 223 iPSC lines with high reprogramming efficiencies from 15 patients with 8 different disease types. Our method allowed the rapid appearance of primary colonies (~ 8 days), all of which were expandable under feeder-free conditions, enabling robust establishment steps with less workload. After thawing, the established iPSC lines were verified to be pluripotency marker-positive and of non-T cell origin. A majority of the iPSC lines were confirmed to be transgene-free, with normal karyotypes. Their trilineage differentiation capability was also verified in a defined in vitro assay.
Conclusion
This robust and highly efficient method enables the rapid and cost-effective establishment of transgene-free iPSC lines from a small volume of PB, thus facilitating the biobanking of patient-derived iPSCs and their use for the modeling of various diseases.
Electronic supplementary material
The online version of this article (10.1186/s13287-019-1273-2) contains supplementary material, which is available to authorized users.
Keywords: Human-induced pluripotent stem cells, Sendai virus vector, SeVdp-302L, Feeder-free, CD34+ hematopoietic stem and progenitor cells, Peripheral blood, Disease modeling, Biobank
Background
In the past decade, thousands of human-induced pluripotent stem cells (hiPSCs) have been generated from healthy donors and from patients afflicted with various diseases [1–3]. The patient-derived hiPSCs have the potential to infinitely produce specialized disease-associated cells and organoids, enabling researchers to recapitulate some pathological aspects in Petri dishes for human disease modeling. In fact, such models have already contributed to the uncovering of molecular mechanisms of pathogenesis, potentially leading to the development of new treatments for some diseases [3]. Therefore, a simple and efficient method to establish patient-derived hiPSC lines in laboratories and biobanks is essential to facilitate the further understanding of disease pathogenesis and the development of safe and effective treatments.
Although many different methods for generating hiPSCs have been developed, they all have huge variations in their characteristics, including efficiency, quality, speed, cost, and robustness. Several non-integrating methods are currently available, using episomal DNAs [4, 5], Sendai virus (SeV) [6], adenovirus [7], PiggyBac transposons [8], minicircles [9], synthesized RNAs [10], and recombinant proteins [11] to transfer reprogramming factors into target cells. Moreover, many additional enhancers for somatic cell reprogramming have been identified and used with or without these technologies [12]. However, these technologies generally remain inefficient overall, with their reprogramming efficiencies being less than 1% in many cases [13–15]. An unstable and/or insufficient transgene expression may be a contributory factor. Prolonged residual exogenous factors may also negatively affect the high-quality generation of transgene-free hiPSCs [4, 6]. Recently, we reported the use of a novel SeV vector, SeVdp-302L, which delivers a single-stranded RNA genome containing multiple transgenes into somatic cells in a manner suitable for hiPSC generation. The SeVdp-302L vector carrying four Yamanaka factors, named SeVdp(KOSM)-302L, induces stable transgene expression and also possesses an auto-erasable feature by responding to the presence of stem cell-specific microRNA-302. Using this vector, we demonstrated the simple and efficient generation of transgene-free hiPSCs without additional reprogramming enhancers [16].
Patient-derived hiPSCs are generated from various somatic cells, the two major sources of which are skin fibroblasts [1] and peripheral blood (PB) [17]. However, skin fibroblasts require some invasive procedures for collection and have a potential risk of accumulated mutations resulting from their exposure to external stressors, such as UV rays. Indeed, undeniably high frequencies of preexisting coding mutations have been identified in both the original fibroblasts and their derived hiPSCs [18]. On the other hand, PB is thought to be a more attractive somatic cell source, since its collection is less invasive and its mutational load is presumably lower than that of skin tissue. PB is also considered useful for hiPSC generation because the cells remain viable enough at room temperature for ~ 48 h after collection [19] and after cryopreservation, enabling long-distance transport of the patient samples and scheduled hiPSC generation. For PB cell reprogramming, either lymphocytes (mostly T cells) or non-lymphocytes (e.g., stem and progenitor cells and myeloid cells) are used to generate hiPSCs. The choice depends on the aim of the studies, where lymphocytes are preferred when the specific immunoreceptor gene rearrangement is required, whereas non-lymphocytes are used for broader applications. Pioneering studies tested PB-derived CD34+ hematopoietic stem and progenitor cells (HSPCs) as a source for generating non-lymphocyte-derived hiPSCs, resulting in the successful establishment of such cells but with very low reprogramming efficiencies [20, 21]. Other studies used mobilized PB-derived CD34+ cells to obtain a sufficient number of target cells [17, 22–25], but the mobilization procedure was not suited to ill-conditioned patients in general. For the use of non-mobilized PB cells, most studies have conducted ex vivo expansion culture of the CD34+ HSPCs either in a way favoring general myeloid cell proliferation or erythroid cell growth, mainly to overcome their scarcity at the steady state or to enhance their overall efficiencies [19, 20, 26–37]. These cultures need a lot of cytokines, making the procedures costly and labor-intensive. Therefore, it is desirable to develop an hiPSC generation protocol that allows the use of non-mobilized PB-derived CD34+ cells without the need for extensive culturing, while achieving high efficiency and robustness.
Previously, we had succeeded in generating disease-specific hiPSCs from patient bone marrow-derived CD34+ cells or from freshly obtained PB-derived CD34+ cells using the SeVdp vector (the one-generation-old version of SeVdp-302L) with on-feeder conditions [38–40]. However, for the purpose of biobanking, we sought to establish a more robust method to fulfill the abovementioned requirements and to allow a feeder-free protocol for simplicity. Consequently, in this present paper, we report the successful establishment of a series of hiPSC lines from 15 patients with 8 different types of diseases, using our new protocol. This simple SeV method allows the robust and rapid establishment of transgene-free hiPSC lines from non-mobilized blood samples, with high reprogramming efficiencies (up to > 5%). The scheduled hiPSC generation was feasible because we could start a CD34+ cell-enrichment step from frozen mononuclear cell stocks without hampering the overall establishment procedures. No extensive expansion culture was necessary. Characterization of the hiPSC lines verified their utility for downstream applications, with their stable pluripotency and intact trilineage differentiation capability. Taken together, the results of this study indicate that this robust and highly efficient method promises the facile establishment of disease-specific hiPSC lines that are compatible with the purpose for biobanking, which will be useful for medical research.
Methods
Preparation of healthy donor and patient PBMC samples
A frozen vial of PB mononuclear cells (PBMCs) derived from a healthy donor was purchased from Lonza Walkersville. The other PB samples described in this report were obtained from 15 patients with 8 different diseases. Informed consent was obtained in an appropriate manner for all the cases according to the Declaration of Helsinki standard ethics procedure, with approval from the Institutional Ethical Committees (see “Ethics approval and consent to participate” section). The non-mobilized fresh PB samples from patients were collected into heparin tubes by venipuncture. Except for 2 cases, all patient PBMCs were purified with Lymphocyte Separation Medium (PromoCell) in 50-mL Leucosep™ Tubes (Greiner Bio-One) within 5 h after PB collection and were then cryopreserved in Bambanker™ freezing medium (NIPPON Genetics) for up to ~ 1 year in liquid nitrogen until use for hiPSC generation (Table 1). In the cases of patients for TkSCR1 and TkS42-1, who resided far from Tokyo, the purification and cryopreservation of their PBMCs were performed on the next day after long-distance transport of the samples to our laboratory.
Table 1.
Information on the healthy donor and patient samples
| Healthy donor and patients (hiPSC line) | Age (year)/gender | No. of cells or blood volume donated | No. of purified PBMCs per 1 mL blood sample |
|---|---|---|---|
| Healthy (TkPP2) | 33/M | 1.0 × 107 cells | N.A. |
| SLE (TkSLE3) | 59/F | 22 mL | 0.5 × 106 cells/mL |
| SLE (TkSLE4) | 50/F | 30 mL | 0.5 × 106 cells/mL |
| SLE (TkSLE5) | 43/F | 30 mL | 1.3 × 106 cells/mL |
| PM (TkSPD3) | 65/M | 30 mL | 0.7 × 106 cells/mL |
| PM (TkSPD4) | 45/F | 30 mL | 2.4 × 106 cells/mL |
| PM (TkSPD5) | 56/M | 30 mL | 1.1 × 106 cells/mL |
| X-CGD (TkSCG3) | 18/M | 38 mL | 0.8 × 106 cells/mL |
| X-CGD (TkSCG4) | 9/F | 20 mL | 1.2 × 106 cells/mL |
| PID (TkSPR1) | 33/F | 30 mL | 1.9 × 106 cells/mL |
| PID (TkSPR2) | 30/F | 22 mL | 1.3 × 106 cells/mL |
| JIA (TkSCR1) | 3/F | 23 mL | 2.4 × 106 cells/mL |
| CMS (TkS42–1) | 11/F | 26 mL | 0.7 × 106 cells/mL |
| MD (TkSmD1) | 59/M | 40 mL | 0.5 × 106 cells/mL |
| MD (TkSmD3) | 32/M | 30 mL | 1.1 × 106 cells/mL |
| KCS2 (TkSKCII2) | 13/F | 22 mL | 1.0 × 106 cells/mL |
The disease/symptom affecting each individual and the name of the derived human induced pluripotent stem cell (hiPSC) line are listed. The age and gender of each donor are also indicated. The number (No.) of peripheral blood mononuclear cells (PBMCs) and total peripheral blood (PB) volume donated are shown for the purchased healthy donor PBMCs and the patient PBMCs, respectively. The right-hand column shows the number of PBMCs purified from 1 mL of patient PB
SLE systemic lupus erythematosus, PM polymyositis, X-CGD X-linked chronic granulomatous disease, PID primary immunodeficiency, JIA juvenile idiopathic arthritis, CMS congenital malformation syndrome, MD mitochondrial diabetes, KCS2 Kenny-Caffey syndrome type 2, N.A not applicable
Preparation of CD34+-enriched cell population
At day − 3, 0.4 × 107 to 1.0 × 107 PBMCs were thawed with ThawSTAR™ (BioCision) and kept overnight in Embryoid Body (EB) medium in 6-well plates at 37 °C with 5% CO2 (Fig. 1a and Table 2). The EB medium consisted of Iscove’s modified Dulbecco’s medium (Sigma) supplemented with 15% fetal bovine serum (Nichirei Biosciences), ITS liquid media supplement (Sigma), penicillin-streptomycin-glutamine (Gibco), 50 μg/mL l-ascorbic acid (Sigma), 0.45 mM 1-thioglycerol (Sigma), and the following six cytokines: 50 ng/mL stem cell factor, 50 ng/mL Fms-related tyrosine kinase 3 ligand, 10 ng/mL interleukin-3, 10 ng/mL interleukin-6, 50 ng/mL thrombopoietin, and 20 ng/mL granulocyte colony-stimulating factor (G-CSF) (all from R&D Systems). At day − 2, enrichment of the CD34+ cells was performed using the CD34 MicroBead Kit (Miltenyi Biotec) according to the manufacturer’s instructions. The CD34+-enriched PBMCs were kept overnight in EB medium in 96-well plates at 37 °C with 5% CO2 to ensure the recovery of truly viable cells for the subsequent reprogramming procedures (Fig. 1a).
Fig. 1.

Healthy donor-derived human-induced pluripotent stem cell (hiPSC) generation from non-mobilized peripheral blood (PB)-derived CD34+ hematopoietic stem and progenitor cells (HSPCs) using SeVdp(KOSM)-302L. a Schematic diagram illustrating the schedule of hiPSC generation. b Percentages of cells expressing CD34, as assessed by FACS analysis of non-enriched peripheral blood mononuclear cells (PBMCs), a flow-through population (flow-through), and the CD34+-selected cells (CD34+ cells). The results demonstrate significant enrichment of the CD34+ cells (up to 60.8%) after immunomagnetic bead selection. c Sequential images of a representative colony derived from SeVdp(KOSM)-302L-transduced CD34+ cells, showing a phase of initial proliferation (day 1–day 4), followed by the formation of spherical colony-like structures (day 5–day 11). Also shown are images of typical hiPSC-like colonies that appeared during the subsequent expansion phase (day 17 and day 37). Magnified images are shown in insets for clarity. P1 and P4 indicate passage 1 and passage 4, respectively. d Colony formation efficiency of each seeded cell type. PBMCs, flow-through cells, and CD34+ cells were tested after infection with SeVdp(KOSM)-302L. Each bar represents the efficiency assessed in each individual well. The mean efficiency values for PBMCs (0.17%), flow-through (0%), and CD34+ PBMCs (5.58%) are shown. e Fluorescent images of live colonies stained with anti-TRA-1-60 antibody (red, top), anti-TRA-1-81 antibody (green, middle), or the CDy1 dye (red, bottom). Nuclei were stained with Hoechst 33342 dye (blue)
Table 2.
Amounts of PB, PBMCs, and CD34+-enriched cells used for reprogramming
| Healthy donor and patients (hiPSC line) | Amounts of PB and cryopreserved PBMCs used for hiPSC generation (% of live PBMCs after thawing) | No. of CD34+-enriched cells for transfection | |
|---|---|---|---|
| Healthy (TkPP2) | – | 1.0 × 107 cells | 15,000 |
| SLE (TkSLE3) | 8.8 mL | 0.4 × 107 cells (33%) | 13,000 |
| SLE (TkSLE4) | 18.4 mL | 1.0 × 107 cells (50%) | 16,000 |
| SLE (TkSLE5) | 7.5 mL | 1.0 × 107 cells (71%) | 39,375 |
| PM (TkSPD3) | 7.5 mL | 0.5 × 107 cells (52%) | 11,500 |
| PM (TkSPD4) | 4.2 mL | 1.0 × 107 cells (61%) | 12,5000 |
| PM (TkSPD5) | 8.8 mL | 1.0 × 107 cells (38%) | 14,000 |
| X-CGD (TkSCG3) | 13.1 mL | 1.0 × 107 cells (53%) | 24,700 |
| X-CGD (TkSCG4) | 8.5 mL | 1.0 × 107 cells (56%) | 9000 |
| PID (TkSPR1) | 5.3 mL | 1.0 × 107 cells (76%) | 18,000 |
| PID (TkSPR2) | 7.7 mL | 1.0 × 107 cells (38%) | 31,000 |
| JIA (TkSCR1) | 4.2 mL | 1.0 × 107 cells (65%) | 33,375 |
| CMS (TkS42–1) | 13.9 mL | 1.0 × 107 cells (21%) | 5500 |
| MD (TkSmD1) | 20.0 mL | 1.0 × 107 cells (12%) | 63,000 |
| MD (TkSmD3) | 4.7 mL | 0.5 × 107 cells (56%) | 55,000 |
| KCS2 (TkSKCII2) | 4.8 mL | 0.5 × 107 cells (68%) | 42,000 |
The amounts of peripheral blood (PB) and peripheral blood mononuclear cells (PBMCs) used in the generation of these human induced pluripotent stem cell (hiPSC) lines are listed. The PB volume shown here was calculated from the corresponding PBMC numbers contained in each frozen vial (cryopreserved PBMCs). The number of PBMCs represents the value before cryopreservation. After thawing, a certain level of cell death occurs; % live PBMCs are thus shown in parentheses, calculated by counting the actual number of living cells recovered after thawing. The number of CD34+-enriched cells used for SeVdp(KOSM)-302L transduction is also indicated
SLE systemic lupus erythematosus, PM polymyositis, X-CGD X-linked chronic granulomatous disease, PID primary immunodeficiency, JIA juvenile idiopathic arthritis, CMS congenital malformation syndrome, MD mitochondrial diabetes, KCS2 Kenny-Caffey syndrome type 2
Analysis of enriched CD34+ cells derived from PBMCs
The purchased healthy donor PBMCs were kept overnight in EB medium after thawing, following which approximately three-quarters of the cells were subjected to CD34+ cell enrichment as described above. Both positively selected cells and a flow-through population were obtained. The remaining one-quarter of PBMCs was left untouched as the non-treated control sample. The three samples were stained with allophycocyanin-conjugated anti-human CD34 antibody (343510, BioLegend), Pacific Blue™ dye-conjugated anti-human CD45 antibody (304022, BioLegend), and a fluorescein isothiocyanate-conjugated anti-human lineage cocktail (CD3, CD14, CD19, CD20, and CD56) (348701, BioLegend). The samples were also stained with propidium iodide to exclude dead cells. Population data were acquired with a fluorescence-activated cell sorting (FACS) Aria II sorter (BD Biosciences) and analyzed using FlowJo X software (Tree Star). A series of serially diluted CD34+ cells was prepared using TkSPD4 donor’s PBMCs by combining the microbead-selected samples and the flow-through population. The immunostaining and flow cytometry analysis were performed in the same way as described above.
hiPSC generation
At day − 1, the CD34+-enriched cells were infected with SeVdp(KOSM)-302L at a multiplicity of infection (MOI) of 3–7 (Fig. 1a). For the experiment shown in Additional file 5: Figure S4, the CytoTune®-iPS2.0 (ID Pharma) was used to infect CD34+-enriched cells at an MOI of > 1, > 3, and > 6, based on the titer described in the instruction manual (> 5.0 × 106 CIU/100 μL). After addition of the viral vector, the cells were left at room temperature for 2 h and then incubated overnight at 37 °C with 5% CO2. At day 0, the SeVdp(KOSM)-302L-infected cells were seeded into iMatrix-511 (Nippi)-coated 6-well plates (TPP) with StemFit® AK02N medium (REPROCELL) containing 10 μM Y-27632 (Fig. 1a). At day 2, half the medium volume was replaced with fresh StemFit AK02N medium, following which the entire medium was changed on every 2 days until colony pickup. At days 8–14, individual colonies with an appropriate size of 250–500 μm in diameter were picked up into iMatrix-511-coated 24-well plates (TPP) containing StemFit AK02N medium supplemented with 10 μM Y-27632 (Fig. 1a). At the medium changes on every 2 days, small interfering RNA (siRNA) treatment against SeVdp(KOSM)-302L was performed 3 times using Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer’s instructions (Fig. 1a). The two siRNAs (siL527 and siL1913), used at 1.6 nM each, were generated from the following single-stranded RNA oligonucleotides (Fasmac): siL527-S, 5′-GGUUCAGCAUCAAAUAUGAAG-3′; siL527-AS, 5′-UCAUAUUUGAUGCUGAACCAU-3′; siL1913-S, 5′-GGUCCAGACAUGAAUUCAAAG-3′; and siL1913-AS, 5′-UUGAAUUCAUGUCUGGACCAU-3′.
hiPSC expansion and maintenance culture for storage
For expansion and maintenance, hiPSCs were cultured on iMatrix-511- or Vitronectin (VTN-N, Gibco)-coated 12- or 6-well plates in StemFit AK02N medium (with iMatrix-511) or Essential 8™ medium (Gibco) (with VTN-N) at 37 °C with 5% CO2. Daily medium change was routinely performed. When passaged, the hiPSCs were mechanically and enzymatically detached from the plates using a scraper, 0.5× TrypLE™ Select (Gibco), 0.5 mM EDTA (Invitrogen), and/or Accutase® (Sigma), and plated on fresh iMatrix-511- or VTN-N-coated plates in the respective medium described above (containing 10 μM Y-27632). Before their use in the experiments, all cells were confirmed to be mycoplasma-free, using the MycoAlert™ Mycoplasma Detection Kit (Lonza) and MycoAlert™ Assay Control Kit (Lonza). Luminescence was measured with a GloMax® Explorer system (Promega). Images of hiPSC colonies were captured with a Leica DMI3000B system and their processing was performed using the ImageJ (NIH) or GNU Image Manipulation Program 2.10 (GIMP2.10). Unless we had a specific need, we generally selected ~ 12 colonies for the subsequent expansion steps that lasted for another 4–5 weeks. The small-scale frozen stocks (2 tubes) of the 12 lines were initially generated as the backup stocks, with the cells kept in the iMatrix-511/StemFit AK02N media (Fig. 1a). Six lines were adapted to culture under the VTN-N/Essential 8 conditions and finally stored as large-scale frozen stocks (10 tubes), where ~ 2.0 × 106 cells per tube were cryopreserved in Bambanker for the hiPSC banking.
RT-PCR verification of SeVdp(KOSM)-302L removal
Total RNA was isolated from the hiPSCs using the AllPrep DNA/RNA Mini Kit (Qiagen), RNeasy® Mini Kit (Qiagen), or Monarch® Total RNA Miniprep Kit (New England Biolabs). Complementary DNA (cDNA) was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The reverse transcription-polymerase chain reaction (RT-PCR) assay for SeVdp(KOSM)-302L cDNA was performed with TaKaRa Ex Taq® Hot Start Version (TaKaRa). GAPDH was used as the internal control. The PCR products were resolved on 2.5% agarose gels. The primers used in this study are listed in Additional file 1: Table S1.
In vitro differentiation of three germ layers
To analyze the capability of the hiPSCs to differentiate into three germ layers, in vitro differentiation was performed with the STEMdiff™ Trilineage Differentiation Kit (STEM CELL Technologies) according to the manufacturer’s instructions.
Quantification of pluripotency and differentiation markers with real-time PCR
cDNA was prepared as described above for quantifying the pluripotency markers octamer-binding transcription factor 4 (OCT4) and NANOG by multiplex TaqMan real-time PCR, using the Eagle Taq Master Mix with ROX (Roche). Universal probes #34 and #69 (Roche) were used for labeling the OCT4 and NANOG products, respectively. The Human G6PD Gene Assay (Roche) was used for quantifying the reference gene. Because the universal probe #69 failed to be incorporated into the NANOG transcripts of the TkPP2 lines owing to the presence of a single nucleotide polymorphism, the SYBR Green method was used with the Power SYBR Green PCR Master Mix (Applied Biosystems) for these cell lines only. The relative expression of OCT4 and NANOG was evaluated with reference to their expression levels in our standard hiPSC line TkDA3-4 previously established from a healthy donor [41].
The gene expression of trilineage differentiation markers was evaluated using a real-time PCR SYBR Green method with the Power SYBR Green PCR Master Mix (Applied Biosystems) or THUNDERBIRD® SYBR qPCR Mix (Toyobo). The marker genes analyzed were paired box 6 (PAX6), SRY-box 1 (SOX1), SRY-box 17 (SOX17), Foxhead box A2 (FOXA2), T-box transcription factor T (or Brachyury) (TBXT), and neural cell adhesion molecule (NCAM). G6PD was used as the reference gene. The primers used in this analysis are listed in Additional file 1: Table S1. All the real-time PCR analyses were performed using a CFX96 C1000™ thermal cycler (Bio-Rad). Relative expression was calculated by the ΔΔCt method with CFX™ Manager software (Bio-Rad).
Detection of pluripotency characteristics with immunostaining and chemical staining
Live hiPSC colonies were directly stained with the NL557-conjugated anti-human TRA-1-60 antibody (NLLC4770R, R&D Systems), the LN493-conjugated anti-human TRA-1-81 antibody (NLLC16581G, R&D Systems), or the fluorescent imaging probe CDy1 [42]. Fluorescence images were captured with a Nikon Eclipse Ti microscope system. Images were edited using ImageJ (NIH) or GNU Image Manipulation Program 2.10 (GIMP2.10).
Karyotyping
hiPSCs were expanded in VTN-N-coated 25-cm2 flasks (TPP) with Essential 8 medium. Standard G-banded karyotyping was performed by the Chromosome Science Laboratory or NIHON Gene Research Laboratories.
TCRγ gene rearrangement analysis
Genomic DNA was isolated using the AllPrep DNA/RNA Mini Kit. Then, the rearrangement of the T cell receptor gamma (TCRγ) gene segment was analyzed using the T Cell Receptor Gamma Gene Rearrangement Assay Kit for Gel Detection (Invivoscribe) according to the manufacturer’s instructions.
Flow cytometry analysis
Detailed information is provided in Additional file 2: Supplemental Methods.
Results
Establishing hiPSC lines from healthy donor PB-derived CD34+ HSPCs
We sought to establish an hiPSC generation method capable of meeting the following requirements: (1) use of non-mobilized PB cells, (2) compatibility with scheduled procedures, (3) integration-free culture, (4) absence of immunoreceptor gene rearrangement, (5) high efficiency, (6) cost-effectiveness, and (7) feeder-free conditions. We finally achieved these goals with the protocol summarized in Fig. 1a.
One example of a series of hiPSC generation processes is shown in Fig. 1b–e. In this attempt, we started from frozen healthy donor PBMCs (1.0 × 107 cells) that were commercially available (Table 1). After thawing at day − 3, the PBMCs were kept overnight in an incubator under cytokine-rich conditions, mainly in order to maximize the recovery of viable HSPCs rather than to aim at cell expansion. At day − 2, CD34+ cell enrichment was performed using immunomagnetic beads, and the level of enrichment was checked in an independent experiment. The ~ 15,000 CD34+ cells enriched in this attempt (Table 2) made up ~ 60% of the final selection product (Fig. 1b). These cells were again kept overnight in a 96-well plate under the cytokine-rich culture condition to obtain only truly viable cells for the subsequent steps (Fig. 1a). At day − 1, SeVdp(KSOM)-302L was added to the cells at an MOI of 3 and the plates were incubated overnight. After these 3-day procedures, the transduced cells were seeded at various cell densities (i.e., 500, 1000, 2000, and 4000 cells/well) onto iMatrix-511-coated 6-well plates containing StemFit AK02N medium. No more hematopoietic cytokines were needed beyond day 0. After seeding, some adherent cells showed initial proliferation on days 1–3 and started forming colony-like structures on days 4–6 (Fig. 1c). Once formed, the growth of these spherical brown colonies was rapid, with their size reaching > 200 μm at day 11 (Fig. 1c). On the basis of the number of colonies counted at day 7, the colony formation efficiency was calculated to be 5.58% per cell input (Fig. 1d and Table 3), which was extremely high compared with that of previous reports (< 1% in many cases) [43, 44]. These primary colonies were confirmed to be TRA-1-60-positive and TRA-1-81-positive (Fig. 1e). They also stained positively with CDy1 dye (Fig. 1e), demonstrating that their characteristics were compatible with those of pluripotent colonies. When the non-enriched PBMCs and flow-through samples (both containing only a few CD34+ cells (Fig. 1b)) were used under the same conditions, colony formation barely occurred (Fig. 1d), suggesting that the CD34+ cells represented a preferable target for highly efficient hiPSC generation in the SeVdp(KOSM)-302L reprogramming system. The idea that the purified CD34+ cells are well-suited to hiPSC establishment was later confirmed by the additional experiment, the results of which showed a strong positive correlation between the proportion of CD34+ cells and the colony formation efficiencies (Additional file 3: Figure S1).
Table 3.
Colony formation efficiencies and number of established lines
| Healthy donor and patients (hiPSC line) | Colony formation efficiencies (No. of colonies/No. of seeded cells) | No. of colonies per 1 mL blood | No. of lines established | Recovery success rate from frozen stock (No. of lines checked) |
|---|---|---|---|---|
| Healthy (TkPP2) | 5.58% (781/14,000) | – | 10 | 100% (2) |
| SLE (TkSLE3) | 2.61% (339/13,000) | 38.5 | 12 | 100% (6) |
| SLE (TkSLE4) | 0.08% (12/16,000) | 0.65 | 4 | 100% (4) |
| SLE (TkSLE5) | 3.26% (1283/39,375) | 171.1 | 12 | 100% (6) |
| PM (TkSPD3) | 1.67% (130/7750) | 17.4 | 12 | 100% (5) |
| PM (TkSPD4) | 3.17% (246/7750) | 59.1 | 14 | 100% (3) |
| PM (TkSPD5) | 0.10% (16/14,000) | 1.8 | 12 | 100% (6) |
| X-CGD (TkSCG3) | 0.29% (23/7750) | 1.7 | 12 | 100% (6) |
| X-CGD (TkSCG4) | 0.26% (23/9000) | 2.7 | 12 | 100% (6) |
| PID (TkSPR1) | 2.43% (438/18,000) | 83.2 | 12 | 100% (5) |
| PID (TkSPR2) | 2.50% (294/11,750) | 38.1 | 12 | 100% (6) |
| JIA (TkSCR1) | 1.59% (530/33,375) | 126.7 | 11 | 100% (6) |
| CMS (TkS42–1) | 0.34% (17/5000) | 1.2 | 14 | 100% (6) |
| MD (TkSmD1) | 1.09% (85/7800) | 4.6 | 26 | 100% (7) |
| MD (TkSmD3) | 1.09% (215/19,763) | 45.9 | 36 | 100% (13) |
| KCS2 (TkSKCII2) | 0.15% (43/27,750) | 8.9 | 12 | 100% (2) |
The colony formation efficiency of each seeded cell line and the estimated number of colonies per 1 mL of peripheral blood are shown. The ratios of the total number of emerged primary colonies/the total number of seeded cells are indicated in parentheses. The number of human induced pluripotent stem cell (hiPSC) lines established from each sample is also shown. The right-hand column shows the success rate of recovering lines from frozen stock, meaning that there were no failures in the freeze–thaw process for all the established stocks tested
SLE Systemic lupus erythematosus, PM Polymyositis, X-CGD X-linked chronic granulomatous disease, PID Primary immunodeficiency, JIA Juvenile idiopathic arthritis, CMS Congenital malformation syndrome, MD Mitochondrial diabetes, KCS2 Kenny-Caffey syndrome type 2
To establish stable hiPSC lines, we tentatively performed a random pickup of 10 colonies larger than the 250-μm diameter on day 11. Their spherical shape under feeder-free conditions allowed for an extremely facile colony pickup procedure. We carried out siRNA treatment against SeVdp-302L during the initial post-pickup culture, with no colonies lost in the subsequent expansion phase. We divided each line into two feeder-free conditions (i.e., iMatrix-511/StemFit AK02N and VTN-N/Essential 8) and eventually established all 10 lines as stable hiPSCs named TkPP2 (Fig. 1c and Table 3). The colonies of the TkPP2 lines showed a typical hiPSC monolayer morphology (day 37, Figs. 1c and 2f) and were stable in culture after their recovery from cryopreservation, with their pluripotency characteristics maintained (discussed below). This trial demonstrated the high success rate of hiPSC generation from non-mobilized PB samples with no need for extensive expansion culture, allowing a rapid (5–6 weeks) and robust protocol that is appropriate for the purpose of biobanking.
Fig. 2.

Establishment of a panel of patient-derived human induced pluripotent stem cell (hiPSC) lines from non-mobilized peripheral blood-derived CD34+ hematopoietic stem and progenitor cells (HSPCs) using SeVdp(KOSM)-302L. a Sequential images of representative primary colony growth observed in the process of TkSLE5 (top) and TkSCR1 cell line establishment (bottom). b Live cell image of a CDy1 dye-stained (red) representative primary colony obtained at day 13 in the course of TkSPR2 cell line generation. c Scatter plots indicating the colony formation efficiency of the seeded cells (y-axis) and the estimated number of hiPSC colonies obtained from 1 mL of blood (x-axis) from 15 patients. d Table showing the failure rates in the colony pickup step (top) and in the subsequent hiPSC expansion process (bottom). e Bar chart showing the step-by-step progress in establishing hiPSC lines from the healthy donor (TkPP2) and 15 patients. For clarity, bars are uniquely colored for each passage number: P0 (passage 0) in blue, P1 (passage 1) in green, P2 (passage 2) in orange, and P3 (passage 3) in yellow. The gray line shows a typical time point (day 11) for colony pickup, but the actual pickup dates varied between day 8 and day 14. f Images of the healthy donor (TkPP2)- and 15 patient-derived hiPSC lines cultured under feeder-free conditions after establishment, showing a typical monolayered colony appearance
Preparation of non-mobilized PB-derived CD34+ HSPCs from patients
We next applied the established method to the generation of patient-derived hiPSC lines. PB samples were obtained from 15 patients of various ages (range 3–65 years) and with different types of diseases (Table 1). Among these 15 patients, there were 3 cases of systemic lupus erythematosus (SLE), 3 cases of polymyositis (PM), 2 cases of X-linked chronic granulomatous disease (X-CGD), 2 cases of primary immunodeficiency (PID), 1 case of juvenile idiopathic arthritis (JIA), 1 case of congenital malformation syndrome (CMS), 2 cases of mitochondrial diabetes (MD), and 1 case of Kenny-Caffey syndrome type 2 (KCS2) (Table 1). The detailed information of their diseases and causative genetic lesions were not provided for this report since they will be reported in more detail by collaborators elsewhere.
Using a gradient separation method, the PBMCs were obtained within a day of collection of 20–40 mL of fresh PB samples from the patients. In the JIA and CMS cases, PBMC purification was performed 1 day after PB collection owing to the need for long-distance transport of the samples. The number of PBMCs obtained ranged between ~ 1.0 × 107 and ~ 7.0 × 107 cells (0.45 × 106 to 2.4 × 106 cells/mL). These were all cryopreserved in aliquots until the start of hiPSC generation (Table 1). At day − 3, a vial containing 0.4 × 107, 0.5 × 107, or 1.0 × 107 PBMCs was thawed for each hiPSC generation attempt, upon which it was shown that ~ 24–88% of PBMCs had been lost during the storage (Table 2). To ensure the survival of CD34+ HSPCs after thawing, the cells were kept overnight in EB medium containing six cytokines. At day − 2, CD34+ cell enrichment was performed as described for TkPP2 (Fig. 1a). The number of recovered cells after enrichment varied significantly among the patient samples, ranging from 5500 to 125,000 cells (Table 2). However, we did not experience any unsuccessful cases with these limited materials (median values; 21,350 cells).
Successful establishment of patient-derived hiPSC lines
According to the established protocol, we infected the patient-derived CD34+-rich HSPCs with SeVdp(KOSM)-302L at an MOI of 3–7 on day − 1 and plated all or a part of them onto iMatrix-511-coated 6-well plates at various cell densities (i.e., 250–20,000 cells/well). Similar to the observations made with TkPP2 establishment, the attached cells gradually formed spherically shaped brown colonies containing pluripotent cells (Fig. 2a, b). The colony formation efficiencies per seeded cells showed some variation, with the average value being 1.38 ± 1.17% (mean ± SD) (Fig. 2c and Table 3). In 9 out of the 15 cases, the efficiencies varied between 1.09% and 3.26%, which translated to yields of 4.6–127.7 colonies per 1 mL of PB (Fig. 2c and Table 3). However, for the other 6 cases, the efficiency values were between 0.08% and 0.34% per seeded cells, translating to 0.65–8.9 colonies per 1 mL of PB (Fig. 2c and Table 3). The lower efficiencies in the latter group may be attributable to the putative low titer of the SeVdp(KOSM)-302L stock used for the period corresponding to the establishment, because when tested later, it was discovered that a higher MOI (i.e., 7 instead of 3) of the same virus was needed to obtain efficiencies above 1%. Nevertheless, the hiPSC generation efficiencies obtained with our method can be considered as being markedly high, even when compared with a previous report that showed an efficient reprogramming (~ 0.02%) of non-mobilized PBMCs using SeVdp(KOSM)-302L in similar feeder-free conditions [45].
Since all colonies that were randomly picked up led to the successful establishment of TkPP2 lines as described above, we performed colony pickup for the establishment of the patient-derived hiPSC lines in a similar manner, mostly on day 11 (based simply on observations under the microscope). Consequently, out of 294 colonies picked up, all but one (0.34% pickup failure) showed initial attachment and growth, enabling subsequent expansion culture. Once subjected to culture for stable hiPSC line establishment, all 223 selected colonies proved eventually to be expandable, leading to the completion of the freezing storage (Fig. 2d). The progress of the establishment processes was similar in all hiPSC lines (Fig. 2e). Furthermore, the overall process was fast, as we were able to end the culture for each line routinely within ~ 5–6 weeks with the storage of a sufficient number of hiPSCs showing typical pluripotent colonies (Fig. 2f). So far, we have not found any cases where we failed to recover viable hiPSC colonies from their frozen stocks (Table 3). Collectively, these results demonstrate that our method enables the scheduled establishment of disease-specific hiPSC lines from patient PB samples with a high success rate.
Confirmation of the transgene-free status and karyotypes
To confirm the removal of SeVdp(KOSM)-302L in the established hiPSC lines, we performed RT-PCR analysis for the presence of vector genome sequences. As shown in Fig. 3a, the amplicons representing the residual existence of the SeV genome were infrequently found. This, however, occurred generally when assessed at an early passage before the initial storage. The same line showed the disappearance of detectable SeV sequences consistently with a few additional passages (Fig. 3a), most likely due to the auto-erasable function equipped in the SeVdp-302L system [16]. Most of the hiPSC lines tested were shown to be transgene-free at the time of storage (81 out of 88 lines, Fig. 3c). Karyotyping analysis has so far been conducted for a few of the patient-derived hiPSC lines and is ongoing for some others, showing mostly normal karyotypes (Fig. 3b, c). Taken together, the results indicate that our method is compatible with the measure to obtain integration-free hiPSC lines with a normal karyotype.
Fig. 3.

Assessment of SeVdp(KOSM)-302L removal and karyotypes. a Gel images showing representative assessment results for the removal of the SeVdp(KOSM)-302L genomes obtained by RT-PCR. The image demonstrates no detection of SeVdp(KOSM)-302L amplicons in all 9 TkPP2 lines, whereas it shows positive results in 4 out of 6 TkSCG4 lines (at passages 5–6). Residual SeVdp(KOSM)-302L was no longer detected in these 6 TkSCG4 lines when assessed a few passages later (additional culture). SeVdp(−), negative control (uninfected hiPSC); SeV(+), low-positive control (hiPSC carrying low copies); SeV(++), high-positive control (hiPSC carrying high copies). GAPDH was used as the internal control. b Representative G-banded karyotyping images for 3 TkPP2 lines and 3 TkSCG4 lines. c Summary of the assessments of SeVdp(KOSM)-302L removal and G-band karyotypes. hiPSC, human induced pluripotent stem cells; N.D., not done
Verification of a non-T lymphoid-cell origin for the hiPSCs
Despite the HSPC enrichment procedure, our target PBMC populations were thought to contain a significant amount of non-CD34+ cells, which should include lymphocytes and monocytes (Fig. 1b). hiPSCs derived from T or B cells would retain the originally rearranged immunoreceptor genome, which may be undesirable for some studies. We therefore investigated the lymphocyte-related genomic rearrangement of the TCRγ locus, using PCR. The TCRγ gene rearrangement was not detected in any of the 38 lines examined (Fig. 4a, b). The corresponding rearrangement was detectable in the T lymphoid cell-derived hiPSC line TkSST2-2, which we reported previously [46], confirming the validity of this assay (Fig. 4a). As B cells were thought to be extremely refractory to reprogramming in our system, necessitating enhancement by the coexpression of other factors (e.g., C/EBPα), an analysis of immunoglobulin genes was not conducted [47, 48]. Although the origin of monocytes cannot be excluded, these results—taken together with the great enhancement in generation efficiencies observed after CD34+ cell enrichment—suggest that the hiPSC lines have likely been derived from HSPCs (Fig. 1d).
Fig. 4.

Analysis of the T cell receptor gamma (TCRγ) gene locus rearrangement. a Electrophoretic images showing the results of detection analysis for the specific TCRγ gene rearrangement, conducted using the T Cell Receptor Gamma Gene Rearrangement Assay kit for Gel Detection. Top: analysis of 9 independent TkPP2 lines. Bottom: analysis of 9 patient-derived human induced pluripotent stem cell (hiPSC) lines (names are indicated). NTC, non-template control. IVS-0009 and IVS-0000 are the monoclonal and polyclonal samples, respectively, provided as positive controls in the kit. Another positive control used was TkSST2-2 (top panel), a monoclonal T cell-derived hiPSC line established previously in our laboratory. Note that there are no single detectable bands in the test sample lanes. b Summary of the results of the TCRγ gene rearrangement analysis. No positive results were seen in the tested samples (38 hiPSC lines in total)
Stable pluripotency and trilineage differentiation capability
All hiPSC lines were in a stable pluripotent state in culture, showing the retention of pluripotency characteristics, i.e., positive staining with the antibodies for TRA-1-60 or TRA-1-81 and the CDy1 dye (Fig. 5a). Expression of the marker genes OCT4 and NANOG was quantified in the 85 hiPSC lines using RT-qPCR to verify their pluripotency features. As shown in Fig. 5b, c, all the lines exhibited the expression of these genes, demonstrating their pluripotent status. Expression of these markers was also confirmed at a single cell level when the samples were assessed by flow cytometry analysis (Additional file 4: Figure S2).
Fig. 5.
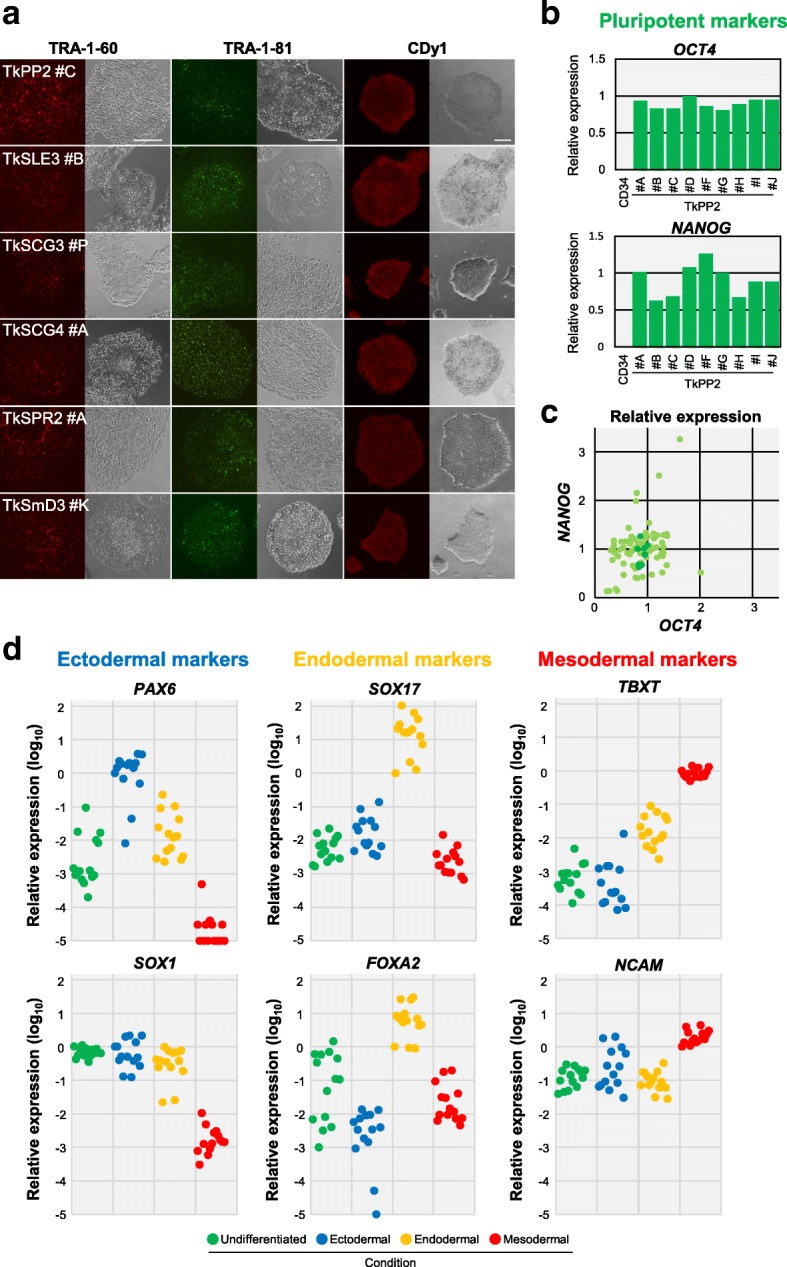
Pluripotency marker expression and trilineage differentiation capability confirmed in the established human induced pluripotent stem cell (hiPSC) lines. a Fluorescent images of live colonies stained with anti-TRA-1-60 antibody (red, left), anti-TRA-1-81 antibody (green, middle), or the CDy1 dye (red, right). Phase-contrast images are also shown in a side-by-side fashion for each picture. Scale bars indicate 200 μm. b Graph demonstrating the quantification results of OCT4 and NANOG transcripts in the TkPP2 lines, assessed by RT-qPCR analysis. The relative expression values for each gene, normalized to the level observed in the standard TkDA3-4 hiPSC sample, are shown. CD34+-enriched cells derived from the TkPP2 donor sample were used as the negative control (CD34). c Scatter plot showing a comprehensive representation of the relative expression values of OCT4 and NANOG in the 85 hiPSC lines examined. The results of 76 patient-derived hiPSCs are shown as light green plots. The results of 9 TkPP2 lines (same data in b) are shown in dark green for comparison. d Graphs showing the relative expression values of each marker gene calculated by the ΔΔCt method with CFX™ Manager software. The plots (14 hiPSC lines) are colored according to the culture condition shown at the bottom. “Undifferentiated” (green) represents hiPSC samples cultured in a pluripotent state. The other samples were cells cultured under differentiation conditions, with each being directed for the corresponding lineage (ectodermal, endodermal, or mesodermal)
Finally, we examined the capability of the 14 selected hiPSC lines to differentiate into three germ layers with an easy-to-use in vitro differentiation assay. After cultivation under different conditions (i.e., either in an undifferentiated state or with differentiation for an ectodermal, an endodermal, or a mesodermal lineage), the hiPSC samples were examined by RT-qPCR for their relative gene expression of three germ-layer markers: PAX6 and SOX1 (ectoderm), SOX17 and FOXA2 (endoderm), and TBXT and NCAM (mesoderm). Selective upregulation patterns were detectable for each marker gene in accordance with the corresponding culture condition in all the hiPSC lines, except for the ectodermal marker SOX1, the expression levels of which were also high in the undifferentiated state (Fig. 5d). Increased expression of lineage-markers was also observed at a protein level in the differentiated TkPP2 samples by flow cytometry analysis, demonstrating the utility of this in vitro differentiation assay system (Additional file 5: Figure S3). These results confirmed that the patient-derived hiPSC lines established by our method could be stably maintained in a pluripotent culture and retain their trilineage differentiation capability, both conditions of which are mandatory for these cell lines to be truly valuable.
Discussion
Herein, we have described a rapid and simple method for the robust establishment of transgene-free hiPSC lines using the auto-erasable SeVdp(KOSM)-302L vector [16]. In this method, we targeted non-mobilized PB-derived CD34+ HSPCs, with no need for cell mobilization and ex vivo expansion culture, and achieved highly efficient cell reprogramming under feeder-free conditions. Primary colonies appeared rapidly, were easily manageable for pickup, and showed robustness in the subsequent expansion steps. Using this method, we succeeded in the scheduled establishment of 223 hiPSC lines from PB samples obtained from 15 patients with 8 different diseases, generating a sufficient number of frozen stocks within 5–6 weeks. These hiPSC lines showed consistent recovery from frozen stocks, a transgene-free status, normal karyotypes, non-T cell derivation, stable pluripotency, and trilineage differentiation capability. This method is therefore valuable to robustly and efficiently generate patient hiPSC lines, allowing reductions in the cost, time, and workload for the entire procedure, which is advantageous for biobanking.
In our hiPSC banking, PB samples were used because they were easily collected from patients, with less invasiveness than that of other sources. PB can also be held at room temperature for ~ 48 h, and the derived PBMCs can be cryopreserved for a long period [19], enabling long-distance sample transport and scheduled hiPSC generation. In fact, we succeeded in generating hiPSCs from all 15 patient-derived PBMC samples that had been cryopreserved for a few months to 1 year. Of note is that despite the TkSCR1 and TkS42-1 PB samples required long-distance transport of over 24 h, they still resulted in the establishment of hiPSC lines without any detectable loss of generation efficiency (Table 3).
Among the hematopoietic cells, CD34+ HSPCs are often the preferred target cells for reprogramming, as they generally allow relatively highly efficient reprogramming [17, 20, 49–51]. Although CD34+ HSPCs can be collected from cord blood and bone marrow as well, PB is the source most suitable for patient hiPSC banking, because of its general availability. Their scarcity, however, has made it difficult to utilize PB-derived CD34+ cells as a regular target. To overcome this limitation, either mobilization or ex vivo expansion culture or both have been adopted for PB-derived HSPC-targeted reprogramming [22–25]. G-CSF-based mobilization, however, is considered too invasive for patient hiPSC generation. When non-mobilized PB samples are used, many protocols include cultivation steps with various combinations of cytokines to increase the target progenitor cells and thus the reprogramming efficiency [19, 21, 26–28, 30–33, 35–37, 52–54]. After expansion culture (typically 4–10 days), approximately 1.0 × 105 to 1.0 × 107 target cells are obtained and used for the reprogramming studies. Considering these facts, it is a marked feature that our protocol eliminates the need for both mobilization and expansion culture (Fig. 1). At present, however, we have not extensively tried to make the protocol completely cytokine-free. After immunomagnetic enrichment, the recovery of CD34+-rich HSPCs should be expected to be very limited because of their extremely small numbers in non-mobilized PB samples. As we continue to use frozen PBMCs for the scheduled hiPSC establishment, the expected yields would become even lower. We therefore included into the culture medium 6 cytokines that favored HSPC survival during the first 3 days. Although this treatment did not lead to a massive expansion of cells, it was nevertheless enough for successful hiPSC generation. It should be noted that our culture scale was small, thus limiting the required volume of medium (3 mL maximum on day − 3, and only 200 μL for culturing CD34+-enriched cells thereafter) and the total amounts of cytokines made available. To cut the cost further, we may be able to modify the cytokine-related steps in the future.
As the number of target cells was limited, our method needed reprogramming efficiencies high enough to produce a sufficient number of pluripotent colonies. To target this small number of cells (< 10,000 cells in some cases), the use of episomal vectors that have shown utility in many applications [5, 28] may not be feasible for the reason of practicality. Viral vectors are advantageous in this context; however, the current methods have revealed low reprogramming efficiencies (< 1%), even when using another type of SeV vector, and yielded some non-expandable primary colonies [19, 24, 31, 34]. In contrast, our method allowed the highly efficient generation of primary colonies (up to 5.58%) and, more importantly, enabled high success rates in the establishment of stable hiPSC lines with almost no fear of failure in the subsequent expansion steps after random pickup (Figs. 1 and 2). The hiPSC lines established showed stable pluripotency and trilineage differentiation capability (Fig. 5). We thus concluded that the special combination of SeVdp(KOSM)-302L and non-mobilized PB-derived CD34+ cells represented the best match, leading to the establishment of the highly successful protocol. This report has further extended the utility of SeVdp(KOSM)-302L to hiPSC generation, which has been demonstrated with other human cellular sources [45, 46, 55–59].
Besides its high efficiency, our protocol reduced the time and workload required for the entire procedure (Figs 1 and 2). Overall, a sufficient number of hiPSCs for cryopreserved stocks—which proved recoverable—were obtained within 5–6 weeks in all cases. More recently, we generally performed colony pickup at day 8 and could end the entire establishment process by day 35 after seeding in many cases. We speculated that the feeder-free condition using iMatrix-511 (Laminin-511-E8 fragment) combined with the StemFit AK02N medium might favor the rapid appearance of primary colonies and subsequent early cell growth, especially due to its great potential in supporting single-cell survival and firm cell adhesion to the plate [28, 60]. We also adapted hiPSCs to Essential 8 medium on VTN-N-coated plates in the late expansion phase for their cryopreservation (Fig. 1). This latter culture could help in the distribution of hiPSCs to other investigators, because we have found that the hiPSCs in stock are compatible with various culture conditions after thawing, irrespective of what culture systems other researchers use. The entire feeder-free protocol also benefits researchers by reducing their workload, eliminating the S23need for feeder cell handling.
Currently, researchers prefer the use of hiPSCs that are free of any genetic materials left behind after reprogramming procedures, because the residual expression of transgenes is known to affect the differentiation capability [61]. We had previously demonstrated the generation of a series of hiPSC lines using an older version of the non-auto-erasable Sendai virus vector (SeVdp), which required routine siRNA treatment to erase the SeV genome in the emerging hiPSCs [38–40]. Although we knew that SeVdp(KSOM)-302L was equipped with an auto-erasable function, we carried out siRNA treatment against SeVdp-302L to maximize the removal of the viral genomes (see the “Methods” section). Overall, the residual SeVdp-302L genome was rarely detected. Even when some residual genomes were detected, they disappeared spontaneously with a few additional cultures (Fig. 3). From these observations, we concluded that the auto-erasable function of SeVdp-302L worked efficiently in our protocol in a similar way as reported previously [16].
In this report, we described the successful establishment of an initial series of 223 hiPSC lines, derived from 15 patients affected by 8 different diseases, which included autoimmune diseases such as SLE and PD, immune deficiencies exemplified by X-CGD, and other diseases such as MD and KCS2 (Table 1). Recently, Gomez Limia et al. [62] reported the successful generation of hiPSCs from a patient, using a very similar method, where non-mobilized PB-derived CD34+ HSPCs with no ex vivo expansion (probably from a fresh, unfrozen sample) were reprogrammed to pluripotency under feeder-free conditions using Geltrex with the SeV vector (CytoTune® 2.0). When tested in our protocol, the CytoTune®-iPS 2.0 also yielded primary hiPSC colonies with very high efficiencies at the given setting (Additional file 6: Figure S4). Although still preliminary, the use of Sendai virus vector systems may be considered commonly compatible with our protocol. By our hands, cell line establishment is currently still ongoing, and the panel of disease-specific hiPSCs is thus expanding. Some of the hiPSC lines established in this study are already being used for disease studies by our collaborators and our research team, and the results will be reported elsewhere in the near future. For example, we have confirmed that neutrophilic cells differentiated from the TkSCG3 and TkSCG4 lines, derived from X-CGD patients, show severe defects in reactive oxygen species production (data not shown), similar to another X-CGD-iPSC line that we reported previously [39]. Such findings indicate the applicability of the established hiPSC lines to disease-modeling studies.
Conclusions
In summary, we have developed an improved feeder-free method for establishing transgene-free hiPSC lines from non-mobilized PB-derived CD34+ HSPCs, using the auto-erasable SeVdp(KOSM)-302L. With this method, we successfully established a panel of disease-specific hiPSC lines from 15 patients with 8 different disease types. The hiPSC generation was feasible with the use of cryopreserved PBMCs, even after long-distance transport of the sampled PB. This method rapidly generated expandable primary colonies with high efficiency, resulting in the establishment of stable hiPSC lines with almost no failure cases. We confirmed that the hiPSC lines were suitable for use in disease modeling studies by showing their efficient transgene removal, normal karyotypes, non-T cell origin, and stable pluripotency and differentiation capability. Overall, this method allows for the scheduled generation of a panel of disease-specific hiPSC lines from patient-derived non-mobilized PB samples in a cost-effective and time- and labor-saving manner.
Additional files
Table S1. List of primers used in this study. (PDF 106 kb)
Supplemental methods. (PDF 76 kb)
Figure S1. Correlation between %CD34+ cells of the source sample and the reprogramming (colony formation) efficiency. (PDF 78 kb)
Figure S2. Flow cytometry analysis of the marker expression in established iPSC clones. (PDF 46 kb)
Figure S3. Flow cytometry analysis of the marker expression in the TkPP2 cells after induction of lineage-oriented differentiation. (PDF 86 kb)
Figure S4. Comparison of CytoTune®-iPS2.0 with SeVdp(KOSM)-302L in the same hiPSC generation protocol. (PDF 87 kb)
Acknowledgements
We thank Drs. Toyoki Nishimura, Yoichiro Oda, and Etsuro Tokuhiro for providing patient care, Dr. Mayuko Tamura for blood sampling, and Hiroko Ohkura and Akiko Kikuchi for technical assistance.
Abbreviations
- BM
Bone marrow
- G-CSF
Granulocyte-colony stimulating factor
- hiPSC
Human-induced pluripotent stem cell
- HSPC
Hematopoietic stem/progenitor cell
- PB
Peripheral blood
- PBMC
Peripheral blood mononuclear cell
- SeV
Sendai virus
Authors’ contributions
TaOku performed the hiPSC establishment, experiments, data analyses, and manuscript preparation. YH established the hiPSC lines, performed the experiments, and edited the manuscript. CYL arranged the hiPSC establishment and supervised the study. HTL discussed the experiments. HS, BN, EK, TsOka, SO, KT, Hka, HH, FM, KK, HM, HN, HW, ST, HT, and SK contributed to the patient recruitment and enrollment. KF, TM, HKo, TS, TY, and TK supported the experiments scientifically. KN, ManOh, and MN prepared the SeVdp(KOSM)-302L. MakOt supervised the study, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the JSPS Grant-in-Aid for Scientific Research (B) Grant Numbers JP18H02778, JSPS KAKENHI, and JP26670474, and the Program for Intractable Disease Research Utilizing Disease-specific iPS Cells (Grant Numbers JP16bm0609006, JP16bm0804004, and JP18bm04004h0102) and the Research Project for Practical Applications of Regenerative Medicine (Grant Number JP17bk0104018h0005) funded by the Japan Agency for Medical Research and Development (AMED).
Availability of data and materials
All experimental data and materials obtained and used in this study were described in this article.
Ethics approval and consent to participate
The research was reviewed and approved by the ethics committee of the IMSUT (#25-3-0701 and #30-8-A0522) and also by all the institutional ethics committees involved in this study. Signed informed consent was obtained from the 10 adult patients and from guardian(s) in the cases of pediatric patients. Informed assent was obtained from the patients themselves, where available. Their samples were de-identified. All experiments were performed with adherence to the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Takashi Okumura, Email: tokumura@ims.u-tokyo.ac.jp.
Yumi Horie, Email: y-horie@ims.u-tokyo.ac.jp.
Chen-Yi Lai, Email: chenyi.lai1014@gmail.com.
Huan-Ting Lin, Email: linht@ims.u-tokyo.ac.jp.
Hirofumi Shoda, Email: shoda-tky@umin.ac.jp.
Bunki Natsumoto, Email: natsumotobunki@gmail.com.
Keishi Fujio, Email: kfujio-tky@umn.ac.jp.
Eri Kumaki, Email: ekumped@tmd.ac.jp.
Tsubasa Okano, Email: tokano.ped@tmd.ac.jp.
Shintaro Ono, Email: onoshin.mail@gmail.com.
Kay Tanita, Email: kay916.vc@gmail.com.
Tomohiro Morio, Email: tmorio.ped@tmd.ac.jp.
Hirokazu Kanegane, Email: hkanegane.ped@tmd.ac.jp.
Hisanori Hasegawa, Email: hasegawa.rheu@tmd.ac.jp.
Fumitaka Mizoguchi, Email: mizoguchi.rheu@tmd.ac.jp.
Kimito Kawahata, Email: kimito.kawahata@marianna-u.ac.jp.
Hitoshi Kohsaka, Email: kohsaka.rheu@tmd.ac.jp.
Hiroshi Moritake, Email: hiroshi_moritake@med.miyazaki-u.ac.jp.
Hiroyuki Nunoi, Email: hnunoi@med.miyazaki-u.ac.jp.
Hironori Waki, Email: wakih-int@h.u-tokyo.ac.jp.
Shin-ichi Tamaru, Email: 16000-all.tamaru@kpe.biglobe.ne.jp.
Takayoshi Sasako, Email: tsasako-tky@umin.ac.jp.
Toshimasa Yamauchi, Email: tyamau-tky@umin.net.
Takashi Kadowaki, Email: kadowaki-3im@h.u-tokyo.ac.jp.
Hiroyuki Tanaka, Email: pnrtn745@yahoo.co.jp.
Sachiko Kitanaka, Email: sachi-tky@umin.ac.jp.
Ken Nishimura, Email: ken-nishimura@md.tsukuba.ac.jp.
Manami Ohtaka, Email: ohtaka@tokiwa-bio.com.
Mahito Nakanishi, Email: mahito-nakanishi@aist.go.jp.
Makoto Otsu, Phone: +81-3-6409-2342, Email: motsu@ims.u-tokyo.ac.jp.
References
- 1.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16:115–130. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 6.Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hämäläinen R, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia F, Wilson KD, Sun N, Gupta DM, Huang M, Li Z, et al. A nonviral minicircle vector for deriving human iPS cells. Nat Methods. 2010;7:197–199. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. 2016;17:183–193. doi: 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- 13.Zhou YY, Zeng F. Integration-free methods for generating induced pluripotent stem cells. Genomics Proteomics Bioinformatics. 2013;11:284–287. doi: 10.1016/j.gpb.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma Riddhi. iPS Cells—The Triumphs and Tribulations. Dentistry Journal. 2016;4(2):19. doi: 10.3390/dj4020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borgohain Manash P., Haridhasapavalan Krishna Kumar, Dey Chandrima, Adhikari Poulomi, Thummer Rajkumar P. An Insight into DNA-free Reprogramming Approaches to Generate Integration-free Induced Pluripotent Stem Cells for Prospective Biomedical Applications. Stem Cell Reviews and Reports. 2018;15(2):286–313. doi: 10.1007/s12015-018-9861-6. [DOI] [PubMed] [Google Scholar]
- 16.Nishimura K, Ohtaka M, Takada H, Kurisaki A, Tran NVK, Tran YTH, et al. Simple and effective generation of transgene-free induced pluripotent stem cells using an auto-erasable Sendai virus vector responding to microRNA-302. Stem Cell Res. 2017;23:13–19. doi: 10.1016/j.scr.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 17.Loh YH, Agarwal S, Park IH, Urbach A, Huo H, Heffner GC, et al. Generation of induced pluripotent stem cells from human blood. Blood. 2009;113:5476–5479. doi: 10.1182/blood-2009-02-204800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agu CA, Soares FA, Alderton A, Patel M, Ansari R, Patel S, et al. Successful generation of human induced pluripotent stem cell lines from blood samples held at room temperature for up to 48 hr. Stem Cell Rep. 2015;5:660–671. doi: 10.1016/j.stemcr.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mack AA, Kroboth S, Rajesh D, Wang WB. Generation of induced pluripotent stem cells from CD34+ cells across blood drawn from multiple donors with non-integrating episomal vectors. PLoS One. 2011;6:e27956. doi: 10.1371/journal.pone.0027956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merling RK, Sweeney CL, Choi U, De Ravin SS, Myers TG, Otaizo-Carrasquero F, et al. Transgene-free iPSCs generated from small volume peripheral blood nonmobilized CD34+ cells. Blood. 2013;121:e98–107. doi: 10.1182/blood-2012-03-420273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou BK, Mali P, Huang X, Ye Z, Dowey SN, Resar LM, et al. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011;21:518–529. doi: 10.1038/cr.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park TS, Huo JS, Peters A, Talbot CC, Verma K, Zimmerlin L, et al. Growth factor-activated stem cell circuits and stromal signals cooperatively accelerate non-integrated iPSC reprogramming of human myeloid progenitors. PLoS One. 2012;7:e42838. doi: 10.1371/journal.pone.0042838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye L, Muench MO, Fusaki N, Beyer AI, Wang J, Qi Z, et al. Blood cell-derived induced pluripotent stem cells free of reprogramming factors generated by Sendai viral vectors. Stem Cells Transl Med. 2013;2:558–566. doi: 10.5966/sctm.2013-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Brzeszczynska J, Samuel K, Black J, Palakkan A, Anderson RA, et al. Efficient episomal reprogramming of blood mononuclear cells and differentiation to hepatocytes with functional drug metabolism. Exp Cell Res. 2015;338:203–213. doi: 10.1016/j.yexcr.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 26.Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, et al. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell. 2010;7:20–24. doi: 10.1016/j.stem.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dowey SN, Huang X, Chou BK, Ye Z, Cheng L. Generation of integration-free human induced pluripotent stem cells from postnatal blood mononuclear cells by plasmid vector expression. Nat Protoc. 2012;7:2013–2021. doi: 10.1038/nprot.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakagawa M, Taniguchi Y, Senda S, Takizawa N, Ichisaka T, Asano K, et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci Rep. 2014;4:3594. doi: 10.1038/srep03594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan HK, Toh CX, Ma D, Yang B, Liu TM, Lu J, et al. Human finger-prick induced pluripotent stem cells facilitate the development of stem cell banking. Stem Cells Transl Med. 2014;3:586–598. doi: 10.5966/sctm.2013-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou BK, Gu H, Gao Y, Dowey SN, Wang Y, Shi J, et al. A facile method to establish human induced pluripotent stem cells from adult blood cells under feeder-free and xeno-free culture conditions: a clinically compliant approach. Stem Cells Transl Med. 2015;4:320–332. doi: 10.5966/sctm.2014-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou H, Martinez H, Sun B, Li A, Zimmer M, Katsanis N, et al. Rapid and efficient generation of transgene-free iPSC from a small volume of cryopreserved blood. Stem Cell Rev. 2015;11:652–665. doi: 10.1007/s12015-015-9586-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Liu T, Van Halm-Lutterodt N, Chen J, Su Q, Hai Y. Reprogramming of blood cells into induced pluripotent stem cells as a new cell source for cartilage repair. Stem Cell Res Ther. 2016;7:31. doi: 10.1186/s13287-016-0290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Táncos Z, Varga E, Kovács E, Dinnyés A, Kobolák J. Establishment of induced pluripotent stem cell (iPSC) line from a 75-year old patient with late onset Alzheimer’s disease (LOAD) Stem Cell Res. 2016;17:81–83. doi: 10.1016/j.scr.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 34.Wen W, Zhang JP, Xu J, Su RJ, Neises A, Ji GZ, et al. Enhanced generation of integration-free iPSCs from human adult peripheral blood mononuclear cells with an optimal combination of episomal vectors. Stem Cell Rep. 2016;6:873–884. doi: 10.1016/j.stemcr.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu H, Huang X, Xu J, Song L, Liu S, Zhang XB, et al. Optimizing the method for generation of integration-free induced pluripotent stem cells from human peripheral blood. Stem Cell Res Ther. 2018;9:163. doi: 10.1186/s13287-018-0908-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor JP, Cash MN, Santostefano KE, Nakanishi M, Terada N, Wallet MA. CRISPR/Cas9 knockout of USP18 enhances type I IFN responsiveness and restricts HIV-1 infection in macrophages. J Leukoc Biol. 2018;103:1225–1240. doi: 10.1002/JLB.3MIA0917-352R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye L, Peng Y, Mo J, Yao Y. MiR-126 enhances VEGF expression in induced pluripotent stem cell-derived retinal neural stem cells by targeting spred-1. Int J Clin Exp Pathol. 2018;11:1023–1030. [PMC free article] [PubMed] [Google Scholar]
- 38.Nishimura K, Sano M, Ohtaka M, Furuta B, Umemura Y, Nakajima Y, et al. Development of defective and persistent Sendai virus vector: a unique gene delivery/expression system ideal for cell reprogramming. J Biol Chem. 2011;286:4760–4771. doi: 10.1074/jbc.M110.183780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin HT, Masaki H, Yamaguchi T, Wada T, Yachie A, Nishimura K, et al. An assessment of the effects of ectopic gp91phox expression in XCGD iPSC-derived neutrophils. Mol Ther Methods Clin Dev. 2015;2:15046. doi: 10.1038/mtm.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kubara K, Yamazaki K, Ishihara Y, Naruto T, Lin HT, Nishimura K, et al. Status of KRAS in iPSCs impacts upon self-renewal and differentiation propensity. Stem Cell Reports. 2018;11:380–394. doi: 10.1016/j.stemcr.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takayama N, Nishimura S, Nakamura S, Shimizu T, Ohnishi R, Endo H, et al. Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J Exp Med. 2010;207:2817–2830. doi: 10.1084/jem.20100844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang NY, Yun SW, Ha HH, Park SJ, Chang YT. Embryonic and induced pluripotent stem cell staining and sorting with the live-cell fluorescence imaging probe CDy1. Nat Protoc. 2011;6:1044–1052. doi: 10.1038/nprot.2011.350. [DOI] [PubMed] [Google Scholar]
- 43.Schlaeger TM, Daheron L, Brickler TR, Entwisle S, Chan K, Cianci A, et al. A comparison of non-integrating reprogramming methods. Nat Biotechnol. 2015;33:58–63. doi: 10.1038/nbt.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye H, Wang Q. Efficient generation of non-integration and feeder-free induced pluripotent stem cells from human peripheral blood cells by Sendai virus. Cell Physiol Biochem. 2018;50:1318–1331. doi: 10.1159/000494589. [DOI] [PubMed] [Google Scholar]
- 45.Trokovic R, Weltner J, Nishimura K, Ohtaka M, Nakanishi M, Salomaa V, et al. Advanced feeder-free generation of induced pluripotent stem cells directly from blood cells. Stem Cells Transl Med. 2014;3:1402–1409. doi: 10.5966/sctm.2014-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iizuka-Koga M, Asashima H, Ando M, Lai CY, Mochizuki S, Nakanishi M, et al. Functional analysis of dendritic cells generated from T-iPSCs from CD4+ T cell clones of Sjögren’s syndrome. Stem Cell Rep. 2017;8:1155–1163. doi: 10.1016/j.stemcr.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bueno C, Sardina JL, Di Stefano B, Romero-Moya D, Muñoz-López A, Ariza L, et al. Reprogramming human B cells into induced pluripotent stem cells and its enhancement by C/EBPα. Leukemia. 2016;30:674–682. doi: 10.1038/leu.2015.294. [DOI] [PubMed] [Google Scholar]
- 48.Muñoz-López Á, van Roon EH, Romero-Moya D, López-Millan B, Stam RW, Colomer D, et al. Cellular ontogeny and hierarchy influence the reprogramming efficiency of human B cells into induced pluripotent stem cells. Stem Cells. 2016;34:581–587. doi: 10.1002/stem.2303. [DOI] [PubMed] [Google Scholar]
- 49.Eminli S, Foudi A, Stadtfeld M, Maherali N, Ahfeldt T, Mostoslavsky G, et al. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat Genet. 2009;41:968–976. doi: 10.1038/ng.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haase A, Olmer R, Schwanke K, Wunderlich S, Merkert S, Hess C, et al. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell. 2009;5:434–441. doi: 10.1016/j.stem.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 51.Ye Z, Zhan H, Mali P, Dowey S, Williams DM, Jang YY, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunisato A, Wakatsuki M, Shinba H, Ota T, Ishida I, Nagao K. Direct generation of induced pluripotent stem cells from human nonmobilized blood. Stem Cells Dev. 2011;20:159–168. doi: 10.1089/scd.2010.0063. [DOI] [PubMed] [Google Scholar]
- 53.Su RJ, Baylink DJ, Neises A, Kiroyan JB, Meng X, Payne KJ, et al. Efficient generation of integration-free ips cells from human adult peripheral blood using BCL-XL together with Yamanaka factors. PLoS One. 2013;8:e64496. doi: 10.1371/journal.pone.0064496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma A, Mücke M, Seidman CE. Human induced pluripotent stem cell production and expansion from blood using a non-integrating viral reprogramming vector. Curr Protoc Mol Biol. 2018;122:e58. doi: 10.1002/cpmb.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawagoe S, Higuchi T, Otaka M, Shimada Y, Kobayashi H, Ida H, et al. Morphological features of iPS cells generated from Fabry disease skin fibroblasts using Sendai virus vector (SeVdp) Mol Genet Metab. 2013;109:386–389. doi: 10.1016/j.ymgme.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 57.Wakao H, Yoshikiyo K, Koshimizu U, Furukawa T, Enomoto K, Matsunaga T, et al. Expansion of functional human mucosal-associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:546–558. doi: 10.1016/j.stem.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 58.Nishimura K, Kato T, Chen C, Oinam L, Shiomitsu E, Ayakawa D, et al. Manipulation of KLF4 expression generates iPSCs paused at successive stages of reprogramming. Stem Cell Rep. 2014;3:915–929. doi: 10.1016/j.stemcr.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamasaki S, Hamada A, Akagi E, Nakatao H, Ohtaka M, Nishimura K, et al. Generation of cleidocranial dysplasia-specific human induced pluripotent stem cells in completely serum-, feeder-, and integration-free culture. In Vitro Cell Dev Biol Anim. 2016;52:252–264. doi: 10.1007/s11626-015-9968-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyazaki T, Futaki S, Suemori H, Taniguchi Y, Yamada M, Kawasaki M, et al. Laminin E8 fragments support efficient adhesion and expansion of dissociated human pluripotent stem cells. Nat Commun. 2012;3:1236. doi: 10.1038/ncomms2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramos-Mejía V, Montes R, Bueno C, Ayllón V, Real PJ, Rodríguez R, et al. Residual expression of the reprogramming factors prevents differentiation of iPSC generated from human fibroblasts and cord blood CD34+ progenitors. PLoS One. 2012;7:e35824. doi: 10.1371/journal.pone.0035824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gomez Limia CE, Devalle S, Reis M, Sochacki J, Carneiro M, Madeiro da Costa R, et al. Generation and characterization of a human induced pluripotent stem (iPS) cell line derived from an acute myeloid leukemia patient evolving from primary myelofibrosis carrying the CALR 52bp deletion and the ASXL1 p.R693X mutation. Stem Cell Res. 2017;24:16–20. doi: 10.1016/j.scr.2017.08.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of primers used in this study. (PDF 106 kb)
Supplemental methods. (PDF 76 kb)
Figure S1. Correlation between %CD34+ cells of the source sample and the reprogramming (colony formation) efficiency. (PDF 78 kb)
Figure S2. Flow cytometry analysis of the marker expression in established iPSC clones. (PDF 46 kb)
Figure S3. Flow cytometry analysis of the marker expression in the TkPP2 cells after induction of lineage-oriented differentiation. (PDF 86 kb)
Figure S4. Comparison of CytoTune®-iPS2.0 with SeVdp(KOSM)-302L in the same hiPSC generation protocol. (PDF 87 kb)
Data Availability Statement
All experimental data and materials obtained and used in this study were described in this article.